
Abstract
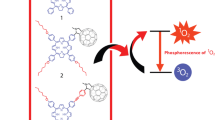
Porphyrin–fullerene dyads, among other donor-acceptor systems, evoke interest as prospective materials for the conversion of solar energy. The principle of their action can be envisioned as depicted in Fig. 20.1
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Porphyrin–fullerene dyads, among other donor-acceptor systems, evoke interest as prospective materials for the conversion of solar energy. The principle of their action can be envisioned as depicted in Fig. 20.1.

Schematic representation of the principle of solar-energy transformation by donor-acceptor dyads
First, the donor-acceptor molecule consumes a photon quantum to form an excited state. From that excited state, it can relax to a specific lower excited state where the electron density is redistributed, resulting in formal relocation of an electron from the donor part of the molecule, which has now a positive charge, to the acceptor part, which acquires a negative charge. If this charge-separated state has lasted enough, these molecules can then interact intermolecularly and act either as a photoredox catalyst or as a photoactive material in organic solar cells.
When we talk about porphyrin–fullerene dyads , in this case, the fullerene is an acceptor fragment while porphyrin is a donor counterpart. A survey of the literature on these compounds presented in our recent work [1] shows that the charge-separated-state lifetime depends strongly on the bridge (denoted in Fig. 20.1 as X) that connects the chromophores. When this bridge is such that it fixes the chromophores together in close contact, i.e., with the porphyrin face oriented towards the C60 sphere, let us call it a face-to-face contact, then the charge-separated state has a very short lifespan of 3.5 ns at the most [2]. A similar picture is observed when the bridge is long and flexible, so that the chromophores can achieve a close face-to-face contact [3]. The reported lifespan of the charge-separated state in this case does not exceed 650 ns. Only when the bridge keeps the chromophores apart, or fixes them in such an alignment that the porphyrin edge (and not the face) is oriented towards C60 sphere, what we shall call a “face-to-edge” alignment, only then does the lifetime of the charge-separated state reach several microseconds. There are only eight examples of such systems with lifetimes of the charge-separated state in the microsecond range [4].
However, it should also be mentioned that the large distance between porphyrin and fullerene fragments is not a sufficient condition to achieve a long lifetime of the charge-separated state. When the bridge enables continuous conjugation of the π-systems of the chromophores, the lifespan of the charge-separated state is significantly reduced.
Based on this analysis, we proposed three criteria to be met to construct a molecule that would be able to form charge-separated state with a long lifetime: the molecule should have fixed and large inter-chromophore distance, conjugation of the chromophores should be avoided, and the alignment of the chromophores should be face-to-edge like.
We proposed porphyrin–fullerene dyad connected by a pyrrolo[3,4-c]–pyrrole bridge (Fig. 20.2) as a compound that should meet all the criteria above, so let us check it. It has a rigid bridge that fixes the chromophores at a large distance, the sp3-carbons that keep the π-systems apart, and N-substitution plus-linear linker that ensures face-to-face orientation of the π-systems. We also added some additional components: the ester groups in the bridge are solubilizing groups. They are added to make the molecule soluble in conventional organic solvents. Another supplement is the aryl groups in the porphyrin core: they are added to enable electronic-structure modulation. Let me refer you to our papers mentioned at the beginning of my talk for the story of the synthesis of these compounds. I will note here that the three compounds we shall discuss now differ only in the aryl substituents, which are phenyl (1a), 4-bromophenyl (1b), and 4-methoxyphenyl (1c). R-substituent is ethyl for every compound.

Designed porphyrin–fullerene dyads for solar-energy conversion
Now, to the photophysical properties. Let us check if these products indeed can form a charge-separated state with a long lifespan.
The absorption spectra of the synthesized porphyrin–fullerene dyads represent the typical porphyrin spectra with Soret band at 415 nm and Q bands in the region of 500–700 nm. Fluorescence spectra with emission close to 650 nm are quite common for porphyrins, as well. So, there is no interaction of the chromophores in the ground state and no charge-separated state so far.
An interesting result was obtained when recording the absorption spectra of a thin film of one of these compounds on a glass support in a dry form (Fig. 20.3, left). On the insert here, you can see a very weak and very broad band that we attributed to the intermolecular charge transfer band. The evidence for that is that this band disappears when we immerse this film in an acetonitrile medium. Still, even should this band correspond to a charge transfer process, this is an intermolecular process, not the intramolecular one we were looking for.

Inter- versus intramolecular charge transfer. Absorption spectra of a thin film of compound 1a on ITO/glass support in dry form and immersed in MeCN (left). Transient absorption 500-ns ∆A spectra (Near-IR range) (right) [1]
So, we proceed further. To locate the charge-separated state resulting from intramolecular charge transfer, we performed a transient-absorption and emission-spectroscopy study of the synthesized dyads (Fig. 20.4). According to this study, when we irradiate the porphyrin-fullerene dyads, they are excited to the first excited-singlet state, which has quite a short lifespan of 2 ns. This excited state can then either relax back by a radiative or irradiative transition, or it can undergo intersystem crossing, giving us a triplet state which has a much longer lifetime of several microseconds. The latter was detected by transient absorption at around 450 nm. Either from this new triplet state or from the initially formed first excited singlet state, another intersystem crossing leads to a lower excited state, which is detectable by transient absorption at ca. 950 nm (Fig. 20.5), which is within the characteristic region of the fullerene radical-anion absorption. The lifespan of the charge-separated state corresponding to this band was found to be 4 μs for methoxy-substituted compound 1c and 2 μs for the benchmark compound 1a. Surprisingly, no charge-separated state (denoted on the scheme as CS) was observed for bromine-substituted compound 1b.

Schematic representation of photo dynamics in porphyrin–fullerene dyads 1 according to transient absorption and emission spectroscopy data

950-nm kinetic traces of the porphyrin–fullerenes 1a–c in deoxygenated (CH2Cl)2 upon 532-nm excitation [1]
We supposed that the reason might lie in the electronic structure of the dyads. To obtain insight into that, we performed DFT calculations (B3LYP 6-31G(d)) on the model dyads 1′ that differ from the real objects by an R-substituent in the ester group, which is methyl instead of ethyl. In Fig. 20.6, you can see the fragment of the energy diagram of molecular orbitals of model compound 1′a. What is interesting is that almost all the orbitals here are mostly located either on fullerene or porphyrin fragments, with HOMO being located on porphyrin and LUMO located on the fullerene part.

Fragment of the energy diagram of molecular orbitals of compound 1′a
Now, we can visualize the photo dynamics occurring in porphyrin–fullerene dyads 1a–c. When the molecule in the ground state is excited with visible light, the electron from, for example, the highest occupied molecular orbital (MO 466) goes to the appropriate level in terms of energy and symmetry-vacant molecular orbital, e.g., to MO 471, giving us the first singlet-excited state, which has a short lifespan as we have already seen, and it can undergo inter-system crossing, giving us the first triplet state. Alternatively, the first singlet-excited state can undergo another process with the electron falling from MO 471 to MO 467. MO 471 is mostly located on porphyrin while MO 467 is mostly located on fullerene. Hence, this transition gives us the charge-separated state. Of course, this is a simplification of the real process, yet it helps to understand what is going on upon interaction of the dyad with light and to formulate the working hypothesis that explains the absence of the charge-separated state in the case of bromine-substituted dyad 1b. We suppose that the processes discussed above compete with each other. The absence of the charge-separated state, in case of the bromine- substituted dyad 1b, means that either formation of the triplet state is accelerated, for example due to the heavy-atom effect, or formation of the charge-separated state is decelerated, or both these effects take place.
To find possible reasons for deceleration of the charge-separated state formation in the case of the bromine-substituted compound 1b, we performed time-dependent DFT calculations that provided us with information about the first lowest 50 excited states; the diagram of the energy of the states is presented on Fig. 20.7. You can see that all the excited states can be separated into several groups: the first two columns are composed of the states that involve the transition between orbitals located mostly on porphyrin moiety, giving us either singlet- (first column) or triplet-excited (second column) states. In the next two columns, there is the same story, but the orbitals are located mostly on the fullerene core. Finally, the last column gives us the formal transitions from the orbitals located on porphyrin to the orbitals located on fullerene. The first four columns are pairs of singlet and triplet states. In the fifth group of states, there are also singlet and triplet states, but we could not see any difference in energy between them, so we combined them into one column.

Energy diagram of the excited states of compounds 1′a–c. The graphics are based on a fragment of the diagram [1]
Now we can discuss the possible reason for deceleration of the process of the charge- separated-state formation in the case of the bromine-substituted compound 1b. As you can see from this diagram, the driving force for the electron transfer, which is the difference between the first excited-singlet state and the charge-separated state, is the lowest one for the bromine-substituted compound 1b and the largest one for methoxy-substituted compound 1c. Yet, the effect that this difference should have on the electron-transfer rate (20.1) depends on the ratio between the Gibbs energy of the reaction (which includes the difference in the electron energy of the two states as the main term) and the reorganizational energy, according to Marcus theory [5].
If the absolute value of the Gibbs energy of the electron transfer is less than the reorganizational energy, then we are in what is called the normal Marcus parabola region where the larger the driving force, the larger the rate constant or the smaller the driving force, the smaller the rate constant. However, if the absolute value of the Gibbs energy is larger than the reorganizational energy, then we turn to the inverted Marcus parabola region where the larger the Gibbs energy the slower the process.
So we have to check in which region we are. We had to estimate the Gibbs energy and the reorganizational energy. The formulae that we used for these estimations are given in (20.2) and (20.3). The one given in (20.2) is based on the Rhem-Weller approach [6], while the assessment in (20.3) estimates Gibbs energy as just the electronic energy difference between the two states. The last estimation can be justified by a notion that the main contribution to the Gibbs energy of a reaction is given by the electronic energy term.
The results are given in Table 20.1. The estimation of Gibbs energy in the fourth column is obtained through the experimental data on cyclic voltammetry and absorption spectrum (20.2), while the estimation given in the seventh column is the one based on the electronic energy of the states (20.3). Though there are some differences between the absolute values, we have the same trend in both cases: the Gibbs energy of the forward electron transfer decreases in the sequence: 1c > 1a > 1b, that is, the Gibbs energy is the largest for the methoxy-substituted compound and the smallest for the bromine-substituted dyad.
Next, we have estimated the reorganizational energy λ. This value has two terms: the outer reorganizational energy λ out and the inner reorganizational energy λ in. The last one is quite difficult to calculate, so we used the literature estimation reported in the work of Williams [7]. According to that report, the inner reorganizational energy falls within 0.2–0.7 electron volt. The outer reorganizational energy (20.4) can be easily calculated [5] with the geometrical parameters of our compounds that we obtained from the DFT optimized structure (Fig. 20.8). Using these data, we calculated reorganizational energy in a number of solvents (Table 20.2) and found that, in a polar solvent like dichloroethane, the reorganizational energy should fall within the range 1.1–1.6 electron volts. Now let us compare it with the estimations for the Gibbs energy:

Structural parameters in DFT (B3LYP/6-31G(d)) optimized structure of compound 1′a
You can see that, in each case, both for estimation of Gibbs energy based on experimental data and on DFT calculations, the absolute value of the Gibbs energy is less than the reorganizational energy. That means we are in the normal Marcus-parabola region, and the trend for the rate constant must be the same as the trend for the driving force. So, unlike compounds with proton and methoxy substituents, the rate of charge-separated state formation in the bromine-substituted compound might not be sufficient for a successful competition with other relaxation processes.
So, we have designed, synthesized, and studied novel porphyrin–fullerene dyads. We found that, in favorable cases, we have a lifespan of the charge-separated state in a microsecond range, a range which is quite good. Yet, we also found that the structural criteria that we formulated were not sufficient, and another factor should be also taken into account: the energy of the charge-separated state should be lowered to reduce the relative rate of the competitive processes.
References
A.S. Konev, A.F. Khlebnikov, P.I. Prolubnikov, A.S. Mereshchenko, A.V. Povolotskiy, O.V. Levin, A. Hirsch, Chem. Eur. J. 21, 1237–1250 (2015)
D.I. Schuster, P. Cheng, P.D. Jarowski, D.M. Guldi, C. Luo, L. Echegoyen, S. Pyo, A.R. Holzwarth, S.E. Braslavsky, R.M. Williams, G. Klihm, J. Am. Chem. Soc. 126, 7257–7270 (2004)
M.E. El-Khouly, J. Hasegawa, A. Momotake, M. Sasaki, Y. Araki, O. Ito, T. Arai, J. Porph. Phthalocyan. 10, 1380 (2006)
Y. Kobori, Y. Shibano, T. Endo, H. Tsuji, H. Murai, K. Tamao. J. Am. Chem. Soc. 131, 1624–1625 (2009)
R.A. Marcus, N. Sutin, Biochim. Biophys. Acta 811, 265–322 (1985)
D. Rehm, A. Weller, Isr. J. Chem. 8, 259–271 (1970)
R.M. Williams, Fullerenes as electron accepting components in supramolecular and covalently linked electron transfer systems. Ph.D. thesis, Faculty of Science, Van’t Hoff Institute for Molecular Sciences, The Univeristy of Amsterdam, Amsterdam, (1996)
A.S. Konev, A.F. Khlebnikov, T.G. Nikiforova, A.A. Virtsev, H. Frauendorf, J. Org. Chem. 78, 2542–2552 (2013)
Acknowledgements
I would like to acknowledge Dr. A. V. Povolotskiy and Dr. A. S. Mereschenko who performed the transient absorption experiments, my colleagues, and collaborators whose contribution to this work is greatly appreciated: Prof. A. F. Khlebnikov, Mr. P.I. Prolubnikov, Dr. O. V. Levin, and Prof. A. Hirsch, and of course thank you for your kind attention.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Konev, A. (2017). Novel Porphyrin–Fullerene Covalent Dyads Capable of Forming Charge-Separated States on a Microsecond Lifetime Scale. In: Yamanouchi, K. (eds) Progress in Photon Science. Springer Series in Chemical Physics, vol 115. Springer, Cham. https://doi.org/10.1007/978-3-319-52431-3_20
Download citation
DOI: https://doi.org/10.1007/978-3-319-52431-3_20
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-52430-6
Online ISBN: 978-3-319-52431-3
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)