
Abstract
The initiation of migraine may be a central or peripheral process. In favor of a peripheral process are the discovery of neuronal pathways from scalp to dura and meninges to cortex, the inhibition in rats of cortical activation by botulinum toxin via Transient Receptor Potential Vanilloid type 1 (TRPV1) and Transient Receptor Potential cation channel, subfamily A, member 1 (TRPA1) receptors, and the profoundly effective migraine prevention seen rapidly with anti-calcitonin gene-related peptide (CGRP) monoclonal antibodies (mABs), which do not cross the blood–brain barrier and therefore have only initial peripheral effects. In favor of a central process are the activation of central neuromodulatory pathways and nuclei, specifically in the dorsal pons. The genesis of aura is activation of N-methyl-D-aspartate receptor (NMDA) glutamate receptors and cortical spreading depression (CSD). The ability to suppress CSD is associated with acute and preventive effects by drugs and neuromodulation devices, but whether this suppression alone is sufficient for either clinical effect is unresolved. Migraine pain is due to both meningeal vasodilation and neurogenic inflammation. Presynaptic release of CGRP, vasoactive intestinal peptide (VIP), substance P (SP), neurokinin A, and likely, pituitary adenylate cyclase activating polypeptide-38 (PACAP-38) results in these processes. Triptans and ergots prevent release of CGRP, reverse CGRP-induced vasodilation, and interfere with return of the pain signal from periphery to brainstem. Anti-CGRP drugs and biologics can terminate migraine acutely (gepants) or prevent migraine (monoclonal antibodies). A trigemino-parasympathetic or trigeminal autonomic reflex arc involves efferents from the superior salivatory nucleus (SSN) synapsing in the sphenopalatine ganglion (SPG), and then post-synaptic neurons proceeding to sinus, ocular, and nasal organs. Activation of this reflex, which involves VIP, results in sinus-like symptoms or cranial autonomic symptoms and signs in migraine, and nociceptive afferents return signals to the brain via the first division of the trigeminal nerve or ophthalmic nerve. The exit of parasympathetic efferents via the SPG allows for targeting of this ganglion for acute and preventive treatment of migraine via blocks, ablation, or neuromodulation.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
Introduction
The anatomy and the pathways in the pathophysiology of migraine, while extensively explored in the past several decades, remain not fully connected. Migraine is generally an inherited neurologic disorder, with both variable genetics and penetrance. The accepted wisdom is that a tendency to neuronal hyperexcitability is what is common to all forms. The onset of an attack of migraine, or the persistence of daily headache in chronic migraine, may be initiated in the brainstem or may begin peripherally in the meninges. The role of cortical activation preceding an attack is also debated.
Migraine is principally a neuronal disorder. Neurons activate the process. The consequence of the excitation can result in vascular, hormonal, and autonomic manifestations, but the initiation is likely neurological. The old idea that migraine was a vascular process has been replaced by the concepts of a neurovascular syndrome, predominantly neuronal.
In favor of a peripheral process at migraine onset are the discovery of neuronal pathways from scalp to dura and meninges to cortex, the inhibition in rats of cortical activation by botulinum toxin via Transient Receptor Potential Vanilloid type 1 (TRPV1) and Transient Receptor Potential cation channel, subfamily A, member 1 (TRPA1) receptors, and the profoundly effective migraine prevention seen rapidly with anti-calcitonin gene-related peptide (CGRP) monoclonal antibodies (mABs) which do not cross the blood–brain barrier and therefore have only initial peripheral effects. In favor of a central process are the activation of central neuromodulatory pathways and nuclei found functionally in migraine, specifically in the dorsal pons.
The genesis of aura is activation of N-methyl-D-aspartate receptor (NMDA) glutamate receptors and cortical spreading depression or depolarization (CSD). The ability to suppress CSD is associated with acute and preventive effects by drugs and neuromodulation devices, but whether this suppression alone is sufficient for either clinical effect is unresolved.
Migraine pain is due to both meningeal vasodilation and neurogenic inflammation. Presynaptic release of CGRP, vasoactive intestinal peptide (VIP), substance P (SP), neurokinin A, and likely, pituitary adenylate cyclase activating polypeptide-38 (PACAP-38) results in these processes. Triptans and ergots prevent release of CGRP, reverse CGRP-induced vasodilation, and interfere with return of the pain signal from periphery to brainstem. Anti-CGRP drugs and biologics can terminate migraine acutely (gepants) or prevent migraine (monoclonal antibodies).
A trigemino-parasympathetic or trigeminal autonomic reflex arc involves efferents from the superior salivatory nucleus (SSN) synapsing in the sphenopalatine ganglion (SPG), and then post-synaptic neurons proceeding to sinus, ocular, and nasal organs. Activation of this reflex, which involves VIP, results in sinus-like symptoms or cranial autonomic symptoms and signs in migraine, and nociceptive afferents return signals to the brain via the first division of the trigeminal nerve or ophthalmic nerve. The exit of parasympathetic efferents via the SPG allows for targeting of this ganglion for acute and preventive treatment of migraine via blocks, ablation, or neuromodulation.
Pain Mechanisms and Anatomy
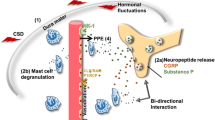
In the case of migraine, it may be reasonable to start with what is clearly known, and then work backwards. The pain mechanisms of migraine are peripheral, in the meninges. Presynaptic activation by serotonin 1D (5-HT 1D) receptors results in the release of CGRP, VIP, substance P (SP), neurokinin A, and likely, PACAP-38 [1, 2].
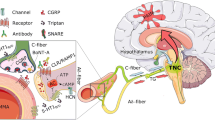
These peptides are neuro-inflammatory, and at least two, CGRP and PACAP-38, vasodilate. Therefore, post-synaptic effects in the meninges include the activation of the arachidonic acid cascade with its attendant inflammation, and vasodilation. The inflammation also includes mast cell degranulation; the vasodilation is associated with vessel fenestration and further release of plasma peptides. These two mechanisms, neurogenic inflammation and vasodilation, stimulate nociceptive afferents which carry pain signals centrally on the first division of the trigeminal nerve, the ophthalmic nerve (V1) (Fig. 2.1).

The neuroanatomy of migraine (see text for details)
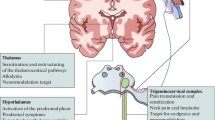
The afferent limb of the trigeminal system has its cell body in the trigeminal ganglion and synapses centrally, in the upper spinal cord and brain stem, in the trigeminal nucleus caudalis (TNC). This complex was named the trigeminocervical complex (TCC) by Goadsby and colleagues [3, 4]. The route from meninges to the TNC is the first-order afferent, and its firing is referred to as peripheral sensitization. Throbbing pain may be the clinical manifestation of peripheral sensitization [5].
In migraine activation, there is also stimulation of a brainstem-parasympathetic reflex, variously called the trigemino-parasympathetic reflex or the trigeminal autonomic reflex, in which parasympathetic efferents with cell bodies in the SSN synapse in the SPG. The exit via the SPG allows for targeting of this ganglion for acute and preventive treatment of migraine via blocks, ablation, or neuromodulation [6].
From the SPG, post-ganglionic parasympathetics travel to both the meninges and sinus-related organs, such as sinuses, eyes, and nose. These are second-order neurons [7]. Activation of this reflex, which involves VIP, results in sinus-like symptoms or cranial autonomic symptoms and signs in migraine. Nociceptive afferents return signals to the brain via the first division of the trigeminal nerve or ophthalmic nerve.
Once the pain mechanisms are engaged peripherally, the central anatomy is in turn sequenced. Pain signals ascend through upper brainstem to thalamus and on to cortex from the TCC. This allows for cortical reception of pain, activation of adjacent brainstem autonomic nuclei, parasympathetic stimulation resulting in sinus-like symptoms and signs, and, probably, a return to the meninges for further neuronal excitation and prolongation of the processes.
In addition, at some point the second-order neurons can start to fire without further peripheral input. This autonomous firing is referred to as central sensitization.
The clinical manifestation of central sensitization may be allodynia. Allodynia is the perception of non-painful stimuli as painful. Thus, at least part of the progression of migraine to central sensitization includes the patient finding touch painful (cutaneous allodynia), and photophonophobia may also be a form of allodynia [7].
Both CGRP and PACAP-38 levels rise during migraine attacks, and at least CGRP falls with treatment. CGRP and PACAP-38 both precipitate migraine attacks when administered intravenously [8,9,10].
There are CGRP and PACAP receptors centrally as well as peripherally, including on cranial parasympathetic ganglion and on the SPG, and these have been shown in both human and mammalian studies [10,11,12,13,14]. Edvinsson, commenting on this stated, “This provides evidence for an interaction between the parasympathetic and sensory systems” [1]. That is, these peptides have action peripherally and centrally, on the pain mechanisms and the processing, and specifically on the parasympathetic manifestations of migraine that clinically present as “sinus symptoms,” ictal ocular and nasal signs and complaints.
The meningeal location of CGRP and some of its receptors raised the question of whether antagonism to its function would be sufficient to terminate migraine attacks or whether central anti-CGRP activity would be necessary for clinical effect. At least seven CGRP receptor antagonists, often referred to as small molecules or gepants, have all been effective in the acute treatment of episodic migraine [15,16,17,18,19,20]. At the time of this writing (December 2016), one gepant, ubrogepant, taken as-needed, is in Phase 3 trials for acute treatment of migraine and another gepant, atogepant, taken daily, is in a Phase 2 trial for migraine prevention.
Triptans and ergots suppress release of CGRP via a 5-HT 1D mechanism. 5-HT 1D receptor agonism also interferes with transduction of pain signals from the periphery to the brainstem. Triptan and ergot agonism at 5-HT 1B receptors results in vasoconstriction, a reversal of CGRP-induced vasodilation.
Four mABs prevented migraine in Phase 2 trials. Three of them are antibodies against the CGRP peptide ligand itself [21,22,23]; one is against the CGRP receptor [24]. Three of four of the mABs have effectively prevented both episodic and chronic migraine. Yet, they are large molecules and do not cross the blood–brain barrier [25, 26]. This means that termination of migraine acutely and prevention of migraine chronically can occur peripherally, at the CGRP receptor in the meninges.
Aura and Migraine
Most auras are defined by the International Classification of Headache Disorders, 3rd Edition, Beta version, as reversible neurologic events, lasting 5–60 min, followed by or accompanied by migraine or non-migrainous headache [27]. The pathophysiology of migraine aura is an excitation of brain that is variously referred to as CSD or cortical spreading depolarization or cortical spreading depression.
CSD occurs with activation of the NMDA glutamate receptors. These receptors gate brain stimulation.
Most commonly, aura is visual and occurs with activation of the occipital cortex. The sequential excitation of the visual dominance columns of Hubel and Wiesel, which mediate edges, curves, movements, and other pre-formed visual percepts, account for the aura often being perceived as positive visual phenomena, although loss of vision, a negative visual phenomenon can also occur [28, 29].
Aura can occur elsewhere in the brain, and sensory and speech and language disturbances are next most common as typical auras. Auras can likely occur in brainstem and cerebellum as well.
In the wake of the depolarization, a wave of post-ictal depression of function occurs, and since this was first to be noted in physiology studies, the term CSD was coined, even though the primary event is excitation [30, 31]. Increased blood flow occurs with the depolarization, decreases with the post-ictal depression, and that decreased blood flow was mistakenly thought to be ischemia, while it is usually a 40% oligemia [32].
The familial hemiplegic migraines (FHM Types 1–3) all result in excess glutamate in the synapse. This excess glutamate is then available to stimulate NDMA receptors, and may account for the severity and duration of these more severe and prolonged auras.
The relationship of aura to migraine pain is not clear. Some migraineurs without clinical aura have CSD on functional imaging [33]. The clinical and pathophysiological questions are linked: does termination of CSD work in stopping migraine with and without aura, or just with aura?
Neuromodulation devices, such as single pulse transcranial magnetic stimulators and noninvasive vagal nerve stimulators, which both terminate CSD, sometimes appear to terminate or prevent migraine with and without aura, but these devices have other actions. A drug, tonabersat, aimed at inhibiting gap junctions or connexins necessary for propagation in CSD did not work to prevent both migraine with and without aura, although it did prevent migraine with aura alone [34]. Topiramate, valproate, propranolol, and amitriptyline significantly reduced CSD propagation speed, and methysergide (a long-acting ergot) showed a strong trend in doing so, but once again, do these medications work by this mechanism primarily, or secondarily [35]? Finally, magnesium and NMDA antagonists show some efficacy in acute and preventive treatment of migraine, but perhaps more so with aura [36, 37].
Burstein and colleagues [5] documented neurons connecting scalp, cortex, and meninges. In essence, they speculate a direct connection between cortical events such as aura and meningeal pain processes. This would account for both effects of anti-CGRP drugs on migraine and aura, and a possible conduit for botulinum toxin to suppress migraine mechanisms and aura.
In addition, activation of CGRP receptors leads to cytosolic calcium-dependent phosphorylation of glutamatergic NMDA-dependent neuronal activation. Thus, CGRP receptor activation indirectly leads to CSD [38, 39].
Zhang, Burstein, and colleagues found in rats that onabotulinumtoxinA inhibits C-type meningeal nociceptors via TRPV1 and TRPA1 activation. Activation of these receptors mediates release of SP and CGRP and modulates peripheral sensitization of nociceptors. There may be a link between the peripheral effects of botulinum toxin and anti-CGRP drugs and the central suppression of migraine and aura through physical and neurotransmitter processes [40, 41].
Genesis of Migraine
It is possible that the initial generation of migraine is central. Two functional MRI (fMRI) studies suggest a central generator in the area of the brainstem containing the dorsal raphe, locus ceruleus, and periaqueductal grey. One posited a central generator in the contralateral dorsal pons, [42] one in the same area in the ipsilateral [43]. The older study, done with Positron Emission Tomography (PET) was not as accurate in localization as the second study done with functional MRI, so the ipsilateral dorsal pons may the correct side.
There is continued controversy as to whether this area is where the migraine starts, or whether it is a central pain modulator. The argument over whether initiation of migraine is central or peripheral has been joined for decades [44].
The Burstein group suggests that migraine could begin peripherally, in the scalp or meninges, and communicate centrally to the cortex. The dramatic effectiveness of the anti-CGRP mAB biologics in migraine prevention, their effectiveness in both episodic and chronic migraine, even refractory chronic migraine with medication overuse headache, and the speed with which they work support this concept [45].
Conclusions
The initiation of migraine may be a central or peripheral process. In favor of a peripheral process are the discovery of neuronal pathways from scalp to dura and meninges to cortex, the inhibition in rats of cortical activation by botulinum toxin via TRPV1 and TRPA1 receptors, and the profoundly effective migraine prevention seen rapidly with anti-CGRP mABs which do not cross the blood–brain barrier and therefore have only initial peripheral effects. In favor of a central process are the activation of central neuromodulatory pathways and nuclei, specifically in the dorsal pons.
The genesis of aura is activation of NMDA glutamate receptors and CSD. CSD is a misnomer in that the activation associated with aura can be non-cortical and is an active depolarization. The ability to suppress CSD is associated with acute and preventive effects by drugs and neuromodulation devices, but whether this suppression alone is sufficient for either clinical effect is unresolved.
Migraine pain is due to both meningeal vasodilation and neurogenic inflammation. Presynaptic release of CGRP, VIP, SP, neurokinin A, and likely, PACAP-38 results in these processes. Triptans and ergots prevent release of CGRP, reverse CGRP-induced vasodilation, and interfere with return of the pain signal from periphery to brainstem. Anti-CGRP drugs and biologics can terminate migraine acutely (gepants) or prevent migraine (mABs).
A trigemino-parasympathetic reflex arc involves efferents from the SSN synapsing in the SPG, and then post-synaptic neurons proceeding to sinus, ocular, and nasal organs. Activation of this reflex, which involves VIP, results in sinus-like symptoms or cranial autonomic symptoms and signs in migraine, and nociceptive afferents return signals to the brain via the first division of the trigeminal nerve or ophthalmic nerve. The exit via the SPG allows for targeting of this ganglion for acute and preventive treatment of migraine via blocks, ablation, or neuromodulation.
References
Edvinsson L. Role of VIP/PACAP in primary headaches. Cephalalgia. 2013;33:1070–2.
Amin F, Hougaard A, Schytz H, Asghar M, Lundholm E, Parvaiz A, de Koning PJ, Andersen MR, Larsson HB, Fahrenkrug J, Olesen J, Ashina M. Investigation of the pathophysiological mechanisms of migraine attacks induced by PACAP38. Brain. 2014;137(Pt 3):779–94.
Goadsby PJ, Zagami AS. Stimulation of the superior sagittal sinus increases metabolic activity and blood flow in certain regions of the brainstem and upper cervical spinal cord of the cat. Brain. 1991;114:1001–11.
Goadsby PJ, Hoskin KL, Knight YE. Stimulation of the greater occipital nerve increases metabolic activity in the trigeminal nucleus caudalis and cervical dorsal horn of the cat. Pain. 1997;73:23–8.
Burstein R, Levy D, Jakubowski M. Effects of sensitization of trigeminovascular neurons to triptan therapy during migraine. Rev Neurol (Paris). 2005;161(6–7):658–60.
Cady R, Saper J, Dexter K, Manley HR. A double-blind, placebo-controlled study of repetitive transnasal sphenopalatine ganglion blockade with Tx360 as acute treatment for chronic migraine. Headache. 2015;55:101–16.
Jakubowski M, Levy D, Goor-Aryeh I, Collins B, Bajwa Z, Burstein R. Terminating migraine with allodynia and ongoing central sensitization using parenteral administration of COX1/COX2 inhibitors. Headache. 2005;45:850–61.
Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol. 1990;28:183–7.
Arulmani U, MaassenVanDenBrink A, Villalon CM, Saxena PR. Calcitonin gene-related peptide and its role in migraine pathophysiology. Eur J Pharmacol. 2004;500:315–30.
Schytz HW, Birk S, Wienecke T, et al. PACAP38 induces migraine-like attacks in patients with migraine without aura. Brain. 2009;132:16–25.
Edvinsson L, Elsa St, Suzuki N, et al. Origin and co-localization of nitric oxide synthase, CGRP, PACAP, and VIP in the cerebral circulation of the rat. Microsc Res Tech. 2001;53:221–8.
Csati A, Tajti J, Tuka B, et al. Calcitonin gene-related peptide and its receptor components in the human sphenopalatine ganglion—interaction with the sensory system. Brain Res. 2012;1435:29–39.
Tuka B, Helyes Z, Markovics A, Bagoly T, Szolcsányi J, Szabó N, Tóth E, Kincses ZT, Vécsei L, Tajti J. Alterations in PACAP-38-like immunoreactivity in the plasma during ictal and interictal periods of migraine patients. Cephalalgia. 2013;33:1085–95.
Vécsei L, Tuka B, Tajti J. Role of PACAP in migraine headaches. Brain. 2014;137:646–53.
Olesen J, Diener HC, Husstedt IW, Goadsby PJ, Hall D, Meier U, Pollentier S, Lesko LM. BIBN 4096 BS clinical proof of concept study group. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med. 2004;350:1104–10.
Connor KM, Shapiro RE, Diener HC, Lucas S, Kost J, Fan X, Fei K, Assaid C, Lines C, Ho TW. Randomized, controlled trial of telcagepant for the acute treatment of migraine. Neurology. 2009;73:970–7.
Diener HC, Barbanti P, Dahlöf C, Reuter U, Habeck J, Podhorna J. BI 44370 TA, an oral CGRP antagonist for the treatment of acute migraine attacks: results from a phase II study. Cephalalgia. 2011;31:573–84.
Hewitt DJ, Aurora SK, Dodick DW, Goadsby PJ, Ge YJ, Bachman R, Taraborelli D, Fan X, Assaid C, Lines C, Ho TW. Randomized controlled trial of the CGRP receptor antagonist MK-3207 in the acute treatment of migraine. Cephalalgia. 2011;31:712–22.
Marcus R, Goadsby PJ, Dodick D, Stock D, Manos G, Fischer TZ. BMS-927711 for the acute treatment of migraine: a double-blind, randomized, placebo controlled, dose-ranging trial. Cephalalgia. 2014;34:114–25.
Voss T, Lipton RB, Dodick DW, Dupre N, Ge JY, Bachman R, Assaid C, Aurora SK, Michelson D. A phase IIb randomized, double-blind, placebo-controlled trial of ubrogepant for the acute treatment of migraine. Cephalalgia. 2016;36:887–98.
Dodick DW, Goadsby PJ, Spierings EL, Scherer JC, Sweeney SP, Grayzel DS. Safety and efficacy of LY2951742, a monoclonal antibody to calcitonin gene-related peptide, for the prevention of migraine: a phase 2, randomised, double-blind, placebo-controlled study. Lancet Neurol. 2014;13:885–92.
Dodick DW, Goadsby PJ, Silberstein SD, Lipton RB, Olesen J, Ashina M, Wilks K, Kudrow D, Kroll R, Kohrman B, Bargar R, Hirman J, Smith J. ALD403 study investigators. Safety and efficacy of ALD403, an antibody to calcitonin gene-related peptide, for the prevention of frequent episodic migraine: a randomised, double-blind, placebo-controlled, exploratory phase 2 trial. Lancet Neurol. 2014;13:1100–7.
Bigal ME, Edvinsson L, Rapoport AM, Lipton RB, Spierings EL, Diener HC, Burstein R, Loupe PS, Ma Y, Yang R, Silberstein SD. Safety, tolerability, and efficacy of TEV-48125 for preventive treatment of chronic migraine: a multicentre, randomised, double-blind, placebo-controlled, phase 2b study. Lancet Neurol. 2015;14:1091–100.
Sun H, Dodick DW, Silberstein S, Goadsby PJ, Reuter U, Ashina M, Saper J, Cady R, Chon Y, Dietrich J, Lenz R. Safety and efficacy of AMG 334 for prevention of episodic migraine: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15:382–90.
Goldberg SW, Silberstein SD. Targeting CGRP: a new era for migraine treatment. CNS Drugs. 2015;29:443–52.
Silberstein S, Lenz R, Xu C. Therapeutic monoclonal antibodies: what headache specialists need to know. Headache. 2015;55:1171–82.
Headache Classification Committee of the International Headache Society (IHS). The international classification of headache disorders, 3rd edition (beta version. Cephalalgia. 2013;33:629–808.
Lashley K. Patterns of cerebral integration indicated by scotomas of migraine. Arch Neurol Psychiatry. 1941;46:331–9.
Hubel DH, Wiesel TN. Receptive fields of single neurones in the cat’s striate cortex. J Physiol. 1959;148:574–91.
Leao AAAP. Spreading depression of activation in the cerebral cortex. J Neurophysiol. 1944;7:359–90.
Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B, Kwong KK, Cutrer FM, Rosen BR, Tootell RB, Sorensen AG, Moskowitz MA. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001;98:4687–92.
Lauritzen M. Pathophysiology of the migraine aura. The spreading depression theory. Brain. 1994;117(Pt 1):199–210.
Woods RP, Iacoboni M, Mazziotta JC. Brief report: bilateral spreading cerebral hypoperfusion during spontaneous migraine headache. N Engl J Med. 1994;331:1689–92.
Hauge AW, Asghar MS, Schytz HW, Christensen K, Olesen J. Effects of tonabersat on migraine with aura: a randomised, double-blind, placebo-controlled crossover study. Lancet Neurol. 2009;8:718–23.
Ayata C, Jin H, Kudo C, Dalkara T, Moskowitz MA. Suppression of cortical spreading depression in migraine prophylaxis. Ann Neurol. 2006;59:652–61.
Noruzzadeh R, Modabbernia A, Aghamollaii V, Ghaffarpour M, Harirchian MH, Salahi S, Nikbakht N, Noruzi N, Tafakhori A. Memantine for prophylactic treatment of migraine without aura: a randomized double-blind placebo-controlled study. Headache. 2016;56:95–103.
Tepper SJ. Nutraceutical and other modalities for the treatment of headache. Continuum (Minneap Minn). 2015;21:1018–31.
Durham PL. CGRP-receptor antagonists–a fresh approach to migraine therapy? N Engl J Med. 2004;350:1073–5.
Tepper SJ, Stillman MJ. Clinical and preclinical rationale for CGRP-receptor antagonists in the treatment of migraine. Headache. 2008;48:1259–68.
Noseda R, Burstein R. Migraine pathophysiology: anatomy of the trigeminovascular pathway and associated neurological symptoms, cortical spreading depression, sensitization, and modulation of pain. Pain. 2013;154(Suppl 1):S44–53.
Zhang X, Strassman AM, Novack V, Brin MF, Burstein R. Extracranial injections of botulinum neurotoxin type A inhibit intracranial meningeal nociceptors’ responses to stimulation of TRPV1 and TRPA1 channels: are we getting closer to solving this puzzle? Cephalalgia. 2016;36:875–86.
Weiller C, May A, Limmroth V, Jüptner M, Kaube H, Schayck RV, Coenen HH, Diener HC. Brain stem activation in spontaneous human migraine attacks. Nat Med. 1995;1:658–60.
Afridi SK, Matharu MS, Lee L, Kaube H, Friston KJ, Frackowiak RS, Goadsby PJ. A PET study exploring the laterality of brainstem activation in migraine using glyceryl trinitrate. Brain. 2005;128(Pt 4):932–9.
Pietrobon D, Striessnig J. Neurobiology of migraine. Nat Rev Neurosci. 2003;4:386–98.
Bigal ME, Dodick DW, Krymchantowski AV, VanderPluym JH, Tepper SJ, Aycardi E, Loupe PS, Ma Y, Goadsby PJ. TEV-48125 for the preventive treatment of chronic migraine: Efficacy at early time points. Neurology. 2016;87:41–8.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Tepper, S.J. (2017). Anatomy and Pathophysiology of Migraine. In: Mehle, M. (eds) Sinus Headache, Migraine, and the Otolaryngologist. Springer, Cham. https://doi.org/10.1007/978-3-319-50376-9_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-50376-9_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-50375-2
Online ISBN: 978-3-319-50376-9
eBook Packages: MedicineMedicine (R0)