
Abstract
Pheochromocytomas and paragangliomas, arising from respective adrenal and extra-adrenal chromaffin tissue, are infrequent causes of hypertension, but important to consider due to their often fatal nature if undiagnosed. Prevalences vary from less than 2% in patients tested due to hypertension and catecholamine-related symptoms to 7% in patients with incidentalomas and up to 40% in patients with specific hereditary syndromes. Germline and somatic mutations of tumor susceptibility genes are increasingly being recognized as important causes of the tumors that influence disease presentation through differences in activated tumorigenic pathways, including those that control catecholamine biosynthetic and secretory machinery. Biochemical diagnosis is now simplified by measurements of plasma-free metanephrines, the O-methylated metabolites of catecholamines. However, inappropriate application of the test hinders its optimal utility, rendering measurements of urinary fractionated metanephrines more suitable for most nonspecialist centers. When appropriately used, the plasma test not only allows accurate diagnosis but also assessments of underlying mutations, presence of malignancy, as well as tumor size and location. Tumor localization is then usually a simple matter, facilitated also by new functional imaging modalities, choice of which can benefit from consideration of underlying mutations. Management and treatment continues to rely on preoperative blockade of the effects of catecholamines, with surgical intervention usually but not always offering cure. Due to risks of postoperative recurrence, including metastatic involvement, long-term follow-up is important, with increasing indications that the nature of this should be personalized according to underling mutations, as well as size, location, and biochemical features of resected primary tumors.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


1 Clinical Presentation
Pheochromocytomas and paragangliomas (PPGLs) are catecholamine-producing neuroendocrine tumors that respectively arise from chromaffin or paraganglial tissue at adrenal and extra-adrenal locations [1]. Paragangliomas usually form from chromaffin cells associated with paravertebral sympathetic ganglia, most usually in the abdomen (e.g., organ of Zuckerkandl), but also in the pelvic areas (e.g., bladder) and less frequently in the thorax (e.g., mediastinum). Paragangliomas may also form at the neck and skull base, but these derive from parasympathetic or carotid body-associated tissue and usually do not produce significant amounts of vasoactive catecholamines.
As a result of tumoral secretion of catecholamines, patients with high blood pressure and symptoms of catecholamine excess are those in whom PPGLs are most frequently suspected. The tumors are also frequently identified among patients who undergo imaging for unrelated purposes and in whom incidental abdominal or adrenal masses (i.e., incidentalomas) are found. Patients with germline mutations of an increasing number of PPGL susceptibility genes represent another group in whom the tumors are now often found in the setting of routine screening due to hereditary risk.
PPGLs usually occur during middle age, but in 10–20% of cases present during childhood [2], the latter usually associated with a hereditary cause reflecting the younger age of presentation of hereditary than sporadic tumors. The clinical presentation of PPGLs can vary enormously from severe cardiovascular emergencies associated with sustained or paroxysmal hypertension and symptoms of catecholamine excess to a completely normotensive and asymptomatic presentation often found among patients screened due the presence of an incidentalomas or an underlying hereditary predisposition [3, 4].
1.1 Prevalence
Autopsy studies have indicated prevalences of PPGLs of 0.05–0.13%, mostly reflecting cases that remained undetected during life [5,6,7]. From reported annual incidences of PPGLs of between two and five cases detected per million per year [8, 9], translating to lifelong prevalences of 0.013–0.033%, it seems clear from the autopsy series that most PPGLs remain undetected throughout life, contributing to premature death. Nevertheless, prevalences at autopsy have dropped from 0.13% in the 50-year period before 1982 [5] to 0.05% in the 20-year period up until the turn of the century [6, 7], suggesting improved diagnosis during life. Presumably there have been further improvements in diagnosis over the subsequent 15 years, so that annual detection rates at some centers might be expected to reach ten or more per million.
Among patients with hypertension, the prevalence of PPGLs has been estimated at 0.6% [10], at least fourfold higher than overall prevalence rates. It can therefore be expected that prevalences are higher, possibly reaching 2% among patients with sustained or paroxysmal hypertension and symptoms of catecholamine excess. This is in agreement with findings that among unselected patients screened for PPGLs, prevalences of PPGLs range from 0.8 to 1.6% [11,12,13]. Among patients with adrenal incidentalomas, prevalences of pheochromocytoma are higher, between 4 and 9% [14, 15], with an overall prevalence of 7% indicated by review of 29 studies performed between 1982 and 2002 [16]. Depending on the mutation, prevalences can be even higher in patients with a hereditary predisposition to PPGLs, reaching 40% in patients with multiple neoplasia type 2 (MEN 2) [17].
1.2 Signs and Symptoms
Most patients with PPGLs have classical symptoms due to the effects of excessive circulating concentrations of catecholamines (Table 31.1). Reported frequencies of symptoms depend however on the studied populations and are often biased by the retrospective nature of studies. Some patients have been reported as normotensive and completely asymptomatic, particularly those in whom tumors are diagnosed based on screening because of hereditary predisposition or of findings of an incidentaloma [4]. In rare cases, tumors may synthesize no or little catecholamines or only produce dopamine [18, 19].
Signs and symptoms of PPGLs usually present paroxysmally, consequent to the hemodynamic and metabolic actions of peak levels of catecholamines (Fig. 31.1). Such episodes usually last between a few to 60 min and can occur spontaneously or may be provoked by drugs (e.g., steroids, antiemetics), anesthesia, tyramine-containing foods, or mechanical factors. Adverse reactions to medications can be particularly problematic (Table 31.2). The frequency of such episodes varies from once daily to only a few times per month.

Paroxysmal hypertension in a patient with a paraganglioma of the urinary bladder. Sonograms of urinary bladder in the patient before and after micturition are shown in panel A. Blood pressure profiles recorded before, during, and after micturition on two separate days are shown in panel B. Blood samples were taken at intervals and plasma concentrations of norepinephrine (NE) measured as shown for the five time points of collections
The hallmark that usually triggers clinicians to consider the diagnosis of PPGL is hypertension. At least 50% of patients have chronic but usually labile hypertension with short-lasting episodes of high surges in blood pressure. Specific blood pressure patterns often feature prominent daytime variability, an absent or blunted diurnal blood pressure rhythm, orthostatic hypotension, and more rarely hypotension [20, 21]. A small percent of patients present with shock, which has been suggested to occur particularly in epinephrine- or dopamine-secreting tumors [22].
Apart from hypertension, there is wide constellation of other established signs and symptoms, dominated by the classic triad of paroxysmal headache, palpitations, and diaphoresis (Table 31.1). In addition many other nonspecific symptoms may be encountered, including nausea, tremulousness, anxiety, panic attacks, weight loss, and gastrointestinal symptoms such as constipation and vomiting. Hyperglycemia and even overt diabetes mellitus in young lean hypertensive subjects should arouse suspicion of the tumor. Due to the nonspecific nature of most symptoms, there are many other clinical conditions, mostly associated with increased sympathetic activity, that mimic presence of a catecholamine-producing tumor [3].
In about 10% of patients with PPGLs, elevations in blood pressure may evolve into a hypertensive crisis with subsequent organ damage involving acute coronary syndrome, left ventricular heart failure, Takotsubo cardiomyopathy, arrhythmias, or stroke [21, 23,24,25]. Occasional patients may also present with multisystem crisis associated with an IL-6-mediated acute inflammatory syndrome, high-grade fever (>40 °C), and leukocytosis [21], evolving into renal failure, pulmonary edema, encephalopathy, and lactic acidosis. Delayed treatment is associated with a high fatality rate [23].
Several factors account for the variable clinical picture of PPGLs, including tumor size, biochemical phenotype, co-secreted peptides, and underlying pathogenic mutations. In general, larger tumors are associated with larger elevations in catecholamines and more prominent signs and symptoms, but this association may be lost when the tumors contain substantial hemorrhage or necrosis. Patients with epinephrine-producing tumors are more likely to display paroxysmal symptoms such as palpitations, tremulousness, and anxiety than tumors producing merely norepinephrine [26,27,28]. This may in part reflect the more prominent beta2-adrenergic effects of epinephrine, but as detailed later may also reflect differences in secretory characteristics. Tumors that produce predominantly dopamine are rare, presenting mainly as paragangliomas, particularly in the head and neck. Patients with such tumors are usually normotensive and asymptomatic [18, 29]. Presence of nausea or orthostatic hypotension has been reported in several patients with dopamine-producing PPGLs [18, 30]. Given the widely varying clinical spectrum of signs and symptoms, a meticulously taken detailed medical history and physical examination are essential for timely diagnosis of this treacherous tumor.
1.3 Incidentalomas
As many as or more than 25% of all pheochromocytomas are now being diagnosed after presentation as an adrenal incidentaloma [31,32,33]. Many cases of abdominal or thoracic paragangliomas are also being discovered after imaging for nonspecific reasons such as abdominal pain or due to other mass effects of tumors [18, 33]. Due to the high prevalence of PPGLs among such patients, combined with the substantial proportion of normotensive and asymptomatic cases, routine screening for PPGLs is widely recommended for all patients with incidentally discovered masses, regardless of the presence or absence of signs and symptoms [16, 33,34,35].
As indicated in one study, biochemical phenotypic features and other tumor characteristics differ between patients presenting with normotensive incidentally discovered adrenal pheochromocytomas and those with similarly sized tumors associated with overt signs and symptoms [36]. Specifically tumors from patients who were normotensive showed lesser increases in both urinary metanephrines and catecholamines than in patients who were hypertensive. This suggested that differences in biochemical features might contribute to discovery of such tumors as incidentalomas rather than more classically secondary to hypertension and symptoms of catecholamine excess.
1.4 Hereditary Disease
At least a third of PPGLs are inherited due to germline mutations of more than 13 tumor susceptibility genes identified to date [37, 38]. The most well-established hereditary causes of PPGLs are those associated with syndromic presentations including neurofibromatosis type 1 (NF 1) due to mutations of the NF1 gene, multiple endocrine neoplasia type 2 (MEN2) due to mutations of the rearranged during transfection (RET) gene, von Hippel-Lindau (VHL) syndrome due to mutations of the VHL gene, and familial paraganglioma syndromes caused by mutations of genes encoding succinate dehydrogenase subunits B and D (SDHB and SDHD). Other less frequent forms of hereditary PPGLs can result from mutations of succinate dehydrogenases A and C (SDHA and SDHC), succinate dehydrogenase complex assembly factor 2 (SDHAF2), transmembrane domain protein 127 (TMEM127), MYC-associated factor X (MAX), prolyl hydroxylase 2 (PHD2), fumarate hydratase (FH), and malate dehydrogenase 2 (MDH2).
Prevalence of PPGLs associated with the above mutations varies according to penetrance of disease and incidence of mutations. For NF1, although relatively common (1:3000), penetrance of pheochromocytoma is low with not more than 5% of patients developing the tumors [39]. In contrast, the penetrance of pheochromocytoma for RET mutation carriers reaches 40–50% [17], while for VHL syndrome it averages 20% with variability according to the particular mutation [40]. For some mutations of the SDHD gene penetrance has been reported to reach up to 100% [41]. Among patients with SDHD, SDHAF2, and MAX mutations, disease is transmitted to offspring paternally, skipping a generation with maternal transmission [42,43,44]. For most of the recently discovered tumor susceptibility genes, penetrance has not yet been precisely established, this requiring long-term follow-up of non-index cases.
Despite the variability and uncertainty of disease risk, it is widely recognized that all patients carrying mutations of PPGL susceptibility genes should undergo periodic screening for the tumors. For patients with VHL syndrome and MEN2, such screening at specialist centers is already well established and clearly results in diagnosis of tumors at an earlier stage when tumors are small and patients are normotensive and asymptomatic [28, 45]. For all there is an emerging need for personalized management according to risk. Patients with SDHB mutations, who carry a high risk for metastatic PPGLs [46], provide an example of those who might particularly benefit from earlier detection of disease before metastatic involvement.
Due to the rich hereditary background of PPGLs, it is recommended by Endocrine Society clinical practice guidelines that all patients with PPGLs receive counseling about possible genetic risk and that genetic testing should be particularly encouraged for several groups of patients [47]: (1) those with a positive family history of PPGLs or tumor susceptibility gene mutations, (2) those with syndromic features, (3) those with PPGLs occurring at a young age, (4) those with multifocal or bilateral adrenal tumors, (5) those with paragangliomas in whom there is high risk of mutations for genes encoding succinate dehydrogenase subunits, and (6) those with metastatic disease in whom there is a high risk of SDHB mutations. The underlying rationale for such testing is that identification of a gene mutation provides a context for routine screening programs that can result in earlier detection of PPGLs and other neoplasms, thereby reducing morbidity and mortality. For patients with aggressive disease in whom surgical intervention is not an option, new developments on the horizon also make it likely that establishing the underlying mutation can provide a rationale for therapies that target downstream signaling pathways [48].
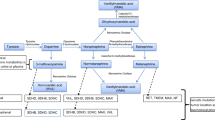
For PPGLs, the pathways leading to tumorigenesis are being rapidly elucidated consequent to their rich hereditary background [37, 49, 50]. As originally indicated by gene expression profiling, the various types of hereditary PPGLs fall into one of two main cluster groups according to the mutation and downstream affected signaling pathways [37]. Mutations of RET, NF1, TMEM127, and MAX, associated with cluster group 2, all involve activation of RAS and kinase signaling pathways and lead to well-differentiated adrenal pheochromocytomas with low susceptibility to malignancy. In contrast, mutations of VHL, SDHB, SDHD, SDHC, SDHA, SDHAF2, PHD2, and FH, associated with the cluster group 1, all result in stabilization of hypoxia-inducible factors (HIFs) leading to activation of hypoxia-angiogenic pathways. Apart from increased expression of hypoxia pathway genes, these tumors are also characterized by increased expression of the HIF2α itself and occur at an earlier age at both adrenal and extra-adrenal locations compared to the more differentiated cluster 2 tumors [51, 52]; this is suggested to reflect importance of transient expression of HIF2α during early differentiation of sympathoadrenal progenitors, with stabilization at the protein level leading to inhibition of both apoptosis and differentiation [53].
Support for involvement of HIF2α in development of cluster 1 PPGLs has come from identification of HIF2α itself as a tumor susceptibility gene, in almost all cases involving somatic mutations [54]. Importantly, HIF2α mutations show mosaicism, indicating occurrence during embryogenesis [55]. Nevertheless, even with the commonality of HIF2α in development of cluster 1 PPGLs, there are additional differences in signaling pathways among cluster 1 PPGLs. For HIF2α, VHL, and PHD2 mutations, stabilization of HIFs reflects the direct central tumorigenic mechanism. In contrast, for mutations of genes encoding succinate dehydrogenase subunits and other Krebs cycle enzymes (e.g., fumarate hydratase), stabilization of HIFs occurs secondary to wider actions of the elevated cellular levels of oncometabolites, succinate, or fumarate [48]. These oncometabolites inhibit not only the prolyl-hydroxylases that facilitate HIF degradation but also other alpha-ketoglutarate-dependent hydroxylases, such as histone and DNA demethylases. As a result, PPGLs resulting from mutations of genes encoding Krebs cycle enzymes exhibit a hypermethylator phenotype, turning off expression of numerous genes involved in restricting growth and controlling differentiation [56]. The result is that among cluster 1 PPGLs, those due to mutations of genes encoding Krebs cycle enzymes, and SDHB in particular, are the most aggressive and poorly differentiated (Fig. 31.2).

Catecholamine phenotypic features in PPGLs according to mutation. Cluster 2 tumors due to RET, NF1, and TMEM1 mutations are well differentiated, metabolizing tyrosine (TYR) to L-dopa (DOPA) by tyrosine hydroxylase, then to dopamine (DA) by aromatic amino acid decarboxylase, then to norepinephrine (NE) by dopamine β-hydroxylase after uptake into noradrenergic vesicles, and then to epinephrine (EPI) by phenylethanolamine N-methyltransferase (PNMT) after leakage of NE into the cytoplasm. DA is metabolized to methoxytyramine (MTY), NE is metabolized to normetanephrine (NMN), and EPI is metabolized to metanephrine (MN), all conversions catalyzed in the cytoplasm by catechol-O-methyltransferase. Cluster 1 tumors due to VHL, HIF2α, and SDHx mutations do not express PNMT so that these tumors do not produce EPI or MN. Also, these tumors, although having lower catecholamine stores compared to cluster 2 tumors, have poorer secretory controls than cluster 2 tumors and secrete catecholamines at higher rates. Tumors due to SDHx mutation are particularly poorly differentiated and often produce large amounts of MTY
2 Biochemistry
2.1 Catecholamine Synthesis, Storage, Secretion, and Metabolism
Synthesis, storage, secretion, and metabolism of catecholamines in PPGLs vary substantially depending on expression of catecholamine biosynthetic and secretory machinery, this as outlined above determined by underlying gene mutations [57, 58] (Fig. 31.2). The well-differentiated cluster 2 type pheochromocytomas all express phenylethanolamine N-methyltransferase (PNMT), which converts norepinephrine to epinephrine. Norepinephrine and epinephrine are stored in electron microscopically distinct secretory vesicles, the presences of which differ in cluster 1 and 2 mutated tumors [28].
Catecholamine secretion primarily involves exocytosis in which storage vesicles fuse with the plasma membrane and extrude their catecholamine contents into the extracellular space. This secretory process is a highly regulated calcium-dependent process responsive to neuronal input or secretogogues for evoked but controlled release of catecholamines. While PPGLs lack the former control, they can be influenced by secretogogues, but vary considerably in expression of the cellular secretory machinery responsive to regulatory control. Cluster 1 mutated tumors not only lack expression of PNMT, but in contrast to well-differentiated cluster 2 tumors also exhibit poorer expression of other key catecholamine biosynthetic enzymes and components responsible for storage and regulated secretion of catecholamines [57] (Fig. 31.2).
As a consequence of the above differences, cluster 1 tumors lack production of epinephrine and also contain lower overall stores of catecholamines [58] (Fig. 31.2). Furthermore, due to their relative lack of regulated secretory pathway machinery, cluster 1 tumors also secrete the limited amounts of catecholamines they produce in a more continuous or constitutive fashion compared to cluster 2 tumors. Thus, in contrast to cluster 1 PPGLs, cluster 2 epinephrine-producing pheochromocytomas, such as those in patients with MEN 2 or NF1, are characterized by highly concentrated stores of catecholamines and relatively low rates of catecholamine secretion. These tumors can, however, be easily provoked to secrete catecholamines in response to secretogogues and other stimuli, which may provide the basis for why epinephrine-producing tumors have been described as more often producing paroxysmal hypertension compared to norepinephrine-producing tumors.
The above differences along with differences in co-secretion of bioactive peptides are likely responsible for some of the highly variable clinical manifestations of chromaffin cell tumors, but as yet there has been no fully prospective study to firmly establish such a link between underlying mutations to differences in presentation of disease. There are, however, forms of undifferentiated PPGLs for which a link seems clear. These in particular involve patients who have PPGLs due to mutations of the SDHB gene which appears to be associated with further downregulated expression of catecholamine phenotypic features due to epigenetic silencing secondary to actions of elevated succinate to inhibit alpha-ketoglutarate-dependent enzymes involved in regulating DNA methylation [56]. As a consequence, tumors in patients with SDHB mutation contain the lowest amounts of catecholamines, among all PPGLs; catecholamine contents are also characterized by relative high proportions of dopamine [58, 59]. The tumors often reach large sizes before their discovery, which may reflect both relative paucity of signs and symptoms and diversion of energy from maintaining chromaffin-like phenotypic features to enhanced growth. Consequently, these tumors carry a high risk of malignancy.
Most other PPGLs contain large amounts of catecholamines, particularly norepinephrine, in about 50% of cases additional epinephrine and a variable amount of dopamine [58]. Tumor contents and secretion of dopamine are impacted by the efficiency of dopamine beta-hydroxylase in converting the dopamine to norepinephrine after the former amine is translocated into secretory vesicles [60]. Importantly, the catecholamines stored in secretory granules exist in a highly dynamic equilibrium with the surrounding cytoplasm, with passive outward leakage into the cytoplasm counterbalanced by inward active transport under the control of vesicular monoamine transporters [61]. For all catecholamines, whether stored in sympathetic neurons, adrenal chromaffin cells, or tumors derived from adrenal or extra-adrenal chromaffin cells, most initial metabolism takes place within the cells where the catecholamines are synthesized [62].
In sympathetic nerves, the presence of monoamine oxidase (MAO), but absence of catecholamine-O-methyltransferase (COMT), leads to deamination of norepinephrine to dihydroxyphenylglycol (DHPG). This primary norepinephrine metabolite is derived largely from norepinephrine leaking from storage vesicles, but is also derived from reuptake of the transmitter back into nerves [62]. Only a small amount of norepinephrine escapes reuptake; some of this is metabolized extraneuronally to normetanephrine or DHPG, while a remaining small proportion (<5%) reaches the circulation as norepinephrine. In adrenal chromaffin cells and tumors of chromaffin cells, the additional presence of COMT leads to production of metanephrine from epinephrine, normetanephrine from norepinephrine, and methoxytyramine from dopamine [63]. This production again depends on leakage of catecholamines from storage vesicles, a continuous process that is independent of exocytotic release of catecholamines, which makes a relatively minor contribution to production of metanephrine. Thus, over 90% of all circulating metanephrine is normally derived from metabolism of epinephrine within adrenal chromaffin cells [64]. These cells also make the single largest contribution to circulating normetanephrine (at least 24%), with the rest from norepinephrine metabolized to normetanephrine in non-neuronal and non-chromaffin extraneuronal cells. Continuous production of the O-methylated metabolites within chromaffin cells and their tumor derivatives explains why measurements of plasma-free and urine deconjugated metanephrines provide advantages over measurements of catecholamines, which can be released by some tumors intermittently or in low amounts. Other catecholamine metabolites are produced in various organs and tissues of the body and also are not as useful as the metanephrines for diagnosis of PPGLs. Vanillylmandelic acid, for example, is almost exclusively formed in the liver as the end product of catecholamine metabolism and derived mainly from DHPG produced in sympathetic nerves [65].
2.2 Biochemical Diagnosis
The superior diagnostic accuracy of measurements of plasma-free metanephrines over other tests has been clearly outlined by several independent studies [66,67,68,69,70,71], with several others confirming the high diagnostic accuracy of either plasma or urinary fractionated metanephrines for identifying patients with PPGLs [72,73,74]. With this evidence at hand, it has been a simple matter for Endocrine Society clinical practice guidelines on PPGLs to stipulate that initial testing for PPGLs should always include measurements of plasma-free or urinary fractionated metanephrines [47]. All other available tests, including urinary or plasma catecholamines, urinary vanillylmandelic acid, urinary total metanephrines (measured by spectrophotometric methods), and chromogranin A, are unnecessary and generally inappropriate for initial screening of PPGLs, but may be employed for follow-up confirmation of positive results of plasma-free or urinary fractionated metanephrines or in specific presentations of disease.
If used correctly with appropriate analytical methods and reference intervals, measurements of plasma-free metanephrines in particular provide diagnostic sensitivity approaching 100% with diagnostic specificity of at least 95% [74]. Over 90% of PPGLs show elevations of normetanephrine, about 50% elevations of metanephrine, and 45% elevations of methoxytyramine, with about 70% showing some combination of increases of normetanephrine with metanephrine or methoxytyramine or both. Moreover, increases are usually well in excess of twofold above the upper cutoffs. Such magnitudes of increases and combinations of increases are rare in patients without PPGLs, providing high positive predictive value in over 80% of patients with PPGLs. For these patients it is simply a matter of locating the tumor. For the other minority of patients with PPGLs in whom false positives are difficult to distinguish from true positives, biochemical diagnosis can be made using the clonidine test when elevations involve normetanephrine or by follow-up to establish increasing concentrations over time.
With measurements of plasma metanephrines by mass spectrometry and employing appropriate preanalytical precautions and reference intervals, diagnosis of PPGLs is simple [75]. Common problems, however, occur with the use of inaccurate analytical methods (e.g., immunoassays), incorrect reference intervals, and inappropriate preparation of patients for blood sampling. For the latter, patients should be sampled after 30 min supine rest and should not be under physiological or mental stress that might increase sympathoadrenal activity. Sampling in the seated position or under any form of stress carries a high likelihood of false positive results. When measurements involve plasma methoxytyramine, sampling should be carried out after an overnight fast.
At many centers, however, clinicians have problems following the above recommendations, and there can also be problems with availability or adequacy of laboratory measurements or reference intervals for plasma-free metanephrines [75]. For this reason, measurements of urinary fractionated metanephrines provide an alternative to plasma-free metanephrines.
2.3 Interpretation of Positive Biochemical Test Results
In addition to indicating the presence of PPGLs, usually with high positive predictive value, patterns of increases in plasma free metanephrines can also be used to predict tumor size, location, underlying mutations, and metastatic involvement [75]. As outlined earlier, mutations of different genes are associated with differences in expression of catecholamine biosynthetic enzymes and thus differences in patterns of increases of normetanephrine, metanephrine, and methoxytyramine [76]. Patients with cluster 2 types mutations, such as those involving RET and NF1 genes, almost all show increases in plasma metanephrine, with or without increases in normetanephrine. In contrast, PPGLs due to cluster 1 type mutations, such as those involving VHL, SDHD, and SDHB genes, do not express PNMT and do not produce epinephrine. Consequently, cluster 1 type tumors are associated with increases of normetanephrine, but not metanephrine. Furthermore, in patients with SDHB and SDHD mutations, there are often increases in methoxytyramine, which reflect the more immature nature of these tumors compared to other PPGLs.
Phenotypic immaturity, as indicated by increases in plasma methoxytyramine, is also associated with higher likelihood of metastatic involvement. In this context increases in plasma methoxytyramine provide the only currently available biomarker for metastatic involvement [59]. Although substantial increases of methoxytyramine are present in only about 60–70% of all cases of metastatic PPGLs, when present they are important to consider as an indicator of malignancy.
Since the extent of increase in plasma metanephrines is dependent on size of vesicular stores of catecholamines, the magnitude of increases in the metabolites can be used to roughly indicate mean tumor diameter [77]. Additionally, increased plasma metanephrine almost always indicates an adrenal pheochromocytoma or recurrence of an adrenal pheochromocytoma, whereas large increases in methoxytyramine relative to normetanephrine are indicative of extra-adrenal tumors. Such biochemical information indicating tumor size and location can be useful for subsequent imaging or interpretation of imaging results.
3 Tumor Localization
3.1 Anatomical Imaging
In general, imaging studies to locate a PPGL should be ordered once there is clear biochemical evidence for the presence of the tumor [47]. Exceptions to this rule are emergency situations where an immediate diagnosis and treatment are required [78]. Imaging studies without an established biochemical diagnosis are not cost-effective and entail a risk for diagnostic confusion with incidentaloma, thus complicating the work-up further. The first choice imaging modality for PPGL is computed tomography scanning (CT) before and after contrast administration of the abdominal and pelvic areas [47]. More than 95% of all PPGLs are located in these areas.
CT is in general preferred over magnetic resonance imaging (MRI) because of its superior spatial resolution. The sensitivity of CT for detecting pheochromocytomas is over 90%, but lower for paragangliomas and for recurrent or metastatic tumors [79, 80]. Since CT has no high reliability for elucidating the nature of a mass, the specificity for correct identification of the mass is moderate, even with consideration of imaging features such as density, contrast enhancement, and washout [81].
Although MRI imaging with or without gadolinium enhancement has a superior sensitivity over CT for detecting paragangliomas [79], again as with CT, specificity of MRI falls short due to features that may impair signal intensity such tumor necrosis or hemorrhage [81]. Apart from extra-adrenal paragangliomas, MRI is also indicated in specific patient groups, such as those with metastases, with intracardiac or head and neck PGLs, with postoperative surgical clips, and with iodine allergy. Patients in whom radiation exposure should be kept to a minimum are also candidates for MRI, including children, pregnant women, and others with known germline mutations who are likely to undergo repeated imaging studies [47].
3.2 Functional Imaging
Since anatomical imaging is insufficient for assessing multifocality or metastatic disease and is not specific for establishing the diagnosis, a complementary functional imaging step is often required after anatomical imaging [82, 83]. The aim of functional imaging is thus to establish the nature of a mass and identify other focal or metastatic lesions. For this purpose several radiolabeled ligands targeting specific cell membrane and/or vesicular catecholamine transport systems are available. Some ligands are employed for single photon emission computed tomography (SPECT), such as 123I-MIBG, while others are used for positron emission tomography (PET), such as 18F-fluorodeoxyglucose (18F-FDG) or 68Ga-DOTATATE.
The most widely available functional imaging ligand is 123I-MIBG. Sensitivity with 123I-MIBG SPECT for detection of pheochromocytoma approaches 100%, but is considerably less for extra-adrenal paragangliomas (56–75%) and metastases, particularly when associated with underlying SDHx mutations (<50%) [84,85,86]. A specific advantage of using 123I-MIBG is the potential to identify patients with metastatic PPGLs who may benefit from treatment with therapeutic doses of 131I-MIBG [83].
PET imaging agents used for functional imaging of PGGLs include 18F-fluorodopamine (18F-FDA), 18F-fluorodeoxyglucose (18F-FDG), 111In-DTPA-pentetreotide, 68Ga-DOTATATE, and 68Ga-DOTATOC [83, 86]. The diagnostic accuracies of these different compounds vary, depending on specific clinical features, such as tumor location, genetic mutation, and metastatic involvement, requiring personalized considerations for best choice of agent as available. Since many PPGLs overexpress subtype 2 somatostatin receptors, radiolabeled somatostatin receptor ligands can be particularly useful for localization. Such ligands developed for PET imaging, including 68Ga-DOTATATE and 68Ga-DOTATOC, have shown particularly excellent diagnostic accuracy for head and neck paragangliomas and SDHB-related metastatic disease [87,88,89].
4 Management
4.1 Preoperative Management
Surgical outcomes of patients with PPGLs have improved considerably over the last 50 years with a current perisurgical mortality rate of less than 1%. This can be attributed to improved presurgical medical preparation of patients as well as emergence of multidisciplinary teams and sophisticated approaches to anesthesiological management. Although most patients are in the long term cured by tumor removal, postsurgical recurrence rates average 16.5% [90].
Elective surgical removal of a catecholamine-producing tumor should be preceded by preoperative medical preparation to prevent or minimize hazardous complications due to massive release of catecholamines from the tumor, particularly provoked by mechanical manipulation during surgery [47, 91]. Such precautions are indicated in all patients in whom elevations of plasma or urinary metanephrines indicate a catecholamine-producing tumor, regardless of the presence or absence of symptoms or whether patients are hypertensive or normotensive. Although some retrospective small series of patients have questioned the need for medical preparation, there are no randomized trials documenting that refraining from medical pretreatment is safe. Since it is impossible to predict the course during surgery in individual patients, a patient-tailored strategy of preoperative blockade provides the safest approach.
The mainstay for successful medical pretreatment remains α-adrenoceptor antagonist therapy using the noncompetitive inhibitor, phenoxybenzamine, or the competitive inhibitor, doxazosin [91, 92]. Evidence from randomized trials showing benefits of one over the other are lacking [47]. Calcium channel blockers are generally not used as first-line blocking agent but do hold a place as an add-on drug to α-adrenoceptor blockade. Similarly, the catecholamine-synthesis inhibitor α-methylparatyrosine (metyrosine) can be employed as an add-on treatment to α-adrenoceptor blockade when required [91, 93]. A β-adrenoceptor antagonist, such as atenolol or metoprolol, can also be included several days before surgery to prevent tachyarrhythmias, but only after α-adrenoceptor blockade [91].
The recommended duration of pharmacological pretreatment in elective patients is 7–14 days, but this is based mainly on expert opinion. The target blood pressure level is less than 130/80 mm Hg in the sitting position with a systolic blood pressure in the upright position higher than 90 mm Hg [47, 91]. A presurgical high-sodium diet and high fluid intake during preparation are helpful to circumvent postsurgical hypotension [91]. Close postsurgical surveillance for at least 24 h is essential for detection and proper treatment of hypotension and hypoglycemia. In specific patients undergoing bilateral or major unique adrenal surgery, the possibility of adrenal insufficiency is the first and most important consideration in situations of postsurgical hypotension.
4.2 Surgical Management
Surgical removal of pheochromocytomas by minimal invasive laparoscopic tumor resection is first choice treatment with the posterior retroperitoneal approach preferred in patients with pheochromocytoma [94]. Paragangliomas may also be resected by laparoscopic surgery depending on tumor size, relation to other organs, and on the experience of the surgeon [95]. Retrospective cohort studies have shown that patients operated by laparoscopy experience less blood loss and have a shorter stay in hospital as compared to conventional open surgery [96, 97]. In patients with underlying mutations such as those with MEN2 and VHL, adrenal-sparing surgery is indicated when technically feasible. Leaving some remnant adrenocortical tissue in situ avoids or postpones need for lifelong steroid replacement therapy for adrenal insufficiency, but this benefit should be balanced against the increased risk of tumor recurrence [98,99,100].
4.3 Follow-Up
Following surgical removal of PPGLs, all patients should undergo follow-up to ascertain whether the tumor has been removed completely [101]. For patients with presurgically elevated plasma or urine metanephrines, measurements can be repeated at 2–6 weeks after surgery. Persistently elevated test results suggest residual disease that should then be confirmed by additional imaging studies.
Since there is a persistent risk of recurrent disease after apparently complete resection of an initially discovered PPGL, it is important that follow-up is continued in the long term well after surgical resection [102]. Risk of recurrence is higher in young patients (<20 years), in those with syndromic presentations, paragangliomas, and patients with large tumors. However, there is no “safe” tumor size below which the risk is zero. Thus, follow-up is recommended in all operated patients annually for at least 10 years, but continuing thereon in high-risk patients, such as those who are young, carry an underlying germline mutation or who present with an extra-adrenal or large tumor. Follow-up should include an annually taken medical history, physical examination including blood pressure, and measurements of plasma or urinary fractionated metanephrines.
References
Tischler AS (2008) Pheochromocytoma and extra-adrenal paraganglioma: updates. Arch Pathol Lab Med 132(8):1272–1284. doi:2007-0731-RAR [pii]
Waguespack SG, Rich T, Grubbs E et al (2010) A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 95(5):2023–2037. doi:10.1210/jc.2009-2830
Manger WM (2009) The protean manifestations of pheochromocytoma. Hom Metab Res 41(9):658–663. doi:10.1055/s-0028-1128139
Mannelli M, Lenders JW, Pacak K et al (2012) Subclinical phaeochromocytoma. Best Pract Res Clin Endocrinol Metab 26(4):507–515. doi:10.1016/j.beem.2011.10.008
Sutton MG, Sheps SG, Lie JT (1981) Prevalence of clinically unsuspected pheochromocytoma. Review of a 50-year autopsy series. Mayo Clin Proc 56(6):354–360
McNeil AR, Blok BH, Koelmeyer TD et al (2000) Phaeochromocytomas discovered during coronial autopsies in Sydney, Melbourne and Auckland. Aust NZ J Med 30(6):648–652
Lo CY, Lam KY, Wat MS, Lam KS (2000) Adrenal pheochromocytoma remains a frequently overlooked diagnosis. Am J Surg 179(3):212–215
Fernandez-Calvet L, Garcia-Mayor RV (1994) Incidence of pheochromocytoma in South Galicia, Spain. J Intern Med 236(6):675–677
Ariton M, Juan CS, AvRuskin TW (2000) Pheochromocytoma: clinical observations from a Brooklyn tertiary hospital. Endocr Pract 6(3):249–252
Omura M, Saito J, Yamaguchi K et al (2004) Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res 27(3):193–202
Hernandez FC, Sanchez M, Alvarez A et al (2000) A five-year report on experience in the detection of pheochromocytoma. Clin Biochem 33(8):649–655
Vaclavik J, Stejskal D, Lacnak B et al (2007) Free plasma metanephrines as a screening test for pheochromocytoma in low-risk patients. J Hypertens 25(7):1427–1431
Brain KL, Kay J, Shine B (2006) Measurement of urinary metanephrines to screen for pheochromocytoma in an unselected hospital referral population. Clin Chem 52(11):2060–2064. doi:10.1373/clinchem.2006.070805
Mantero F, Terzolo M, Arnaldi G et al (2000) A survey on adrenal incidentaloma in Italy. Study group on adrenal Tumors of the Italian Society of endocrinology. J Clin Endocrinol Metab 85(2):637–644
Terzolo M, Bovio S, Pia A et al (2009) Management of adrenal incidentaloma. Best Pract Res Clin Endocrinol Metab 23(2):233–243. doi:10.1016/j.beem.2009.04.001
Mansmann G, Lau J, Balk E et al (2004) The clinically inapparent adrenal mass: update in diagnosis and management. Endocrine Rev 25(2):309–340
Howe JR, Norton JA, Wells SA Jr (1993) Prevalence of pheochromocytoma and hyperparathyroidism in multiple endocrine neoplasia type 2A: results of long-term follow-up. Surgery 114(6):1070–1077
Eisenhofer G, Goldstein DS, Sullivan P et al (2005) Biochemical and clinical manifestations of dopamine-producing paragangliomas: utility of plasma methoxytyramine. J Clin Endocrinol Metab 90:2068–2075
Timmers HJ, Pacak K, Huynh TT et al (2008) Biochemically silent abdominal Paragangliomas in patients with mutations in the Sdhb Gene. J Clin Endocrinol Metab 93:4826–4832
Zelinka T, Strauch B, Petrak O et al (2005) Increased blood pressure variability in pheochromocytoma compared to essential hypertension patients. J Hypertens 23(11):2033–2039. doi:00004872-200511000-00018 [pii]
Prejbisz A, Lenders JW, Eisenhofer G, Januszewicz A (2011) Cardiovascular manifestations of phaeochromocytoma. J Hypertens 29(11):2049–2060. doi:10.1097/HJH.0b013e32834a4ce9
Bergland BE (1989) Pheochromocytoma presenting as shock. Am J Emerg Med 7(1):44–48
Brouwers FM, Eisenhofer G, Lenders JW, Pacak K (2006) Emergencies caused by pheochromocytoma, neuroblastoma, or ganglioneuroma. Endocrinol Metab Clin N Am 35(4):699–724
Giavarini A, Chedid A, Bobrie G et al (2013) Acute catecholamine cardiomyopathy in patients with phaeochromocytoma or functional paraganglioma. Heart ;99(19):1438–1444. doi: 10.1136/heartjnl-2013-304073
Riester A, Weismann D, Quinkler M et al (2015) Life-threatening events in patients with pheochromocytoma. Eur J Endocrinol 173(6):757–764. doi:10.1530/EJE-15-0483
Lance JW, Hinterberger H (1976) Symptoms of pheochromocytoma, with particular reference to headache, correlated with catecholamine production. Arch Neurol 33(4):281–288
Ito Y, Fujimoto Y, Obara T (1992) The role of epinephrine, norepinephrine, and dopamine in blood pressure disturbances in patients with pheochromocytoma. World J Surg 16(4):759–763. discussion 763-754
Eisenhofer G, Walther MM, Huynh TT et al (2001) Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab 86(5):1999–2008
Proye C, Fossati P, Fontaine P et al (1986) Dopamine-secreting pheochromocytoma: an unrecognized entity? Classification of pheochromocytomas according to their type of secretion. Surgery 100(6):1154–1162
Awada SH, Grisham A, Woods SE (2003) Large dopamine-secreting pheochromocytoma: case report. South Med J 96(9):914–917
Lenders JW, Eisenhofer G, Mannelli M, Pacak K (2005) Phaeochromocytoma. Lancet 366(9486):665–675
Amar L, Servais A, Gimenez-Roqueplo AP et al (2005) Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. J Clin Endocrinol Metab 90(4):2110–2116
Kopetschke R, Slisko M, Kilisli A et al (2009) Frequent incidental discovery of phaeochromocytoma: data from a German cohort of 201 phaeochromocytoma. Eur J Endocrinol 161(2):355–361. doi:10.1530/EJE-09-0384
Terzolo M, Pia A, Ali A et al (2002) Adrenal incidentaloma: a new cause of the metabolic syndrome? J Clin Endocrinol Metab 87(3):998–1003
Kirshtein B, Pagliarello G, Yelle JD, Poulin EC (2007) Incidence of pheochromocytoma in trauma patients during the management of unrelated illness: a retrospective review. Int J Surg 5(5):332–335. doi:10.1016/j.ijsu.2007.04.015
Haissaguerre M, Courel M, Caron P et al (2013) Normotensive incidentally discovered pheochromocytomas display specific biochemical, cellular, and molecular characteristics. J Clin Endocrinol Metab 98(11):4346–4354. doi:10.1210/jc.2013-1844
Dahia PL (2014) Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer 14(2):108–119. doi:10.1038/nrc3648
Flynn A, Benn D, Clifton-Bligh R et al (2015) The genomic landscape of phaeochromocytoma. J Pathol 236(1):78–89. doi:10.1002/path.4503
Walther MM, Herring J, Enquist E et al (1999) von Recklinghausen’s disease and pheochromocytomas. J Urol 162(5):1582–1586
Walther MM, Keiser HR, Choyke PL et al (1999) Management of hereditary pheochromocytoma in von Hippel-Lindau kindreds with partial adrenalectomy. J Urol 161(2):395–398
Peczkowska M, Erlic Z, Hoffmann MM et al (2008) Impact of screening kindreds for SDHD p.Cys11X as a common mutation associated with paraganglioma syndrome type 1. J Clin Endocrinol Metab 93(12):4818–4825. doi:10.1210/jc.2008-1290
Baysal BE (2004) Genomic imprinting and environment in hereditary paraganglioma. Am J Med Genet C Semin Med Genet 129C(1):85–90
Comino-Méndez I, Gracia-Aznárez FJ, Schiavi F et al (2011) Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet 43:663–667
Hoekstra AS, Devilee P, Bayley JP (2015) Models of parent-of-origin tumorigenesis in hereditary paraganglioma. Semin Cell Dev Biol 43:117–124. doi:10.1016/j.semcdb.2015.05.011
van Duinen N, Steenvoorden D, Bonsing BA et al (2010) Pheochromocytomas detected by biochemical screening in predisposed subjects are associated with lower prevalence of clinical and biochemical manifestations and smaller tumors than pheochromocytomas detected by signs and symptoms. Eur J Endocrinol 163(1):121–127. doi:10.1530/EJE-10-0114
Amar L, Bertherat J, Baudin E et al (2005) Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol 23(34):8812–8818
Lenders JWM, Duh QY, Eisenhofer G et al (2014) Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 99:1915–1942
Favier J, Amar L, Gimenez-Roqueplo AP (2015) Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nature Rev Endocrinol 11(2):101–111. doi:10.1038/nrendo.2014.188
Jochmanova I, Yang C, Zhuang Z, Pacak K (2013) Hypoxia-inducible factor signaling in pheochromocytoma: turning the rudder in the right direction. J Natl Canc Inst 105(17):1270–1283. doi:10.1093/jnci/djt201
Qin N, de Cubas AA, Garcia-Martin R et al (2014) Opposing effects of HIF1alpha and HIF2alpha on chromaffin cell phenotypic features and tumor cell proliferation: insights from MYC-associated factor X. Int J Cancer 135(9):2054–2064. doi:10.1002/ijc.28868
Eisenhofer G, Huynh TT, Pacak K et al (2004) Distinct gene expression profiles in norepinephrine- and epinephrine-producing hereditary and sporadic pheochromocytomas: activation of hypoxia-driven angiogenic pathways in von Hippel-Lindau syndrome. Endocr Relat Cancer 11:897–911
Eisenhofer G, Timmers H, Lenders JW et al (2011) Age at diagnosis of pheochromocytoma differs according to catecholamine phenotype and tumor location. J Clin Endocrinol Metab 96:375–384
Richter S, Qin N, Pacak K, Eisenhofer G (2013) Role of hypoxia and HIF2a in development of the sympathoadrenal cell lineage and chromaffin cell tumors with distinct catecholamine phenotypic features. Adv Pharmacol 68:285–317
Zhuang Z, Yang C, Lorenzo F et al (2012) Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. NEJM 367(10):922–930. doi:10.1056/NEJMoa1205119
Buffet A, Smati S, Mansuy L et al (2014) Mosaicism in HIF2A-related polycythemia-paraganglioma syndrome. J Clin Endocrinol Metab 99(2):E369–E373. doi:10.1210/jc.2013-2600
Letouze E, Martinelli C, Loriot C et al (2013) SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23(6):739–752. doi:10.1016/j.ccr.2013.04.018
Eisenhofer G, Huynh TT, Elkahloun A et al (2008) Differential expression of the regulated catecholamine secretory pathway in different hereditary forms of pheochromocytoma. Am J Physiol Endocrinol Metab 295(5):E1223–E1233
Eisenhofer G, Pacak K, Huynh TT et al (2011) Catecholamine metabolomic and secretory phenotypes in phaeochromocytoma. Endocr Relat Cancer 18(1):97–111. doi:ERC-10-0211 [pii]. doi:10.1677/ERC-10-0211
Eisenhofer G, Lenders JW, Siegert G et al (2012) Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer 48:1739–1749
Feldman JM (1985) Increased dopamine production in patients with carcinoid tumors. Metab Clin Exp 34(3):255–260
Eisenhofer G, Kopin IJ, Goldstein DS (2004) Leaky catecholamine stores: undue waste or a stress response coping mechanism? Ann N Y Acad Sci 1018:224–230
Eisenhofer G, Kopin IJ, Goldstein DS (2004) Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev 56(3):331–349
Eisenhofer G, Keiser H, Friberg P et al (1998) Plasma metanephrines are markers of pheochromocytoma produced by catechol-O-methyltransferase within tumors. J Clin Endocrinol Metab 83(6):2175–2185
Eisenhofer G, Rundquist B, Aneman A et al (1995) Regional release and removal of catecholamines and extraneuronal metabolism to metanephrines. J Clin Endocrinol Metab 80(10):3009–3017
Eisenhofer G, Aneman A, Hooper D et al (1996) Mesenteric organ production, hepatic metabolism, and renal elimination of norepinephrine and its metabolites in humans. J Neurochem 66(4):1565–1573
Lenders JW, Pacak K, Walther MM et al (2002) Biochemical diagnosis of pheochromocytoma: which test is best? JAMA 287(11):1427–1434
Raber W, Raffesberg W, Bischof M et al (2000) Diagnostic efficacy of unconjugated plasma metanephrines for the detection of pheochromocytoma. Arch Intern Med 160(19):2957–2963
Unger N, Pitt C, Schmidt IL et al (2006) Diagnostic value of various biochemical parameters for the diagnosis of pheochromocytoma in patients with adrenal mass. Eur J Endocrinol 154(3):409–417
Hickman PE, Leong M, Chang J et al (2009) Plasma free metanephrines are superior to urine and plasma catecholamines and urine catecholamine metabolites for the investigation of phaeochromocytoma. Pathology 41(2):173–177. doi:907871493 [pii]. doi:10.1080/00313020802579284
Grouzmann E, Drouard-Troalen L, Baudin E et al (2010) Diagnostic accuracy of free and total metanephrines in plasma and fractionated metanephrines in urine of patients with pheochromocytoma. Eur J Endocrinol 162(5):951–960. doi:EJE-09-0996 [pii]. doi:10.1530/EJE-09-0996
Pussard E, Chaouch A, Said T (2014) Radioimmunoassay of free plasma metanephrines for the diagnosis of catecholamine-producing tumors. Clin Chem Lab Med 52(3):437–444. doi:10.1515/cclm-2013-0406
Procopiou M, Finney H, Akker SA et al (2009) Evaluation of an enzyme immunoassay for plasma-free metanephrines in the diagnosis of catecholamine-secreting tumors. Eur J Endocrinol 161(1):131–140. doi:EJE-09-0172 [pii]. doi:10.1530/EJE-09-0172
Christensen TT, Frystyk J, Poulsen PL (2011) Comparison of plasma metanephrines measured by a commercial immunoassay and urinary catecholamines in the diagnosis of pheochromocytoma. Scan J Clin Lab Invest 71(8):695–700. doi:10.3109/00365513.2011.622410
Darr R, Pamporaki C, Peitzsch M et al (2014) Biochemical diagnosis of phaeochromocytoma using plasma-free normetanephrine, metanephrine and methoxytyramine: importance of supine sampling under fasting conditions. Clin Endocrinol 80(4):478–486. doi:10.1111/cen.12327
Eisenhofer G, Peitzsch M (2014) Laboratory evaluation of pheochromocytoma and paraganglioma. Clin Chem 60(12):1486–1499. doi:10.1373/clinchem.2014.224832
Eisenhofer G, Lenders JW, Timmers H et al (2011) Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem 57(3):411–420. doi:clinchem.2010.153320 [pii]. doi:10.1373/clinchem.2010.153320
Eisenhofer G, Lenders JW, Goldstein DS et al (2005) Pheochromocytoma catecholamine phenotypes and prediction of tumor size and location by use of plasma free metanephrines. Clin Chem 51(4):735–744
Amar L, Eisenhofer G (2015) Diagnosing phaeochromocytoma/paraganglioma in a patient presenting with critical illness: biochemistry versus imaging. Clin Endocrinol 83(3):298–302. doi:10.1111/cen.12745
IliasI, SahdevA, ReznekRHet al (2007) The optimal imaging of adrenal tumours: a comparison of different methods. Endocr Relat Cancer 14 (3):587-599. doi:14/3/587[pii]10.1677/ERC-07-0045
Sahdev A, Sohaib A, Monson JP et al (2005) CT and MR imaging of unusual locations of extra-adrenal paragangliomas (pheochromocytomas). Eur Radiol 15(1):85–92
Leung K, Stamm M, Raja A, Low G (2013) Pheochromocytoma: the range of appearances on ultrasound, CT, MRI, and functional imaging. AJR Am J Roentgenol 200(2):370–378. doi:10.2214/AJR.12.9126
Pacak K, Eisenhofer G, Goldstein DS (2004) Functional imaging of endocrine tumors: role of positron emission tomography. Endocr Rev 25(4):568–580
Taieb D, Timmers HJ, Hindie E et al (2012) EANM 2012 guidelines for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging 39(12):1977–1995. doi:10.1007/s00259-012-2215-8
Wiseman GA, Pacak K, O’Dorisio MS et al (2009) Usefulness of 123I-MIBG scintigraphy in the evaluation of patients with known or suspected primary or metastatic pheochromocytoma or paraganglioma: results from a prospective multicenter trial. J Nucl Med 50(9):1448–1454. doi:jnumed.108.058701 [pii]. doi:10.2967/jnumed.108.058701
Fiebrich HB, Brouwers AH, Kerstens MN et al (2009) 6-[F-18]Fluoro-L-dihydroxyphenylalanine positron emission tomography is superior to conventional imaging with (123)I-metaiodobenzylguanidine scintigraphy, computer tomography, and magnetic resonance imaging in localizing tumors causing catecholamine excess. J Clin Endocrinol Metab 94(10):3922–3930. doi:10.1210/jc.2009-1054
Timmers HJ, Chen CC, Carrasquillo JA et al (2012) Staging and functional characterization of pheochromocytoma and paraganglioma by 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography. J Natl Canc Inst 104(9):700–708. doi:10.1093/jnci/djs188
Maurice JB, Troke R, Win Z et al (2012) A comparison of the performance of (6)(8)Ga-DOTATATE PET/CT and (1)(2)(3)I-MIBG SPECT in the diagnosis and follow-up of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging 39(8):1266–1270. doi:10.1007/s00259-012-2119-7
Janssen I, Blanchet EM, Adams K et al (2015) Superiority of [68Ga]-DOTATATE PET/CT to other functional imaging modalities in the localization of SDHB-associated metastatic Pheochromocytoma and Paraganglioma. Clin Cancer Res 21(17):3888–3895. doi:10.1158/1078-0432.CCR-14-2751
Janssen I, Chen CC, Taieb D et al (2016) 68Ga-DOTATATE PET/CT in the localization of head and neck Paragangliomas compared with other functional imaging modalities and CT/MRI. J Nucl Med 57(2):186–191. doi:10.2967/jnumed.115.161018
Amar L, Fassnacht M, Gimenez-Roqueplo AP et al (2012) Long-term postoperative follow-up in patients with apparently benign pheochromocytoma and paraganglioma. Hom Metab Res 44(5):385–389
Pacak K, Eisenhofer G, Ahlman H et al (2007) Pheochromocytoma: recommendations for clinical practice from the first international symposium. Nat Clin Pract Endocrinol Metab 3(2):92–102
Weingarten TN, Cata JP, O’Hara JF et al (2010) Comparison of two preoperative medical management strategies for laparoscopic resection of pheochromocytoma. Urology 76(2):508e506–511. doi: 10.1016/j.urology.2010.03.032
Steinsapir J, Carr AA, Prisant LM, Bransome ED Jr (1997) Metyrosine and pheochromocytoma. Arch Int Med 157(8):901–906
Walz MK, Alesina PF, Wenger FA et al (2006) Laparoscopic and retroperitoneoscopic treatment of pheochromocytomas and retroperitoneal paragangliomas: results of 161 tumors in 126 patients. World J Surg 30(5):899–908. doi:10.1007/s00268-005-0373-6
Goers TA, Abdo M, Moley JF et al (2013) Outcomes of resection of extra-adrenal pheochromocytomas/paragangliomas in the laparoscopic era: a comparison with adrenal pheochromocytoma. Surg Endosc 27(2):428–433. doi:10.1007/s00464-012-2451-9
Shen WT, Grogan R, Vriens M et al (2010) One hundred two patients with pheochromocytoma treated at a single institution since the introduction of laparoscopic adrenalectomy. Arch Surg 145(9):893–897. doi:10.1001/archsurg.2010.159
Agarwal G, Sadacharan D, Aggarwal V et al (2012) Surgical management of organ-contained unilateral pheochromocytoma: comparative outcomes of laparoscopic and conventional open surgical procedures in a large single-institution series. Langenbeck’s Arch Surg 397(7):1109–1116. doi:10.1007/s00423-011-0879-3
Asari R, Scheuba C, Kaczirek K, Niederle B (2006) Estimated risk of pheochromocytoma recurrence after adrenal-sparing surgery in patients with multiple endocrine neoplasia type 2A. Arch Surg 141(12):1199–1205.; discussion 1205. doi:10.1001/archsurg.141.12.1199
Benhammou JN, Boris RS, Pacak K et al (2010) Functional and oncologic outcomes of partial adrenalectomy for pheochromocytoma in patients with von Hippel-Lindau syndrome after at least 5 years of followup. J Urol 184(5):1855–1859. doi:10.1016/j.juro.2010.06.102
Grubbs EG, Rich TA, Ng C et al (2013) Long-term outcomes of surgical treatment for hereditary pheochromocytoma. J Am Coll Surg 216(2):280–289. doi:10.1016/j.jamcollsurg.2012.10.012
Plouin PF, Amar L, Dekkers OM et al (2016) European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol 174(5):G1–G10. doi:10.1530/EJE-16-0033
Amar L, Lussey-Lepoutre C, Lenders J et al (2016) Management of endocrine disease: recurrence or new tumors after complete resection of phaeochromocytomas and paragangliomas. A systematic review and meta-analysis. Eur J Endocrinol. 175(4):R135–R145. doi: 10.1530/EJE-16-0189
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter
Cite this chapter
Eisenhofer, G., Lenders, J.W.M. (2018). Paroxysmal Hypertension: Pheochromocytoma. In: Berbari, A., Mancia, G. (eds) Disorders of Blood Pressure Regulation. Updates in Hypertension and Cardiovascular Protection. Springer, Cham. https://doi.org/10.1007/978-3-319-59918-2_31
Download citation
DOI: https://doi.org/10.1007/978-3-319-59918-2_31
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-59917-5
Online ISBN: 978-3-319-59918-2
eBook Packages: MedicineMedicine (R0)