
Abstract
Renal biopsy interpretation demands clinicopathologic correlation, which is particularly challenging in cases of endocarditis-associated glomerulonephritis. Not only can the clinical diagnosis of endocarditis be challenging, the morphologic spectrum of endocarditis-associated glomerulonephritis is unique among infection-associated glomerulonephritides in that it can mimic other diseases, and importantly, those that require a vastly different therapy. Though much of the available literature pertaining to endocarditis-associated glomerulonephritis originated from autopsy specimens obtained during the pre-antibiotic era, it is critical for the clinician and pathologist alike to be familiar with the current era of endocarditis-associated glomerulonephritis literature described in recent renal biopsy and autopsy series and as well as case reports, and to maintain a high index of suspicion.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Endocarditis
- Acute bacterial endocarditis
- Subacute bacterial endocarditis
- Crescentic glomerulonephritis
- Endocapillary proliferative glomerulonephritis
- Mesangial proliferative glomerulopathy
- Infection-related glomerulonephritis
- Infective endocarditis
- Cardiac infection
- Cardiac valve vegetation
- ANCA antibody
Introduction Overview
Renal biopsy interpretation demands clinicopathologic correlation, which is particularly challenging in cases of endocarditis-associated glomerulonephritis. Not only can the clinical diagnosis of endocarditis be challenging, the morphologic spectrum of endocarditis-associated glomerulonephritis is unique among infection-associated glomerulonephritides in that it can mimic other diseases, and importantly, those that require a vastly different therapy. Though much of the available literature pertaining to endocarditis-associated glomerulonephritis originated from autopsy specimens obtained during the pre-antibiotic era, it is critical for the clinician and pathologist alike to be familiar with the current era of endocarditis-associated glomerulonephritis literature described in recent renal biopsy and autopsy series and as well as case reports, and to maintain a high index of suspicion.
Infective Endocarditis Terminology
Historically, infection of the heart valves has been classified as either acute or subacute bacterial endocarditis on the basis of clinical grounds. This division not only reflected severity of disease and clinical course but also was influenced by virulence of the infecting microorganism and presence of underlying cardiac disease. Acute bacterial endocarditis usually involves a virulent bacterial organism infecting a previously normal heart. The classic example of this is Staphylococcus aureus infection in intravenous drug abusers. In subacute bacterial endocarditis, a bacterial organism of low virulence infects a previously damaged heart, such as the case in a rheumatic heart infected by Streptococcus viridans. The virulent microorganisms of acute bacterial endocarditis can lead to necrotizing valvular infections that are difficult to cure with antibiotics and may require surgery, whereas the lower virulence microorganisms in subacute bacterial endocarditis cause less destructive disease and a protracted clinical course typically with a better outcome. Other causative bacteria include coagulase negative Staphylococci (Staphylococcus epidermidis), known to infect prosthetic valves, enterococci, and the HACEK group of oral cavity commensals (Haemophilus, Actinobacillus, Cardiobacterium, Eikenella, and Kingella) [1]. There have also been reports of Gonococcus and gram-negative bacteria such as Coxiella burnetii, Bartonella henselae, and Brucella [2,3,4,5]. Although bacteria are the most common cause of endocarditis, infections are also caused by viruses, fungi, rickettsiae, and chlamydiae [6]. Given the numerous potential organisms underlying this disease, the preferred term today is infective endocarditis.
Furthermore, the glomerulonephritis due to infective endocarditis is not a postinfectious glomerulonephritis in that there is no latent period between eradication of the infection and onset of the glomerulonephritis, but is rather the result of an ongoing infection. Hence, the term endocarditis-associated or -related glomerulonephritis is preferred. In some patients with infective endocarditis, identification of the glomerulonephritis coincides with the first clinical recognition of infection [7].
Renal Disease Due to Infective Endocarditis
Renal disease due to infective endocarditis is well established with the earliest reports published over 100 years ago [8, 9]. The earliest literature on endocarditis-associated glomerulonephritis originated from autopsy specimens during the pre-antibiotic era. Though renal infarction and abscess formation were the most common findings, Löhlein in 1910, Baehr in 1912, and then in the 1930s Bell each described glomerular lesions associated with endocarditis [8,9,10]. All emphasized the presence of “embolic lesions.” These lesions were thought to be caused by small infected emboli from the infected cardiac valve that lodged within glomerular capillaries. Given that septic emboli leading to micro- and macro-abscess formation was a very common finding in these autopsy studies, this was a seemingly sensible explanation for microscopic focal proliferative glomerular lesions with mild exudation. However, after prolonged searching only Baehr was able to demonstrate bacteria within these glomerular lesions in rare cases [9]. Building upon the work of others, Bell characterized two forms of glomerulitis found in association with endocarditis. The diffuse form he described as an increase in number and size of the endothelial cells with thickening of the capillary basement membranes. The embolic or focal form included the presence of two lesions, “fresh and fibrotic.” The “fresh hyaline” lesion he described as thrombosis and necrosis of capillary loops and the “fibrous lesion” was described as a segmental or global fibrous obliteration of glomeruli. Analysis of his data reveals what appears to be the first description of epithelial crescents in the context of infective endocarditis [10]. Although not a point of emphasis, epithelial crescents were found in 31% of cases studied with subacute bacterial endocarditis. Even his well-known description of the “hyaline lesion,” thought to be the result of the ‘lodgement’ of bacteria into glomerular capillaries, appears to be a segmental necrotizing lesion in the photomicrographs [10]. Illustrations of crescents appeared in publications as early as the 1870s–1880s by Langhans and Purdy [11, 12]. Recognition that glomerular crescents correlated with poor outcome began to occur in the early 1900s by investigators including Volhard and Fahr and others [8, 13, 14]. However, perhaps Bell’s lack of emphasis on the presence of crescents and failure to recognize the necrotizing lesions was due to the fact that this was written in an era before the full significance of these findings were well recognized. One should also keep in mind that these earlier studies were on autopsy specimens and that they all occurred during the pre-antibiotic era.
Historical Evolution of Glomerular Injury Pattern in Endocarditis-Associated Glomerulonephritis
Based on these early studies and the many reports that followed, it was thought that the most common form of glomerulonephritis associated with infective endocarditis is a focal, segmental, or diffuse proliferative glomerulonephritis consisting of the presence of endocapillary proliferation with occasional infiltrating leukocytes [15,16,17]. This is the endocarditis-associated glomerulonephritis previously discussed in the major renal medicine [18, 19] and renal pathology [20,21,22,23] textbooks and was said to be the major pattern seen in more than 80% of cases of infective endocarditis with a glomerulonephritis. However, the literature supporting this view in these reference works was largely derived from autopsy studies from the pre- and post-antibiotic era or early renal biopsy studies from the 1970s. Renal involvement related to infective endocarditis previously described in the literature was also in part based on clinical observations that lacked histologic confirmation.
The advent of antibiotics has drastically altered the clinical course and prognosis of infective endocarditis. Data by Spain and King [24] proved the decreased incidence of renal complications of infective endocarditis with the use of antibiotic therapy. In time, several observations argued against the embolic nature of renal injury in infective endocarditis, and a circulating immune complex mechanism was proposed [25,26,27,28]. The use of immunofluorescent microscopy for the evaluation of glomerular immunoglobulin and complement deposition has been pivotal in shifting this paradigm. Supporting the concept of circulating immune complex injury, the finding of granular glomerular basement membrane and mesangial deposition of immunoglobulins and complement was documented [25]. In contrast, support for activation of the alternate complement pathway has been shown in cases of S. aureus infective endocarditis [29]. There have also been reports of endocarditis-associated glomerulonephritis that show no immunoglobulin or complement positivity by immunofluorescence, and a single report of “full house” immunostaining [30,31,32].
However, though insight into the mechanism of infective endocarditis-associated glomerulonephritis is better understood, the most common histologic pattern related to infective endocarditis was until recently still thought to be the classic description of infection-associated glomerulonephritis: a focal, segmental, or diffuse global proliferative glomerulonephritis consisting of endocapillary hypercellularity with the conspicuous presence of inflammatory cells by light microscopic examination, and immunofluorescence showing granular immune complex deposition positive for C3 and IgG [15,16,17]. In these cases, large subepithelial “hump-like” deposits are typically seen by electron microscopy. These findings are prototypical of post-Streptococcal glomerulonephritis and were the pattern most commonly seen in a recent large series of post-infectious glomerulonephritis in the elderly in which Staphylococcus was the most common infectious agent [33].
Fernandez Guerrero et al. [16] published a large case series of infective endocarditis in 2012. It was derived entirely from autopsy study of 68 patients from 1970 through 2008 with emphasis on cardiac and brain pathology but they did also examine for glomerulonephritis. Although renal infarcts and abscess formation were the most often described renal manifestations (in 30–36 and 18–19% of cases, respectively), still, glomerulonephritis was noted in 15% of cases between 1970 and 1985 and in 7% of cases between 1986 and 2008, with the most common pattern focal proliferative glomerulonephritis, with only one case of diffuse proliferative glomerulonephritis mentioned and no other patterns described. Interestingly, in another autopsy study of 82 cases with infective endocarditis from 1972 through 1986, Toth et al. noted 8 cases (10%) of crescentic glomerulonephritis [34]. Of interest, dating back to 1995, Montseny et al. studied 76 patients with infection-associated glomerulonephritis, of which 10 were related to endocarditis. Of these patients with endocarditis-associated glomerulonephritis, 3 had an endocapillary proliferative pattern and the majority, 7 patients, were crescentic. In comparison, glomerulonephritis related to all other sites of infection (including upper respiratory track, lung/pleura, skin, and teeth) showed an endocapillary proliferative pattern in the majority of cases, and in only a minority of cases a crescentic pattern [35]. Additionally, over the last twenty years, there have been case reports and mostly small case series describing the less familiar association of infective endocarditis with crescentic glomerulonephritis rather than focal or diffuse endocapillary proliferative glomerulonephritis [7, 17, 36–47].
One such series from the modern era was published in 2000 by Majumdar et al., with the majority of cases studied from post-mortem samples. They found that two-thirds of patients with endocarditis-associated glomerulonephritis showed a pauci-immune crescentic pattern of glomerular injury [48]. This long history of endocarditis-associated glomerulonephritis was built on by our study of 49 patients in 2015, which was the largest cohort of endocarditis-associated glomerulonephritis in the current era (2001–11) from nonautopsy cases studied exclusively by renal biopsy [49]. In this book chapter, this cohort has been further built on since that publication to now include 62 patients with endocarditis-associated glomerulonephritis. Of these 62 patients that fulfilled the modified Duke criteria [50] for diagnoses of infective endocarditis and underwent renal biopsies during the active phase of their illnesses, crescentic glomerulonephritis was the most common pattern of glomerular injury (47%) (Fig. 4.1), followed by focal or diffuse endocapillary proliferative glomerulonephritis (43%) (Fig. 4.2), and mesangial proliferative glomerulonephritis (10%) (Fig. 4.3). Of the endocarditis-associated crescentic glomerulonephritis cases, 41% were pauci-immune.

Two glomeruli with segmental necrosis and one with a cellular crescent (glomerulus on the left) in a 62-year-old male with crescentic glomerulonephritis associated with mitral valve Streptococcus viridans infective endocarditis. The uninvolved portions of the glomerular tufts appear normal, with no mesangial expansion or endocapillary hypercellularity (Jones methenamine silver; ×200)

Endocapillary hypercellularity in a patient with focal proliferative glomerulonephritis associated with aortic valve methicillin-sensitive Staphylococcus aureus infective endocarditis (periodic acid-Schiff; ×400)

Mild mesangial hypercellularity in a patient with infective endocarditis (periodic acid-Schiff; ×400)
Therefore, endocarditis-associated glomerulonephritis is unique among infection-associated glomerulonephritides in that more recent studies demonstrate an evolution in awareness to a pauci-immune necrotizing and crescentic glomerulonephritis as the most commonly manifested pattern [49, 51]. Cases with immune complex deposition still occur, and various patterns are noted by light microscopy including the more familiar pattern of endocapillary proliferative glomerulonephritis. Mesangial proliferative glomerulonephritis also occurs, though least commonly and consequently receives little attention in the literature. Thus, the pattern of glomerular injury related to infective endocarditis is a spectrum, both in terms of light and immunofluorescence microscopy findings. The true incidence of glomerulonephritis associated with infective endocarditis is unknown, with autopsy studies reporting up to 22–26% [17, 48].
Clinical Presentation and Laboratory Data
Clinical Evolution of Endocarditis-Associated Glomerulonephritis
Just as the morphologic spectrum of endocarditis-associated glomerulonephritis has evolved, our own findings in infective endocarditis [49] confirm and extend observations emphasized in recent reviews documenting the evolution in clinical findings in bacterial infection-related GN in adults over the past three decades [51–53]. This evolution occurring in recent decades includes the change in demographics from younger to older patients, the frequency of comorbidities such as diabetes and HIV, and the change in predominance of infectious agents from primarily Streptococcal to a broader array of organisms with predominance of Staphylococci [33, 51, 54, 55].
Infective endocarditis carries a mortality rate of 40–50% [56]. Over the past decades, infective endocarditis outcomes have not improved, and infection rates are steadily increasing [56]. Recent case series and reviews of infective endocarditis have compared findings from current and previous eras, confirmed similar changes in the demographics of the disease and updated the clinical and pathologic features in both adults and children [16, 57]. However, few of these recent reports have focused primarily on infective endocarditis-related renal lesions, and much of the data currently available still includes predominately autopsy-derived information [16, 48].
Clinical Presentation
In keeping with the overall trends in infection-related glomerulonephritis, findings in infective endocarditis in the current era involve predominately adult males with a 2.6:1 male predominance, older mean age at biopsy (mean age 47 years) with 25% elderly patients, and increased prevalence of Staphylococcal rather than Streptococcal infection (Tables 4.1 and 4.2) [49]. In general, postinfectious and infection-associated glomerulonephritis typically present with the acute nephritic syndrome and hypocomplementemia [23]. The most common presentation of infective endocarditis-associated glomerulonephritis is acute renal failure in which there was doubling of the serum creatinine (82%) (Table 4.1) [49]. This observation that the most common presentation is acute renal failure rather than acute nephritic syndrome differs from overall findings in infection-related glomerulonephritis [23, 51] and may be unique to this patient population with compromised cardiac function. In our material, only 8% presented with the typical acute nephritic syndrome of hematuria, hypertension, and renal failure and only about sixty percent with low serum complement levels. Other clinical syndromes at presentation include rapidly progressive glomerulonephritis (5%), and nephrotic syndrome with nephrotic range proteinuria (>3.5 g/day), hypoalbuminemia (serum albumin <3 g/dL), and peripheral edema (5%) (Table 4.1). The unique manifestations of endocarditis-associated glomerulonephritis are possibly related to the fact that these infections are often persistent and ongoing at the time of the kidney biopsy rather than being a classic postinfectious phenomenon [51]. Furthermore, the diagnosis of glomerulonephritis could prompt investigations that lead to a diagnosis of infective endocarditis. Indeed, cases of rapidly progressive ANCA-positive glomerulonephritis have been reported as the presenting feature of infective endocarditis [58, 59].
Predisposing States or Coexisting Conditions
Conditions favoring endocarditis are noted in a majority (64%) of our patients, including intravenous drug use (37%), prosthetic valves (16%), and prior valvular disease (11%), yet this leaves over 50% of patients with no known prior cardiac disease (Table 4.1) [49]. A minority of patients had associated comorbid conditions, with the most common being hepatitis C infection (24%) and diabetes mellitus (18%) (Fig. 4.4). Less common predisposing states or coexisting conditions included coronary artery disease, chronic obstructive pulmonary disease, congestive heart failure, autoimmune disease, recent surgery, and malignancy [49].

Cellular crescent in a 54-year-old diabetic male with Streptococcus agalactiae tricuspid valve endocarditis who presented with nephrotic syndrome (periodic acid-Schiff; ×400)
Laboratory Data and Serologic Studies
In general, bacterial infections can trigger the production of various autoantibodies, such as antinuclear antibodies (ANA), anticardiolipin antibodies, cryoglobulins, rheumatoid factor, and anti-neutrophil cytoplasmic antibodies (ANCA) [37, 60]. In our renal biopsy study of endocarditis-associated glomerulonephritis [49], the average serum creatinine was 3.8 mg/dL (range 1.0–12.0) (Table 4.1). Hematuria was present in almost all cases. Daily proteinuria averaged 2.1 g (range 0.5–15). Twenty-eight patients had an ANA test, 86% of which were negative. ANCA testing was carried out in over half of patients and was positive in 25%. ANCA specificities included both pANCA and cANCA, as well as cases with dual positivity (Table 4.1) (Fig. 4.5) [49]. In general, ANCA specificity associated with endocarditis was initially thought to be anti-PR3, but cases with dual ANCA positivity and MPO-ANCA positivity have also now been reported in association with endocarditis [37, 49, 61–63]. Testing for cryoglobulins have varied reports of positivity from 17 to 95% positive, though many of these studies have limited renal histologic correlation [60] and the cryoglobulin test is frequently false negative. Similarly, large amounts of serum immunoglobulins and circulating immune complexes may be formed as a result of bacteremia, but this does not necessarily imply deposition within the kidney by immunofluorescence [47]. Just over half of patients (60%) had hypocomplementemia in our renal biopsy series, which was most commonly (35%) low C3 (complement component 3) with normal C4 (complement component 4); since only a few patients had reduction in C4 this suggests most had activation of the alternative complement pathway.

Necrosis in a glomerulus from a patient with a prosthetic pulmonic valve and Bartonella pulmonic valve infective endocarditis in which the immunofluorescence showed 2+ IgG, 2–3+ IgM, and 2–3+ C3. ANCA serologies were positive for both MPO and PR3. The patient was treated with antibiotics and steroids, then after surgical treatment with pulmonic valve replacement, the ANCA titers decreased. (hematoxylin and eosin; ×400)
Cardiac Involvement
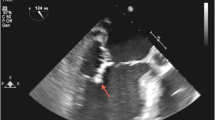
Infective endocarditis can involve any one of the four cardiac valves. In our current expanded study of 62 patients, endocarditis leading to glomerulonephritis most commonly involved the tricuspid valve (44%), followed by the mitral (38%), aortic (24%), and pulmonic (4%) valves (Table 4.2); infection of more than one valve was seen in 10% of patients (Fig. 4.6). In our study, 84% represented patients with community-acquired infective endocarditis in native valves, 94% of which had positive blood cultures compared to 90% positive blood cultures in the patients with prosthetic valve endocarditis [49]. One of the major Duke’s criteria to the diagnosis of infective endocarditis is vegetations noted by echocardiogram; these were noted in greater than two-thirds of patients in our renal biopsy study [49]. Of note, because transthoracic echocardiogram may not be able to detect small vegetations, transesophageal echocardiogram may be needed [64]. The most commonly noted sign of cardiac involvement in patients without vegetations on echocardiogram was new valvular regurgitation/murmur; the most common other criteria for diagnosis of infective endocarditis in these patients included fever, septic pulmonary emboli, and predisposing heart condition or injection drug use. For the entire cohort, the most common vascular phenomena was septic pulmonary infarcts, with only a minority of patients with the finding of intracranial hemorrhage, and rare patients with findings including conjunctival hemorrhages, nail splinter hemorrhages, or evidence of mycotic aneurysm [49].

Global endocapillary hypercellularity in a 47-year-old female intravenous drug user with diffuse proliferative glomerulonephritis associated with tricuspid and pulmonic valve methicillin-sensitive Staphylococcus aureus infective endocarditis. Immunofluorescence microscopy showed trace IgG, negative IgA, negative IgM, and 2–3+ C3 in a granular mesangial and capillary wall pattern. The patient had recurrent infective endocarditis two years following the initial biopsy. (PAS; ×400)
Infectious Agents
Several studies note a similar rate of culture-negative endocarditis at about 8–9% [49, 65, 66]. Over half of patients with culture positive endocarditis are classified as having acute rather than subacute endocarditis. In our experience, the agent found on culture in the acute group is most often S. aureus (58%), with methicillin resistance in almost half (44%); the second most common pathogens found are Streptococcus species (21%) (Table 4.2) [49]. Less common causes of endocarditis noted include Gemella species, Gonococcus, and gram-negative bacteria such as C. burnetii, B. henselae, and Brucella [2–5, 49], as well as the HACEK group of oral cavity commensals (Haemophilus, Actinobacillus, Cardiobacterium, Eikenella, and Kingella) [1]. The most common cause of endocarditis in patients with history of IV drug abuse is Staphylococcal infection (86%), affecting the tricuspid valve or tricuspid and pulmonic valves in 74%, followed by mitral or aortic valves in 26% [49].
Pathologic Findings and Clinicopathologic Correlation
Light Microscopy
Glomerular Findings
The patterns of glomerular injury described associated with infective endocarditis predominately include focal or diffuse necrotizing and crescentic glomerulonephritis, focal or diffuse endocapillary proliferative glomerulonephritis, and mesangial proliferative glomerulonephritis. Rare reports of endocarditis-associated glomerulonephritis with cryoglobulinemia with an MPGN pattern have also been described [59]. Designation as focal versus diffuse is made by applying the typical cut-off value of 50%, with focal meaning <50% of nonsclerotic glomeruli are involved and diffuse meaning ≥50% of nonsclerotic glomeruli are involved [67, 68]. Glomerular endocapillary proliferation in biopsies with focal or diffuse proliferative patterns is defined as endocapillary hypercellularity and occlusion of capillary lumens by endothelial cells, mesangial cells, and/or white blood cells from the peripheral circulation. In our study, cases of endocarditis-associated glomerulonephritis with a crescentic pattern do not show proliferative changes in portions of the glomerular tufts uninvolved by necrosis or crescent formation. Glomeruli with an increase in mesangial matrix and cells without closure of capillary lumens are included in the mesangial proliferative group. Proliferation in biopsies with the mesangial proliferative pattern of glomerular injury is defined as ≥4 cells per mesangial region in more than 50% of glomeruli without occlusion of capillary loops [69].
The most common pattern of glomerulonephritis associated with infection in general is typically that of endocapillary proliferation. However, infective endocarditis-associated glomerulonephritis is unique in that the most common pattern recently recognized is a crescentic glomerulonephritis (in 47% of patients) (Fig. 4.7); in a majority of patients these glomerular inflammatory changes are diffuse (59%) and necrotizing lesions are frequent (79%) (Table 4.3) [49]. Diffuse endocapillary proliferative glomerulonephritis is the second most common pattern (37%) (Fig. 4.8). Of the patients with proliferative glomerulonephritis, some also had focal crescent formation. Only two cases in our renal biopsy study of 49 patients published in 2015 had the previously classically described pattern of focal proliferative glomerulonephritis without crescents or necrosis (4%) [49]. Over 20 years prior, case reports and small case series have also documented the association of infective endocarditis with crescentic glomerulonephritis rather than focal or diffuse proliferative glomerulonephritis [7, 17, 34, 36–47]. In 2000, Majumdar et al. [48] found that two-thirds of patients with endocarditis-associated glomerulonephritis showed a pauci-immune crescentic pattern of glomerular injury.

Diffuse necrotizing and crescentic glomerulonephritis with numerous red blood cell casts in a 31-year-old male with culture-negative aortic valve endocarditis involving 88% of glomeruli. ANCA serology was negative. The patient was treated with antibiotics, steroids, and cytoxan, and had persistent renal dysfunction at 23 months follow-up (Jones methenamine silver; ×100)

Diffuse endocapillary proliferative glomerulonephritis in a patient with methicillin-resistant Staphylococcus aureus infective endocarditis (hematoxylin and eosin; ×100)
Mild mesangial hypercellularity is the third major finding after crescentic and endocapillary proliferative glomerulonephritis and account for 10% of cases [49]. All of these cases showed only mild and often segmental mesangial hypercellularity without endocapillary proliferation or crescent formation.
In our study of 62 patients with endocarditis-associated glomerulonephritis, glomerulonephritis with membranoproliferative pattern or membranous glomerulopathy was not seen. Specifically, no cases of membranoproliferative glomerulonephritis with or without cryoglobulinemic features or cases of thrombotic microangiopathy were found. In our study, a mean of 10% of glomeruli were globally sclerotic (range, 0–53%) [49].
Tubulointerstitial and Vascular Findings
Acute tubular injury is present in the background in the majority of cases, typically manifested by thinning of the tubular epithelium (Fig. 4.9). In part, this may be the result of obstructed blood flow through glomeruli and thus impaired perfusion of the tubules by way of the peritubular capillaries. Red blood cell casts are noted histologically in more than half of the cases. Almost all cases have interstitial inflammation (Fig. 4.10), which is most often focal, but abundant interstitial neutrophils are present in a minority of cases. Large numbers of eosinophils are usually not seen.

Normal appearing glomerulus and surrounding tubular injury manifested by cytoplasmic thinning and mild luminal ectasia, from a 45-year-old male with history of rheumatic fever as a child and mitral valve insufficiency. He developed mitral valve Coxiella burnetii infective endocarditis with acute kidney injury and a renal biopsy showed focal crescentic glomerulonephritis (not shown) involving 15% of glomeruli. The patient was treated with antibiotics, and had persistent renal dysfunction at follow-up (hematoxylin and eosin; ×200)

Glomerulus with endocapillary proliferation including neutrophils, and surrounding mild interstitial inflammation in an 84-year-old male with infective endocarditis-associated diffuse proliferative glomerulonephritis (hematoxylin and eosin; ×200)
Though infarcts and micro-abscesses are noted most commonly in autopsy studies, no micro-abscesses or cortical necrosis were present in our renal biopsy material of 62 cases. The degree of tubular atrophy and interstitial fibrosis present was most often mild (<25% of estimated cortical involvement) (40%) or absent (42%). Similarly, arteriosclerosis and arteriolar hyalinosis were most often absent (33%) or mild (32%). Vasculitis in the form of necrotizing arteritis was not noted.
Immunofluorescence
One has to pay attention to and look for and evaluate glomeruli with no or small crescents to avoid over-interpreting nonspecific staining secondary to glomerular necrosis and crescent formation. Deposits by immunofluorescence appear granular, with the location most often either a combination of mesangial and capillary loop (53%) (Fig. 4.11) or within the mesangial region only (39%) (Fig. 4.12) (Table 4.3). Though completely negative staining by immunofluorescence for immunoglobulins and complement is rare (5% of biopsies), up to 44% of biopsies in our study of 62 patients met criteria for pauci-immune staining intensity of immunoglobulins. Almost half of these (12 patients) had crescentic glomerulonephritis by light microscopy (Table 4.4). Of these 12 patients, ANCA was positive in 3, negative in 5, and not done in 4. In 2000, Majumdar et al. [48] also found that two-thirds of patients with endocarditis-associated glomerulonephritis have a pauci-immune pattern. Pauci-immune was defined as staining 0–2+ or less intensity for all immunoglobulins (IgG, IgM, and IgA) on a scale of 0–4+ [70]. Most pathologists using a 0–3+ scale, pauci-immune is usually defined as positivity of 1+ or less. Though the staining properties of C3 can be controversial and inconsistently interpreted as immune complex type or not, in our study of endocarditis-associated glomerulonephritis, the definition of pauci-immune disease is defined by immunoglobulin staining only, and does not account for the intensity of complement staining in glomeruli. This also seems prudent given that large case series have shown glomerular C3 deposition is not uncommon in pauci-immune, ANCA-associated glomerulonephritis [70, 71].

Glomerulus with coarsely granular mesangial and capillary wall staining for C3 (direct immunofluorescence; ×400)

Glomerulus with granular predominantly mesangial staining for C3 (direct immunofluorescence; ×400)
Immunofluorescence examination in cases of endocarditis-associated glomerulonephritis will most likely show C3 (95% of cases show positivity) (Table 4.3) [49]. C3 also has the highest mean intensity compared to other immunoreactants when positive (Tables 4.3 and 4.4). Cases with positivity for at least one subclass of immunoglobulin will typically also show complement staining. In our study of 62 patients, IgA was the least common immunoglobulin to be positive (29%), whereas IgG and IgM were both positive in 34% (Table 4.3) [49]. Interestingly, just over half of the cases with an endocapillary proliferative pattern by light microscopy had positive IgG (56%), and biopsies with a crescentic pattern had IgG in only 21% of cases. In this study, nine cases had IgA-dominant staining and an additional two were codominant for IgA and IgG (total 18%). A “full house” pattern with IgG, IgM, IgA, and complement positivity was seen in only 3% of cases. C3 only staining was present in 37% of cases (Table 4.3).
It is worth mentioning that the definition of pauci-immune necrotizing and crescentic glomerulonephritis is arbitrary. Mostly, “pauci-immune” is defined based on immunofluorescence findings, and some base this only on the presence or absence of immunoglobulins, disregarding complement (particularly C3). To some, immunofluorescence with C3 only staining better fits into the category “pauci-immune” because C3 only is not technically not part of an “immune complex” (meaning, complement together with immunoglobulin). However, strong C3 staining despite lack of immunoglobulin, especially together with well-defined electron dense deposits by ultrastructural examination, still suggests an immune-mediated process that should raise the possibility of an infection-related etiology; this is the case with many of the cases of endocarditis-associated glomerulonephritis described herein. The classic designations of immunofluorescent findings are primarily the subdivisions of “immune complex type” versus “pauci-immune.” In reality, C3-predomiant staining could be a third and separate category in itself because when C3 only or predominant staining is detected, one must make the sometimes arduous decision as to which category to place these findings. For example, in that sense, many cases of poststreptococcal glomerulonephritis could in theory be classified as pauci-immune because there is only C3 deposition, even though there are plenty of subepithelial “humps” by electron microscopy. Many cases of infection-associated glomerulonephritis have C3 deposits only or C3-dominant deposits with only relatively weak immunoglobulin staining. The key is that pauci-immune is not synonymous with not being immune-mediated, it is just not associated with large clumps of immune complexes. For this reason, electron microscopy, if possible, should be performed in every case because, if there is a well-perfused glomerulus with open capillaries present for examination, and if there are no or very few electron dense immune-type deposits present, that finding is more consistent with a pauci-immune process such as that noted in the majority of ANCA-mediated disease.
Electron Microscopy
Consistent with immunofluorescence findings, electron dense deposits by ultrastructural examination are most commonly present within the mesangium (Fig. 4.13). In our renal biopsy series of 62 patients with endocarditis-associated glomerulonephritis, mesangial electron dense deposits were noted in 87% of cases, subendothelial electron dense deposits in 47% (Fig. 4.14), and subepithelial electron dense deposits in 34% of cases (Table 4.3); but only the minority of cases (18%) showed the classic infection-related large subepithelial humps [49]. Interestingly, in more than one series of IgA-dominant Staphylococcal infection-associated glomerulonephritis in the literature, large subepithelial humps were similarly rare or were not seen [72, 73]. In contrast, in a study of 109 elderly patients with postinfectious glomerulonephritis from various etiologies combined, subepithelial electron dense deposits were seen in 92% of cases and in most cases exhibited a “hump-shaped” appearance [33]. Therefore, while it is helpful when large “hump-like” subepithelial or hinge region electron dense deposits are noted, their absence does not exclude an infectious etiology. The degree of foot process effacement in endocarditis-associated glomerulonephritis ranges from none to severe, with approximately equal proportions of none, mild, moderate, and severe [49].

Small electron dense deposits within the glomerular mesangium (arrows) (osmium tetroxide, ×12,000)

Subendothelial electron dense deposit in a patient with diffuse proliferative glomerulonephritis associated with prosthetic aortic valve Streptococcus viridans infective endocarditis (osmium tetroxide, ×12,000)
Clinicopathologic Correlation
Infectious Agent and Biopsy Findings
Interestingly, although there was no statistical difference between the occurrences of staphylococcal or streptococcal species on blood cultures between pauci-immune cases and those with immune complex deposition in our series, all cases with Bartonella, Coxiella, or Cardiobacterium on culture had immunoglobulin and C3 deposition by immunofluorescence [49]. We did not find significant associations between the bacterial agent on culture and the various light microscopic patterns of glomerulonephritis except that most cases with Bartonella, Coxiella, Cardiobacterium, or Gemella had crescentic glomerulonephritis and 3/4 cases of culture-negative endocarditis patients had crescentic glomerulonephritis. Similarly, Bookman et al. [4] and Liapis (referenced in [23]) presented 4 cases of B. henselae endocarditis-associated necrotizing and crescentic glomerulonephritis which mimicked vasculitis by light microscopy, with C3 staining by immunofluorescence and mesangial and subendothelial deposits by electron microscopy [4, 23].
Immunopathology
The immunopathology of endocarditis-associated glomerulonephritis has not been well characterized previously beyond identification of IgG and C3 deposition in an immune complex pattern [25–28, 48]. Recent biopsy series suggest that more complex pathogenic mechanisms are involved. Although C3 staining was positive in virtually the entire cohort in our large renal biopsy series, staining for IgG was present in only 34% and in fewer than 21% of those with the most severe crescentic lesions [49]; in fact, IgM was equal to IgG as the most commonly noted immunoglobulin (34%), and showed higher mean staining intensity when positive (2.0) compared to IgG (1.8) (Fig. 4.15, Tables 4.3 and 4.4). A lack of immunoglobulin staining in crescentic endocarditis-associated glomerulonephritis has been noted in more than one study [48, 49]. The finding of prominent C3 staining and the presence of readily detectable immune deposits by EM are more consistent with the C3-dominant pattern of immune deposition commonly seen in infection-related GN in general [51]. Some C3 deposition can also be seen in ANCA-associated vasculitis in 33–85% of cases; however, electron microscopy usually shows no or only few deposits [70, 71, 74].

Glomeruli with necrosis and cellular crescent formation from a biopsy with crescentic glomerulonephritis involving 75% of glomeruli, associated with tricuspid valve Streptococcus mitis infective endocarditis in a 43-year-old female intravenous drug user. ANCA serology was negative. Immunofluorescence microscopy showed 2–3+ IgM and C3 in a granular mesangial and capillary wall pattern. The patient was treated with antibiotics and had a full renal recovery at 6 months (Jones methenamine silver; ×200)
Furthermore, there is as much inconsistency in the literature as there is controversy regarding the classification of glomerulonephritis as immune complex-type versus pauci-immune. In theory, the term “immune complex” would refer to complexes of both immunoglobulins together with complement components identified by tissue immunofluorescence study. One must observe when the term “immune complex-type” is reported yet immunofluorescence reveals C3 only without immunoglobulin staining. Though large amounts of serum immunoglobulins and circulating immune complexes may be formed as a result of bacteremia, this does not necessarily imply deposition within the kidney by immunofluorescence.
Interestingly, despite the now known association of IgA-dominant infection-associated glomerulonephritis occurring with staphylococcal infections in both diabetic patients and nondiabetics [72, 73] and the fact that staphylococcal infections are now the most common causative agent for endocarditis-associated glomerulonephritis, IgA is present in less than one third of cases of endocarditis-associated glomerulonephritis by immunofluorescence.
Crescentic Glomerulonephritis and Differential Diagnosis of Vasculitis
Initiating mechanisms that lead to crescent formation have been simplified to antibodies (including ANCA via activating neutrophils and anti-GBM) and immune complexes, however, more complex and heterogeneous mechanisms are likely triggers to glomerular injury. Today, when a pauci-immune crescentic glomerulonephritis is present, the emphasis is on ANCA-associated glomerulonephritis and it can be easily forgotten that glomerular injury of various etiologies can result in crescent formation. Indeed, crescentic glomerulonephritis has been recognized by others as a final and fatal pathway of several etiologically diverse glomerular disease processes [75]. The common initiating mechanism is rupture or compromise of glomerular capillary walls, allowing inflammatory mediators to enter Bowman’s space and stimulate epithelial proliferation. The presence of fibrin is an indication that plasma constituents have entered as well. In time, the cells of the crescent are replaced by collagen as evidenced by the evolution of cellular crescents to fibrocellular and then fibrous crescents. Rather than being a specific disease, necrotizing and crescentic glomerulonephritis is the most severe form of glomerular inflammation observed histologically [76]. Today, we know the aggressive nature of this lesion and the importance of excluding ANCA-associated disease when a crescentic glomerulonephritis is present. After all, ANCA-associated disease is the most common cause of pauci-immune crescentic glomerulonephritis [74, 77].
However, endocarditis-associated glomerulonephritis is an important entity to consider in the differential diagnosis given the significant morphologic and clinical overlap (Fig. 4.16). Importantly, the presence of a positive ANCA serology does not exclude the possibility of endocarditis-associated glomerulonephritis, as 25% of patients tested for ANCA were positive in our series. In another study, 20% of cases with endocarditis-associated pauci-immune necrotizing and crescentic glomerulonephritis were ANCA positive [48]. There have also been several recent case reports detailing this pitfall as well [37, 61, 78–83]. Of note, the forms of small vessel vasculitis that can accompany glomerulonephritis or that can occur associated with ANCA disease in the kidney, including necrotizing arteritis, necrotizing arteriolitis, and leukocytoclastic medullary angiitis were not present in any of the 62 patients in our renal biopsy series with endocarditis-associated glomerulonephritis. However, the skin manifestations of endocarditis including Osler’s nodes, Janeway lesions, and splinter hemorrhages can mimic cutaneous vasculitis associated with ANCA. In a study by Chirinos et al. of eight ANCA-positive patients with subacute bacterial endocarditis, seven had skin manifestations, most commonly purpura [78].

Necrosis and circumferential cellular crescent in a 30-year-old male with ANCA-negative diffuse crescentic glomerulonephritis associated with methicillin-resistant Staphylococcus aureus infective endocarditis (Masson’s trichrome stain; ×400)
Given that infectious organisms have long been thought to play a significant role in both the development and the activation of ANCA, the finding of a significant number of patients with both infective endocarditis and pauci-immune crescentic glomerulonephritis should perhaps not be surprising [74, 84, 85]. Renal biopsies with pauci-immune crescentic glomerulonephritis associated with strong C3 staining should raise the possibility of endocarditis. However, even though C3 staining is very common in biopsies with a crescentic pattern (that is, it is sensitive), it is not specific in that renal biopsy case series from documented ANCA-associated glomerulonephritis show glomerular C3 staining in 33–85% of cases [70, 71, 74]. Of course, the best preserved glomeruli should be evaluated and interpreted by both immunofluorescence and electron microscopy, as C3 may be entrapped within areas of scarring, and even immunoglobulins can become entrapped within areas of fibrinoid necrosis. Therefore, it is important for the clinician and renal pathologist alike to always interpret biopsy findings in the context of clinicopathologic correlation and to maintain a high index of suspicion for the changing face of infective endocarditis-associated glomerulonephritis, especially considering the potential adverse outcome if a patient with endocarditis was mistakenly treated for ANCA-associated glomerulonephritis with cytotoxic agents in lieu of antibiotics.
Diagnostic Challenges of Endocarditis and Endocarditis-Associated Glomerulonephritis
The clinical identification of infective endocarditis can be very difficult. In one report, infective endocarditis was unrecognized in almost 20% of cases at the time of nephrology consult [48]. Also, in the most recent large autopsy series, infective endocarditis was not diagnosed until autopsy in 38.2% of cases [16]. Though they also examined their cases pre- and post-echocardiography availability, the introduction of echocardiography did not reduce the undetected diagnosis rate in their autopsy series. These studies are in disagreement with current reliance on either the original or the modified Duke criteria for the diagnosis of endocarditis [50, 86, 87]. Fernandez Guerrero et al. [16] attribute this to the common absence of fever, cardiac murmurs, and other clinical features considered characteristic of infective endocarditis. Transthoracic echocardiogram is less sensitive than transesophageal. If transthoracic echocardiogram is negative and there is suspicion for endocarditis, transesophageal echocardiogram has to be performed [64, 88]. Additionally, the prevalence of negative blood cultures among patients with endocarditis ranges from 2.5 to 31% [65] and, in one report, 19% of patients with culture-negative endocarditis were afebrile [66]. Although knowledge of both the clinical and pathologic spectrum of glomerulonephritis in patients with infective endocarditis in the current era is expanding, including the frequency of acute kidney injury and of crescentic glomerulonephritis, these clinical diagnostic challenges suggest that the immunologic mechanisms that underlie endocarditis-associated glomerulonephritis are more complex than previously appreciated. Perhaps the spectrum of pathological findings is in part due to the spectrum of infectious agents and pathophysiology as well.
As previously mentioned, another challenge the pathologist and clinician alike encounter is the morphologic overlap between renal biopsy findings in ANCA-associated glomerulonephritis and infective endocarditis-associated glomerulonephritis, and the fact that 20–25% of patients with infective endocarditis-associated glomerulonephritis can have positive ANCA serology [48, 49]. Furthermore, noninfective ANCA-associated endocarditis is yet another complicating factor when considering the differential diagnosis of bacterial endocarditis [78].
Pathogenesis
Several questions regarding the pathogenesis of the glomerulonephritis in patients with infective endocarditis have been raised. Initially, the glomerulonephritis was believed to be embolic in nature. Subsequently for many years, an underlying immune complex pathogenesis was assumed based on immunofluorescence findings of granular IgG and C3 deposits in glomeruli [25–28]. However, our largest renal biopsy series to date supports a primary immune complex mechanism in only a minority of patients, a conclusion also reached by others [48]. Several possibilities may explain when glomerular immune complex (IC) formation does occur; these include passive trapping of ICs from the circulation, formation of ICs in situ following prior localization of exogenous cationic bacterial antigens, or reactivity of an IgG antibody with endogenous components of the glomerulus itself as occurs in membranous nephropathy or anti-glomerular basement membrane antibody disease [89]. In the latter case, molecular mimicry between glomerular and bacterial constituents would likely be involved, thus making the process autoimmune in nature [52]. In a majority of cases, it is likely that formation of ICs in glomeruli is not the principal pathogenic event given the paucity of IgG deposition found in cases of severe glomerulonephritis and the probable alternate pathway mechanism of complement activation.
Several potential mechanisms could explain how glomerular tissue injury occurs in patients with infective endocarditis without IgG deposition. Bacterial antigens could localize in glomeruli independently of antibody and cause injury through initiation of activating the plasmin system or direct activation of the alternate complement pathway via mannose-binding lectin, thus producing a C3-dominant nephropathy. This is the case of the Streptococcal pyogenic exotoxin B antigen incriminated in post-Streptococcal glomerulonephritis [52, 90]. Staphylococcal super-antigens are also capable of causing direct tissue injury in the absence of immune deposits, especially to endothelial cells [91]. No studies of biologic activity or localization of bacterial antigenic proteins in infective endocarditis have yet to be performed.
Another possible mechanism that has been reported from several sources could be formation of the associated ANCA antibody in patients with infection [61, 78]. Bacterial infections that are well-known to lead to ANCA-positive serology include suppurative lung disease, and infections with Pseudomonas, Klebsiella, Escherichia Coli, and Ross River virus [74, 84, 85, 92]. High levels of cytokines secondary to the infection may prime neutrophils and monocytes to be activated by ANCA when present, therefore result in a synergistic inflammatory process [93]. This concept is supported by worsening glomerulonephritis and increased levels of circulating tumor necrosis factor-alpha in mice with anti-myeloperoxidase (MPO)-related glomerulonephritis after injection of bacterial lipopolysaccharide [94]. Induction of antibodies to complementary peptides of the target antigen (auto-antigen complementarity) leading to anti-idiotypic antibodies that react with self-proteins such as proteinase 3 (PR3) has been postulated for infectious agents such as Staphylococci, which then can produce autoimmune tissue injury without depositing in glomeruli [95]. If these ANCA antibodies are pathogenic in these patients rather than a secondary phenomenon, they are believed to damage glomeruli indirectly by activating neutrophils in the microvasculature. The activated neutrophils then release complement-activating factors which lead to alternative pathway activation involving the C5a receptor [84, 85, 96]. Another consideration is the consequence of coinfection by hepatitis C virus (HCV) in some of these patients. Chronic HCV infection can lead to prolonged antigen stimulation and severe autoimmune manifestations including induction of ANCA against MPO, PR3, and bactericidal permeability increasing protein and cathepsin G [83, 97, 98].
Lastly, the recent explosion of interest in glomerulopathies with a dominance of C3 deposition has clarified the role of both inherited and acquired abnormalities in complement-regulatory proteins, such as complement factor H (CFH), in contributing to unregulated activation of the alternative complement pathway and thus deposition of complement proteins in glomeruli [99]. Initiation of complement activation by infections in the presence of inherited or acquired abnormalities in complement regulation has been documented to lead to persistent, chronic C3 nephropathies, with similar pathologic appearances to many of the patients with documented infective endocarditis-associated glomerulonephritis [100]. Therefore, some of the lesions seen in endocarditis-associated glomerulonephritis could reflect an underlying complement-regulatory protein dysfunction.
Another unique feature of endocarditis-associated glomerulonephritis is that these occur during the course of infection rather than a latent reaction seen weeks after as in other etiologies of infection-associated glomerulonephritis. Perhaps this in part has to do with the protracted course that can occur in endocarditis or that when the glomerulonephritis is detected the infection had already been going on for some time.
Treatment and Outcome
Treatment
The presence of both a serious infection and a serious glomerulonephritis produces a challenging therapeutic dilemma. Certainly treatment of the infection is paramount, though no clear guidelines exist as to whether the addition of steroids with or without cytotoxic agents is helpful or harmful. There are case reports of successful use of plasmapheresis in endocarditis-associated glomerulonephritis [39, 101]. One report of an ANCA-negative S. viridans endocarditis-associated diffuse crescentic glomerulonephritis with C3 and C1q staining by immunofluorescence showed dramatic improvement with plasmapheresis [42]. Others have reported using plasmapheresis plus immunosuppression [44], while some report therapeutic success with antibiotics alone [45]. Also reported is a case of ANCA-positive Streptococcus bovis and Neisseria subflava infective endocarditis in a patient with vasculitic purpura showing resolution of skin lesions and renal recovery with antibiotic therapy alone [102].
Treatment data was obtained from 48 of 62 patients with endocarditis-associated glomerulonephritis in our study, and consisted of antibiotics in 71% of patients and antibiotics plus immunosuppressive therapy in 29%, with the latter comprised of combinations of prednisone, methylprednisone, and/or Cytoxan. Only one patient was treated with antibiotics plus prednisone and also received plasma exchange. Surgical treatment was performed in 21% of patients including seven with valve replacement and three with valve repair. More details of the treatment are provided in Chap. 5.
Follow-up and Outcome
Ultimately, the prognosis of a patient with endocarditis-associated glomerulonephritis most likely has more to do with the various extra renal manifestations, such as brain and lung involvement, than with the renal findings. In our renal biopsy study of 62 patients with endocarditis-associated glomerulonephritis, follow-up and outcome data were available in 45 patients with an average follow-up term of 21 months (range 0.5–84 months). For outcome analysis, end-stage renal disease was defined as requiring renal replacement therapy, persistent renal dysfunction was defined by elevation of serum creatinine 0.2 mg/dL above baseline levels or follow-up creatinine >1.2 mg/dL (for those in whom baseline levels were unavailable), and complete recovery was defined as normalization of serum creatinine to baseline levels or to creatinine ≤1.2 mg/dL (for those patients in whom baseline creatinine were unavailable). Of these 45 patients, eleven died (25%); 5 progressed to end-stage renal disease (11%), 15 had persistent renal dysfunction (33%) and 14 had complete renal recovery (31%) (Table 4.5).
Of the eleven patients that died, one was a three-year-old child and ten were adults (age range 31–79 years, mean 61); seven of these deaths occurred within two months of biopsy. The valve involved by endocarditis was the aortic valve in five patients, tricuspid valve in three, mitral valve in two, and combined tricuspid and mitral valves in one. Four patients had a prosthetic cardiac valve. Common clinical findings in all eleven patients that died include fever and vegetations by echocardiogram, as well as a combination of various other clinical findings. The organisms on culture included C. burnetii, Gemella species, Bartonella, and S. viridans, with the remainder Staphylococcal species. Over half were treated with antibiotics alone (60%) and less than half (40%) with antibiotics and immunosuppression. There were no clinicopathologic trends useful in differentiating the patients that died versus surviving patients (Figs. 4.17 and 4.18). Among surviving patients, those with higher percentages of globally sclerotic glomeruli, more interstitial fibrosis, and higher average serum creatinine at biopsy had worst outcomes.

Global endocapillary hypercellularity in a 66-year-old male with aortic valve Coxiella burnetii endocarditis. The patient was c-ANCA positive and had normal serum complement levels. The patient succumbed to his illness and died 1.5 months after the biopsy was performed (hematoxylin and eosin; ×400)

Diffuse endocapillary hypercellularity in a 31-year-old male with methicillin-sensitive Staphylococcus aureus infective endocarditis. The patient was ANCA negative, and was treated with antibiotics, prednisone, and plasma exchange, leading to a full renal recovery (periodic acid-Schiff; ×100)
While a second attack of infection-associated glomerulonephritis may be unusual as is the case with post-Streptococcal glomerulonephritis, in our study, two patients (3%) were found to have recurrent attacks of endocarditis-associated glomerulonephritis (data not previously reported). Both patients were females in their late 40s with history of intravenous drug use and hepatitis C virus infection. One had methicillin sensitive S. aureus (MSSA) pulmonic and tricuspid valve endocarditis treated with antibiotics leading to full recovery of renal function, followed by recurrent MSSA endocarditis two years later requiring tricuspid valve replacement. The other patient had Enterococcus mitral valve endocarditis treated with antibiotics and five months later mitral valve replacement and AV fistula, followed by recurrent infective endocarditis and infected shunt with fever, methicillin resistant S. aureus bacteremia, seizures, and stroke.
Abbreviations
- IC:
-
Immune complex
- MPGN:
-
Membranoproliferative glomerulonephritis
- ANA:
-
Anti-nuclear antibody
- ANCA:
-
Anti-neutrophil cytoplasmic antibody
- C3:
-
Complement component 3
- C4:
-
Complement component 4
- GN:
-
Glomerulonephritis
- Ig:
-
Immunoglobulin
- MPO:
-
Myeloperoxidase
- MRSA:
-
Methicillin-resistant Staphylococcus aureus
- MSSA:
-
Methicillin-sensitive Staphylococcus aureus
- PR3:
-
Proteinase-3
References
Schoen FJ, Mitchell RN. The heart. In: Kumar V, Abbas AK, Fausto N, Robbins SL, Cotran RS, editors. Robbins and cotran pathologic basis of disease. Vol. 1. 7th ed. Philadelphia, PA: Saunders Elsevier; 2005. p. 555–618.
Ebright JR, Komorowski R. Gonococcal endocarditis associated with immune complex glomerulonephritis. Am J Med. 1980;68:793–6.
Perez-Fontan M, Huarte E, Tellez A, Rodríguez-Carmona A, Picazo ML, Martinez-Ara J. Glomerular nephropathy associated with chronic Q fever. Am J Kidney Dis. 1988;11(4):298–306.
Bookman I, Scholey JW, Jassal SV, Lajoie G, Herzenberg AM. Necrotizing glomerulonephritis caused by “Bartonella henselae” endocarditis. Am J Kidney Dis. 2004;43(2):e25–30.
Elzouki AY, Akthar M, Mirza K. Brucella endocarditis associated with glomerulonephritis and renal vasculitis. Pediatr Nephrol. 1996;10(6):748–51.
Burch GE, Colcolough HL. Progressive coxsackie viral pancarditis and nephritis. Ann Intern Med. 1969;71(5):963–70.
Nasr SH, Markowitz GS, Stokes MB, Said SM, Valeri AM, D’Agati VD. Acute postinfectious glomerulonephritis in the modern era. Medicine. 2008;87(1):21–32.
Lohlein M. Ueber hāmorrhagische nierenaffektioned bei chronischer ulzerözer endokarditis. Med Klin. 1910;6:375–9 (German).
Baehr G. Glomerular lesions of subacute bacterial endocarditis. J Exp Med. 1912 4/1/1912;15(4):330–47. PubMed PMID: 19867526. Pubmed Central PMCID: PMCID: PMC2124924.
Bell ET. Glomerular lesions associated with endocarditis. Am J Pathol. 1932;8(6):639–69.
Langhans T. Ueber die Veränderungen der Glomeruli bei der Nephritis nebst einigen Bemerkungen über die Entstehung der Fibrincylinder. Archiv für pathologische Anatomie und Physiologie und für klinische Medicin. 1879;76(1):85–118.
Purdy CW. Bright’s disease and allied affections of the kidneys. Philadelphia: Lea Brothers; 1886.
Oertel H. The anatomic histological processes of Bright’s disease and their relation to the functional changes. Cal State J Med. 1914;12(8):351.
Volhard F, Fahr T. Die Brightsche Nierenkrankheit. Berlin: Springer; 1914.
Eknoyan G, Lister BJ, Kim HS, Greenberg SD. Renal complications of bacterial endocarditis. Am J Nephrol. 1985;5:457–69.
Fernandez Guerrero ML, Alvarez B, Manzarbeitia F, Renedo G. Infective endocarditis at autopsy: a review of pathologic manifestations and clinical correlates. Medicine. 2012;91(3):152–64. PubMed PMID: 22543628. Epub 2012/05/01. eng.
Neugarten J, Baldwin DS. Glomerulonephritis in bacterial endocarditis. Am J Med. 1984;77(2):297–304.
Rodriquez-Iturbe B, Burdmann EA, Barsoum RS. Glomerular diseases associated with infection. In: Floege J, Johnson RJ, Feehally J, editors. Comprehensive clinical nephrology. 4th ed. St. Louis: Elsevier Saunders; 2010. p. 662–74.
Appel GB, Radhakrishnan J, D’Agati VD. Secondary glomerular disease. In: Taal MW, Chertow GM, Marsden PA, Skorecki K, Yu ASL, Brenner BM, editors. Brenner & Rector’s the kidney. Vol. 1. 9th ed. Philadelphia: Elsevier; 2012. p. 1246–7.
Rogers TE, Rakheja D, Zhou XJ. Glomerular diseases associated with nephritic syndrome and/or rapidly progressive glomerulonephritis. In: Zhou XJ, Laszik Z, Nadasdy T, D’Agati VD, Silva FG, editors. Silva’s diagnostic renal pathology. New York: Cambridge University Press; 2009. p. 178–228.
D’Agati VD, Jennette JC, Silva FG. Non-neoplastic kidney diseases. Silver Springs: American Registry of Pathology; 2005.
Farris III BA. Endocarditis. In: Colvin RB, editor. Diagnostic pathology: kidney diseases. Manitoba: Amirsys; 2011. p. Section 2 108–13.
Nadasdy T, Silva FG. Acute post-infectious glomerulonephritis and glomerulonephritis caused by persistent bacterial infection. In: Jennette JC, Olson JL, Schwartz MM, Silva FG, editors. Heptinstall’s pathology of the kidney. Vol. 1. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2007. p. 372–80.
Spain DM, King DW. The effect of penicillin on the renal lesions of subacute bacterial endocarditis. Ann Intern Med. 1952;36(4):1086–9.
Cordeiro A, Costa H, Laginha F. Immunologic phase of subacute bacterial endocarditis a new concept and general considerations. Am J Cardiol. 1965;16(4):477–81.
Bayer AS, Theofilopoulos AN. Immunopathogenetic aspects of infective endocarditis. Chest. 1990;97(1):204–12.
Gutman RA, Striker GE, Gilliland BC, Cutler RE. The immune complex glomerulonephritis of bacterial endocarditis. Medicine. 1972;51(1):1–25.
Keslin MH, Messner RP, Williams RC. Glomerulonephritis with subacute bacterial endocarditis. Immunofluorescent studies. Arch Intern Med. 1973;132(4):578–81.
O’Connor DT, Weisman MH, Fierer J. Activation of the alternate complement pathway in S. aureus infective endocarditis and its relationship to thrombocytopenia, coagulation abnormalities, and acute glomerulonephritis. Clin Exp Immunol. 1978;34(2):179–87. PubMed PMID: 737901. Pubmed Central PMCID: PMC1537484.
Morel-Maroger L, Sraer JD, Herreman G, Godeau P. Kidney in subacute endocarditis pathological and immunofluorescence findings. Arch Path. 1972;94(3):205–13.
Boulton-Jones JM, Sissons JGP, Evans DJ, Peters DK. Renal lesions of subacute infective endocarditis. Br Med J. 1974;2(5909):11–4. PubMed PMID: 4595180. Pubmed Central PMCID: PMC1610141.
Lee LC, Lam KK, Lee CT, Chen JB, Tsai TH, Huang SC. “Full house” proliferative glomerulonephritis: an unreported presentation of subacute infective endocarditis. J Nephrol. 2007;20(6):745–9.
Nasr SH, Fidler ME, Valeri AM, Cornell LD, Sethi S, Zoller A, et al. Postinfectious glomerulonephritis in the elderly. J Am Soc Nephrol. 2011;22(1):187–95.
Toth T. Crescentic involved glomerulonephritis in infective endocarditis. Int Urol Nephrol. 1990;22(1):77–8.
Montseny J, Meyrier A, Kleinknecht D, Callard P. The current spectrum of infectious glomerulonephritis: experience with 76 patients and review of the literature. Medicine. 1995;74(2):63–73.
Miyata E, Nakayama M, Amano K, HIrano T, Uesugi N. A case of infectious endocarditis-associated crescentic glomerulonephritis with intracranial hemorrhage. J Nephrol. 2010;23(6):738–42. PubMed PMID: 20155718.
Bonaci-Nikoloic B, Andrejevic S, Pavlovic M, Dimcic Z, Ivanovic B, Nikolic M. Prolonged infections associated with antineutrophil cytoplasmic antibodies specific to proteinase 3 and myeloperoxidase: diagnostic and therapeutic challenge. Clin Rheumatol. 2010;29(8):893–904.
Sadikoglu B, Bilge I, Kilicaslan I, Gokce MG, Emre S, Ertugrul T. Crescentic glomerulonephritis in a child with infective endocarditis. Pediatr Nephrol. 2006;21:867–9.
Couzi L, Morel D, Deminiere C, Merville P. An unusual endocarditis-induced crescentic glomerulonephritis treated by plasmapheresis. Clin Nephrol. 2004;62(6):461–5.
Kannan S, Mattoo TK. Diffuse crescentic glomerulonephritis in bacterial endocarditis. Pediatr Nephrol. 2001;16:423–8.
Osafune K, Takeoka H, Kanamori H, Koshiyama H, Hirose K, Hanada M, et al. Crescentic glomerulonephritis associated with infective endocarditis: renal recovery after immediate surgical intervention. Clin Exp Nephrol. 2000;4(4):329–34.
Daimon S, Mizuno Y, Fujji S, Mukai K, Hanakawa H, Otsuki N, et al. Infective endocarditis-induced crescentic glomerulonephritis dramatically improved by plasmapheresis. Am J Kidney Dis. 1998;32(2):309–13.
Ades L, Akposso K, Costa de Beauregard MA, Haymann JP, Mougenot B, Rondeau E, et al. Bacterial endocarditis associated with crescentic glomerulonephritis in a kidney transplant patient: first case report. Transplantation. 1998;66(5):653–4.
Rovzar MA, Logan JL, Ogden DA, Graham AR. Immunosuppressive therapy and plasmapheresis in rapidly progressive glomerulonephritis associated with bacterial endocarditis. Am J Kidney Dis. 1986;7(5):428–33.
Orfila C, Lepert JC, Modesto A, Goudable C, Suc JM. Rapidly progressive glomerulonephritis associated with bacterial endocarditis: efficacy of antibiotic therapy alone. Am J Nephrol. 1993;13(3):218–22.
Gao GW, Lin SH, Lin YF, Diang LK, Lu KC, Yu FC, et al. Infective endocarditis complicated with rapidly progressive glomerulonephritis. Chin Med J. 1996;57(6):438–42.
Agarwal A, Clements J, Sedmak DD, Imler D, Nahman JNS, Orsinelli DA, et al. Subacute bacterial endocarditis masquerading as type III essential mixed cryoglobulinemia. J Am Soc Nephrol. 1997;8:1971–6.
Majumdar A, Chowdhary S, Ferreira MA, Hammond LA, Howie AJ, Lipkin GW, et al. Renal pathological findings in infective endocarditis. Nephrol Dial Trans. 2000;15:1782–7.
Boils CL, Nasr SH, Walker PD, Couser WG, Larsen CP. Update on endocarditis-associated glomerulonephritis. Kidney Int. 2015;87(6):1241–9.
Li JS, Sexton DJ, Mick N, Nettles R, Fowler VGJ, Ryan T, et al. Proposed modifications to the Duke criteria for the diagnosis of infective endocarditis. Clin Infect Dis. 2000;30(4):633–8.
Nasr SH, Radhakrishnan J, D’Agati VD. Bacterial infection–related glomerulonephritis in adults. Kidney Int. 2013;83(5):792–803.
Couser WG, Johnson RJ. The etiology of glomerulonephritis: roles of infection and autoimmunity. Kidney Int. 2014;86(5):905–14.
Nast CC. Infection-related glomerulonephritis: changing demographics and outcomes. Adv Chronic Kidney Dis. 2012;19(2):68–75. PubMed PMID: 22449343. Epub 2012/03/28. eng.
Tang SC, Lai KN. The pathogenic role of the renal proximal tubular cell in diabetic nephropathy. Nephrol Dial Transplant. 2012;27(8):3049–56. PubMed PMID: 22734110. Epub 2012/06/27. eng.
Nadasdy T, Hebert LA. Infection-related glomerulonephritis: understanding mechanisms. Semin Nephrol. 2011;31(4):369–75. PubMed PMID: 21839370. Epub 2011/08/16. eng.
Bor DH, Woolhandler S, Nardin R, Brusch J, Himmelstein DU. Infective endocarditis in the U.S., 1998–2009: a nationwide study. PLoS One. 2013;8(3):e60033. PubMed PMID: 23527296. Pubmed Central PMCID: 3603929.
Johnson JA, Boyce TG, Cetta F, Steckelberg JM, Johnson JN. Infective endocarditis in the pediatric patient: a 60-year single-institution review. Mayo Clin Proc. 2012;87(7):629–35. PubMed PMID: 22766082. Pubmed Central PMCID: Pmc3497940. Epub 2012/07/07. eng.
Hanf W, Serre JE, Salmon JH, Fabien N, Ginon I, Dijoud F, et al. Rapidly progressive ANCA positive glomerulonephritis as the presenting feature of infectious endocarditis. Rev Med Interne. 2011;32(12):e116–8. PubMed PMID: 21277658 French.
Griffin KA, Schwartz MM, Korbet SM. Pulmonary-renal syndrome of bacterial endocarditis mimicking Goodpasture’s syndrome. Am J Kidney Dis. 1989;14(4):329–32.
Hurwitz D, Quismorio FP, Friou GJ. Cryoglobulinaemia in patients with infectious endocarditis. Clin Exp Immunol. 1975;19(1):131–41.
Tiliakos AM, Tiliakos NA. Dual ANCA positivity in subacute bacterial endocarditis. J Clin Rheum. 2008;14(1):38–40.
Hellmich B, Ehren M, Lindstaedt M, Meyer M, Pfohl M, Schatz H. Anti-MPO-ANCA-positive microscopic polyangiitis following subacute bacterial endocarditis. Clin Rheumatol. 2001;20(6):441–3.
Miranda-Filloy JA, Veiga JA, Juarez Y, Gonzalez-Juanatey C, Gonzalez-Gay MA, Garcia-Porrua C. Microscopic polyangiitis following recurrent Staphylococcus aureus bacteremia and infectious endocarditis. Clin Exp Rheumatol. 2006;24(6):705–6.
Shively BK, Gurule FT, Roldan CA, Leggett JH, Schiller NB. Diagnostic value of transesophageal compared with transthoracic echocardiography in infective endocarditis. J Am Coll Cardiol. 1991;18(2):391–7.
Fournier PE, Thuny F, Richet H, Lepidi H, Casalta JP, Arzouni JP, et al. Comprehensive diagnostic strategy for blood culture-negative endocarditis: a prospective study of 819 new cases. Clin Infect Dis. 2010;51:131–40.
Hoen B, Selton-Suty C, Lacassin F, Etienne J, Briançon S, Leport C, et al. Infective endocarditis in patients with negative blood cultures: analysis of 88 cases from a one-year nationwide survey in France. Clin Infect Dis. 1995;20(3):501–6. Pubmed Central PMCID: 7756467.
Berden AE, Ferrario F, Hagen EC, Jayne DR, Jennette JC, Joh K, et al. Histopathologic classification of ANCA-associated glomerulonephritis. J Am Soc Nephrol. 2010;21(10):1628–36. PubMed PMID: 20616173. Epub 2010/07/10. eng.
Churg J, Bernstein J, Glassock RJ. Renal disease: classification and atlas of glomerular diseases. 2nd ed. New York: Igaku-Shoin; 1995.
Roberts ISD, Cook HT, Troyanov S, Alpers CE, Amore A, Barratt J, et al. The Oxford classification of IgA nephropathy: pathology definitions, correlations and reproducibility. Kidney Int. 2009;76(5):546–56.
Vizjak A, Rott T, Koselj-Kajtna M, Rozman B, Kaplan-Pavlovcic S, Ferluga D. Histologic and immunohistologic study and clinical presentation of ANCA-associated glomerulonephritis with correlation to ANCA antigen specificity. Am J Kidney Dis. 2003;41(3):539–49.
Chen M, Xing G-Q, Yu F, Liu G, Zhao M-H. Complement deposition in renal histopathology of patients with ANCA-associated pauci-immune glomerulonephritis. Nephrol Dial Transplant. 2009;24:1247–52.
Satoskar AA, Nadasdy G, Plaza JA, Sedmak D, Shidham G, Hebert L, et al. Staphylococcus infection-associated glomerulonephritis mimicking IgA nephropathy. Clin J Am Soc Nephrol. 2006 Received March 28, 2006. Accepted September 3, 2006;1(6):1179–86. Epub Epub 2006 Oct 11.
Nasr SH, Markowitz GS, Whelan JD, Albanese JJ, Rosen RM, Fein DA, et al. IgA-dominant acute poststaphylococcal glomerulonephritis complicating diabetic nephropathy. Hum Pathol. 2003;34(12):1235–41.
Kambham N. Crescentic glomerulonephritis: an update on pauci-immune and anti-GBM diseases. Adv Anat Pathol. 2012;19(2):111–24.
Olsen S. Extracapillary glomerulonephritis. A semiquantitative lightmicroscopical study of 59 patients. Acta Pathol Microbiol Scandinavica Suppl. 1974;Suppl 249:7–19.
Harris AA, Falk RA, Jennette JC. Crescentic glomerulonephritis with a paucity of glomerular immunoglobulin localization. Am J Kidney Dis. 1998;32(1):179–84. PubMed PMID: 0272-6386/98/3201.
Tarzi RM, Cook HT, Pusey CD. Crescentic glomerulonephritis: new aspects of pathogenesis. Semin Nephrol. 2011;31(4):361–8. PubMed PMID: 21839369. Epub 2011/08/16. eng.
Chirinos JA, Corrales-Medina VF, Garcia S, Lichtstein DM, Bisno AL, Chakko S. Endocarditis associated with antineutrophil cytoplasmic antibodies: a case report and review of the literature. Clin Rheumatol. 2007;26(4):590–5.
Choi HK, Lamprecht P, Niles JL, Gross WL, Merkel PA. Subacute bacterial endocarditis with positive cytoplasmic antineutrophil cytoplasmic antibodies and anti-proteinase 3 antibodies. Arthritis Rheum. 2000;43(1):226–31.
Haseyama T, Imai H, Komatsuda A, Hamai K, Ohtani H, Kibira S, et al. Proteinase-3-antineutrophil cytoplasmic antibody (PR3-ANCA) positive crescentic glomerulonephritis in a patient with Down’s syndrome and infectious endocarditis. Nephrol Dial Transplant. 1998;13(8):2142–6.
Subra JF, Michelet C, LaPorte J, Carrere F, Reboul P, Cartier F, et al. The presence of cytoplasmic antineutrophil cytoplasmic antibodies (C-ANCA) in the course of subacute bacterial endocarditis with glomerular involvement, coincidence or association? Clin Nephrol. 1998;49(1):15–8.
Bauer A, Jabs WJ, Süfke S, Maass M, Kreft B. Vasculitic purpura with antineutrophil cytoplasmic antibody-positive acute renal failure in a patient with Streptococcus bovis case and Neisseria subflava bacteremia and subacute endocarditis. Clin Nephrol. 2004;62(2):144–8.
Lamprecht P, Schmitt WH, Gross WL. Mixed cryoglobulinaemia, glomerulonephritis, and ANCA: essential cryoglobulinaemic vasculitis or ANCA-associated vasculitis? Nephrol Dial Transplant. 1998;13(1):213–21.
Falk RJ, Jennette JC. ANCA disease: Where is the field heading? J Am Soc Nephrol. 2010;21(5):745–52.
Cartin-Ceba R, Peikert T, Specks U. Pathogenesis of ANCA-associated vasculitis. Curr Rheumatol Rep. 2012;14(6):481–93.
Mylonakis E, Calderwood SB. Infective endocarditis in adults. N Engl J Med. 2001;345(18):1318–30.
Bashore TM, Cabell C, Fowler V. Update on infective endocarditis. Curr Probl Cardiolo. 2006;31(4):274–352.
Nishimura RA, Otto CM, Bonow RO, Carabello BA, Erwin JPr, Guyton RA, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(22):2438–88.
Couser WG. Basic and translational concepts of immune-mediated glomerular diseases. J Am Soc Nephrol. 2012;23(3):381–99. PubMed PMID: 22282593. Epub 2012/01/28. eng.
Rodriguez-Iturbe B, Musser JM. The current state of poststreptococcal glomerulonephritis. J Am Soc Nephrol. 2008;19:1855–64.
Salgado-Pabon W, Breshears L, Spaulding AR, Merriman JA, Stach CS, Horswill AR, et al. Superantigens are critical for Staphylococcus aureus Infective endocarditis, sepsis, and acute kidney injury. mBio. 2013;4(4) (Ahead of print). PubMed PMID: 23963178. Pubmed Central PMCID: Pmc3747586. Epub 2013/08/22. eng.
Savige J, Pollock W, Trevisin M. What do antineutrophil cytoplasmic antibodies (ANCA) tell us? Best practice and research. Clin Rheumatol. 2005;19(2):263–76. PubMed PMID: 15857795. Epub 2005/04/29. eng.
Jennette JC, Falk RJ. Pathogenesis of the vascular and glomerular damage in ANCA-positive vasculitis. Nephrol Dial Transplant. 1998;13(Suppl 1):16–20.
Huugen D, Tervaert JW, Heeringa P. TNF-alpha bioactivity-inhibiting therapy in ANCA-associated vasculitis: clinical and experimental considerations. Clin J Am Soc Nephrol CJASN. 2006;1(5):1100–7. PubMed PMID: 17699331. Epub 2007/08/19. eng.
Pendergraft WF, Preston GA, Shah RR, Tropsha A, Carter CW Jr, Jennette JC, et al. Autoimmunity is triggered by cPR-3(105-201), a protein complementary to human autoantigen proteinase-3. Nat Med. 2004 Jan;10(1):72-9. PubMed PMID: 14661018. Epub 2003/12/09. eng.
Jennette JC, Falk RJ, Gasim AH. Pathogenesis of antineutrophil cytoplasmic autoantibody vasculitis. Curr Opin Nephrol Hypertens. 2011;20(3):263–70. PubMed PMID: 21422922. Epub 2011/03/23. eng.
Ramos-Casals M, Jara LJ, Medina F, Rosas J, Calvo-Alen J, Mana J, et al. Systemic autoimmune diseases co-existing with chronic hepatitis C virus infection (the HISPAMEC Registry): patterns of clinical and immunological expression in 180 cases. J Intern Med. 2005;257(6):549–57.
Wu YY, Hsu TC, Chen TY, Liu TC, Liu GY, Lee YJ, et al. Proteinase 3 and dihydrolipoamide dehydrogenase (E3) are major autoantigens in hepatitis C virus (HCV) infection. Clin Exp Immunol. 2016;128(2):347–52.
Bomback AS, Appel GB. Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nat Rev Nephrol. 2012;8(11):634–42. PubMed PMID: 23026947. Epub 2012/10/03. eng.
Sethi S, Fervenza FC, Zhang Y, Zand L, Meyer NC, Borsa N, et al. Atypical postinfectious glomerulonephritis is associated with abnormalities in the alternative pathway of complement. Kidney Int 2013;83(2):293–9. PubMed PMID: 23235567. Pubmed Central PMCID: 3561505. Epub 2012/12/14. eng.
McKenzie PE, Taylor AE, Woodroffe AJ, Seymour AE, Chan YL, Clarkson AR. Plasmapheresis in glomerulonephritis. Clin Nephrol. 1979;12(3):97–108.
Bauer A, Jabs WJ, Süfke S, Maass M, Kreft B. Vasculitic purpura with antineutrophil cytoplasmic antibody-positive acute renal failure in a patient with Streptococcus bovis and Neisseria subflava bacteremia and subacute endocarditis. Clin Nephrol. 2004;62(2):144–8.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Boils, C.L. (2017). Endocarditis-Associated Glomerulonephritis. In: Satoskar, A., Nadasdy, T. (eds) Bacterial Infections and the Kidney. Springer, Cham. https://doi.org/10.1007/978-3-319-52792-5_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-52792-5_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-52790-1
Online ISBN: 978-3-319-52792-5
eBook Packages: MedicineMedicine (R0)