
Abstract
The role of mitochondrial membranes in oxidative phosphorylation, energy production, calcium signaling, and mitochondrial metabolism is well established, especially with regard to lipid and steroid homeostasis, communication with other organelles, apoptosis, and human disease. Here we address the complementary role of the mitochondrial outer membrane (MOM) as a platform for cytosolic proteins to function as potent regulators of the MOM permeability through their direct functional interaction with the voltage-dependent anion channel (VDAC), the major MOM channel. Two abundant cytosolic proteins, dimeric tubulin and α-synuclein (α-syn), modulate fluxes of ATP/ADP and other water-soluble mitochondrial metabolites through VDAC. Their interaction with VDAC is a multistep process in which the first step is the lipid-sensitive binding to the MOM and the second is the voltage-dependent blockage of the VDAC pore. Both proteins induce characteristic rapid reversible blockages of VDAC reconstituted into planar lipid membranes with nanomolar efficiency and are thus able to effectively regulate ATP/ADP fluxes through VDAC in a lipid-dependent manner. A proposed general model of VDAC inhibition explains satisfactorily how two very different and otherwise unrelated proteins induce qualitatively similar blockages of reconstituted VDAC. In in vitro biophysical experiments, tubulin and α-syn display similar lipid-dependent membrane-binding properties, which make these proteins well suited for targeting the MOM and strongly enhance the availability of these molecules for the voltage-sensitive interaction with VDAC. As the physiological significance of the association of tubulin and α-syn with mitochondrial membranes in cells is only beginning to be appreciated, we propose a new role of mitochondrial lipids in the control of MOM permeability. Overall, the emerging data indicate that the physiological conditions in the cell, such as the expression level of α-syn, the ratio between dimeric and polymerized tubulin in the cytosol, cytosolic pH, MOM lipid composition, level of lipid peroxidation, and potential across the MOM, strongly determine the efficiency with which both cytosolic proteins regulate normal metabolite exchange through VDAC or cause mitochondrial dysfunction.
This chapter was created within the capacity of an US governmental employment. US copyright protection does not apply.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Voltage-Dependent Anion Channel
- VDAC
- Tubulin
- α-Synuclein
- Synucleinopathies
- Peripheral proteins
- Lipid-protein interaction
1 Introduction
Mitochondrial functions extend far beyond energy production in the form of ATP. Mitochondria play a crucial role in Ca2+ homeostasis, in steroid biosynthesis, and in programmed cell death and as a source of the endogenous toxic reactive oxygen species (ROS) in the cell. The involvement of mitochondrial membranes in all these processes is well recognized, especially with regard to lipid synthesis and trafficking or communication with other organelles. The two mitochondrial membranes, the mitochondrial inner membrane (MIM) and the mitochondrial outer membrane (MOM) , play rather different roles in mitochondrial function, which are intimately related to their different lipid compositions. The MIM’s signature is a high cardiolipin (CL) content (up to 20%), while the MOM has high cholesterol content (up to 10%) (de Kroon et al. 1997; Flis and Daum 2013). The MIM’s prominent roles in oxidative phosphorylation, energy production, Ca2+ signaling, and mitochondrial metabolism are well established. By contrast, the role of the MOM was previously thought to be the simple confinement of proteins and metabolites in the crowded intermembrane space. This viewpoint changed drastically in the mid-1990s with the discovery of the MOM permeabilization (MOMP) process at the early stage of apoptosis. Since then, the role of the MOM as a platform for apoptosis execution by Bcl-2 family proteins has been well documented (e.g., see Chipuk et al. 2006). The involvement of MOM lipids in formation and promotion of supramolecular openings induced by the pro-apoptotic proteins Bax, Bak, Bid, and others has been demonstrated (Basanez et al. 2002; Kuwana et al. 2002; Terrones et al. 2004; Yethon et al. 2003), leading to the proposal that together with the MOM lipids, the pro-apoptotic Bax/Bak oligomeric complex is directly involved in the formation of the so-called lipidic pores in the MOM (Landeta et al. 2011; Schafer et al. 2009; Terrones et al. 2004). Furthermore, lipid ceramide was itself shown to form large, cytochrome c-permeable pores in the MOM and thus be sufficient to induce MOMP and promote apoptosis (Colombini 2010; Siskind et al. 2002). The unique MIM phospholipid CL is associated with all of the major players in oxidative phosphorylation. Due to its characteristic nonlamellar shape, with four poly-unsaturated acyl chains, CL also plays a pivotal role in maintaining the curvature of the highly bent cristae. In addition to its structural and protein-lipid interaction functions in the MIM, CL is essential for anchoring pro-apoptotic tBid and thus promoting MOMP (Gonzalvez et al. 2005; Shamas-Din et al. 2015). The formation of the apoptotic “activation platform” composed of CL, caspase-8, and full-length Bid at the mitochondrial contact sites has been suggested (Gonzalvez et al. 2008), directly connecting mitochondrial lipids with Barth syndrome, a chromosome X-linked cardioskeletal myopathy and neutropenia that is often fatal in infancy and early childhood due to heart failure and bacterial infections (Schlame and Ren 2006). This is just one of numerous cases in which mitochondrial lipids are involved in human diseases.
The few examples mentioned above represent the conventional understanding of the functional and structural roles of the MOM lipids in the mitochondrial apoptotic pathway. In the current review, we discuss a different, complementary view of the MOM as a platform for cytosolic proteins to regulate MOM permeability by their direct functional interaction with the voltage-dependent anion channel (VDAC). We focus particularly on the role of the mitochondrial lipids in this regulation.
2 VDAC Voltage-Gating and Regulation of ATP Flux
As the major channel and most abundant protein in the MOM, VDAC is responsible for most of the metabolite flux in and out of mitochondria. VDAC was shown to be involved in a wide variety of mitochondria-associated pathologies, from various forms of cancer to neurodegeneration (Shoshan-Barmatz et al. 2010). It is therefore not surprising that VDAC has emerged as a promising pharmacological target (Shoshan-Barmatz and Ben-Hail 2012). The uniqueness of this large, passive, weakly selective β-barrel channel arises mainly from its location at the interface between the mitochondria and the cytosol (Colombini 2004), where it serves as a pathway for all mitochondrial water-soluble respiratory substrates, such as ATP, ADP, and small ions (Colombini et al. 1996; DeHart et al. 2014; Rostovtseva et al. 2005). By contrast, there is a large variety of highly specified carriers and exchangers to translocate these substrates across the MIM. The weak anionic selectivity of the VDAC pore favors transport of negatively charged mitochondrial metabolites (Colombini et al. 1996). It is generally believed that VDAC regulates metabolite fluxes using its conserved ability to “gate” or adopt different conducting and, crucially, ion-selective states (Colombini 2004; Hodge and Colombini 1997; Lemasters and Holmuhamedov 2006; Lemasters et al. 2012; Rostovtseva and Colombini 1996). However, it is not yet known with certainty whether VDAC gating occurs in vivo, since all electrophysiological data on VDAC function have thus far been obtained using an in vitro system of VDAC reconstituted into planar lipid bilayers, where gating is induced by the applied voltage (Colombini 1989).
One major dilemma is that the very existence of a significant potential across MOM in vivo is uncertain. Conventional thinking holds that this potential is essentially zero due to the high abundance of VDAC in the MOM (e.g., see discussions in Colombini 2004; Lemeshko 2006; Rostovtseva and Bezrukov 2012 and Chap. 9 of the current volume). In this view, the main source of the potential across the MOM is the so-called Donnan potential , which arises from the large difference in the concentrations of VDAC-impermeable charged macromolecules (e.g., the 12-kDa cytochrome c protein, which carries nine positive charges) in the mitochondrial intermembrane space and in the cytosol (Colombini 2004). The few available estimates of the MOM transmembrane potential range from 10 mV (Lemeshko 2006) to as high as 46 mV (Porcelli et al. 2005) and even higher (Lemeshko 2014a, b, 2016). Recent work suggests that other biochemical processes can enhance the Donnan potential considerably. Lemeshko (2014a, b, 2016 and Chap. 9 of the current volume) proposes that VDAC complexes with hexokinases at the cytosolic side and/or with mitochondrial creatine kinase in the intermembrane space could play the role of “batteries,” generating a potential across the MOM as high as 50 mV. In this picture, the Gibbs free energy of kinase reactions is the driving force. Importantly, the cytoplasmic side of the MOM is negative relative to the potential of the intermembrane space. Regardless of the source and magnitude of the MOM potential in vivo, it has been experimentally demonstrated using reconstituted VDAC that ATP passes readily through the open, weakly anion-selective VDAC state, but essentially does not pass through its low-conducting, weakly cation-selective states induced by the applied potential in the voltage-clamp mode (Rostovtseva and Colombini 1996, 1997). Furthermore, using current noise analysis on a single VDAC channel in the presence of relatively high molecular weight negatively charged metabolites, it was found that VDAC could discriminate between different adenine mono- and dinucleotides and also between similarly charged molecules, such as ATP and UTP (Rostovtseva et al. 2002a, b). These findings suggest that VDAC has been evolutionary selected to regulate fluxes of metabolites by dynamically adapting the electrostatic environment of its pore in favor of adenine nucleotides (Noskov et al. 2016). It is then natural to expect the existence of endogenous cytosolic (and perhaps mitochondrial also) proteins that are able to efficiently regulate VDAC permeability for the major mitochondrial metabolites, especially ATP and ADP.
It was only a matter of time before such endogenous VDAC regulators were found. Two apparently unrelated cytosolic proteins, dimeric tubulin and α-synuclein (α-syn), were discovered to reversibly block reconstituted VDAC with nanomolar (nmol/l) efficiency and thereby control the fluxes of ATP, ADP, and other metabolites through VDAC (Rostovtseva et al. 2008, 2015). Notably, while both α-syn and tubulin are abundant cytosolic proteins with other functions (well known for tubulin, but not as clear for α-syn, as discussed below), they appear to double as membrane-associated proteins, displaying high affinity to mitochondrial membranes. In this second role, they are potent regulators of VDAC.
3 VDAC Regulation by Tubulin and α-Synuclein
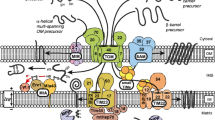
Tubulin is an abundant cytosolic protein known primarily for its role as the building block of microtubules. It is a heterodimer with α- and β-subunits forming an acidic, water-soluble, compactly folded 110-kDa protein with a well-defined crystal structure (Nogales et al. 1998). Each subunit has an unstructured C-terminal tail (CTT) composed of 11–20 amino acids (Westermann and Weber 2003) exposed at the protein surface (Fig. 8.1a). Both α- and β-tubulin CTTs are highly negatively charged and are essentially poly-Glu peptides. In microtubules, dimeric tubulin is assembled in such a way that the exposed, growing end of the microtubule is always the β-subunit. As a result, microtubule-targeting agents (MTAs) used in chemotherapy bind exclusively to the β-subunit (Field et al. 2014). Until recently, any physiological role of unassembled, cytosol-solubilized dimeric tubulin, other than as a supply for polymerization into microtubules, remained unknown.

Both VDAC blockers, the α/β-tubulin heterodimer and α-synuclein, possess extended disordered anionic C-terminal tails. Tubulin structure is adapted from (Nogales et al. 1998)
α-Syn, by contrast, is a small, 14-kDa, 140-amino acid, intrinsically disordered protein (Fig. 8.1b) highly expressed in the central nervous system and constituting up to 1% of total cytosolic protein in normal brain cells (Kruger et al. 2000). It is the major component of the Lewy bodies characteristically observed in the brains of Parkinson’s disease (PD) patients (Spillantini et al. 1997). Based on the observation that Lewy bodies consist primarily of fibrillary α-syn, most studies have focused on the pathological role of α-syn aggregates. However, the precise role of α-syn in normal brain cells and in α-synucleinopathies , particularly in its monomeric form, remains largely mysterious.
What do these two genetically, structurally, and physiologically unrelated proteins have in common? In this review, we discuss the following similarities: both proteins (1) possess a disordered highly negative charged C-terminus; (2) induce characteristic rapid reversible blockages of VDAC reconstituted into planar lipid membranes with nanomolar efficiency; (3) are “amphitropic” proteins, i.e., water-soluble proteins that are reversibly associated with cellular membranes under certain physiological conditions; (4) specifically bind to mitochondrial membranes in live cells; and (5) modulate mitochondrial metabolism and respiration by regulating VDAC permeability in a lipid-dependent manner.
3.1 Regulation of VDAC Permeability by Dimeric Tubulin and Monomeric α-Synuclein
In these experiments, a single VDAC channel spans a planar bilayer lipid membrane that separates and electrically isolates two buffer-filled compartments (Rostovtseva and Bezrukov 2015). The ionic current through the VDAC channel is generated by an applied transmembrane potential and is monitored for changes induced by the addition of dimeric tubulin or α-syn. Single-channel recordings of VDAC reconstituted into planar lipid bilayers yield strikingly similar blockage events induced by addition of dimeric tubulin and α-syn to the membrane-bathing buffer solution at nanomolar concentrations (Fig. 8.2a, b). Two representative experiments in which 50 nM tubulin or α-syn added to one side of the membrane induce fast time-resolved transitions (insets at a finer time scale) between VDAC’s high-conductance open state and low-conductance blocked state are presented in Fig. 8.2a, b. Without these inhibitors, the VDAC conductance at low applied potentials (<30 mV) is very stable and essentially lacks transitions to the low-conductance state (left traces in Fig. 8.2a, b). The channel can maintain the open state for up to a few hours at low potentials under the experimental conditions represented by Fig. 8.2a, b. The conductance of the open state in the presence of either inhibitor remains the same as in the control, and the conductance of the blocked state is ~0.4 of the open-state conductance. This is in striking contrast to the typical VDAC voltage-induced gating, which is characterized by long (10–100 s) closures to multiple low-conductance (“closed”) states (Fig. 8.2c).

Dimeric tubulin (a) and α-synuclein (b) induce fast, reversible, partial blockages of VDAC reconstituted into planar lipid bilayers, with kinetics that are dramatically different from those of VDAC closure induced by applied voltage (voltage gating) (c). (a, b) Representative current records of single VDAC channels before and after addition of 50 nM tubulin (a) or α-syn (b) to one side of the membrane at −25 mV applied voltage (potential is more negative at the side of tubulin or α-syn addition). The channel conductance fluctuates between the high-conducting, “open,” and low-conducting, “blocked”, state with well time-resolved blockage events shown in insets at the finer time scales. Long-dashed lines indicate the zero-current level, and short-dashed lines show the open and blocked states. (c) Typical VDAC voltage-induced gating under −50 mV applied voltage. Under the applied potential, the channel conductance moves from a single open state (dash-dot-dot lines) to various low-conducting or “closed” states (dotted lines). The medium was 1 M KCl buffered with 5 mM HEPES at pH 7.4. VDAC was isolated from mitochondria of N. crassa (a, c) or rat liver (b)
Both tubulin and α-syn induce VDAC blockage in a concentration-dependent manner. The number of blockage events increases with the blocker protein’s bulk concentration (Rostovtseva et al. 2012, 2015). The concentration dependence of the blockage on-rate is described by a simple ligand-binding equation with K d in the 10–50 nM range for both inhibitors, depending on the experimental conditions such as the membrane lipid composition or pH. At low (<50 nM) tubulin or α-syn concentrations, the rates of interaction with VDAC can, to a first approximation, be described by a first-order reaction, where the rate of blockage events (the “on-rate”) linearly increases with blocker concentration, while the characteristic duration of the blockages (the inverse “off-rate”) is concentration independent.
3.2 A General Model of VDAC Blockage, Requiring a Channel Inhibitor to Have an Anionic Disordered Domain and Lipid-Binding Domain
Figure 8.1 illustrates that the existence of a highly negatively charged unstructured CTT is a common feature of tubulin and α-syn. Both tubulin and α-syn block VDAC measurably only when a negative potential is applied from the side of protein addition (Rostovtseva and Bezrukov 2015; Rostovtseva et al. 2015). When the sign of the potential is reversed, no blockage events are observed, and the channel open-state conductance is as steady as in a control experiment without protein addition (see Fig. 1.5 in (Rostovtseva and Bezrukov 2015). This observation, and the fact that tubulin with proteolytically cleaved CTTs does not induce characteristic VDAC blockages even at micromolar concentrations (Rostovtseva et al. 2008), suggest that the negatively charged CTTs are responsible for the channel blockage by tubulin. On the other hand, synthetic peptides of α- and β-tubulin CTTs do not measurably block the channel, up to 10 μM concentration (Rostovtseva et al. 2008). Experiments with a peptide consisting of the 45 C-terminal amino acids of α-syn likewise did not reveal interaction with VDAC (Rostovtseva et al. 2015). Altogether, these observations suggest that while the disordered acidic C-terminal peptide of either tubulin or α-syn is required for VDAC blockage, it should be attached to an “anchor” that holds it at the channel entrance (Fig. 8.3), preventing free translocation through the pore, which is expected to occur at time scales too fast to be experimentally observed. Tubulin’s bulky body is about twice as large as the VDAC pore entrance and cannot translocate; in the case of α-syn, its membrane-binding domain is responsible for this anchoring function . In both cases, the increased applied negative voltage keeps the anionic CTT inside the positively charged pore longer (Fig. 8.4c, d). Indeed, experiments show that the dwell time of tubulin- and α-syn-induced blockages is highly voltage dependent and increases exponentially with the applied voltage (Fig. 8.4c) (Rostovtseva et al. 2012). The steep voltage dependence of the open probability yields the effective gating charge of 10–13 elementary charges depending on the salt concentration of the membrane-bathing buffer (Gurnev et al. 2012). Such a high gating charge is characteristic of the most sensitive voltage-gated channels of neurophysiology to date (Swartz 2008).

Models of VDAC blockage by tubulin (a) and α-syn (b), in which the negatively charged C-terminal tails of tubulin or α-syn partially block the channel by entering the net positive VDAC pore in its open state under negative applied potentials. (a) Hypothetical structural models of dimeric tubulin binding to the membrane. (a) For tubulin bound by the α-subunit, both CTTs of the α- and β-subunits can reach the VDAC pore, while the vinblastine-binding site remains exposed. (b) For tubulin bound to the membrane by the β-subunit, only the β-subunit CTT is able to block the channel, while the vinblastine-binding site is inaccessible. Intermediate binding configurations, including the case where both subunits are responsible for membrane binding, are also possible and would in general present both CTTs to the VDAC pore (not shown). (b) Structural model of membrane-bound α-syn with its α-helical N-terminal domain bound to the membrane and the acidic disordered CTT blocking the VDAC pore. The VDAC β-barrel (3D model of mouse VDAC1 is adapted from Ujwal et al. (2008)) is shown embedded into lipid bilayer)

VDAC blockages by tubulin or α-syn are highly voltage dependent. (a, b) Log-binned distribution of the time of blockage events induced by tubulin (a) or α-syn (b) obtained from statistical analysis of current records such as those shown in Fig. 8.2a, b. Distributions of the times Fig. 8.4 (continued) spent by the channel in the tubulin-blocked state, τ b, require at least two exponents for fitting, with characteristic times τ (1) off and τ (2) off shown by the solid line (a). The distribution of blockage events induced by α-syn, on the other hand, is satisfactorily described by single exponential fitting (solid line) (b). Applied voltages were −25 mV (a) and −35 mV (b). (c) Both characteristic tubulin-induced blockage times exponentially depend on the applied voltage and do not depend on the membrane lipid composition (indicated in the graph). (d) Voltage dependence of the blockage time induced by α-syn has a biphasic character. (e) The turnover potential V* separates the blocking regime, where the absolute applied potential |V| ≤ V*, from the translocation regime, where |V| ≥ V*. At relatively low voltages, when the CTT (red) is captured in the pore, increasing the electrical field keeps it inside the pore longer, while the membrane-bound N-terminal domain (green) prevents translocation of the molecule. This results in reversible capture of the CTT and an exponential increase in the characteristic blockage time τ off. Voltages higher than V* detach the N-terminal domain from the membrane surface and allow the whole molecule to translocate through the channel, resulting in a decrease of τ off (Adapted from Rostovtseva et al. (2015))
Additional evidence that the tubulin CTT partitions into the VDAC pore was obtained in experiments designed to probe the tubulin-blocked state using different approaches (Gurnev et al. 2011) in combination with molecular dynamics (MD) simulations (Noskov et al. 2013). It was shown that the small-ion selectivity switches from anionic to cationic when the channel moves from the open state to the tubulin-blocked state (Gurnev et al. 2011). These results indicate that the highly negatively charged tubulin CTT, by entering a net positive pore, reverses the net charge of the pore interior. By measuring nonelectrolyte polymer partitioning into the ion channel, it was found that the dimensions of the blocked pore are significantly smaller than those of the open state (Gurnev et al. 2011). MD simulations of the α-tubulin-VDAC1 complex demonstrate that in the presence of the unstructured CTT of α-tubulin in the VDAC1 pore, the pore conductance decreases by about 60% and switches its selectivity from anion- to cation-preferring channels (Noskov et al. 2013), thus confirming electrophysiological data obtained on reconstituted VDAC. The negatively charged C-terminus of the bound α-tubulin molecule is complemented by the VDAC pore providing matching basic residues that form stable salt bridges involving Arg15, Lys20, Lys12, and Lys32 of VDAC (Noskov et al. 2013). Finally, and most importantly, channel experiments and MD simulations demonstrate that ATP is excluded from the tubulin-blocked state (Gurnev et al. 2011; Noskov et al. 2013). Altogether, these results provide strong evidence that tubulin is a potent regulator of VDAC, which could efficiently modulate fluxes of nucleotides through the channel and thus control mitochondria respiration. Experiments with isolated mitochondria (Monge et al. 2008; Rostovtseva et al. 2008) and human hepatoma HepG2 cells (Maldonado et al. 2011, 2013) confirm that the VDAC-tubulin interaction is functionally important for regulating mitochondrial respiration .
For intrinsically disordered α-syn, where the whole molecule can, in principle, translocate through the channel because a single polypeptide strand fits comfortably into the ~2.5 to 2.7 nm diameter VDAC pore, the relationship between dwell times and applied voltages becomes more complex than for tubulin. In contrast to the tubulin-induced blockages , the dwell time of α-syn-induced blockages exhibits a biphasic dependence on the applied potential (Rostovtseva et al. 2015) (Fig. 8.4d). At voltages lower than a certain value, the so-called turnover potential, (indicated as V* in Fig. 8.4e) dwell time increases with voltage in a manner characteristic for the blockage regime (Hoogerheide et al. 2016). At voltages higher than the turnover potential, the dwell time decreases with voltage, signifying a predominance of translocation (Fig. 8.4e). Based on these data, a model of α-syn interaction with VDAC was proposed, where the anionic C-terminus partially blocks the positively charged VDAC pore and the membrane-bound N-terminus holds it at the membrane surface preventing complete translocation through the channel at voltages smaller than 30 mV (Fig. 8.4e) (Rostovtseva et al. 2015). Therefore, under certain conditions, such as the applied potential, lipid composition, and ionic strength of buffer solution, α-syn can either block VDAC in qualitatively similar manner to tubulin or translocate through the channel (Fig. 8.4e). The latter process, if confirmed by future experiments in live cells, implies that VDAC serves as a pathway for α-syn into the mitochondrial space, where it could directly target electron transport chain complexes of the inner membrane as in vivo experiments suggest (Devi et al. 2008; Elkon et al. 2002; Ellis et al. 2005; Liu et al. 2009; Luth et al. 2014) as well as other intermembrane space proteins.
Importantly, the proposed model of VDAC inhibition (Fig. 8.3) is quite general and does not require any specific interaction between the VDAC pore and inhibitor . This model explains satisfactorily how two such different proteins as tubulin and α-syn induce qualitatively similar blockages of VDAC and suggests that other cytosolic proteins may also be involved. Generalizing based on these two examples, the architecture of a VDAC inhibitor should fulfil two main requirements: have a negatively charged disordered C- or N-terminus and a membrane-binding domain. Then, it is natural to expect that the membrane lipid composition is an important factor in interaction of both tubulin and α-syn with reconstituted VDAC.
4 Tubulin and α-Synuclein Interactions with VDAC Strongly Depend on Membrane Lipid Composition
4.1 Effect of Membrane Lipid Composition on VDAC-Tubulin Interaction
The characteristic on-rate constant, k on, of VDAC-tubulin binding depends on both the hydrophobic and polar parts of the phospholipid (Fig. 8.5a–c). The effect of lipid charge is observed primarily at the physiologically low salt concentrations: addition of the negatively charged diphytanoyl-phosphatidylserine (DPhPS) to the neutral diphytanoyl-phosphatidylcholine (DPhPC) (PS:PC = 4:1) resulted in ~200 times decrease of k on in 0.1 M KCl (Fig. 8.5a) (Rostovtseva et al. 2012), which may suggest that the acidic regions of the tubulin globule may be sufficiently close to the negatively charged membrane surface to be electrostatically repelled. At a high salt concentration of 1.5 or 1 M KCl, the presence of DPhPS does not affect the on-rate (Fig. 8.5a). Salt concentration affects the on-rate of the blockage in both neutral and charged membranes: in 0.1 M KCl, the k on was ~5 times higher than in 1.5 M KCl for the neutral DPhPC membranes but ~50 times lower than in 1.5 M KCl in the negatively charged DPhPC/DPhPS membranes (Fig. 8.5a). The type of lipid hydrocarbon acyl chains also significantly affects the blockage kinetics: when oleoyl chains in DOPC have been replaced with phytanoyl in DPhPC, the on-rate of tubulin blockage increased ~70 times (Fig. 8.5b). Surprisingly, the on-rates appeared to be consistently different when planar bilayers were formed from the same lipid composition but using different organic solvents, such as hexadecane or petroleum jelly (Rostovtseva et al. 2012). A comparison of the k on values obtained in DPhPC membranes, formed after pretreatment of the orifice across which a planar membrane is made from two opposing lipid monolayers (Rostovtseva and Bezrukov 2015), with hexadecane (Fig. 8.5a) (see details in Bezrukov and Vodyanoy 1993) or petroleum jelly (Fig. 8.5b) (see details in Schein et al. 1976) is shown in Fig. 8.5a, b. These data indicate that tubulin-VDAC interaction is sensitive to the hydrophobic core of planar lipid bilayer, thus pointing out to a presence of the non-electrostatic component in this interaction. Additional evidence of the involvement of a hydrophobic component in tubulin-VDAC binding is that k on increases gradually with the DOPE content of DOPC membranes and is ~200 times higher in a pure DOPE membrane than in a pure DOPC membrane (Fig. 8.5c) (Rostovtseva et al. 2012). These observations point to tubulin binding to the membrane as an important requirement for VDAC blockage , in which the on-rate depends on the surface concentration of tubulin.

Effect of lipid composition on the on-rate constants of VDAC blockage by tubulin. (a) Effect of charged lipids on the on-rate constant of VDAC blockage by tubulin depends on salt concentration. Membranes were formed from pure DPhPC or a DPhPC/DPhPS (1:4) mixture in 0.1 or 1.5 M KCl buffered with 5 mM HEPES at pH 7.4. (b) Acyl chain composition affects the k on of VDAC blockage. Membranes were formed from pure DPhPC and DPhPC/DPhPS (1:1), from pure DOPC and DOPC/CL (1:0.08), and from PLE with and without cholesterol (10:1). (c) The k on of blockages induced by tubulin increases with PE content in DOPC/DOPE mixture. The data were obtained at –25 mV of applied voltage. Medium consisted of 1 M KCL and 5 mM HEPES, pH 7.4 (b, c). VDAC was isolated from N. crassa mitochondria
The natural lipid composition of the rat liver MOM is a mixture of ~44 and 35% of phosphatidylcholine (PC) and phosphatidylethanolamine (PE), respectively, with ~20% of charged lipids consisting primarily of ~14% CL and ~5% of phosphatidylinositol (PI) (de Kroon et al. 1997) with cholesterol (~10% of total MOM lipid). The soybean polar lipid extract (PLE) closely resembles rat liver MOM lipid composition and therefore has been tested in tubulin-VDAC experiments (Rostovtseva et al. 2012). In PLE membranes, tubulin blocked VDAC even less effectively than in DOPC (Fig. 8.5b), perhaps due to the presence of ~25% of the negatively charged PI and PA lipids in the mixture. Addition of 10% of cholesterol to the PLE mixture did not affect the on-rate constant significantly (Fig. 8.5b). CL also did not change VDAC-tubulin binding when added to the DOPC membranes in 1 M KCl (Fig. 8.5b).
For all studied lipid compositions, the off-rate of VDAC blockage by tubulin was not affected (Fig. 8.4c) (Rostovtseva et al. 2012). This is consistent with the picture of VDAC-tubulin binding as a first-order reaction in which the on-rate is concentration dependent and the off-rate is not. In this view, the on-rate depends on the effective concentration of tubulin close to the channel entrance and thus on tubulin binding to the membrane surface, which in turn depends on membrane lipid composition (see Sect. 8.5.2 below). The role of lipid composition, particularly charged headgroups, on the concentration-independent off-rate of capture of the tubulin CTT into the VDAC pore has not yet been determined. When the tubulin CTT enters the VDAC pore, the strength of the binding, in terms of its residence time in the pore, does not depend on the lipid composition. Thus, at a constant applied voltage the equilibrium of VDAC-tubulin binding is predominantly defined by the on-rate. Consistent with the proposed lipid-dependent step of VDAC blockage by tubulin, we can suggest that any modifications of tubulin association with mitochondrial membrane induced by substantial lipid homeostasis in mitochondria in vivo would result in the different on-rates of VDAC-tubulin binding and consequently in different VDAC permeability to ATP or ADP.
4.2 Effect of Membrane Lipid Composition on VDAC-α-syn Interaction
When α-syn blocks VDAC (Fig. 8.2b), the lipid membrane composition affects the on-rates of VDAC blockage in a manner qualitatively similar to what was shown for tubulin. Using a lipid mixture mimicking the MOM lipid composition, it was found that the on-rate of α-syn-induced VDAC blockage is higher in non-lamellar DOPE than in lamellar DOPC (Jacobs et al. 2016). In particular, the on-rate increases up to tenfold with the increase of PE content in PC membranes. Remarkably, the off-rate at high transmembrane potentials was also lipid dependent, as seen by a 5 mV increase in the “turnover potential” which separates regimes of blockage and translocation with the increase of PE content (Jacobs et al. 2016). This lipid sensitivity supports a model shown in Fig. 8.3b in which the binding of α-syn’s N-terminal domain to the membrane is followed by C-terminal blockage of the VDAC pore governed by transmembrane potential (Rostovtseva et al. 2015). Thus, similar to tubulin blockage of VDAC, the lipid dependence of α-syn-VDAC binding can be primarily attributed to the lipid sensitivity of α-syn molecule binding to the membrane, a phenomenon that has been demonstrated by various in vitro biophysical experiments (see Sect. 8.5.1 below).
At physiologically low salt concentrations of 150 mM KCl, the on-rates increase more than tenfold compared to experiments performed in high salt of 1 M KCl, while translocation of α-syn through VDAC is impeded (Jacobs et al. 2016). The effect of ionic strength on the on-rate of VDAC blockages observed in neutral membranes made of DOPE/DOPC mixture suggests that both electrostatic and hydrophobic components of α-syn-membrane association are involved in α-syn-VDAC binding, thus confirming the previously suggested participation of electrostatic forces in α-syn-membrane binding (Davidson et al. 1998).
It must be noted that despite the striking phenomenological similarity between VDAC blockages by α-syn and tubulin, there are a number of significant quantitative and qualitative differences between the two. For instance, the presence of PE has ~10 times higher effect on the on-rate of tubulin-VDAC binding than on α-syn-induced blockage; the characteristic time of blockage by α-syn is at least ten times smaller than the shortest one for tubulin, τ (1) off (compare Fig. 8.4c with Fig. 8.4d); anionic lipids significantly increase the on-rate of VDAC blockage by α-syn in low and high salts but not by tubulin (Fig.8.5a, b).
Overall, these findings provide an example of lipid-controlled protein-protein interactions where the choice of lipid species is able to change the equilibrium binding constant by orders of magnitude. They also suggest a new regulatory role of both charged and neutral mitochondrial lipids in the control of MOM permeability, and hence mitochondria respiration, by modulating VDAC sensitivity to blockage by tubulin and α-syn. Considering the substantial lipid homeostasis in mitochondria during morphological changes such as fusion and fission (Furt and Moreau 2009), under apoptotic stress (Crimi and Esposti 2011), or lipid oxidation by ROS produced by mitochondria (Kagan et al. 2004; Pamplona 2008; Paradies et al. 2011), lipids could be potent regulators of VDAC and therefore of MOM permeability in vivo.
5 Membrane-Binding Properties of α-Synuclein and Tubulin
5.1 α-Synuclein-Membrane Binding
There are three distinctive regions of α-syn: the slightly net positively charged N-terminal domain (residues 1–60), the central nonpolar “nonamyloid-β component” (NAC) domain (residues 61–95), and the highly acidic C-terminal domain containing 15 negative charges (residues 96–140) (Rochet et al. 2012) (Fig. 8.1b). In solution, α-syn exists in the disordered form but can adopt an α-helical structure in its N-terminal domain upon binding to lipid membranes. The structure of membrane-bound α-syn is the subject of intense investigation and has been comprehensively reviewed (Pfefferkorn et al. 2012b). One of the primary techniques used to study adsorption of α-syn onto membrane surfaces has been circular dichroism (CD). CD reveals that α-syn, while disordered in bulk media, adopts a secondary structure with different helical content upon association with different lipid membranes (Davidson et al. 1998). In particular, anionic lipids stabilize an amphipathic helical structure in bound α-syn (Jiang et al. 2015). CD also shows an increase in helicity for non-lamellar zwitterionic lipids such as PE (Jo et al. 2000).
Complementary techniques find that there is measurable association between α-syn and membranes composed of lipids that do not induce helices. Specifically, fluorescence correlation spectroscopy (FCS) uses fluorescently labeled α-syn to determine the fractions of unbound α-syn , which diffuses freely in bulk solution, and liposome-bound α-syn, which has a diffusion constant similar to the liposomes to which it is bound (Rhoades et al. 2006). By this technique, it has been determined that α-syn does in fact associate with PC lipids, and there is a strong variation in binding to different anionic lipid species (Middleton and Rhoades 2010; Rhoades et al. 2006).
These results suggest a complex correlation between the secondary structure adopted by membrane-bound α-syn and the measured binding affinity. In particular, lipids with small and/or negatively charged headgroups enhance both binding affinity and helix formation; these enhancements are highest for phosphatidic acid (PA) headgroups and weakest for PC. Neutron reflectometry , which reports on membrane structure, protein penetration depth, and protein extension outside the membrane, is a promising technique for examining the structure of α-syn and the membranes to which it binds. Studies on anionic lipids have demonstrated that α-syn resides in the membrane headgroup region without significant penetration into the hydrophobic core of the membrane (Jiang et al. 2015; Pfefferkorn et al. 2012a).
α-Syn has also been demonstrated to show strong curvature sensitivity (Middleton and Rhoades 2010) and in fact appears to have a strong effect on the morphology of membrane surfaces to which it binds (Braun et al. 2014; Jiang et al. 2013; Shi et al. 2015). This is not surprising given α-syn’s affinity for binding nonlamellar lipids in planar bilayers; indeed, the two phenomena are difficult to separate conceptually (Cornell 2015). In highly curved micelles, α-syn forms a broken helix (Ulmer et al. 2005); on small unilamellar vesicles, it is thought that the helix can be either broken or continuous (Lokappa and Ulmer 2011). Intriguingly, in membranes with lipid compositions mimicking real systems, a multiplicity of membrane conformations was confirmed in nuclear magnetic resonance (NMR) and computational studies (Bodner et al. 2009; Fusco et al. 2014). Together, these observations suggest that the degree of helix formation depends strongly on the lipid composition and other factors in vivo and may be a mechanism to fine-tune α-syn-lipid-binding affinities.
The propensity of α-syn to target anionic lipids with headgroups that are small relative to the fatty acid chains and as a result to demonstrate strong curvature sensitivity has significant in vivo implications. The mitochondrial outer membrane of Saccharomyces cerevisiae is composed of 45% PC lipids; the rest are all small headgroup (PE at 33%, CL at 6%, PA at 4%) and/or charged lipids (CL, PA, phosphoinositol (PI) at 10%, phosphatidylserine (PS) at 1%) (Zinser et al. 1991). In addition, the mitochondrial network is highly dynamic; the fusion/fission processes necessarily create regions of high curvature in MOM that may additionally enhance (or be enhanced by) α-syn binding. In the absence of significant inhibition by proteinaceous membrane components, α-syn can be expected to have substantial interactions with the membranes of mitochondrial composition and topology.
5.2 Tubulin-Membrane Association
Tubulin has been known to bind to liposomes and membrane extracts for several decades. These early results have been exhaustively compiled and reviewed (Wolff 2009). Unlike α-syn, however, few definitive statements can be made about the structure of membrane-bound tubulin. CD experiments suggest that both the helical character and the hydrophobic environment of membrane-bound tubulin increase relative to solubilized protein (Kumar et al. 1981). While these results have been interpreted as suggesting tubulin to be integrated into the hydrophobic region of the membrane, they are intriguingly similar to observations for α-syn and may simply indicate the formation of additional secondary structure upon association with the membrane. Other features of tubulin-membrane association, particularly its reversibility (Caron and Berlin 1980) and well-defined binding constant (Bernier-Valentin et al. 1983), support the notion of a peripheral attachment.
The lipid specificity of tubulin binding is not well studied. PE lipids appear to increase microtubule assembly (Hargreaves and McLean 1988), possibly due to a tubulin concentrating effect at surfaces containing PE lipids . This is supported by fluorescence studies showing an increase in tubulin binding to PE-containing large unilamellar vesicles (Rostovtseva et al. 2012). The effect of anionic lipids on microtubule assembly appears to have more to do with microtubule associated proteins than with tubulin itself (Wolff 2009).
Despite the known crystal structure of dimeric tubulin (Nettles et al. 2004; Nogales et al. 1998), the binding mechanism of the tubulin dimer to the membrane surface remains unknown, including the fundamental question of whether the α- or β-subunit (or both) is proximal to the membrane (Fig. 8.3a). The most indicative clue is the unchanged β-tubulin/vinblastine-binding affinity between membrane-bound and solubilized tubulin (Bhattacharyya and Wolff 1975), suggesting that the vinblastine-binding site remains exposed in membrane-bound tubulin (Fig. 8.3a, a).
The identification of the membrane-binding surface of tubulin is of critical importance for two reasons. First, MTAs overwhelmingly bind to the β-subunit (Field et al. 2014). If the β-subunit is responsible for membrane binding (Fig. 8.3a, b), one might expect that β-tubulin-bound MTAs would interfere with tubulin-membrane binding (see below discussion in 8.6.1). If, on the other hand, the α-subunit binds to membranes (Fig. 8.3a, a), new opportunities arise for drug development targeting the membrane-binding surface of tubulin. Second, the detailed mechanism of tubulin’s interaction with VDAC depends on the structure and orientation of membrane-bound tubulin on the membrane surface. The relatively short α-tubulin CTT (11 amino acids, ≈4.4 nm long) is attached near one end of the dimer (Fig. 8.1b) and would be unable to reach the membrane surface and be captured into the VDAC pore for a variety of surface configurations, including nearly all in which β-tubulin is bound (Fig. 8.3a, b). Conversely, the longer β-tubulin CTT (17 amino acids, ≈6.8 nm long), which is attached near the dimer interface (Fig. 8.1b), is likely to be surface accessible for all reasonable binding configurations, including binding via α-tubulin (Fig. 8.3a, a). Binding configurations for which both subunits simultaneously interact with the membrane are also possible. One would expect both CTTs to be accessible to the VDAC pore in this case.
The multiple dwell times of the CTTs in the channel (Rostovtseva et al. 2008) (Fig. 8.4a) argue for a distribution of either CTT isotypes (see discussion in part 8.6.1 below) or of the orientation of surface-bound tubulin dimer (Fig. 8.3a). The simplest explanation is that both the α- and β-CTTs are captured into the channel; however, the wide variety of mammalian CTT isotypes and post-translational modifications which predominantly occur in the CTT (Westermann and Weber 2003), such as polyglutamation (Sirajuddin et al. 2014), as well as the multiplicity of subunits and isoforms of each subunit, may as well be responsible.
The disambiguation of the role of the two tubulin subunits in binding to mitochondrial membranes would be an important step forward in understanding the physiology of the tubulin-mitochondrial interaction and regulation of VDAC in particular. The recent development of systems producing recombinant tubulin (Minoura et al. 2013; Sirajuddin et al. 2014; Vemu et al. 2016) is likely to stimulate research in this direction. Biophysical studies of the lipid composition dependence of tubulin binding to mitochondrial membranes, such as those described for α-syn, would be particularly useful. The large, elongated structure of the tubulin dimer makes it particularly suitable for in vitro structural techniques such as neutron reflectometry (Heinrich and Losche 2014).
To summarize, the membrane-binding properties of tubulin and α-syn have a number of similar features. Both are strongly sensitive to lipid-packing defects caused by the presence of nonlamellar lipids in planar bilayers (PE vs PC). The helical content of both proteins increases with binding, with α-syn’s amphipathic helical binding domains stabilized by anionic lipids. These properties are well suited for targeting the MOM and enhancing the availability of these molecules for voltage-induced interaction with VDAC. In addition, if tubulin is shown to bind peripherally, they appear to form a new and interesting class of peripheral membrane proteins that target lipid-packing defects and/or curvature without (as best we know) being directly involved in lipid-trafficking pathways (Cornell 2015). It remains to be seen if this is a general property of peripheral membrane proteins associated with the MOM.
6 Physiological Relevance of Tubulin and α-Synuclein Interaction with Mitochondrial Membranes
6.1 Tubulin Association with Mitochondrial Membranes
The year 2017 marks the 50th anniversary of the discovery of tubulin , yet some topics related to this protein remain underappreciated. One such topic is the nature of tubulin association with cell membranes. As pointed out in Sect. 8.5.2, the presence of tubulin in membranes isolated from various types of cells and tissues has been known since the 1970s (Bhattacharyya and Wolff 1975; Feit and Barondes 1970) followed a few years later by the demonstration of tubulin’s association with mitochondrial membranes (Bernier-Valentin et al. 1983). A clear co-localization of β-tubulin with MOM has been shown by (Saetersdal et al. 1990). Since then, only a few studies have been dedicated to establish the nature of this “mitochondrial” tubulin.
Tubulin is found to be associated with mitochondrial membranes from a wide range of cancerous and noncancerous human cell lines, namely, neuroblastoma (SK-N-SH and IMR32), lung carcinoma (A549), breast adenocarcinoma (MCF-7), nasal septum adenocarcinoma (RPMI 2650), cervix carcinoma (HeLa), ovarian carcinoma (A2780 and OVCAR-3, Cicchillitti et al. 2008), and noncancerous breast cells (HBL-100) (Carre et al. 2002). β-Tubulin was also found associated with mitochondria from rat cardiomyocytes (Guzun et al. 2011). While the amount of mitochondria-associated tubulin appears to differ between cell lines from which mitochondria were isolated, Carre et al. estimated that it represents ~2% of total cellular tubulin. Interestingly, in this study α- and β-tubulins were present at comparable levels in mitochondria, suggesting that mitochondrial tubulin is a heterodimer. Noteworthy differences were found between cytosolic and mitochondrial tubulins: mitochondria contain more tyrosinated and acetylated α-tubulin and also more βIII isotype compared to cytosolic tubulin. In rat cardiomyocytes, βII-tubulin was associated with mitochondria, while βIII-tubulin was colocalized with sarcomeric Z-lines and βIV with polymerized microtubules (Guzun et al. 2011). Interestingly, Cicchillitti et al. (2008) analyzed the migration pattern of βIII-tubulin from ovarian cancer cells on two-dimensional gels and observed that βIII isotype exists under two distinct isoforms that differ in their isoelectric point (pI). While one βIII isoform was detectable only in the mitochondrial compartment, the other βIII isoform was present in both cytoskeletal and mitochondrial preparations. Further characterization of βIII isoforms by separation of polymerized and free tubulin fractions allowed finding the “mitochondrial” isoform in the pool of free tubulin, while the “cytoskeletal” isoform was found mainly associated with the pellet containing assembled tubulin. Altogether these data suggest that tubulin associated with mitochondria is a dimer consisting of α-tubulin and one of the β-tubulin isotypes, depending on cellular tissue. Whether there is βIII isotype specificity for mitochondria has yet to be proven.
The fact that tubulin and VDAC could be co-immunoprecipitated (Carre et al. 2002) is of particular relevance for the in vitro findings described in previous sections. Tubulin interaction with mitochondrial membranes and VDAC is a promising direction for chemotherapy and, more specifically, for understanding MTA’s mechanism of action. MTAs like vinorelbine and paclitaxel have been shown to have a direct effect on mitochondria isolated from human neuroblastoma. They notably induce cytochrome c release, PTP opening, and mitochondria swelling (Andre et al. 2000). Mitochondria-associated tubulin has been proposed to be responsible for the mitochondrial dysfunctions induced by both stabilizing and destabilizing MTAs. Yet, the exact underlying mechanisms remain to be elucidated, requiring a clear depiction of how MTAs bind to dimeric mitochondrial tubulin and whether this binding modifies tubulin dimer conformation, as suggested by in vitro experiments (Arnal and Wade 1995), and consequently affects tubulin-membrane association (Fig. 8.3a). Another related question to explore is the impact of MTA-tubulin binding on tubulin interaction with VDAC. This would bring a better understanding of MTA direct targets in cancer cells and also in neuronal cells, since MTA treatment is associated with the development of peripheral neuropathies (Ballatore et al. 2012).
Mitochondrial membrane composition and its maintenance are essential for normal mitochondrial function, structure, and biogenesis. Thus, changes in the phospholipid composition affect mitochondrial functions and dynamics and have been linked to a variety of human diseases such as Barth syndrome (Schlame and Ren 2006), heart failure (Sparagna et al. 2007), neurodegenerative diseases (Aufschnaiter et al. 2016), and, more recently, cancer (Ribas et al. 2016). While lipid metabolism in apoptosis and cancer together with lipid replacement therapies are emerging fields (Huang and Freter 2015; Monteiro et al. 2013; Nicolson and Ash 2014), the way mitochondrial tubulin could be affected by MOM lipid content changes is still an open avenue to be explored. Nowadays, profiling of mitochondrial lipids in pathological conditions is made possible through “lipidomics” studies. This term, coined in the early 2000s (Wilson 2003), relates to novel mass spectrometry analytical approaches to quantitatively define lipid compositions from many different sources. Search for reliable procedures to standardize mitochondria isolation and minimize contaminations by membranes from other organelles is now a point of focus (Kappler et al. 2016). A future perspective is to discern whether tubulin binding to mitochondria is affected by MTA treatment in cancer and neuronal cells.
Potentially, the key factor in modulating tubulin binding to mitochondrial membranes is the increased generation of ROS, a common feature in aging and cancer (Brieger et al. 2012; Minelli et al. 2009). Mitochondria are the source and also the first target of oxidative products. Oxidation of mitochondrial phospholipids and associated proteins triggers structural changes in lipid bilayer organization and consequently alters membrane fluidity and permeability, leading inevitably to mitochondrial dysfunction (Paradies et al. 2014).
Future research will show if lipid peroxidation affects tubulin binding to mitochondria. So far, it is known that products of lipid peroxidation and especially 4-hydroxy-2-nonenal (HNE) disrupt the soluble/polymer tubulin equilibrium in favor of free tubulin (Kokubo et al. 2008; Neely et al. 1999; Olivero et al. 1990). Chemotherapy treatments, including taxanes and vinca alkaloids to a lesser extent, generate oxidative stress and lipid peroxidation products (Conklin 2004; Panis et al. 2012). Cellular data confirmed a mitochondrial origin of ROS after MTAs treatment (Le Grand et al. 2014) as well as an increased level of lipid peroxidation in an animal model of the chemotherapy-induced peripheral neuropathy (Greeshma et al. 2015). βIII-tubulin lacks the highly oxidizable Cys-239 found in βI, βII, and βIV isotypes (Joe et al. 2008), thus making βIII more resistant to free radicals. This could be one of the plausible explanations why mitochondria are enriched for this specific isotype. βIII-tubulin is of high interest in the cancer research field since this isotype has been associated with tumor development and aggressiveness and also in resistance to chemotherapy in tumors with poor prognosis (Mariani et al. 2015; Seve and Dumontet 2010). In this context, it would be beneficial to investigate if MTA-induced lipid peroxidation could regulate VDAC-tubulin interaction and thus participate in affecting physiopathological conditions.
6.2 α-Synuclein Association with Mitochondrial Membranes
Since the identification of dominant mutations in the SNCA gene, α-syn became the subject of numerous investigations. It has been reported that α-syn localizes at both the MOM and MIM from diverse models of dopaminergic neurons (Cole et al. 2008; Devi et al. 2008; Li et al. 2007; Parihar et al. 2008; Shavali et al. 2008) and that accumulation of α-syn in mitochondria impairs complex I resulting in increased oxidative stress (Devi et al. 2008; Parihar et al. 2008; Pennington et al. 2010). However, other group showed the absence of the inhibition effect on complex I by α-syn accumulated in mitochondria isolated from mouse brain (Banerjee et al. 2010). While most of the studies agree on the inhibitory effect of α-syn on the oxidative phosphorylation capacity and on the promotion of oxidative stress, the exact mechanism(s) of α-syn effect on the mitochondrial bioenergetics remains largely unknown. There is still no consensus about the molecular identity of the pathway for α-syn to cross the MOM. A few studies suggested the translocase of the outer membrane complex 40 (TOM40) as the α-syn pathway (Bender et al. 2013; Devi et al. 2008). However, it seems very unlikely that acidic protein without a mitochondria-specific precursor would translocate through the highly substrate-specific and cation-selective Tom40 pore (Kuszak et al. 2015). On the contrary, VDAC would nicely account for the translocation pathway of α-syn into the mitochondria and its access to complex I and probably to other electron transport complexes (Fig. 8.6). Toxicity associated with such interaction has been shown in a yeast PD model (Rostovtseva et al. 2015) and is currently under investigation in a neuronal cell model.

Proposed physiological implications of α-syn blockage and translocation through VDAC. By reversibly blocking the VDAC pore, α-syn temporarily disrupts ATP/ADP fluxes between mitochondria and the cytosol and thus regulates them. By translocating through VDAC, α-syn reaches complexes of the electron transport chain (cI, cII, cIII, and cIV) in the MIM and impairs their function. This leads to the loss of mitochondrial potential ΔΨ and enhanced ROS production. ROS induces mitochondrial lipid peroxidation, which may modulate α-syn binding to the MOM as well as monomeric α-syn oxidation, leading to amplification of toxic fibrillary α-syn (Fα-syn) in the cytosol (Reprinted with permission from Rostovtseva et al. (2015))
α-Syn’s connection to mitochondrial lipid homeostasis in cells was proposed in a study showing that a decreased level of mitochondrial PE found in yeast and worm models of PD was correlated with accumulation of α-syn into cytoplasmic foci (Wang and Witt 2014). An earlier study showed a decreased level of the total PE and PC content in brains from PD patients (Riekkinen et al. 1975). These results are consistent with the in vitro data and model of α-syn-VDAC interaction discussed above in Sect. 8.4.2. The role of CL in α-syn-induced mitochondrial dysfunctions is now quite well documented (Ghio et al. 2016). In mouse models, the lack of α-syn was associated with decrease in CL (Ellis et al. 2005). Remarkably, both interactions between CL and α-syn and changes in CL content have been reported to have deleterious effects on mitochondria. In addition, the direct association of α-syn and CL has been correlated with disruption of mitochondrial dynamics in favor of fragmentation (Bueler 2009). On the other hand, altered CL content and expression levels of α-syn have generated seemingly contradictory results. Some data report that overexpression of the N-terminal domain of α-syn in neuronal models of PD is correlated with a decline in mitochondrial CL content, alterations in mitochondrial morphology, bioenergetics deficits, and decrease in mitochondrial membrane potential (Shen et al. 2014), while another study reported that α-syn knockout mice showed a decrease in mitochondrial CL and CL precursors and consequently reduction of complex I/III activity (Ellis et al. 2005). The number of similarities between the effects produced by the decrease of CL content and changes of α-syn expression levels points to an intricate relationship between α-syn and CL that requires further investigation.
There is strong evidence that oxidative stress is associated with PD pathology (Borza 2014) (Dias et al. 2013) as well as with numerous other pathologies. Several cellular models showed that oligomeric but not monomeric α-syn significantly increases the rate of ROS production, subsequently inducing lipid peroxidation in both primary co-cultures of neurons and astrocytes. Since inhibition of lipid peroxidation protects cells from cell death induced by oligomeric α-syn, the authors concluded that lipid peroxidation induced by misfolding of α-syn may play an important role in the cellular mechanism of neuronal cell loss observed in PD (Angelova et al. 2015). But so far, details about the specific phospholipid species undergoing oxidation and leading to mitochondrial dysfunction in PD are lacking. Given the high vulnerability of CL to peroxidation and the overall role of CL oxidation products in apoptosis and metabolic signaling (Kagan et al. 2014), CL is a promising candidate for further investigation in a PD context. Recently, Tyurina and coauthors (Tyurina et al. 2015) used a rat model of PD and lipidomics approach to identify oxygenated molecular species of CL formed in dysfunctional mitochondria. This particular study showed an increase in oxidized CL; therefore, a crucial point that remains unanswered is to what extent α-syn still binds to a modified CL. More in vitro data are needed to unravel the connections between α-syn and lipid peroxidation products in regard to α-syn conformational states and their relative binding to mitochondrial membranes.
The cartoon presented in Fig. 8.6 (Rostovtseva et al. 2015) illustrates how the previously discussed array of data on ROS production, mitochondrial dysfunction, lipid peroxidation, α-syn oxidation and fibrillation, and α-syn expression level can be reconciled within a model of MOM permeability regulation by α-syn interaction with VDAC and translocation through VDAC. By crossing the MOM through VDAC , α-syn is able to directly target complexes of the electron transport chain in the MIM, which could lead to the loss of the mitochondrial potential, enhanced ROS production, and mitochondrial lipid peroxidation followed by mitochondrial dysfunction. ROS in turn could oxidize monomeric α-syn in the cytosol (and most likely mitochondrial bound α-syn), causing α-syn oligomerization (Hashimoto et al. 1999) and consequently amplification of α-syn toxicity. ROS-induced mitochondrial lipid peroxidation could change monomeric α-syn binding to mitochondria (Ruiperez et al. 2010) and thus affect the α-syn-induced mitochondrial toxicity cycle. Depending on physiological conditions in the cell, such as α-syn expression level (Devi et al. 2008), cytosolic pH (Cole et al. 2008), MOM lipid composition (especially its CL and PE content Ellis et al. 2005), and potential across MOM, α-syn could regulate a normal ATP/ADP exchange through VDAC or cause mitochondrial dysfunction. In general, the model in Fig. 8.6 illustrates how mitochondrial lipids could be intimately involved in regulation of MOM permeability and mitochondrial function by cytosolic proteins as long as these proteins have the characteristics identified in Sect. 8.3.
7 Future Perspectives
We have attempted to show that the voltage-activated interaction between VDAC and charged cytosolic proteins is not specific in the traditional sense of ion channel regulation. Rather, the complexity of the dependences of the interaction rate on salt concentration, lipid composition, and protein concentration appears to arise entirely from the crucial involvement of peripheral binding of the cytosolic proteins to the lipid membrane. Thus, a complete characterization of this phenomenon requires a dissection of each step of interaction by applying a number of biophysical and biochemical techniques that report on various aspects of peripheral protein binding. As we have shown here, the VDAC channel itself is a sensitive single-molecule probe of the membrane-bound tubulin and α-syn. The planar bilayer approach is also well suited for study of protein binding using the nonlinear electrical properties (e.g. “second harmonics” techniques) of lipid bilayers (Peterson et al. 2002; Sokolov and Kuzmin 1980). Due to the absence of intrinsic curvature, planar bilayers are complementary to the liposome-based techniques (CD, FCS, gravimetric, electrokinetic mobility). Other useful platforms include solid supported (Castellana and Cremer 2006) or tethered (He et al. 2005; Lang et al. 1994) bilayer lipid membrane systems. Though these membranes have the disadvantages of steric hindrances and membrane stresses due to proximity to a planar substrate, they nonetheless feature unparalleled stability and have led to significant advances using neutron reflectometry, surface plasmon resonance, and optical spectroscopy. We expect these experimental techniques to be especially informative when complemented by in silico platforms, including both atomistic and coarse-grained (e.g., MARTINI MD simulation Marrink et al. 2007; Monticelli et al. 2008) models for protein binding to MOM-mimicking membranes. Visualization of proteins interacting with VDAC in biological samples through the use of a dual-color super-resolution microscopy of protein distribution in the MOM would provide a translation of in vitro data and models into a live-cell context. Another important element to assay in cells is the mitochondrial lipidomics and its modifications (remodeling, variation of content, oxidative forms) since lipid changes have been related to numerous pathological conditions and are even considered in some cases as biomarkers. Altogether, the combination of electrophysiology with an array of biophysical, biochemical, and computational methods is needed to reveal the role(s) of lipids in regulation of MOM permeability and, consequently, in mitochondrial function.
References
Andre N, Braguer D, Brasseur G, Goncalves A, Lemesle-Meunier D, Guise S, Jordan MA, Briand C (2000) Paclitaxel induces release of cytochrome c from mitochondria isolated from human neuroblastoma cells. Cancer Res 60(19):5349–5353
Angelova PR, Horrocks MH, Klenerman D, Gandhi S, Abramov AY, Shchepinov MS (2015) Lipid peroxidation is essential for alpha-synuclein-induced cell death. J Neurochem 133(4):582–589
Arnal I, Wade RH (1995) How does taxol stabilize microtubules? Curr Biol 5(8):900–908
Aufschnaiter A, Kohler V, Diessl J, Peselj C, Carmona-Gutierrez D, Keller W, Buttner S (2016) Mitochondrial lipids in neurodegeneration. Cell Tissue Res 367(1):125–140
Ballatore C, Brunden KR, Huryn DM, Trojanowski JQ, Lee VM, Smith AB 3rd (2012) Microtubule stabilizing agents as potential treatment for Alzheimer’s disease and related neurodegenerative tauopathies. J Med Chem 55(21):8979–8996
Banerjee K, Sinha M, Pham Cle L, Jana S, Chanda D, Cappai R, Chakrabarti S (2010) Alpha-synuclein induced membrane depolarization and loss of phosphorylation capacity of isolated rat brain mitochondria: implications in Parkinson’s disease. FEBS Lett 584(8):1571–1576
Basanez G, Sharpe JC, Galanis J, Brandt TB, Hardwick JM, Zimmerberg J (2002) Bax-type apoptotic proteins porate pure lipid bilayers through a mechanism sensitive to intrinsic monolayer curvature. J Biol Chem 277(51):49360–49365
Bender A, Desplats P, Spencer B, Rockenstein E, Adame A, Elstner M, Laub C, Mueller S, Koob AO, Mante M et al (2013) TOM40 mediates mitochondrial dysfunction induced by alpha-synuclein accumulation in Parkinson’s disease. PLoS ONE 8(4):e62277
Bernier-Valentin F, Aunis D, Rousset B (1983) Evidence for tubulin-binding sites on cellular membranes – plasma-membranes, mitochondrial-membranes, and secretory granule membranes. J Cell Biol 97(1):209–216
Bezrukov SM, Vodyanoy I (1993) Probing alamethicin channels with water-soluble polymers. Effect on conductance of channel states. Biophys J 64(1):16–25
Bhattacharyya B, Wolff J (1975) Membrane-bound tubulin in brain and thyroid tissue. J Biol Chem 250(19):7639–7646
Bodner CR, Dobson CM, Bax A (2009) Multiple tight phospholipid-binding modes of α-synuclein revealed by solution NMR spectroscopy. J Mol Biol 390(4):775–790
Borza LR (2014) A review on the cause-effect relationship between oxidative stress and toxic proteins in the pathogenesis of neurodegenerative diseases. Rev Med Chir Soc Med Nat Iasi 118(1):19–27
Braun AR, Lacy MM, Ducas VC, Rhoades E, Sachs JN (2014) Alpha-synuclein-induced membrane remodeling is driven by binding affinity, partition depth, and interleaflet order asymmetry. J Am Chem Soc 136(28):9962–9972
Brieger K, Schiavone S, Miller FJ Jr, Krause KH (2012) Reactive oxygen species: from health to disease. Swiss Med Wkly 142:w13659
Bueler H (2009) Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson’s disease. Exp Neurol 218(2):235–246
Caron JM, Berlin RD (1980) Reversible adsorption of microtubule protein to phospholipid-vesicles. J Cell Biol 87(2):A255–A255
Carre M, Andre N, Carles G, Borghi H, Brichese L, Briand C, Braguer D (2002) Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. J Biol Chem 277(37):33664–33669
Castellana ET, Cremer PS (2006) Solid supported lipid bilayers: from biophysical studies to sensor design. Surf Sci Rep 61(10):429–444
Chipuk JE, Bouchier-Hayes L, Green DR (2006) Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ 13(8):1396–1402
Cicchillitti L, Penci R, Di Michele M, Filippetti F, Rotilio D, Donati MB, Scambia G, Ferlini C (2008) Proteomic characterization of cytoskeletal and mitochondrial class III beta-tubulin. Mol Cancer Ther 7(7):2070–2079
Cole NB, Dieuliis D, Leo P, Mitchell DC, Nussbaum RL (2008) Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp Cell Res 314(10):2076–2089
Colombini M (1989) Voltage gating in the mitochondrial channel, VDAC. J Membr Biol 111(2):103–111
Colombini M (2004) VDAC: the channel at the interface between mitochondria and the cytosol. Mol Cell Biochem 256(1–2):107–115
Colombini M (2010) Ceramide channels and their role in mitochondria-mediated apoptosis. Biochim Biophys Acta 1797(6–7):1239–1244
Colombini M, Blachly-Dyson E, Forte M (1996) VDAC, a channel in the outer mitochondrial membrane. In: Narahashi T (ed) In ion channels. Plenum Press, New York, pp 169–202
Conklin KA (2004) Chemotherapy-associated oxidative stress: impact on chemotherapeutic effectiveness. Integr Cancer Ther 3(4):294–300
Cornell RB (2015) Membrane lipid compositional sensing by the inducible amphipathic helix of CCT. Biochim Biophys Acta. doi:10.1016/j.bbalip.2015.1012.1022
Crimi M, Esposti MD (2011) Apoptosis-induced changes in mitochondrial lipids. Biochim Biophys Acta 1813(4):551–557
Davidson WS, Jonas A, Clayton DF, George JM (1998) Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem 273(16):9443–9449
de Kroon AI, Dolis D, Mayer A, Lill R, de Kruijff B (1997) Phospholipid composition of highly purified mitochondrial outer membranes of rat liver and Neurospora crassa. Is cardiolipin present in the mitochondrial outer membrane? Biochim Biophys Acta 1325(1):108–116
DeHart DN, Gooz M, Rostovtseva TK, Sheldon KL, Lemasters JJ, Maldonado EN (2014) Antagonists of the inhibitory effect of free tubulin on VDAC induce oxidative stress and mitochondrial dysfunction. Biophys J 106(2):591a–591a
Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK (2008) Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem 283(14):9089–9100
Dias V, Junn E, Mouradian MM (2013) The role of oxidative stress in Parkinson’s disease. J Park Dis 3(4):461–491
Elkon H, Don J, Melamed E, Ziv I, Shirvan A, Offen D (2002) Mutant and wild-type alpha-synuclein interact with mitochondrial cytochrome C oxidase. J Mol Neurosci 18(3):229–238
Ellis CE, Murphy EJ, Mitchell DC, Golovko MY, Scaglia F, Barcelo-Coblijn GC, Nussbaum RL (2005) Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein. Mol Cell Biol 25(22):10190–10201
Feit H, Barondes SH (1970) Colchicine-binding activity in particulate fractions of mouse brain. J Neurochem 17(9):1355–1364
Field JJ, Waight AB, Senter PD (2014) A previously undescribed tubulin binder. Proc Natl Acad Sci U S A 111(38):13684–13685
Flis VV, Daum G (2013) Lipid transport between the endoplasmic reticulum and mitochondria. Cold Spring Harb Perspect Biol 5(6):1–22
Furt F, Moreau P (2009) Importance of lipid metabolism for intracellular and mitochondrial membrane fusion/fission processes. Int J Biochem Cell B 41(10):1828–1836
Fusco G, De Simone A, Gopinath T, Vostrikov V, Vendruscolo M, Dobson CM, Veglia G (2014) Direct observation of the three regions in alpha-synuclein that determine its membrane-bound behaviour. Nat Commun 5:3827
Ghio S, Kamp F, Cauchi R, Giese A, Vassallo N (2016) Interaction of alpha-synuclein with biomembranes in Parkinson’s disease – role of cardiolipin. Prog Lipid Res 61:73–82
Gonzalvez F, Pariselli F, Dupaigne P, Budihardjo I, Lutter M, Antonsson B, Diolez P, Manon S, Martinou JC, Goubern M et al (2005) tBid interaction with cardiolipin primarily orchestrates mitochondrial dysfunctions and subsequently activates Bax and Bak. Cell Death Differ 12(6):614–626
Gonzalvez F, Schug ZT, Houtkooper RH, MacKenzie ED, Brooks DG, Wanders RJ, Petit PX, Vaz FM, Gottlieb E (2008) Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol 183(4):681–696
Greeshma N, Prasanth KG, Balaji B (2015) Tetrahydrocurcumin exerts protective effect on vincristine induced neuropathy: behavioral, biochemical, neurophysiological and histological evidence. Chem Biol Interact 238:118–128
Gurnev PA, Rostovtseva TK, Bezrukov SM (2011) Tubulin-blocked state of VDAC studied by polymer and ATP partitioning. FEBS Lett 585(14):2363–2366
Gurnev PA, Queralt-Martin M, Aguilella VM, Rostovtseva TK, Bezrukov SM (2012) Probing tubulin-blocked state of VDAC by varying membrane surface charge. Biophys J 102(9):2070–2076
Guzun R, Karu-Varikmaa M, Gonzalez-Granilo M, Kuznetsov A, Michel L, Cottet-Rousselle C, Saaremae M, Kaam T, Metsis M, Grimm M et al (2011) Mitochondria-cytoskeleton interaction: distribution of β-tubulins in cardiomyocytes and HL-1 cells. Biochim Biophys Acta 1807:458–469
Hargreaves AJ, McLean WG (1988) The characterization of phospholipids associated with microtubules, purified tubulin and microtubule associated proteins in vitro. Int J Biochem 20(10):1133–1138
Hashimoto M, Hsu LJ, Xia Y, Takeda A, Sisk A, Sundsmo M, Masliah E (1999) Oxidative stress induces amyloid-like aggregate formation of NACP/alpha-synuclein in vitro. Neuroreport 10(4):717–721
He L, Robertson JW, Li J, Karcher I, Schiller SM, Knoll W, Naumann R (2005) Tethered bilayer lipid membranes based on monolayers of thiolipids mixed with a complementary dilution molecule. 1. Incorporation of channel peptides. Langmuir 21(25):11666–11672
Heinrich F, Losche M (2014) Zooming in on disordered systems: neutron reflection studies of proteins associated with fluid membranes. Biochim Biophys Acta 1838(9):2341–2349
Hodge T, Colombini M (1997) Regulation of metabolite flux through voltage-gating of VDAC channels. J Membr Biol 157(3):271–279
Hoogerheide DP, Gurnev PA, Rostovtseva TK, Bezrukov SM (2016) Mechanism of alpha-synuclein translocation through a VDAC nanopore revealed by energy landscape modeling of escape time distributions. Nanoscale 9(1):183–192
Huang C, Freter C (2015) Lipid metabolism, apoptosis and cancer therapy. Int J Mol Sci 16(1):924–949
Jacobs D, Hoogerheide DP, Rovini A, Gurnev PA, Bezrukov SM, Rostovtseva TK (2016) Membrane lipid composition regulates alpha-synuclein blockage of and translocation through the mitochondrial voltage-dependent anion channel. Biophys J 110(3):19a–20a
Jiang Z, de Messieres M, Lee JC (2013) Membrane remodeling by alpha-synuclein and effects on amyloid formation. J Am Chem Soc 135(43):15970–15973
Jiang Z, Hess SK, Heinrich F, Lee JC (2015) Molecular details of alpha-synuclein membrane association revealed by neutrons and photons. J Phys Chem B 119(14):4812–4823
Jo E, McLaurin J, Yip CM, St George-Hyslop P, Fraser PE (2000) Alpha-synuclein membrane interactions and lipid specificity. J Biol Chem 275(44):34328–34334
Joe PA, Banerjee A, Luduena RF (2008) The roles of cys 124 and ser239 in the functional properties of human betaIII tubulin. Cell Motil Cytoskeleton 65(6):476–486
Kagan VE, Borisenko GG, Tyurina YY, Tyurin VA, Jiang JF, Potapovich AI, Kini V, Amoscato AA, Fujii Y (2004) Oxidative lipidomics of apoptosis: redox catalytic interactions of cytochrome C with cardiolipin and phosphatidylserine. Free Radic Biol Med 37(12):1963–1985
Kagan VE, Chu CT, Tyurina YY, Cheikhi A, Bayir H (2014) Cardiolipin asymmetry, oxidation and signaling. Chem Phys Lipids 179:64–69
Kappler L, Li J, Haring HU, Weigert C, Lehmann R, Xu G, Hoene M (2016) Purity matters: a workflow for the valid high-resolution lipid profiling of mitochondria from cell culture samples. Sci Rep 6:21107
Kokubo J, Nagatani N, Hiroki K, Kuroiwa K, Watanabe N, Arai T (2008) Mechanism of destruction of microtubule structures by 4-hydroxy-2-nonenal. Cell Struct Funct 33(1):51–59
Kruger R, Muller T, Riess O (2000) Involvement of alpha-synuclein in Parkinson’s disease and other neurodegenerative disorders. J Neural Transm (Vienna) 107(1):31–40
Kumar N, Klausner RD, Weinstein JN, Blumenthal R, Flavin M (1981) Interaction of tubulin with phospholipid-vesicles. 2. Physical changes of the protein. J Biol Chem 256(11):5886–5889
Kuszak AJ, Jacobs D, Gurnev PA, Shiota T, Louis JM, Lithgow T, Bezrukov SM, Rostovtseva TK, Buchanan SK (2015) Evidence of distinct channel conformations and substrate binding affinities for the mitochondrial outer membrane protein translocase pore Tom40. J Biol Chem 290(43):26204–26217
Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD (2002) Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111(3):331–342
Landeta O, Landajuela A, Gil D, Taneva S, Di Primo C, Sot B, Valle M, Frolov VA, Basanez G (2011) Reconstitution of proapoptotic BAK function in liposomes reveals a dual role for mitochondrial lipids in the BAK-driven membrane permeabilization process. J Biol Chem 286(10):8213–8230
Lang H, Duschl C, Vogel H (1994) A new class of thiolipids for the attachment of lipid bilayers on gold surfaces. Langmuir 10(1):197–210
Le Grand M, Rovini A, Bourgarel-Rey V, Honore S, Bastonero S, Braguer D, Carre M (2014) ROS-mediated EB1 phosphorylation through Akt/GSK3beta pathway: implication in cancer cell response to microtubule-targeting agents. Oncotarget 5(10):3408–3423
Lemasters JJ, Holmuhamedov E (2006) Voltage-dependent anion channel (VDAC) as mitochondrial governator – thinking outside the box. Biochim Biophys Acta 1762(2):181–190
Lemasters JJ, Holmuhamedov EL, Czerny C, Zhong Z, Maldonado EN (2012) Regulation of mitochondrial function by voltage dependent anion channels in ethanol metabolism and the Warburg effect. Biochim Biophys Acta 1818(6):1536–1544
Lemeshko VV (2006) Theoretical evaluation of a possible nature of the outer membrane potential of mitochondria. Eur Biophys J 36(1):57–66
Lemeshko VV (2014a) VDAC electronics: 1. VDAC-hexo(gluco)kinase generator of the mitochondrial outer membrane potential. Biochim Biophys Acta 1838(5):1362–1371
Lemeshko VV (2014b) VDAC electronics: 2. A new, anaerobic mechanism of generation of the membrane potentials in mitochondria. Biochim Biophys Acta 1838(7):1801–1808
Lemeshko VV (2016) VDAC electronics: 3. VDAC-creatine kinase-dependent generation of the outer membrane potential in respiring mitochondria. BBA Biomembr 1858(7):1411–1418
Li WW, Yang R, Guo JC, Ren HM, Zha XL, Cheng JS, Cai DF (2007) Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport 18(15):1543–1546
Liu G, Zhang C, Yin J, Li X, Cheng F, Li Y, Yang H, Ueda K, Chan P, Yu S (2009) Alpha-synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci Lett 454(3):187–192
Lokappa SB, Ulmer TS (2011) α-synuclein populates both elongated and broken helix states on small unilamellar vesicles. J Biol Chem 286(24):21450–21457
Luth ES, Stavrovskaya IG, Bartels T, Kristal BS, Selkoe DJ (2014) Soluble, prefibrillar alpha-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J Biol Chem 289(31):21490–21507
Maldonado EN, Patnaik J, Mullins MR, Lemasters JJ (2011) Free tubulin modulates mitochondrial membrane potential in cancer cells. Cancer Res 70(24):10192–10201
Maldonado EN, Sheldon KL, DeHart DN, Patnaik J, Manevich Y, Townsend DM, Bezrukov SM, Rostovtseva TK, Lemasters JJ (2013) Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: regulation by free tubulin and erastin. J Biol Chem 288(17):11920–11929
Mariani M, Karki R, Spennato M, Pandya D, He S, Andreoli M, Fiedler P, Ferlini C (2015) Class III beta-tubulin in normal and cancer tissues. Gene 563(2):109–114
Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH (2007) The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B 111(27):7812–7824
Middleton ER, Rhoades E (2010) Effects of curvature and composition on alpha-synuclein binding to lipid vesicles. Biophys J 99(7):2279–2288
Minelli A, Bellezza I, Conte C, Culig Z (2009) Oxidative stress-related aging: a role for prostate cancer? Biochim Biophys Acta 1795(2):83–91
Minoura I, Hachikubo Y, Yamakita Y, Takazaki H, Ayukawa R, Uchimura S, Muto E (2013) Overexpression, purification, and functional analysis of recombinant human tubulin dimer. FEBS Lett 587(21):3450–3455
Monge C, Beraud N, Kuznetsov AV, Rostovtseva T, Sackett D, Schlattner U, Vendelin M, Saks VA (2008) Regulation of respiration in brain mitochondria and synaptosomes: restrictions of ADP diffusion in situ, roles of tubulin, and mitochondrial creatine kinase. Mol Cell Biochem 318(1–2):94–1562
Monteiro JP, Oliveira PJ, Jurado AS (2013) Mitochondrial membrane lipid remodeling in pathophysiology: a new target for diet and therapeutic interventions. Prog Lipid Res 52(4):513–528
Monticelli L, Kandasamy SK, Periole X, Larson RG, Tieleman DP, Marrink SJ (2008) The MARTINI coarse-grained force field: extension to proteins. J Chem Theory Comput 4(5):819–834
Neely MD, Sidell KR, Graham DG, Montine TJ (1999) The lipid peroxidation product 4-hydroxynonenal inhibits neurite outgrowth, disrupts neuronal microtubules, and modifies cellular tubulin. J Neurochem 72(6):2323–2333
Nettles JH, Li HL, Cornett B, Krahn JM, Snyder JP, Downing KH (2004) The binding mode of epothilone A on alpha, beta-tubulin by electron crystallography. Science 305(5685):866–869
Nicolson GL, Ash ME (2014) Lipid replacement therapy: a natural medicine approach to replacing damaged lipids in cellular membranes and organelles and restoring function. Biochim Biophys Acta 1838(6):1657–1679
Nogales E, Wolf SG, Downing KH (1998) Structure of the alpha beta tubulin dimer by electron crystallography. Nature 391(6663):199–203
Noskov SY, Rostovtseva TK, Bezrukov SM (2013) ATP transport through VDAC and the VDAC-tubulin complex probed by equilibrium and nonequilibrium MD simulations. Biochemistry 52(51):9246–9256
Noskov SY, Rostovtseva TK, Chamberlin AC, Teijido O, Jiang W, Bezrukov SM (2016) Current state of theoretical and experimental studies of the voltage-dependent anion channel (VDAC). Biochim Biophys Acta 1858(7 Pt B):1778–1790
Olivero A, Miglietta A, Gadoni E, Gabriel L (1990) 4-hydroxynonenal interacts with tubulin by reacting with its functional -SH groups. Cell Biochem Funct 8(2):99–105
Pamplona R (2008) Membrane phospholipids, lipoxidative damage and molecular integrity: a causal role in aging and longevity. Biochim Biophys Acta 1777(10):1249–1262
Panis C, Herrera AC, Victorino VJ, Campos FC, Freitas LF, De Rossi T, Colado Simao AN, Cecchini AL, Cecchini R (2012) Oxidative stress and hematological profiles of advanced breast cancer patients subjected to paclitaxel or doxorubicin chemotherapy. Breast Cancer Res Treat 133(1):89–97
Paradies G, Petrosillo G, Paradies V, Ruggiero FM (2011) Mitochondrial dysfunction in brain aging: role of oxidative stress and cardiolipin. Neurochem Int 58(4):447–457
Paradies G, Paradies V, Ruggiero FM, Petrosillo G (2014) Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J Gastroenterol 20(39):14205–14218
Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P (2008) Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol Life Sci 65(7–8):1272–1284
Pennington K, Peng J, Hung CC, Banks RE, Robinson PA (2010) Differential effects of wild-type and A53T mutant isoform of alpha-synuclein on the mitochondrial proteome of differentiated SH-SY5Y cells. J Proteome Res 9(5):2390–2401
Peterson U, Mannock DA, Lewis RN, Pohl P, McElhaney RN, Pohl EE (2002) Origin of membrane dipole potential: contribution of the phospholipid fatty acid chains. Chem Phys Lipids 117(1–2):19–27
Pfefferkorn CM, Heinrich F, Sodt AJ, Maltsev AS, Pastor RW, Lee JC (2012a) Depth of α-synuclein in a bilayer determined by fluorescence, neutron reflectometry, and computation. Biophys J 102(3):613–621
Pfefferkorn CM, Jiang Z, Lee JC (2012b) Biophysics of alpha-synuclein membrane interactions. Biochim Biophys Acta 1818(2):162–171
Porcelli AM, Ghelli A, Zanna C, Pinton P, Rizzuto R, Rugolo M (2005) pH difference across the outer mitochondrial membrane measured with a green fluorescent protein mutant. Biochem Biophys Res Commun 326(4):799–804
Rhoades E, Ramlall TF, Webb WW, Eliezer D (2006) Quantification of alpha-synuclein binding to lipid vesicles using fluorescence correlation spectroscopy. Biophys J 90(12):4692–4700
Ribas V, Garcia-Ruiz C, Fernandez-Checa JC (2016) Mitochondria, cholesterol and cancer cell metabolism. Clin Transl Med 5(1):22
Riekkinen P, Rinne UK, Pelliniemi TT, Sonninen V (1975) Interaction between dopamine and phospholipids. Studies of the substantia nigra in Parkinson disease patients. Arch Neurol 32(1):25–27
Rochet JC, Hay BA, Guo M (2012) Molecular insights into Parkinson’s disease. Prog Mol Biol Transl Sci 107:125–188
Rostovtseva TK, Bezrukov SM (2012) VDAC inhibition by tubulin and its physiological implications. Biochim Biophys Acta 1818(6):1526–1535
Rostovtseva TK, Bezrukov SM (2015) Function and regulation of mitochondrial voltage-dependent anion channel. In: Delcour AH (ed) Electrophysiology of unconventional channels and pores. Springer, Switzerland, pp 3–31
Rostovtseva T, Colombini M (1996) ATP flux is controlled by a voltage-gated channel from the mitochondrial outer membrane. J Biol Chem 271(45):28006–28008
Rostovtseva T, Colombini M (1997) VDAC channels mediate and gate the flow of ATP: implications for the regulation of mitochondrial function. Biophys J 72(5):1954–1962
Rostovtseva TK, Komarov A, Bezrukov SM, Colombini M (2002a) Dynamics of nucleotides in VDAC channels: structure-specific noise generation. Biophys J 82(1):193–205
Rostovtseva TK, Komarov A, Bezrukov SM, Colombini M (2002b) VDAC channels differentiate between natural metabolites and synthetic molecules. J Membr Biol 187(2):147–156
Rostovtseva TK, Tan WZ, Colombini M (2005) On the role of VDAC in apoptosis: fact and fiction. J Bioenerg Biomembr 37(3):129–142
Rostovtseva TK, Sheldon KL, Hassanzadeh E, Monge C, Saks V, Bezrukov SM, Sackett DL (2008) Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc Natl Acad Sci U S A 105(48):18746–18751
Rostovtseva TK, Gurnev PA, Chen MY, Bezrukov SM (2012) Membrane lipid composition regulates tubulin interaction with mitochondrial voltage-dependent anion channel. J Biol Chem 287(35):29589–29598
Rostovtseva TK, Gurnev PA, Protchenko O, Hoogerheide DP, Yap TL, Philpott CC, Lee JC, Bezrukov SM (2015) Alpha-synuclein shows high affinity interaction with voltage-dependent anion channel, suggesting mechanisms of mitochondrial regulation and toxicity in Parkinson disease. J Biol Chem 290(30):18467–18477
Ruiperez V, Darios F, Davletov B (2010) Alpha-synuclein, lipids and Parkinson’s disease. Prog Lipid Res 49(4):420–428
Saetersdal T, Greve G, Dalen H (1990) Associations between beta-tubulin and mitochondria in adult isolated heart myocytes as shown by immunofluorescence and immunoelectron microscopy. Histochemistry 95(1):1–10
Schafer B, Quispe J, Choudhary V, Chipuk JE, Ajero TG, Du H, Schneiter R, Kuwana T (2009) Mitochondrial outer membrane proteins assist bid in Bax-mediated lipidic pore formation. Mol Biol Cell 20(8):2276–2285
Schein SJ, Colombini M, Finkelstein A (1976) Reconstitution in planar lipid bilayers of a voltage-dependent anion-selective channel obtained from paramecium mitochondria. J Membr Biol 30(2):99–120
Schlame M, Ren M (2006) Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett 580(23):5450–5455
Seve P, Dumontet C (2010) Class III beta tubulin expression in nonsmall cell lung cancer. Rev Mal Respir 27(4):383–386
Shamas-Din A, Bindner S, Chi X, Leber B, Andrews DW, Fradin C (2015) Distinct lipid effects on tBid and Bim activation of membrane permeabilization by pro-apoptotic Bax. Biochem J 467(3):495–505
Shavali S, Brown-Borg HM, Ebadi M, Porter J (2008) Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci Lett 439(2):125–128
Shen J, Du T, Wang X, Duan C, Gao G, Zhang J, Lu L, Yang H (2014) Alpha-synuclein amino terminus regulates mitochondrial membrane permeability. Brain Res 1591:14–26
Shi Z, Sachs JN, Rhoades E, Baumgart T (2015) Biophysics of alpha-synuclein induced membrane remodelling. Phys Chem Chem Phys 17(24):15561–15568
Shoshan-Barmatz V, Ben-Hail D (2012) VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 12(1):24–34
Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N (2010) VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Asp Med 31(3):227–285
Sirajuddin M, Rice LM, Vale RD (2014) Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat Cell Biol 16(4):335–344
Siskind LJ, Kolesnick RN, Colombini M (2002) Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J Biol Chem 277(30):26796–26803
Sokolov VS, Kuzmin VG (1980) Study of surface-potential difference in bilayer-membranes according to the 2nd harmonic response of capacitance current. Biofizika 25(1):170–172
Sparagna GC, Chicco AJ, Murphy RC, Bristow MR, Johnson CA, Rees ML, Maxey ML, McCune SA, Moore RL (2007) Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J Lipid Res 48(7):1559–1570
Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388(6645):839–840
Swartz KJ (2008) Sensing voltage across lipid membranes. Nature 456(7224):891–897
Terrones O, Antonsson B, Yamaguchi H, Wang HG, Liu JH, Lee RM, Herrmann A, Basanez G (2004) Lipidic pore formation by the concerted action of proapoptotic BAX and tBID. J Biol Chem 279(29):30081–30091
Tyurina YY, Polimova AM, Maciel E, Tyurin VA, Kapralova VI, Winnica DE, Vikulina AS, Domingues MR, McCoy J, Sanders LH et al (2015) LC/MS analysis of cardiolipins in substantia nigra and plasma of rotenone-treated rats: implication for mitochondrial dysfunction in Parkinson’s disease. Free Radic Res 49(5):681–691
Ujwal R, Cascio D, Colletier J-P, Faham S, Zhang J, Toro L, Ping P, Abramson J (2008) The crystal structure of mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into metabolite gating. Proc Natl Acad Sci U S A 105(46):17742–17747
Ulmer TS, Bax A, Cole NB, Nussbaum RL (2005) Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem 280(10):9595–9603
Vemu A, Atherton J, Spector JO, Szyk A, Moores CA, Roll-Mecak A (2016) Structure and dynamics of single-isoform recombinant neuronal human tubulin. J Biol Chem:12907–12915
Wang S, Witt SN (2014) The Parkinson’s disease-associated protein α-synuclein disrupts stress signaling–a possible implication for methamphetamine use? Microb Cell 1(4):131
Westermann S, Weber K (2003) Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol 4(12):938–947
Wilson JF (2003) Long-suffering lipids gain respect: technical advances and enhanced understanding of lipid biology fuel a trend toward lipidomics. (Lab consumer). Scientist 17(5):34–37
Wolff J (2009) Plasma membrane tubulin. Biochim Biophys Acta 1788(7):1415–1433
Yethon JA, Epand RF, Leber B, Epand RM, Andrews DW (2003) Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J Biol Chem 278(49):48935–48941
Zinser E, Sperka-Gottlieb CD, Fasch EV, Kohlwein SD, Paltauf F, Daum G (1991) Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomyces cerevisiae. J Bacteriol 173(6):2026–2034
Acknowledgments
These studies were supported by the Intramural Research Program of the NIH, Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Rostovtseva, T.K., Hoogerheide, D.P., Rovini, A., Bezrukov, S.M. (2017). Lipids in Regulation of the Mitochondrial Outer Membrane Permeability, Bioenergetics, and Metabolism. In: Rostovtseva, T. (eds) Molecular Basis for Mitochondrial Signaling. Biological and Medical Physics, Biomedical Engineering. Springer, Cham. https://doi.org/10.1007/978-3-319-55539-3_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-55539-3_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-55537-9
Online ISBN: 978-3-319-55539-3
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)