
Abstract
Blastic plasmacytoid dendritic cell neoplasm is an aggressive malignant neoplasm originating from precursor plasmacytoid dendritic cells. Blastic plasmacytoid dendritic cell neoplasm shows a characteristic immunophenotype in which the tumor cells express CD4, CD56, TCL1, and bright CD123, and lack expression of lineage-specific myeloid and lymphoid antigens. Although cytogenetic and molecular aberrations are common, diagnostic recurrent abnormalities have not been identified. In this chapter, we summarize the clinical, pathologic, and genetic abnormalities identified to date. We explore the current concepts in treatment and the potential role of targeted therapies in the management of this rare disorder.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Blastic plasmacytoid dendritic cell neoplasm
- Plasmacytoid dendritic cells
- Acute myeloid leukemia
- CD123
- Interleukin–3 receptor α–chain
Introduction
First described in 1994 [1], blastic plasmacytoid dendritic cell neoplasm (BPDCN) is a rare , aggressive hematopoietic neoplasm. It has been variably known as blastic NK-cell lymphoma , agranular CD4+ natural killer cell leukemia , and agranular CD4+/CD56+ hematodermic neoplasm .
The normal cellular counterpart to BPDCN is the precursor plasmacytoid dendritic cell (pDCs). These cells play central roles in infectious and inflammatory conditions , primarily through secretion of type I interferons which stimulate T-cells and B-cells, resulting in effective augmentation of anti-viral immune responses, or in the case of autoimmune conditions, generation of abnormally autoreactive T-cells and B-cells, via overstimulated antigen presenting cells [2]. Reactive pDCs are increased in the lymph nodes of patients with inflammatory disorders such as Kikuchi-Fujimoto lymphadenitis and hyaline vascular Castleman disease [3, 4].
Given its relationship to normal plasmacytoid dendritic cells, BPDCN is classified as a precursor neoplasm related to acute myeloid leukemia (AML) in the World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues [5].
Epidemiology
In large retrospective reviews, BPDCN accounts for less than 1% of all acute leukemias and lymphomas, although, until recently, markers that help to distinguish BPDCN from AML were not widely available. There is a slight male predominance (3:1), with no ethnic or racial predisposition [6]. Mean age at presentation is 60–70 years, but a large age distribution is observed, including rare congenital cases. A pre-existing myeloid neoplasm, such as myelodysplastic syndrome or chronic myelomonocytic leukemia, has been noted in 5–10% of patients [6].
Etiology
The pathogenesis of BPDCN is poorly understood, and as such, no known environmental exposures or genetic predispositions have been described.
Clinical Features
The majority of patients have cutaneous involvement at the time of diagnosis, characterized by nodules or purpuric plaque-like skin lesions (Fig. 18.1). BPDCN may also present as a leukemic infiltrate in the blood or marrow, show involvement of the lymph nodes or spleen, or demonstrate simultaneous tissue and blood/marrow involvement. In cases where the neoplasm appears confined to the skin, dissemination to peripheral blood and bone marrow occurs shortly thereafter. Thrombocytopenia, anemia, and absolute neutropenia are commonly found on peripheral blood evaluation. Clinical features are summarized in Table 18.1.

Cutaneous manifestations of blastic plasmacytoid dendritic cell neoplasm (BPDCN). (a) Violaceous nodules can be seen with skin infiltration by BPDCN. (b) Plaques are also commonly seen (Images courtesy of Dr. Youn Kim, Stanford University)
Morphology and Immunophenotyping
Morphologic Features
The morphologic features of neoplastic cells of BPDCN can be quite variable, but in the most classic cases, cells are medium-sized with rounded to slightly irregular nuclei, fine chromatin, absent or inconspicuous small nucleoli, and scant to moderate amounts of cytoplasm; small cytoplasmic vacuoles may be present (Figs. 18.2 and 18.3). In cutaneous tissues, BPDCN may show perivascular, periadnexal, or sheet-like pattern of growth in the dermis and subcutis, with sparing of the overlying epidermis (Fig. 18.2). Bone marrow evaluation may reveal patchy involvement or complete effacement .

Morphologic and immunohistochemical features of skin infiltration by blastic plasmacytoid dendritic cell neoplasm (BPDCN). (a) Sheet-like infiltration of mononuclear cells with sparing of the epidermis (H&E, 1×) (b) With higher power magnification showing neoplastic cells to have blastic chromatin with small to indistinct nucleoli (H&E, 400×). Positive immunohistochemical stains for (c) CD123 (40×), and (d) TCL1 (40×) are shown

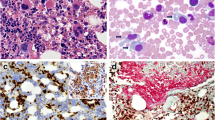
Features of bone marrow infiltration by blastic plasmacytoid dendritic cell neoplasm (BPDCN). (a) Morphologic evaluation of a bone marrow aspirate shows scattered mononuclear cells with fine/blastic chromatin and small to indistinct nucleoli, and eccentric nuclei (red arrows; Wright-Giemsa, 1000×). (b) An H&E stained bone marrow core biopsy shows BPDCN cells (yellow arrows; H&E, 400×). Positive immunohistochemical stains for (c) TCL1 (400×) and (d) CD123 are shown (400×)
Immunophenotype
Immunophenotypic analysis is critical in order to make the diagnosis of BPDCN and distinguish it from other entities. These proliferations consistently show expression of CD4 and CD56; however, this combination is nonspecific, as it can also be seen in the setting of NK/T-cell malignancies and AML , particularly those with monocytic differentiation. CD45 intensity can be dim or moderate by flow cytometry, also raising the possibility of AML or lymphoblastic leukemia/lymphoma. Bright CD123 expression is a hallmark of BPDCN and a useful feature in distinguishing it from AML . While expression of the myeloid-associated antigens CD13 and CD33 can be present, other markers of the myelomonocytic lineage (CD14, CD163, myeloperoxidase, lysozyme) are uniformly absent. Expression of CD303/BDCA-2, CLA/CD162, TCL1, and TdT is variable, which some investigators speculate may reflect the stage of maturation of the pDC that gives rise to the neoplastic clone [7]. T-cell-associated antigens CD2 and CD7 were found to be expressed in 37% and 11% of cases studied in the largest series [8]. There are isolated reports of cytoplasmic CD3 expression in BPDCN; however, it has been attributed to the use of a polyclonal antibody and consensus guidelines suggest that the presence of CD3 should exclude a diagnosis of BPDCN [9]. Expression of B-lineage-associated antigens (CD19, CD20, PAX5) has not been described. CD22, a B-lineage marker, has been documented in a few cases when using the s-HCL-1 antibody clone; however, evaluation of 5 additional anti-CD22 clones showed no significant staining, indicating a clone-specific phenomenon [10].
Because of the clinical, morphologic, and immunophenotypic overlap with AML , a panel of antibodies is commonly performed to arrive at the correct diagnosis. Several groups have attempted to define the optimal antibody panel for distinguishing AML from BPDCN. Sangle and coworkers determined that a panel comprising CD4, CD56, CD123 , lysozyme, myeloperoxidase, TCL1, and MxA proved useful in discriminating AML from BPDCN [11]. BPDCN was strongly associated with positive staining for CD4, CD56, CD123, TCL1, and MxA expression. Further, the expression of MPO and lysozyme confirmed the diagnosis of AML . CLA/CD162 and CD303 expression showed no significant association with the reference diagnosis, although only 7 of 17 cases of BPDCN were studied with these two markers. Julia et al. studied the immunoprofile of 91 BPDCN cases and propose that a diagnosis of BPDCN can be established when at least 4 of 5 markers (CD4, CD56, CD123, CD303, and TCL1) are expressed, in agreement with a prior study by Cronin and colleagues [8, 12]. None of the cases showed simultaneous absence of CD4 and CD56. CD303, a specific marker of plasmacytoid dendritic cells, was found to be expressed in 63% of the 85 cases tested. Recently, myeloid cell nuclear differentiation antigen (MNDA) expression was found to be expressed in the majority of AML and uniformly negative BPDCN, providing another marker to help sort out the differential diagnosis. [13]. Immunohistochemical markers useful in differentiating BPDCN from mimics are summarized in Table 18.2.
Cytogenetics and Molecular Features
Karyotypic abnormalities are a common finding, with up to 80% of BPDCN showing complex (≥3) clonal abnormalities [14]. A few recurring chromosomal loci are deleted in BPDCN, namely 5q, 12p, 13q, 6q, 15q, and 9 (Table 18.3) [14,15,16]. In a handful of cases, a t(6;8)(p21;q24) translocation involving MYC has been identified [14, 17,18,19].
Sapienza and investigators subjected 27 BPDCN samples from untreated patients to gene expression profiling (GEP) , which revealed that BPDCN appears significantly more related to normal myeloid precursors than lymphoid precursors, and closely resembles resting pDCs [20]. Further analysis showed that BPDCN tended to look more similar to AML on the molecular level, but shared patterns of gene deregulation that overlapped with both AML and ALL. GEP studies have demonstrated altered expression of tumor suppressors (RB1, LATS2, CDC14B, DBC1, SYK, KPNA3) and oncogenes (HES6, RUNX2, FLT3) [21].
Recently, whole-exome sequencing of BPDCN by Menezes identified 38 genes of interest [22]. Interrogation of 28 cases yielded mutations in a number of genes with known pathogenic effects in myeloid malignancies, including genes involved in DNA methylation (TET2, DNMT3A, IDH1, IDH2), chromatin remodeling (ASXL1), cell proliferation (NRAS, KRAS), transcription factors (ETV6, IKZF1/2/3, RUNX1), splicing machinery (SF3B1, SRSF2, U2AF1, ZRSR2), protein kinases (FLT3, JAK2, KIT), tumor suppressors (TP53), and ubiquitination (CBLB, CBLC, UBE2G2). Subsequently, 33 additional cases of BPDCN were subjected to massively paralleled sequencing, identifying many of the same molecular aberrations [23]. However, none of these mutations is specific for BPDCN and the prognostic significance of these mutations remains to be determined. A summary of pathogenic mutations detected by sequencing methods is presented in Table 18.4. Below we review in detail some of the more commonly described abnormalities and possible pathogenetic mechanisms.
Cell Cycle Genes (RB1, CDK Inhibitors, IKZF1, TP53)
Loss of gene loci important in the normal function of the cell cycle is common in BPDCN. RB1 is a cell cycle gene located at 13q13.1-q14.3 and deletion or downregulation of RB1 has been identified in approximately half of cases studied (13/26, 50%) [14, 21, 24]. Normally, Rb1 prevents cells from transitioning from the gap 1 (G1) phase (G1) of the cell cycle into the synthesis (S) phase. Thus, in the case of BPDCN, with loss of Rb1, the deletion or downregulation of its activities is believed to alleviate the block from G1 to S phases.
CDK inhibitors are also frequently (23/30, 77%) disrupted in BPDCN, resulting in unimpeded entry into the cell cycle [15, 24]. CDKN1B (p27Kip1), CDKN2A (p16INK4A), and CDKN2B (p15INK4B) are CDK inhibitors that each play a role in controlling the G1/S-phase transition in the cell cycle. Deletion of the 9p21.3 locus (including CDKN2A/CDKN2B) and 12p13.2-p13.1 locus (including CDKN1B) was discovered in 67% and 57% of BPDCN studied by array-based comparative genomic hybridization (aCGH) [24]. Although this was a small series of nonuniformly treated patients, multivariate analysis suggested that the presence of homozygous 9p21.3 deletion was an independent prognostic factor. Jardin et al. identified loss of CDKN2A/CDKN2B and CDKN1B loci in a similar proportion of cases, suggesting that these alterations are important in the pathogenesis of BPDCN.
In about 20% of BPDCN cases, a locus on 7p12.2 that contains IKZF1 is deleted [24]. Furthermore, Menezes and coworkers found frame shift and missense mutations in the IKZF gene family in an additional 20% of cases [22]. IKZF1 encodes Ikaros, a DNA- and protein-binding transcription factor with zinc finger binding motifs. Ikaros plays a crucial role in the cell cycle regulation and cell differentiation, including an important role in lymphocyte development [25]. The significance of IKZF1 mutations and deletions in BPDCN is still unknown.
Mutations of TP53, or loss of the chromosome region, 17p13, encompassing TP53 are seen in many cases of BPDCN [15, 26]. The protein product of TP53, p53, is a tumor suppressor, and is activated during cellular stress and exerts anti-proliferative effects at the G1/S and G2/M checkpoints in the cell cycle primarily through activating the CDK inhibitor p21. P53 also functions as a pro-apoptotic protein by activating BAX. Germline TP53 mutations are seen in Li-Fraumeni syndrome (LFS). In LFS, patients have a 25-fold increased risk of developing cancer by age 50. Breast and adrenal carcinomas, gliomas, sarcoma, and leukemia are the most common neoplasms encountered in this setting.
Genes Involved in Hematopoiesis (ETV6, TET2, FLT3, ASXL1)
In slightly more than half of the BPDCN cases studied, Leroux et al. found deletions in the locus surrounding ETV6, suggesting that loss of transcriptional repression by ETV6 may play an important role in BPDCN pathogenesis [14]. In addition, a rare case of BPDCN harboring an ETV6 rearrangement with an unknown partner gene has also been reported [27].
ETV6 encodes a protein that mediates cell proliferation and differentiation, and is required for establishing embryonic hematopoiesis in the bone marrow [28]. How deletions and mutations affect the pathogenesis of BPDCN is still unknown, though the ETV6 locus is frequently translocated in cases of AML and B-lymphoblastic leukemia (B-LBL) . ETV6 functional effects in translocations are dependent on its partner and ultimately result in oncogenesis.
TET2 (Ten-Eleven Translocation-2) is part of a family of dioxygenases that promote DNA demethylation and mutations have been found in approximately 50% of the BPDCN cases interrogated [22, 26, 29]. These mutations are heterozygous, with most occurring in exons 3 and 11 as frame shift or nonsense mutations. Changes in TET2 expression and function can lead to alterations in posttranscriptional modification of histones and ultimately changes in gene expression.
Yet another gene mutated in BPDCN, FLT3, encodes the protein FMS-like tyrosine kinase 3, which is a receptor tyrosine kinase that transmits signals important for cellular proliferation and survival. FLT3 is critical for normal development of the hematopoietic systems, including pDCs [30,31,32,33]. A significant proportion of cases of AML harbor FLT3 mutations, either in the form of internal tandem duplications (ITD) involving the juxtamembrane domain or tyrosine kinase domain (TKD) mutations and affect prognosis [34]. While BPDCN shows some morphologic, immunophenotypic, and genetic overlap with AML, only rare published cases have demonstrated FLT3 mutations, including both ITD and TKD mutations [22, 26]. FLT3 inhibitors are being evaluated in the treatment of AML and may be of utility in FLT3-mutated BPDCN [35].
ASXL1 is mutated in roughly one-third of the BPDCN cases interrogated [22]. ASXL1 mutations are not unique to BPDCN, as they are found in a number of myeloid malignancies , including chronic myelomonocytic leukemia , myelodysplastic syndrome , and AML. ASXL1 can act independently and in concert with BRCA1-associated protein (BAP1) to promote deubiquitination of histone proteins, some of which are involved in cell proliferation. Although this pathway is thought to be important for regulating myelopoiesis, specific mechanisms of tumorigenesis in the setting of ASXL1 mutations are largely unknown [36,37,38,39,40].
DNA Repair Genes (HINT1, EWSR1, NPM1)
Studies of the 5q commonly deleted region in BPDCN have yielded a few known cancer-related genes, including HINT1 [41]. HINT1 (histidine triad nucleotide-binding protein 1) encodes a purine phosphoramidase that inhibits transcriptional activity of activation protein-1 (AP-1), α-catenin, MITF, and USF2 (upstream transcription factor 2, c-fos interacting). HINT1 deficiency impairs acetylation of the ATM protein, which inhibits DNA repair mechanisms [42]; its functional role in BPDCN is unknown.
EWSR1 is a member of the TET2 family of genes; it binds DNA/RNA and has a general role in gene expression and cell signaling. Additionally, EWSR1 plays a role in controlling DNA-damage-induced alternative splicing of some oncogenic proteins, such as BRCA1 [43]. EWSR1 gene fusions are common in sarcomas, including the Ewing sarcoma family of tumors. A single case of BPDCN has shown EWSR1 rearrangement with an unidentified translocation partner, suggesting that this locus may play a role in the pathogenesis of some cases of BPDCN [44].
NPM1 encodes the protein nucleophosmin, which mediates a number of different cellular processes, including DNA repair, regulation of the TP53 tumor suppressor pathway, and cell cycle events. NPM1 mutations have been identified in 20% of the cases of BPDCN analyzed, and include frame shift, nonsense, and missense mutations [22]. NPM1 is familiar in the context of AML , where it is mutated in approximately 50% of cases, and can affect prognosis [45]; however, the role of NPM1 mutations in BPDCN is unclear .
Therapy and Prognosis
As a rare entity, a uniform approach to therapy for BPDCN has not been devised. Non-Hodgkin lymphoma (hyper-CVAD, CHOP, or CHOP-like), AML , and ALL-type regimens have been employed [46,47,48,49]. While the majority of patients are able to achieve complete remission (CR), nearly all relapse, regardless of therapy, with a median overall survival (OS) of approximately 12 months [5].
Pagano et al. retrospectively identified 43 patients with BPDCN [50]. Of those treated with induction therapy, 60% were treated with an AML regimen and 35% were treated with an ALL regimen. CR was obtained in 17 patients, and though ALL-directed therapies showed significantly better initial remission rates than those that received AML-directed treatment (p = 0.02), patients treated with ALL therapy were more likely to experience eventual relapse. Hypomethylating agents (e.g., 5-azacitidine), which are commonly used in the treatment of myeloid malignancies, have been used in the treatment of a few BPDCN patients; however, despite good initial responses, the patients showed dismal outcomes [51]. Hyper-CVAD has also been shown to have some efficacy with median OS of 18 months and median CR of 21 months in at least one study [49].
The role of allogeneic and autologous stem cell transplant is still not well understood but in some instances, prolonged survival can be seen in patients [49, 50]. In the study by Pagano et al., the median OS of 6 allogeneic hematopoietic stem cell transplant recipients was 23 months, compared to 7 months in the 35 patients who did not undergo transplant. In a separate study by Pemmaraju et al., allogeneic and autologous stem cell transplant (SCT) patients had an overall similar median OS (18 months), compared to non-SCT patients treated with hyper-CVAD, CHOP, and other therapies (23 months).
Given the expression of CD123 and CD56 in BPDCN, clinical trials with anti-CD123 therapy (SL-401) and anti-CD56 (lorvotuzamab) are underway. In a prospective study of SL-401 therapy, Frankel et al. found that 7/9 patients had objective response, with 3 patients alive and in remission (3, 7, and 20 months follow-up) [52]. Clinical trials using lorvotuzamab for BPDCN are ongoing [49].
Conclusion
BPDCN is a rare, aggressive malignancy derived from immature plasmacytoid dendritic cells that shows a characteristic CD4+/CD56+/CD123 + immunophenotype. Even with aggressive therapy, including stem cell transplant, the prognosis is dismal. Although there is significant morphologic, immunophenotypic, mutational overlap with AML , recent gene expression profiling studies have shown distinct differences between these two entities, possibly providing new avenues for developing targeted therapies. While there are notable differences, the spectrum of gene alterations discovered by sequencing methods suggests at least a partial overlap with AML and implies that therapeutic strategies targeting aberrant methylation, chromatin remodeling, and splicing machinery in AML should also be investigated in BPDCN. Early results with anti-CD123 therapy have shown promising results; however, additional investigation will be required to determine whether this provides improved recurrence-free or overall survival. Increased awareness of this entity and further investigation of pathogenic mechanisms using modern techniques will serve to better define and devise optimal treatment strategies for BPDCN.
References
Adachi M, Maeda K, Takekawa M, Hinoda Y, Imai K, Sugiyama S, et al. High expression of CD56 (N-CAM) in a patient with cutaneous CD4-positive lymphoma. Am J Hematol. 1994;47(4):278–82.
Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25(3):383–92.
Pilichowska ME, Pinkus JL, Pinkus GS. Histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto disease): lesional cells exhibit an immature dendritic cell phenotype. Am J Clin Pathol. 2009;131(2):174–82.
Rollins-Raval MA, Marafioti T, Swerdlow SH, Roth CG. The number and growth pattern of plasmacytoid dendritic cells vary in different types of reactive lymph nodes: an immunohistochemical study. Hum Pathol. 2013;44(6):1003–10.
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: IARC Press; 2008.
Facchetti F, Cigognetti M, Fisogni S, Rossi G, Lonardi S, Vermi W. Neoplasms derived from plasmacytoid dendritic cells. Mod Pathol. 2016;29(2):98–111.
Martín-Martín LLA, Vidriales B, Caballero MD, Rodrigues AS, Ferreira SI, Lima M, Almeida S, Valverde B, Martínez P, Ferrer A, Candeias J, Ruíz-Cabello F, Buadesa JM, Sempere A, Villamor N, Orfao A, Almeida J. Classification and clinical behavior of blastic plasmacytoid dendritic cell neoplasms according to their maturation-associated immunophenotypic profile. Oncotarget. 2015;6(22):19204–16.
Julia F, Dalle S, Duru G, Balme B, Vergier B, Ortonne N, et al. Blastic Plasmacytoid dendritic cell neoplasms: Clinico-immunohistochemical correlations in a series of 91 patients. Am J Surg Pathol. 2014;38(5):673–80.
Herling M, Jones D. CD4+/CD56+ hematodermic tumor: the features of an evolving entity and its relationship to dendritic cells. Am J Clin Pathol. 2007;127(5):687–700.
Reineks EZ, Osei ES, Rosenberg A, Auletta J, Meyerson HJ. CD22 expression on blastic plasmacytoid dendritic cell neoplasms and reactivity of anti-CD22 antibodies to peripheral blood dendritic cells. Cytometry B Clin Cytom. 2009;76((4):237–48.
Sangle NA, Schmidt RL, Patel JL, Jeffrey Medeiros L, Agarwal AM, Perkins SL, et al. Optimized immunohistochemical panel to differentiate myeloid sarcoma from blastic plasmacytoid dendritic cell neoplasm. Mod Pathol (An Official Journal of the United States and Canadian Academy of Pathology, Inc). 2014;27(8):1137–43.
Cronin DM, George TI, Reichard KK, Sundram UN. Immunophenotypic analysis of myeloperoxidase-negative leukemia cutis and blastic plasmacytoid dendritic cell neoplasm. Am J Clin Pathol. 2012;137(3):367–76.
Johnson RCKJ, Natkunam Y, Sundram U, Freud AG, Gammon B, Cascio MJ. Myeloid cell nuclear differentiation antigen (MNDA) expression distinguishes Extramedullary presentations of myeloid leukemia from Blastic Plasmacytoid dendritic cell neoplasm. Am J Surg Pathol. 2016;40(4):502–9.
Leroux DMF, Callanan M, Radford-Weiss I, Dastugue N, Feuillard J, Le Mée F, Plessis G, Talmant P, Gachard N, Uettwiller F, Pages MP, Mozziconacci MJ, Eclache V, Sibille C, Avet-Loiseau H, Lafage-Pochitaloff M. CD4(+), CD56(+) DC2 acute leukemia is characterized by recurrent clonal chromosomal changes affecting 6 major targets: a study of 21 cases by the Groupe Français de Cytogénétique Hématologique. Blood. 2002;99(11):4154–9.
Jardin F, Callanan M, Penther D, Ruminy P, Troussard X, Kerckaert JP, et al. Recurrent genomic aberrations combined with deletions of various tumour suppressor genes may deregulate the G1/S transition in CD4+CD56+ haematodermic neoplasms and contribute to the aggressiveness of the disease. Leukemia. 2009;23(4):698–707.
Wiesner T, Obenauf AC, Cota C, Fried I, Speicher MR, Cerroni L. Alterations of the cell-cycle inhibitors p27(KIP1) and p16(INK4a) are frequent in blastic plasmacytoid dendritic cell neoplasms. J Invest Dermatol. 2010;130(4):1152–7.
Nakamura Y, Kayano H, Kakegawa E, Miyazaki H, Nagai T, Uchida Y, et al. Identification of SUPT3H as a novel 8q24/MYC partner in blastic plasmacytoid dendritic cell neoplasm with t(6;8)(p21;q24) translocation. Blood cancer journal. 2015;5:e301.
Takiuchi YMH, Aoki K, Kato A, Ono Y, Nagano S, Arima H, Inoue D, Mori M, Tabata S, Yanagita S, Matsushita A, Nishio M, Imai Y, Imai Y, Ito K, Fujita H, Kadowaki N, Ishikawa T, Takahashi T. Leukemic manifestation of blastic plasmacytoid dendritic cell neoplasm lacking skin lesion : a borderline case between acute monocytic leukemia. J Clin Exp Hematop. 2012;52(2):107–11.
Momoi ATK, Kawai K, Tsuchiyama J, Suzuki N, Yano T, Uesugi Y, Takahashi M, Aizawa Y. Cutaneous lymphoblastic lymphoma of putative plasmacytoid dendritic cell-precursor origin: two cases. Leuk Res. 2002;26(7):693–8.
Sapienza MR, Fuligni F, Agostinelli C, Tripodo C, Righi S, Laginestra MA, et al. Molecular profiling of blastic plasmacytoid dendritic cell neoplasm reveals a unique pattern and suggests selective sensitivity to NF-kB pathway inhibition. Leukemia. 2014;28(8):1606–16.
Dijkman R, van Doorn R, Szuhai K, Willemze R, Vermeer MH, Tensen CP. Gene-expression profiling and array-based CGH classify CD4+CD56+ hematodermic neoplasm and cutaneous myelomonocytic leukemia as distinct disease entities. Blood. 2007;109(4):1720–7.
Menezes J, Acquadro F, Wiseman M, Gomez-Lopez G, Salgado RN, Talavera-Casanas JG, et al. Exome sequencing reveals novel and recurrent mutations with clinical impact in blastic plasmacytoid dendritic cell neoplasm. Leukemia. 2014;28(4):823–9.
Stenzinger AEV, Pfarr N, Andrulis M, Jöhrens K, Klauschen F, Siebolts U, Wolf T, Koch PS, Schulz M, Hartschuh W, Goerdt S, Lennerz JK, Wickenhauser C, Klapper W, Anagnostopoulos I, Weichert W. Targeted ultra-deep sequencing reveals recurrent and mutually exclusive mutations of cancer genes in blastic plasmacytoid dendritic cell neoplasm. Oncotarget. 2014;5(15):6404–13.
Lucioni M, Novara F, Fiandrino G, Riboni R, Fanoni D, Arra M, et al. Twenty-one cases of blastic plasmacytoid dendritic cell neoplasm: focus on biallelic locus 9p21.3 deletion. Blood. 2011;118(17):4591–4.
Georgopoulos K, Winandy S, Avitahl N. The role of the Ikaros gene in lymphocyte development and homeostasis. Annu Rev Immunol. 1997;15:155–76.
Jardin FRP, Parmentier F, Troussard X, Vaida I, Stamatoullas A, Leprêtre S, Penther D, Duval AB, Picquenot JM, Courville P, Capiod JC, Tilly H, Bastard C, Marolleau JP. TET2 and TP53 mutations are frequently observed in blastic plasmacytoid dendritic cell neoplasm. Br J Haematol. 2011;153(3):413–6.
Gao NA, Wang XX, Sun JR, WZ Y, Guo NJ. Blastic plasmacytoid dendritic cell neoplasm with leukemic manifestation and ETV6 gene rearrangement: a case report. Exp Ther Med. 2015;9(4):1109–12.
Wang LCSW, Fujiwara Y, Davidson L, Visvader J, Kuo F, Alt FW, Gilliland DG, Golub TR, Orkin SH. The TEL/ETV6 gene is required specifically for hematopoiesis in the bone marrow. Genes Dev. 1998;12(15):2392–402.
Alayed K, Patel KP, Konoplev S, Singh RR, Routbort MJ, Reddy N, et al. TET2 mutations, myelodysplastic features, and a distinct immunoprofile characterize blastic plasmacytoid dendritic cell neoplasm in the bone marrow. Am J Hematol. 2013;88(12):1055–61.
Watowich SS, Liu YJ. Mechanisms regulating dendritic cell specification and development. Immunol Rev. 2010;238(1):76–92.
Lyman SD, Jacobsen SE. C-kit ligand and Flt3 ligand: stem/progenitor cell factors with overlapping yet distinct activities. Blood. 1998;91(4):1101–34.
Adolfsson J, Borge OJ, Bryder D, Theilgaard-Mönch K, Astrand-Grundström I, Sitnicka E, et al. Upregulation of Flt3 expression within the bone marrow Lin(−)Sca1(+)c-kit(+) stem cell compartment is accompanied by loss of self-renewal capacity. Immunity. 2001;15(4):659–69.
Maraskovsky E, Daro E, Roux E, Teepe M, Maliszewski CR, Hoek J, et al. Vivo generation of human dendritic cell subsets by Flt3 ligand. Blood. 2000;96(3):878–84.
Berman E, Maloy M, Devlin S, Jhanwar S, Papadopoulos E, Jakubowski A. Stem cell transplantation in adults with acute myelogenous leukemia, normal cytogenetics, and the FLT3-ITD mutation. Leuk Res. 2016;40:33–7.
Wander S, Levis M, Fathi A. The evolving role of FLT3 inhibitors in acute myeloid leukemia: quizartinib and beyond. Ther Adv Hematol. 2014;5(3):65–77.
Abdel-Wahab O, Dey A. The ASXL-BAP1 axis: new factors in myelopoiesis, cancer and epigenetics. Leukemia. 2013;27(1):10–5.
Daou S, Hammond-Martel I, Mashtalir N, Barbour H, Gagnon J, Iannantuono NV, et al. The BAP1/ASXL2 histone H2A Deubiquitinase complex regulates cell proliferation and is disrupted in cancer. J Biol Chem. 2015;290(48):28643–63.
LaFave LM, Béguelin W, Koche R, Teater M, Spitzer B, Chramiec A, et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med. 2015;21(11):1344–9.
Micol JB, Abdel-Wahab O. The role of additional sex combs-like proteins in cancer. Cold Spring Harb Perspect Med. 2016;6:a026526.
Sahtoe DD, van Dijk WJ, Ekkebus R, Ovaa H, Sixma TK. BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nat Commun. 2016;7:10292.
Fu Y, Fesler M, Mahmud G, Bernreuter K, Jia D, Batanian JR. Narrowing down the common deleted region of 5q to 6.0 Mb in blastic plasmacytoid dendritic cell neoplasms. Cancer Genet. 2013;206(7–8):293–8.
Li H, Balajee AS, Su T, Cen B, Hei TK, Weinstein IB. The HINT1 tumor suppressor regulates both gamma-H2AX and ATM in response to DNA damage. J Cell Biol. 2008;183(2):253–65.
Paronetto MP, Miñana B, Valcárcel J. The Ewing sarcoma protein regulates DNA damage-induced alternative splicing. Mol Cell. 2011;43(3):353–68.
Cao QLF, Niu G, Xue L, Han A. Blastic plasmacytoid dendritic cell neoplasm with EWSR1 gene rearrangement. J Clin Pathol. 2014;67(1):90–2.
Oelschlaegel U, Mohr B, Schaich M, Schakel U, Kroschinsky F, Illmer T, et al. HLA-DRneg patients without acute promyelocytic leukemia show distinct immunophenotypic, genetic, molecular, and cytomorphologic characteristics compared to acute promyelocytic leukemia. Cytometry B Clin Cytom. 2009;76((5):321–7.
Feuillard J, Jacob MC, Valensi F, Maynadie M, Gressin R, Chaperot L, et al. Clinical and biologic features of CD4(+)CD56(+) malignancies. Blood. 2002;99(5):1556–63.
Gruson B, Vaida I, Merlusca L, Charbonnier A, Parcelier A, Damaj G, et al. L-asparaginase with methotrexate and dexamethasone is an effective treatment combination in blastic plasmacytoid dendritic cell neoplasm. Br J Haematol. 2013;163(4):543–5.
Gilis L, Lebras L, Bouafia-Sauvy F, Espinouse D, Felman P, Berger F, et al. Sequential combination of high dose methotrexate and L-asparaginase followed by allogeneic transplant: a first-line strategy for CD4+/CD56+ hematodermic neoplasm. Leuk Lymphoma. 2012;53(8):1633–7.
Pemmaraju N, Kantarjian HM, Cortes JE, Duvic M, Khoury JD, Patel K, Daver N, O'Brien S, Pierce S, Garcia-Manero G, Jabbour E, Jain N, Faderl S, Thomas D, Frankel AE, Qazilbash MH, Konopleva M. Blastic Plasmacytoid dendritic cell neoplasm (BPDCN): a large single-center experience: analysis of clinical and molecular characteristics and patient outcomes. Blood. 2015;126(23):3746.
Pagano L, Valentini CG, Pulsoni A, Fisogni S, Carluccio P, Mannelli F, et al. Blastic plasmacytoid dendritic cell neoplasm with leukemic presentation: an Italian multicenter study. Haematologica. 2013;98(2):239–46.
Laribi K, Denizon N, Ghnaya H, Atlassi M, Besancon A, Pineau-Vincent F, et al. Blastic plasmacytoid dendritic cell neoplasm: the first report of two cases treated by 5-azacytidine. Eur J Haematol. 2014;93(1):81–5.
Frankel AE, Woo JH, Ahn C, Pemmaraju N, Medeiros BC, Carraway HE, et al. Activity of SL-401, a targeted therapy directed to interleukin-3 receptor, in blastic plasmacytoid dendritic cell neoplasm patients. Blood. 2014;124(3):385–92.
Herling M, Teitell MA, Shen RR, Medeiros LJ, Jones D. TCL1 expression in plasmacytoid dendritic cells (DC2s) and the related CD4+ CD56+ blastic tumors of skin. Blood. 2003;101(12):5007–9.
Marafioti TPJ, Ballabio E, Reichard KK, Tedoldi S, Hollowood K, Dictor M, Hansmann ML, Pileri SA, Dyer MJ, Sozzani S, Dikic I, Shaw AS, Petrella T, Stein H, Isaacson PG, Facchetti F, Mason DY. Novel markers of normal and neoplastic human plasmacytoid dendritic cells. Blood. 2008;111(7):3778–92.
Boiocchi L, Lonardi S, Vermi W, Fisogni S, Facchetti F. BDCA-2 (CD303): a highly specific marker for normal and neoplastic plasmacytoid dendritic cells. Blood. 2013;122(2):296–7.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter
Cite this chapter
Cascio, M.J., Ohgami, R.S. (2018). Blastic Plasmacytoid Dendritic Cell Neoplasm. In: Chang, CC., Ohgami, R. (eds) Precision Molecular Pathology of Myeloid Neoplasms. Molecular Pathology Library, vol 12. Springer, Cham. https://doi.org/10.1007/978-3-319-62146-3_18
Download citation
DOI: https://doi.org/10.1007/978-3-319-62146-3_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-62144-9
Online ISBN: 978-3-319-62146-3
eBook Packages: MedicineMedicine (R0)