
Abstract
Migraine is a strongly disabling disease characterized by a unilateral throbbing headache lasting for up to 72 h for each individual attack. There have been many theories on the pathophysiology of migraine throughout the years. Currently, the neurovascular theory dominates, suggesting clear involvement of the trigeminovascular system. The most recent data show that a migraine attack most likely originates in the hypothalamus and activates the trigeminal nucleus caudalis (TNC). Although the mechanisms are unknown, activation of the TNC leads to peripheral release of calcitonin gene-related protein (CGRP), most likely from C-fibers. During the past year monoclonal antibodies against CGRP or the CGRP receptor have emerged as the most promising targets for migraine therapy, and at the same time established the strong involvement of CGRP in the pathophysiology of migraine. The viewpoint presented here focuses further on the activation of the CGRP receptor on the sensory Aδ-fiber, leading to the sensation of pain. The CGRP receptor activates adenylate cyclase, which leads to an increase in cyclic adenosine monophosphate (cAMP). We hypothesize that cAMP activates the hyperpolarization-activated cyclic nucleotide-gated (HCN) channels, triggering an action potential sensed as pain. The mechanisms behind migraine pain on a molecular level, particularly their importance to cAMP, provide clues to potential new anti-migraine targets. In this article we focus on the development of targets related to the CGRP system, and further include novel targets such as the pituitary adenylate cyclase-activating peptide (PACAP) system, the serotonin 5-HT1F receptor, purinergic receptors, HCN channels, adenosine triphosphate-sensitive potassium channels (KATP), and the glutaminergic system.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
A migraine attack most likely originates in the hypothalamus, leading to activation of the trigeminovascular system. |
The hallmark of trigeminovascular activation is release of calcitonin gene-related protein from trigeminal C-fibers, which we postulate sensitizes and activates Aδ-fibers via a cyclic adenosine monophosphate (cAMP) pathway. |
Current anti-migraine drugs in development target the trigeminovascular system, with particular focus on the cAMP signaling pathway. |
1 Introduction
A migraine attack is typically initiated by premonitory symptoms, followed by a unilateral throbbing headache lasting for up to 72 h [1]. There have been several hypotheses attempting to explain the pathophysiology of migraine over the years. Initially the vascular theory dominated, followed by a theory in which neurological aspects were in focus [2]. The current view integrates both of these, with general understanding of the importance of the trigeminovascular system. Recent advances both in preclinical and clinical research allow us to integrate the findings in a model that offers a potential explanation regarding the mechanism of pathophysiology of migraine (Fig. 1). The understanding of the mechanisms behind migraine and the migraine pain, particularly on a molecular level, provides hints about the areas where potential new targets can be found (Fig. 2). In the current article we present our view of the mechanism of migraine, from the trigger to the sensation of pain.

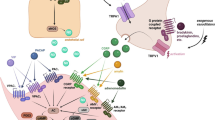
Current view of migraine pathophysiology and potential mechanisms of available specific treatments. The migraine attack is initiated with premonitory symptoms and activation of the hypothalamus. Following hypothalamic activation, the trigeminus nucleus caudalis (TNC) is activated. This leads to activation of the trigeminal ganglion (TG), most likely one-sided, and calcitonin gene-related peptide (CGRP) release. The CGRP release, here exemplified at the middle meningeal artery (MMA), leads to vasodilation. Furthermore, CGRP activates the calcitonin receptor-like receptor/receptor activity-modifying protein (CLR/RAMP1, the CGRP receptor) on the Aδ-fiber. The CGRP receptor activates adenylate cyclase (AC), increasing intracellular cyclic adenosine monophosphate (cAMP). The increase in cAMP leads to a hyper-excitability and a hypothesized activation of hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. cAMP increases the open-probability giving an action potential from the Aδ-fiber, which travels back to the TNC and is further sensed as pain. Sensitization of Aδ-fibers might, in addition, lead to normal stimuli, such as touch, being sensed as pain. The current specific treatment for migraine is the triptans, which prevent CGRP release, induce vasoconstriction on the MMA, and lead to hypo-excitability of the Aδ-fiber. The novel monoclonal antibodies bind either the CGRP receptors or CGRP directly and prevent the effects of CGRP. CGRP vesicular fusion is dependent on three exocytotic Soluble N-ethylmaleimide sensitive fusion Attachment Protein REceptor (SNAREs), which include SNAP25, syntaxin 1, and synaptobrevin. SNAP25 is cleaved by botulinum toxin serotype A (BoNT-A, Botox®), which prevents exocytosis. ATP adenosine triphosphate

Novel anti-migraine targets. The nerve fiber endings are exemplified here at the middle meningeal artery (MMA). Potential targets at the C-fibers lead to reduced calcitonin gene-related peptide (CGRP) release, most likely thorough reducing cyclic adenosine monophosphate (cAMP). Potential targets for agonists are ditans at the serotonin 5-HT1F receptor or purinergic receptors such as the P2Y13 receptor. Both these targets will also lead to hypo-excitability when expressed in the Aδ-fiber. Targets at the Aδ-fiber links to the same intracellular cAMP pathways. Further targets at the Aδ-fiber include the pituitary adenylate cyclase-activating peptide (PACAP, yellow) or PACAP receptor 1 (PAC1). One hypothesized target of cAMP is the hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. Increase in cAMP leads to hyperexcitability and action potentials through the HCN channel. Furthermore, increasing the activity of phosphodiesterase (PDE) will lead to breakdown of cAMP. Activating of adenosine triphosphate (ATP)-dependent potassium channels (KATP) leads to prolonged hyperpolarization and could trigger the HCN channel. Inhibiting these channels could have anti-migraine potential. AC adenylate cyclase, CLR/RAMP1 calcitonin receptor-like receptor/receptor activity-modifying protein
2 Triggering Migraine
A migraine attack is divided into four phases: (1) premonitory symptoms; (2) the aura (not in all patients); (3) the headache; and (4) the postdrome phase [3]. Referring to their migraine pain, patients will typically name triggers of their attacks such as stress, cheese, chocolate, wine, bright light, or lack of sleep [4, 5], but there is little evidence that these are the actual triggers of migraine. Experimental provocation using self-reported triggers only caused migraine with aura in a very small subgroup of patients [6]. These triggers are now considered part of the actual migraine attack [7]. As an example, the sensation of craving (e.g., for chocolate) might be a part of the migraine attack. Following this the migraineur will eat chocolate and report that the migraine attack was triggered by chocolate consumption. The craving of a special food might instead be due to preparations for an incoming attack. As the premonitory symptoms precede the headache phase, this stage has been widely studied in order to understand the real migraine trigger.
2.1 Central Triggering Mechanisms
In the search for the migraine trigger, the brainstem was proven to be crucially involved in migraine attacks [8, 9]. Although still highly relevant in understanding the migraine physiology, it is now currently accepted that the migraine trigger is further upstream. Based on the current understanding of the premonitory systems, the most likely modulator/initiator of migraine pain has been suggested to be the hypothalamus [10]. The most important study leading in this direction was performed by Schulte and May [11]. In their study, a single migraine patient was scanned for 30 days, and they found that the hypothalamus was more active in the final 24 h preceding the migraine attack [11]; a study with a larger cohort of patients is underway from the same authors. This activity further coupled strongly to the brainstem and down to the trigeminal nucleus caudalis (TNC) during the ictal stage. Although Raffaelli and Menon [12] might have been the first to suggest a link between the limbic system and migraine in 1975, this has gained renewed interest in light of the recent functional magnetic resonance imaging (fMRI) scans showing hypothalamic involvement. The limbic system, particularly the responses to stress and reward, has been suggested to play an important modulatory role in the hypothalamus [13]. We believe that the link between the limbic system and hypothalamus will be highly relevant for further understanding the underlying neurological origins of a migraine attack. An important part of the limbic system, the nucleus accumbens, has been linked to the placebo effect [14], which could explain this recurrent issue in clinical anti-migraine studies.
The fMRI scans in Schulte and May’s [11] study showed that hypothalamic activity precedes the migraine attack, with connections to the thalamus and different brainstem regions followed by activation of the TNC. TNC activation could further lead to increased activity of neurons that facilitate trigeminovascular pain transmission. However, we do not believe that the origin of migraine pain sensation lies in the TNC. First and foremost, this is because the TNC is out of reach of current anti-migraine medications [15, 16] (which we return to in Sect. 4.2.2). We therefore speculate that changes in the TNC cause further activation of the trigeminal ganglion (TG). A further mismatch in the communication or tuning of the TG might lead to sensitization and possibly triggering of calcitonin gene-related protein (CGRP) release from only one of the ganglia. From that point forward, the migraine attack is no longer only central, and we continue to postulate on the origin of this peripheral pain.
2.2 Peripheral Mechanism of Pain Sensation
Following the altered activity of the TNC, we postulate that the TG is activated. This appears to be associated with increased release of CGRP. Goadsby et al. [17] showed that trigeminal activation in cats caused CGRP release. Subsequently, a similar mechanism was seen in humans during a migraine attack and an increased release of CGRP was found in blood collected from the jugular vein [18]. The enhanced secretion of CGRP must originate from outside of the blood–brain barrier (BBB) as it is not permeable to the CGRP peptide [19]. Furthermore, a migraine treatment that was in the final stages of development at that time (sumatriptan) not only prevented migraine pain but simultaneously reduced the CGRP levels in blood [18]. Later, both in vitro and in vivo experiments on CGRP release showed the inhibitory effects of triptans [20, 21]. Therefore, it is clear that the migraine pain is associated with the release of CGRP, which is known to originate from the trigeminal C-fibers [22].
CGRP binds to calcitonin receptor-like receptor/receptor activity-modifying protein (CLR/RAMP1; the CGRP receptor), both of which are widely distributed in the trigeminovascular system [22]. Potential targets for CGRP include the middle meningeal artery [23, 24], mast cells [25], satellite glial cells [26], and, importantly, Aδ-neurons [27]. Regarding mast cells, it is worth mentioning that the full CGRP receptor appears to only be expressed in rodent mast cells and not in human mast cells [22]. When CGRP is released either locally or as a result of antidromic stimulation of the TG, one will observe middle meningeal vasodilation (origin of the vascular theory), vasodilation of arteries in sensory organs (which might lead to sensitization), mast cell degranulation (in rats), and possible activation of satellite glial cells (surrounding TG neurons). The increased release of CGRP could therefore explain most of the phenotypical changes observed during a migraine attack. Although CGRP can be linked to the symptoms observed, these changes cannot directly explain the migraine pain. This is exemplified by the fact that not all vasodilators cause migraine pain [28, 29].
CGRP being released from the C-fibers can directly activate the Aδ-neurons, and there is recent experimental evidence of this occurring [27, 30]. The Aδ-fibers transmit pain signals from the periphery and could very well be the origin of the migraine pain signal. With the current success of the novel migraine medications directly targeting the CGRP/CGRP receptors, we suggest that the pain must originate from the Aδ-fibers as these pain-transmitting neurons are the only nerve fibers containing CGRP receptors in the periphery [16, 22]. However, binding of CGRP to CLR/RAMP1 on the Aδ-fibers most likely causes an increase in cyclic adenosine monophosphate (cAMP), cAMP response element-binding protein (CREB), and p38 (mitogen-activated protein kinase), similar to the data on human and rat trigeminal neuronal bodies [31], which cannot itself lead to excitation of the neuron.
3 Generation of Pain
There are now strong data on CGRP being involved in triggering delayed migraine pain in migraine patients. Work from Ashina et al. [32] has nicely demonstrated the involvement of several molecules and pathways in migraine pain. The triggering of pain was originally linked to the vasodilatory effects of the migraine triggers [33, 34]. Although all known triggers are vasodilators, not all vasodilators trigger a delayed migraine attack [28, 29]. A review of the migraine triggers shows that they all link to activation of cAMP or cyclic guanosine monophosphate (cGMP) pathways. The current data show that CGRP and other neuropeptides such as pituitary adenylate cyclase-activating peptide (PACAP) can trigger a migraine attack in slightly more than 50% of migraineurs [35]. Furthermore, both cAMP [36, 37] (downstream of the CGRP receptor) and cGMP induction trigger migraine in around 80% of patients [38]. The most recent data show that the potassium channel opener levcromakalim causes migraine pain in 100% of the patients (preliminary results reported at the Migraine Trust International Symposium [MTIS] in London in 2018 [39]; ClinicalTrials.gov identifier NCT03228355 [40]). This order indicates that as we travel down the signaling pathway the success rate of triggering a migraine increases. Of note, cilostazol is not completely phosphodiesterase (PDE) 3 specific as it also inhibits adenosine uptake and increases extracellular adenosine [41]. Adenosine causes middle meningeal vasodilation ex vivo and in vivo [42, 43].
As mentioned earlier, cAMP activation does not cause pain per se. When CGRP was injected peripherally in the skin, there was no mention of pain generation (only vasodilation) [44]. Similar data were obtained from injection into the facial skin when CGRP alone caused no pain; this is in contrast to CGRP co-injection with Substance P or Substance P alone, which was painful [45, 46]. In addition, CGRP does not cause pain or migraine in healthy volunteers, only a weak headache associated with the vasodilation [47]. Therefore, a mechanism must exist that transmits the increased CGRP release, CGRP receptor activation, and cAMP into the sensation of pain, particularly in migraineurs. Here, we speculate on the origin of migraine pain. The peripheral fibers from the TG contain C-fibers that store CGRP and Aδ-fibers have CGRP receptors [22]. As the Aδ-fibers might be stimulated with CGRP, or artificially with cAMP/cGMP breakdown inhibitors, this initiates intracellular increase in these cyclic nucleotides. One channel group that is activated both by increases in cAMP and in cGMP is the hyperpolarization-activated cyclic nucleotide-gated (HCN) channels [48], which are known to be involved in neuropathic pain [49]. Increase in cAMP/cGMP increases the open-probability, and hence increases neuronal excitability and firing of the neurons [50, 51].
HCN channels are expressed in the TG, but most of the current knowledge originates from studies on the dorsal root ganglion (DRG). HCN channels are expressed in the DRG neurons and are involved in neuropathic pain generation from Aδ-fibers. The HCN expression profile is similar when comparing DRG and TG [52]. Furthermore, inflammation (Freund’s complete adjuvant applied to the dura) increases the expression of HCN channels in the TG, and Cho et al. [53] suggest that HCN1 and HCN2 channels are involved in inflammation-induced sensory neuron hyperexcitability. We hypothesize that CGRP leads to an increase in cAMP in the Aδ-fibers, leading to an increase in the open-probability of HCN channels, which induces triggering of spontaneous firing and generation of an action potential. The action potential is transmitted to the TNC and further to the central pain centers, and the perception of migraine pain occurs. This is exemplified by the signal after application of inflammatory soup at the dura, which travels through the TNC and increases the glutamate [54]. The increase in glutamate correlates with sensory thresholds on the face. In addition, it has been shown that p38, one of the downstream signaling molecules following CGRP receptor activation in trigeminal neurons [55], also modulates HCN function [56].
The recent review and hypothesis from Ashina et al. on the involvement of K+ channels in migraine pathophysiology [57], might seem counter-intuitive as the opening of K+ channels leads to neuronal hypo-excitability, exemplified by the effect in intracardiac neurons [58]. It has therefore been suggested by this group that the pain most likely originates from the induced arterial dilation [57]. We propose a different view that integrates the opening of K+ channels and the sensation of pain. HCN channels are, as the name suggests, activated by hyperpolarization. Adenosine triphosphate-sensitive potassium channel (KATP) openers (such as levcromakalim) increase the K+ permeability of the Aδ-fibers, the fibers hyperpolarize, and then the HCN channels will open. After a delay, the HCN channels will trigger an action potential as a response to prolonged hyperpolarization, which has been demonstrated in other systems [59]. The action potential generated by the HCN channel is sensed as pain [59, 60].
HCN channels might not be the only origin of pain. When CGRP is increasing the cAMP in the Aδ-fibers, they could also become hyper-excitable for other stimuli [30]. Aδ-fibers are known to be high-threshold receptors; they are only activated by severe mechanical stimulation or extreme heat (above + 45 °C) or cold (below + 15 °C). However, after being activated by CGRP, the threshold for these responses might be lowered and normal touch or hot/cold stimulation could now be perceived as pain. Preventing sensitizing of Aδ neuronal synapses was recently shown to be a possible mechanism of action of the CGRP antibodies [27]. We therefore believe that the search for new targets should be focused on modulators of neuropeptide signaling and cAMP/cGMP targets in the Aδ fibers. It is worth noting that cGMP inhibits breakdown of intracellular cAMP [61]; hence, the full pathophysiology could be explained by cAMP increases alone.
4 Novel Approaches to Known Targets
4.1 Serotoninergic Targets
The most used acute treatment for migraine is agonists for the serotonin (5-hydroxytryptamine [5-HT]) receptors, particularly those targeted against 5-HT1B/1D [62]. The triptans have been used as the first line for the acute treatment of migraine for nearly 30 years. They were originally developed as vascular constrictors, with high craniovascular selectivity. Indeed, the triptans constrict the middle meningeal artery, particularly through 5-HT1B receptors [63]. Advances in the understanding of migraine have expanded on the hypothesis behind the mechanisms of actions responsible for the therapeutic effects of the triptans. One of the first clinical experiments to illustrate a potential second mechanism of action showed that the anti-migraine effect of the triptans correlated with normalization of CGRP levels in migraine patients [18]. The authors therefore suggested that triptans might not only lead to vasoconstriction but also reduce the secretion of CGRP, and expanded on this hypothesis in several preclinical studies [64]. In line with this, the current understanding of the mechanism of action behind the triptans is that the activation of Gi-coupled 5-HT1B/1D receptors reduces the intracellular cAMP [65]. This would reduce CGRP release from C-fibers [20] and potentially reduce neuronal excitability in Aδ-fibers [66].
Although the triptans were developed as cranio-selective vasoconstrictors, they still cause vasoconstriction in other parts of the vasculature, particularly the coronary arteries [67, 68]. Therefore, there has been a focus on developing serotoninergic agonists without vasoconstrictive properties. Several of the triptans bind to the 5-HT1F receptor [69], and it is believed that the vasoconstrictive properties are linked mainly to 5-HT1B [63]. The 5-HT1F receptor is also expressed in the trigeminovascular system [70, 71]. Further development of targeted 5-HT1F led to development of LY344864 and LY334370; both drugs were developed as specific 5-HT1F antagonists that prevented dural protein extravasation [72, 73]. These agonists did not cause vascular constriction [72, 74] and LY334370 was proven to be effective in migraine [75]. Unfortunately, it caused liver toxicity in long-term use experiments in beagle dogs [76]. Further 5-HT1F agonist development led to LY573144, which was investigated by CoLucid (COL-144), now acquired by Eli-Lilly [77].
LY573144/COL-144 has been renamed lasmiditan, and very recently a phase III trial was completed with positive results [78]. Compared with placebo, more patients administered lasmiditan 200 mg were free of headache pain at 2 h after dosing (32.2% vs. 15.3%). Furthermore, compared with placebo, more patients administered lasmiditan 200 mg (40.7% vs. 29.5%) were free of their most bothersome symptom at 2 h after dosing. Interestingly, this study included more than 75% of patients with one or more cardiovascular risk factors. Adverse events were mostly mild or moderate in intensity, with the most common adverse events (≥ 2% than placebo) being dizziness, fatigue, lethargy, nausea, paresthesia, and somnolence [78]. This promising study provides class I evidence that lasmiditan increased the proportion of adult patients with migraine who were headache pain free at 2 h after treating an acute migraine attack. Hence, there is a promising future for the ‘ditans’—agonists targeting the 5-HT1F receptor.
Returning to the intracellular signaling pathway, the 5-HT1F receptor is also a Gi protein-coupled receptor [79] but, unlike the triptans, it is devoid of vasoconstrictive properties [74]. The newest ditan, lasmiditan, further prevents the release of CGRP from structures of the trigeminovascular system [80], suggesting that vasoconstrictive properties are not necessary for mitigation of migraine pain. In contrast, reduction of CGRP levels might be one of the hallmarks of potential migraine treatments [81].
4.2 Calcitonin Gene-Related Protein (CGRP) System
2018 was a remarkable year for the current treatment options for migraine as patients received the first treatment developed to directly modulate the CGRP-ergic part of the trigeminovascular system [81]. So far, all the anti-migraine medications targeting the CGRP system have been proven efficient [82]. In the early 2000s the first small-molecule antagonist against the CGRP receptor created huge enthusiasm for a novel treatment for migraine patients not responding to triptans [83]. Unfortunately, long-term studies on the oral compound telcagepant showed liver toxicity in some patients when used on a daily basis, and despite the migraine-preventive effect, its development was terminated [84]. The current anti-migraine treatments targeted against CGRP can be divided in two groups: gepants and monoclonal antibodies (mAbs).
4.2.1 Gepants
Although telcagepant failed to reach the market, the receptor target remains relevant, and currently there are two small-molecule gepants being investigated in acute migraine. Ubrogepant (MK-1602) and rimegepant (BMS-927711) are both chemically distinct from telcagepant, and both have positive results from a phase II clinical trial [85, 86]. The results from the phase III trials are not published yet but Tfelt-Hansen and Loder [87] beautifully summarize the data released from the trials so far. The major concern raised in their commentary is the effect size of these compounds, as they are not superior to the triptans. However, they might be very effective in a subset of patients. Atogepant (AGN-241689) is a third gepant with similar structure to ubrogepant, which is currently in trials as a prophylactic anti-migraine treatment with promising results (NCT02848326 [88]).
4.2.2 Monoclonal Antibodies
The mAb treatments for migraine have been a huge market hit, and hundreds of thousands of patients are currently being treated (mainly in the USA). The mAbs were recently approved in European countries but await political decisions on reimbursement policies. All the mAb trials for galcanezumab, fremanezumab, and eptinezumab, targeting CGRP itself, and erenumab, which targets the CLR/RAMP-1 receptor, were successful and surprisingly well-tolerated with few adverse effects [82]. This lack of adverse effects is partially due to the lack of permeability of antibodies in general through the BBB [89]. There is no clear proof that the BBB is altered in migraine attacks [15, 81]. Further, there must be body-wide compensatory mechanisms at play. In conclusion, the anti-migraine effect of the specific targeting of CGRP is very strong evidence for the involvement of CGRP in migraine pathology [81].
The safety aspects of CGRP antibodies over triptans have been discussed elsewhere [90]. In general, mAbs do not cause any adverse effects on blood pressure and do not lead to vasoconstriction. Whether preventing CGRP signaling during an ischemic event could lead to a worsened outcome of said event is not known [91]. Nevertheless, with the view of developing new treatments, one might question whether CGRP antagonists (either mAbs or gepants) target the same mechanisms as the triptans. In addition, does targeting CGRP signaling directly only lead to fewer adverse events, or does it further improve migraine outcome? A protocol used to study the effect of fremanezumab allowed up to 30% of patients using a stable dose of one migraine-preventive medication to continue these medications [92]. The fremanezumab trial showed similar reduction in migraine days as the other clinical trials on CGRP/CGRP receptor mAbs (summarized in Lambru et al. [93]), despite the inclusion of patients already using medications. The paper describing the results from the fremanezumab clinical trial does not report data that separates the patients using other medications. We believe that any difference in outcomes between patients using and not using preventing medications would have been reported (if observed), although the sample size may have been too low to observe such differences. It is our belief that both triptans and CGRP/CGRP receptor mAbs would target the same system in a majority of patients. Are there patients in whom either triptans or mAbs might be superior, or even in whom a combination could be favorable?
Triptans prevent the release of CGRP [18, 20] and, working through the same mechanism [65], would also theoretically prevent the release of other neuropeptides (such as PACAP), which has been shown to be clinically relevant in humans [94]. Hence, if CGRP is not the only neuropeptide involved in migraine, adding triptans might be beneficial. In addition, activation of 5-HT1B/1D/1F receptors will lead to a generally reduced neuronal excitability [66], which could be beneficial in patients treated with mAbs. Nevertheless, we believe that the group that will benefit most from mAbs treatment is patients in whom triptans do not lead to reduced CGRP release. The expression of 5-HT1B/1D/1F receptors might vary between patients (which is also observed in coronary arteries, with some standard error of the mean values being greater than 50% of the mean [67]), and this might be the cause of the low effect of triptans in some patients. To date, finding these patients can only be achieved by testing the effect of mAbs as there are currently no molecular explanations or markers available.
4.3 Botulinum toxin serotype A (Botox®)
Botulinum toxin serotype A (BoNT-A, Botox®) is a neurotoxin that is rapidly taken up in peripheral nerve endings by binding to the cell membrane [95]. Once present in the cytosol, BoNT-A is cleaved and the light chain proceeds to cleave, a Soluble N-ethylmaleimide sensitive fusion Attachment Protein REceptor (SNARE), which is important in vesicle docking. This prevents release of neurotransmitters [96], mainly within the cholinergic system. Regarding migraine, the exact site and mode of action is still unclear. Because the trigeminovascular system holds a key position in head pain, it is hypothesized that preventing CGRP release is the most likely/desirable mechanism of action [97]. BoNT-A has been approved in migraine following the positive outcome of two multicenter trials using subcutaneous administration into facial, temporal, and neck skin [98, 99]. Specific administration to tissues outside the calvaria (extracranial site of administration) in rats showed that BoNT-A could inhibit C-fiber stimulation induced by the transient receptor potential cation channel subfamily V member 1 (TRPV1) agonist capsaicin. BoNT-A had no effect on Aδ-fibers [100]. Combined, this indicates that the effect of BoNT-A is to lower the activity of the nociceptive C-fibers, putatively at an extracranial site. We postulate that although BoNT-A might not target the sensory nerves that are directly involved in the trigeminovascular dysregulation in migraine, it might lower their overall activity.
Novel BoNT-A treatments are currently on the way, with the most novel approach involving the development of various BoNT-A chimeras. BoNT-A and BoNT-E chimeras act more transiently and BoNT-E is a superior inhibitor of mediated neuropeptide release [101]. A second example, binary toxin (BiTox), is a synthetic recombinant BoNT-A chimera that appears to lack paralytic effects [102], while it suppresses evoked action potentials in trigeminovascular neurons [103]. Further developments include a substance P-conjugated BoNT-A protein (which targets neurokinin [NK]-1-positive neurons), which can be endocytosed by TG neurons in culture while retaining its activity [104]. A more efficient version of this molecule inhibited inflammatory and neuropathic pain [105]. However, NK-1 antagonists had no effect in migraine prevention [106], and therefore this might not be the right approach in migraine. In contrast, targeting BoNT-A towards TRPV1-positive fibers could be a more specific mode of action in preventing CGRP release. A secondary approach could be targeting BoNT-A to CGRP receptor-positive Aδ-neurons. This would prevent Aδ-fiber activity by reducing glutamate release, in a similar way as for the kynurenic pathway (see Sect. 5.4). Full characterization of the protein expression within the nociceptive cranial sensory system could uncover potential targets for novel selective BoNT-A.
5 Novel Targets
5.1 The Pituitary Adenylate Cyclase-Activating Peptide (PACAP) System
In addition to CGRP, the neuropeptide PACAP has been in focus as a potential anti-migraine target [107]. Initial investigations on neuropeptides and migraine showed that only CGRP was elevated during a migraine attack [18]. In a later study, elevated PACAP levels were detected in the ictal period relative to the attack-free period (where PACAP levels were actually significantly lower than in healthy volunteers) [94, 108]. Injection of both CGRP and PACAP causes migraine-like symptoms in migraine patients [35]. However, in the same study only PACAP significantly induced premonitory symptoms [35]. Therefore, PACAP might precede CGRP and be important in the early phase of a migraine attack.
Unlike PACAP, vasoactive intestinal peptide (VIP) does not lead to migraine when injected in migraine patients [28, 29]. Both PACAP and VIP can bind VPAC1 (VIP and PACAP receptor 1) and VPAC2 receptors, with similar affinity, but the PACAP receptor 1 (PAC1) binds PACAP with much higher affinity than for VIP [109]. This suggest that the induction of migraine by PACAP most likely occurs through the PAC1 receptor. As a side note, in dural vessel studies there seems only to be VPAC1/2 receptors mediating dilatation [110], suggesting that the PACAP migraine mechanisms are likely not related to vasodilation. In line with findings from the in vivo patient studies, Amgen produced and started clinical testing a mAb against the PAC1 receptors (AMG301), which is currently in a phase II trial (NCT03238781 [111]); no data are publicly available at this time. Similar to the CGRP-ergic system, a mAb directed against PACAP-38 itself has been developed and is in clinical trials by Alder (ALD1910). Data from preclinical studies on ALD1910 have just been published [112], showing positive results in an umbellulone-induced rat model of neurogenic vasodilation and parasympathetic lacrimation. Details of upcoming clinical studies have not been disclosed as yet.
5.2 Targeting the Cyclic Adenosine Monophosphate (cAMP) System
When reviewing the current literature, there is overwhelming evidence that the cAMP pathway is significantly involved in the pathophysiology of migraine. Both triptans and ditans target receptors that generally decrease cAMP levels [65, 79]. Known inducers of migraine are CGRP and PACAP, both binding to receptors that result in increasing cAMP levels in the cells where they are expressed [31, 113], and increasing cAMP directly (by inhibiting PDEs) leads to a migraine attack in migraineurs [36]. In addition, there is evidence that an increase in cGMP also leads to migraine-like attacks in migraineurs [38]. We believe that the molecular pathways activated in migraine are essential to understanding new treatments, at least from the perspective of treating peripheral symptoms.
There are three potential ways of targeting the cAMP system: (1) application of agonists to receptors that couple to Gi; (2) inhibition of adenylate cyclase, leading to prevention of cAMP production; and (3) enhancing breakdown of cAMP by stimulating PDE.
5.2.1 Purinergic Receptors
There are several potentially interesting receptors that can result in decreased cAMP. Here we focus on the purinergic receptors, which are largely understudied. Despite the receptor family comprising 19 receptors, only one drug (clopidogrel and its analogs) has made it to the market [114], where its effect is antithrombic. Three receptors, the P2Y12, P2Y13 and P2Y14 receptors, couple through Gi and thereby will decrease intracellular cAMP levels [115]. Agonists of these receptors could potentially be interesting anti-migraine targets. Inhibition of presynaptic transmission of sympathetic [116] and cholinergic nerves [117] has been reported following adenosine diphosphate-induced activation of the P2Y12 or P2Y13 receptors, respectively. P2Y12 receptors are most likely not a suitable candidate, as they are known to sensitize platelet aggregation, e.g., to thrombin [118]. Regarding the P2Y13 receptors, there are some interesting preliminary data demonstrating that a P2Y13 receptor agonist shows very similar results to sumatriptan in preclinical models of migraine [119]. Further studies are needed to make conclusions regarding a potential anti-migraine effect.
5.2.2 cAMP/Cyclic Guanosine Monophosphate (cGMP) Phosphodiesterase Activators
Sildenafil (PDE5 inhibitor) and cilostazol (PDE3 inhibitor) are both known to induce migraine [36, 38], further strengthening the view that cAMP (and cGMP) accumulation could be part of an important pathway that triggers migraine. Therefore, activation of cAMP-selective PDEs (PDE3 or PDE5) should be considered as a possible new target in migraine treatment. To our knowledge, no such activator has been developed for PDE3 or PDE5. Interestingly, this does exist for PDE4, illustrating a first proof of concept [120]. Despite the potential promising effect, the risk of serious adverse events is likely as these systems are widely distributed in the body.
5.3 Membrane Channels
As discussed in Sect. 3, neither CGRP nor cAMP activation can trigger pain directly. For pain to develop, nociceptive nerve fibers need to be activated and depolarized. Preventing such a depolarization could be a potential migraine target. Here we have chosen to focus on one specific and novel pain target, the HCN channels. The noted activation of cAMP/cGMP increases the open-probability, and hence the neuronal excitability and firing of the neurons [48]. Preventing the open-probability by applying an HCN antagonist is an interesting target in many types of neuropathic pain [53], but also for migraine. In an inflammatory migraine model, expression of HCN channels increases in the TG, and the authors suggest that HCN1 and HCN2 channels are involved in inflammation-induced sensory neuron hyperexcitability [53]. Furthermore, in data presented at MTIS London 2018 [121], Professor McNaughton showed that ivabradine prevented both glyceryl trinitrate (GTN)- and medication overuse (repeated exposure to sumatriptan)-induced hyperalgesia. Unfortunately, in clinical use ivabradine causes strong bradycardia with no therapeutic window [122]. This is caused by the repetitive firing in the cardiac action potential being driven by the HCN4 channel [123]. The structure of a HCN channel was recently determined [124]—this might lead to a more specific antagonist, devoid of cardiac effects [125].
Levcromakalim opens KATP channels, leading to hyperpolarization, and KATP channels have been proposed as potential antimigraine targets [57]. KATP channel activators or activation in vivo leads to hyperpolarization [58]. After a delay, the HCN channels could trigger an action potential as a response to a prolonged hyperpolarization, as has been demonstrated in other systems [59], which could be sensed as pain [59, 60]. There are few data on these mechanisms and we can only speculate that prolonged hyperpolarization might trigger migraine pain. If this is the case, potassium channel antagonists might be potential anti-migraine channels as they prevent long-term hyperpolarization.
5.4 Glutamate/Kynurenate
Signals travelling through the Aδ-fibers are further transmitted to the second-order neurons of the TNC, where glutamate is the main excitatory neurotransmitter [126]. Targeting the glutaminergic system might therefore prevent the pain signal from ever reaching the brain [127]. However, targeting this system must be approached with utmost care due to potential adverse effects. A small number of clinical trials exist targeting the glutaminergic system. Tezampanel (LY293558) targets the GluK5 subunit of the kainate receptor. One phase II trial showed positive results in the primary endpoint of 2 h pain freedom but this has not been investigated further [128]. Similarly, adx10059, a negative allosteric modulator for mGlu5, has also been tested for efficacy in migraine. The study had a positive result for the primary outcome, but liver enzyme elevation stopped further development [129, 130]. Preventing communication between the TG and TNC is still an interesting target, as a potential modulator might have an anti-nociceptive effect.
Furthermore, regulating the connection between the TG and TNC might be affected by the kynurenic pathway. Kynurenate has been shown to be involved in the pathophysiology of migraine [131]. Its effect is most likely mediated by inhibitory effects on ionotropic glutamate receptors [132]. There have been some preclinical studies on kynurenate. For example, GTN infusion led to expressional changes in the kynurenate pathway in one study [133]. In addition, in a model of trigeminovascular inflammation [134], kynurenate and its analogs have been shown to suppress nociceptive activation of the trigeminal pathway and reduce the release of glutamate [135].
6 Concluding Remarks
The current line of treatments and those in development all target the peripheral symptoms of migraine. The availability of receptors and ease of modulating signaling targets in the peripheral trigeminovascular system has made it a preferred target over the central nervous system [15]. At the same time, central adverse effects are less likely if peripheral structures are the treatment target.
We are now much closer to understanding the pathophysiology of migraine. Nevertheless, discovering the mechanism behind the initial trigger and a migraine cure (unlike mitigating pain) still seems far away. Preventing peripheral pain will therefore most likely be the main target in the near future. When the signaling and modulation of the hypothalamus is better understood, there might be hope for a true migraine cure.
The question therefore remains, can we in the meantime treat all patients and provide pain relief? The current best approach—both CGRP/CGRP receptor mAbs and triptans [81, 136]—is to find the right combination of neuropeptide release inhibitors and prevent activation of their targets. However, in some patients, other neuropeptides such as PACAP might be involved in the origin of migraine pain [108]. In addition, there may be neurons for which triptans are not the best agonist to modulate neuronal excitability. The need for novel modulators will hopefully be met as research focus turns to the cAMP and cGMP systems and their activation in migraine. We also believe that the hypothesized involvement of HCN channels offers an interesting explanation for the actual sensation of pain, which deserves future focus.
References
Headache Classification Committee of the International Headache Society (IHS). The International classification of headache disorders, 3rd edition. Cephalalgia. 2018;38:1–211.
Tfelt-Hansen PC, Koehler PJ. One hundred years of migraine research: major clinical and scientific observations from 1910 to 2010. Headache. 2011;51:752–78.
Charles A. The evolution of a migraine attack—a review of recent evidence. Headache. 2013;53:413–9.
Schoonman GG, Evers DJ, Terwindt GM, van Dijk JG, Ferrari MD. The prevalence of premonitory symptoms in migraine: a questionnaire study in 461 patients. Cephalalgia. 2006;26:1209–13.
Quintela E, Castillo J, Munoz P, Pascual J. Premonitory and resolution symptoms in migraine: a prospective study in 100 unselected patients. Cephalalgia. 2006;26:1051–60.
Hougaard A, Amin FM, Hauge AW, Ashina M, Olesen J. Provocation of migraine with aura using natural trigger factors. Neurology. 2013;80:428–31.
Karsan N, Goadsby PJ. Biological insights from the premonitory symptoms of migraine. Nat Rev Neurol. 2018;14:699–710.
Stankewitz A, Aderjan D, Eippert F, May A. Trigeminal nociceptive transmission in migraineurs predicts migraine attacks. J Neurosci. 2011;31:1937–43.
Weiller C, May A, Limmroth V, Juptner M, Kaube H, Schayck RV, et al. Brain stem activation in spontaneous human migraine attacks. Nat Med. 1995;1:658–60.
Schulte LH, Allers A, May A. Hypothalamus as a mediator of chronic migraine: evidence from high-resolution fMRI. Neurology. 2017;88:2011–6.
Schulte LH, May A. The migraine generator revisited: continuous scanning of the migraine cycle over 30 days and three spontaneous attacks. Brain. 2016;139:1987–93.
Raffaelli E Jr, Menon AD. Migraine and the limbic system. Headache. 1975;15:69–78.
Rajmohan V, Mohandas E. The limbic system. Indian J Psychiatry. 2007;49:132–9.
Benedetti F, Carlino E, Pollo A. How placebos change the patient’s brain. Neuropsychopharmacology. 2011;36:339–54.
Lundblad C, Haanes KA, Grande G, Edvinsson L. Experimental inflammation following dural application of complete Freund’s adjuvant or inflammatory soup does not alter brain and trigeminal microvascular passage. J Headache Pain. 2015;16:91.
Eftekhari S, Salvatore CA, Johansson S, Chen TB, Zeng Z, Edvinsson L. Localization of CGRP, CGRP receptor, PACAP and glutamate in trigeminal ganglion. Relation to the blood-brain barrier. Brain Res. 2015;1600:93–109.
Goadsby PJ, Edvinsson L, Ekman R. Release of vasoactive peptides in the extracerebral circulation of humans and the cat during activation of the trigeminovascular system. Ann Neurol. 1988;23:193–6.
Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol. 1990;28:183–7.
Edvinsson L, Nilsson E, Jansen-Olesen I. Inhibitory effect of BIBN4096BS, CGRP(8-37), a CGRP antibody and an RNA-Spiegelmer on CGRP induced vasodilatation in the perfused and non-perfused rat middle cerebral artery. Br J Pharmacol. 2007;150:633–40.
Gupta S, Akerman S, van den Maagdenberg AM, Saxena PR, Goadsby PJ, van den Brink AM. Intravital microscopy on a closed cranial window in mice: a model to study trigeminovascular mechanisms involved in migraine. Cephalalgia. 2006;26:1294–303.
Amrutkar DV, Ploug KB, Hay-Schmidt A, Porreca F, Olesen J, Jansen-Olesen I. mRNA expression of 5-hydroxytryptamine 1B, 1D, and 1F receptors and their role in controlling the release of calcitonin gene-related peptide in the rat trigeminovascular system. Pain. 2012;153:830–8.
Eftekhari S, Warfvinge K, Blixt FW, Edvinsson L. Differentiation of nerve fibers storing CGRP and CGRP receptors in the peripheral trigeminovascular system. J Pain. 2013;14:1289–303.
Gupta S, Mehrotra S, Avezaat CJ, Villalon CM, Saxena PR, MaassenVanDenBrink A. Characterisation of CGRP receptors in the human isolated middle meningeal artery. Life Sci. 2006;79:265–71.
Khan S, Amin FM, Christensen CE, Ghanizada H, Younis S, Olinger ACR, et al. Meningeal contribution to migraine pain: a magnetic resonance angiography study. Brain. 2019;142:93–102.
Ottosson A, Edvinsson L. Release of histamine from dural mast cells by substance P and calcitonin gene-related peptide. Cephalalgia. 1997;17:166–74.
Thalakoti S, Patil VV, Damodaram S, Vause CV, Langford LE, Freeman SE, et al. Neuron-glia signaling in trigeminal ganglion: implications for migraine pathology. Headache. 2007;47:1008–23.
Melo-Carrillo A, Strassman AM, Nir RR, Schain AJ, Noseda R, Stratton J, et al. Fremanezumab-a humanized monoclonal anti-CGRP antibody-inhibits thinly myelinated (Aδ) but not unmyelinated (C) meningeal nociceptors. J Neurosci. 2017;37:10587–96.
Rahmann A, Wienecke T, Hansen JM, Fahrenkrug J, Olesen J, Ashina M. Vasoactive intestinal peptide causes marked cephalic vasodilation, but does not induce migraine. Cephalalgia. 2008;28:226–36.
Hansen JM, Sitarz J, Birk S, Rahmann AM, Oturai PS, Fahrenkrug J, et al. Vasoactive intestinal polypeptide evokes only a minimal headache in healthy volunteers. Cephalalgia. 2006;26:992–1003.
Hurley JH, Kunkler PE, Zhang L, Knopp KL, Oxford GS. Role of intraganglionic transmission in the trigeminovascular pathway. Mol Pain. 2019;15:1744806919836570.
Walker CS, Raddant AC, Woolley MJ, Russo AF, Hay DL. CGRP receptor antagonist activity of olcegepant depends on the signalling pathway measured. Cephalalgia. 2018;38:437–51.
Ashina H, Schytz HW, Ashina M (2018) CGRP in Human Models of Migraine. In: Handbook of experimental pharmacology. Springer, Berlin. https://doi.org/10.1007/164_2018_128
Brennan KC, Charles A. An update on the blood vessel in migraine. Curr Opin Neurol. 2010;23:266–74.
Asghar MS, Hansen AE, Amin FM, van der Geest RJ, Koning P, Larsson HB, et al. Evidence for a vascular factor in migraine. Ann Neurol. 2011;69:635–45.
Guo S, Vollesen AL, Olesen J, Ashina M. Premonitory and nonheadache symptoms induced by CGRP and PACAP38 in patients with migraine. Pain. 2016;157:2773–81.
Guo S, Olesen J, Ashina M. Phosphodiesterase 3 inhibitor cilostazol induces migraine-like attacks via cyclic AMP increase. Brain. 2014;137:2951–9.
Birk S, Kruuse C, Petersen KA, Tfelt-Hansen P, Olesen J. The headache-inducing effect of cilostazol in human volunteers. Cephalalgia. 2006;26:1304–9.
Kruuse C, Thomsen LL, Birk S, Olesen J. Migraine can be induced by sildenafil without changes in middle cerebral artery diameter. Brain. 2003;126:241–7.
Ashina M (2018) Human models of migraine - short-term pain for long-term gain. MTIS London, Migraine Trust Lecture, 7th September.
Danish Headache Center. Headache inducing effect of cromakalim in migraine patients [ClinicalTrials.gov identifier NCT03228355]. National Institutes of Health, ClinicalTrials.gov. 2019. https://clinicaltrials.gov. Accessed 2 Apr 2019.
Liu Y, Shakur Y, Yoshitake M, Kambayashi JJ. Cilostazol (pletal): a dual inhibitor of cyclic nucleotide phosphodiesterase type 3 and adenosine uptake. Cardiovasc Drug Rev. 2001;19:369–86.
Haanes KA, Labastida-Ramirez A, Chan KY, de Vries R, Shook B, Jackson P, et al. Characterization of the trigeminovascular actions of several adenosine A2A receptor antagonists in an in vivo rat model of migraine. J Headache Pain. 2018;19:41.
Haanes KA, Edvinsson L. Expression and characterization of purinergic receptors in rat middle meningeal artery-potential role in migraine. PLoS One. 2014;9:e108782.
Fuller RW, Conradson TB, Dixon CM, Crossman DC, Barnes PJ. Sensory neuropeptide effects in human skin. Br J Pharmacol. 1987;92:781–8.
Pedersen-Bjergaard U, Nielsen LB, Jensen K, Edvinsson L, Jansen I, Olesen J. Calcitonin gene-related peptide, neurokinin A and substance P: effects on nociception and neurogenic inflammation in human skin and temporal muscle. Peptides. 1991;12:333–7.
Pedersen-Bjergaard U, Nielsen LB, Jensen K, Edvinsson L, Jansen I, Olesen J. Algesia and local responses induced by neurokinin A and substance P in human skin and temporal muscle. Peptides. 1989;10:1147–52.
Hansen JM, Hauge AW, Olesen J, Ashina M. Calcitonin gene-related peptide triggers migraine-like attacks in patients with migraine with aura. Cephalalgia. 2010;30:1179–86.
Emery EC, Young GT, McNaughton PA. HCN2 ion channels: an emerging role as the pacemakers of pain. Trends Pharmacol Sci. 2012;33:456–63.
Young GT, Emery EC, Mooney ER, Tsantoulas C, McNaughton PA. Inflammatory and neuropathic pain are rapidly suppressed by peripheral block of hyperpolarisation-activated cyclic nucleotide-gated ion channels. Pain. 2014;155:1708–19.
Momin A, Cadiou H, Mason A, McNaughton PA. Role of the hyperpolarization-activated current Ih in somatosensory neurons. J Physiol. 2008;586:5911–29.
Tu H, Deng L, Sun Q, Yao L, Han JS, Wan Y. Hyperpolarization-activated, cyclic nucleotide-gated cation channels: roles in the differential electrophysiological properties of rat primary afferent neurons. J Neurosci Res. 2004;76:713–22.
Manteniotis S, Lehmann R, Flegel C, Vogel F, Hofreuter A, Schreiner BS, et al. Comprehensive RNA-Seq expression analysis of sensory ganglia with a focus on ion channels and GPCRs in trigeminal ganglia. PLoS One. 2013;8:e79523.
Cho HJ, Staikopoulos V, Furness JB, Jennings EA. Inflammation-induced increase in hyperpolarization-activated, cyclic nucleotide-gated channel protein in trigeminal ganglion neurons and the effect of buprenorphine. Neuroscience. 2009;162:453–61.
Oshinsky ML, Luo J. Neurochemistry of trigeminal activation in an animal model of migraine. Headache. 2006;46(Suppl 1):S39–44.
Vause CV, Durham PL. CGRP stimulation of iNOS and NO release from trigeminal ganglion glial cells involves mitogen-activated protein kinase pathways. J Neurochem. 2009;110:811–21.
Poolos NP, Bullis JB, Roth MK. Modulation of h-channels in hippocampal pyramidal neurons by p38 mitogen-activated protein kinase. J Neurosci. 2006;26:7995–8003.
Al-Karagholi MA, Hansen JM, Severinsen J, Jansen-Olesen I, Ashina M. The KATP channel in migraine pathophysiology: a novel therapeutic target for migraine. J Headache Pain. 2017;18:90.
Hogg RC, Adams DJ. An ATP-sensitive K(+) conductance in dissociated neurones from adult rat intracardiac ganglia. J Physiol. 2001;534:713–20.
Emery EC, Young GT, Berrocoso EM, Chen L, McNaughton PA. HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science. 2011;333:1462–6.
Takasu K, Ono H, Tanabe M. Spinal hyperpolarization-activated cyclic nucleotide-gated cation channels at primary afferent terminals contribute to chronic pain. Pain. 2010;151:87–96.
Manganiello VC, Degerman E. Cyclic nucleotide phosphodiesterases (PDEs): diverse regulators of cyclic nucleotide signals and inviting molecular targets for novel therapeutic agents. Thromb Haemost. 1999;82:407–11.
Humphrey PP. The discovery of a new drug class for the acute treatment of migraine. Headache. 2007;47(Suppl 1):S10–9.
Razzaque Z, Heald MA, Pickard JD, Maskell L, Beer MS, Hill RG, et al. Vasoconstriction in human isolated middle meningeal arteries: determining the contribution of 5-. Br J Clin Pharmacol. 1999;47:75–82.
Goadsby PJ, Edvinsson L. Joint 1994 Wolff Award Presentation. Peripheral and central trigeminovascular activation in cat is blocked by the serotonin (5HT)-1D receptor agonist 311C90. Headache. 1994;34:394–9.
Adham N, Romanienko P, Hartig P, Weinshank RL, Branchek T. The rat 5-hydroxytryptamine1B receptor is the species homologue of the human 5-hydroxytryptamine1D beta receptor. Mol Pharmacol. 1992;41:1–7.
Storer RJ, Goadsby PJ. Microiontophoretic application of serotonin (5HT)1B/1D agonists inhibits trigeminal cell firing in the cat. Brain. 1997;120(Pt 12):2171–7.
MaassenVanDenBrink A, van den Broek RW, de Vries R, Bogers AJ, Avezaat CJ, Saxena PR. Craniovascular selectivity of eletriptan and sumatriptan in human isolated blood vessels. Neurology. 2000;55:1524–30.
Longmore J, Hargreaves RJ, Boulanger CM, Brown MJ, Desta B, Ferro A, et al. Comparison of the vasoconstrictor properties of the 5-HT1D-receptor agonists rizatriptan (MK-462) and sumatriptan in human isolated coronary artery: outcome of two independent studies using different experimental protocols. Funct Neurol. 1997;12:3–9.
Rubio-Beltran E, Labastida-Ramirez A, Villalon CM, MaassenVanDenBrink A. Is selective 5-HT1F receptor agonism an entity apart from that of the triptans in antimigraine therapy? Pharmacol Ther. 2018;186:88–97.
Classey JD, Bartsch T, Goadsby PJ. Distribution of 5-HT(1B), 5-HT(1D) and 5-HT(1F) receptor expression in rat trigeminal and dorsal root ganglia neurons: relevance to the selective anti-migraine effect of triptans. Brain Res. 2010;1361:76–85.
Frederiksen SD, Warfvinge K, Ohlsson L, Edvinsson L. Expression of pituitary adenylate cyclase-activating peptide, calcitonin gene-related peptide and headache targets in the trigeminal ganglia of rats and humans. Neuroscience. 2018;393:319–32.
Shepheard S, Edvinsson L, Cumberbatch M, Williamson D, Mason G, Webb J, et al. Possible antimigraine mechanisms of action of the 5HT1F receptor agonist LY334370. Cephalalgia. 1999;19:851–8.
Phebus LA, Johnson KW, Zgombick JM, Gilbert PJ, Van BK, Mancuso V, et al. Characterization of LY344864 as a pharmacological tool to study 5-HT1F receptors: binding affinities, brain penetration and activity in the neurogenic dural inflammation model of migraine. Life Sci. 1997;61:2117–26.
Cohen ML, Schenck K. Contractile responses to sumatriptan and ergotamine in the rabbit saphenous vein: effect of selective 5-HT(1F) receptor agonists and PGF(2alpha). Br J Pharmacol. 2000;131:562–8.
Goldstein DJ, Roon KI, Offen WW, Ramadan NM, Phebus LA, Johnson KW, et al. Selective seratonin 1F (5-HT(1F)) receptor agonist LY334370 for acute migraine: a randomised controlled trial. Lancet. 2001;358:1230–4.
Ramadan NM, Skljarevski V, Phebus LA, Johnson KW. 5-HT1F receptor agonists in acute migraine treatment: a hypothesis. Cephalalgia. 2003;23:776–85.
Nelson DL, Phebus LA, Johnson KW, Wainscott DB, Cohen ML, Calligaro DO, et al. Preclinical pharmacological profile of the selective 5-HT1F receptor agonist lasmiditan. Cephalalgia. 2010;30:1159–69.
Kuca B, Silberstein SD, Wietecha L, Berg PH, Dozier G, Lipton RB. Lasmiditan is an effective acute treatment for migraine: a phase 3 randomized study. Neurology. 2018;91:e2222–32.
Adham N, Kao HT, Schecter LE, Bard J, Olsen M, Urquhart D, et al. Cloning of another human serotonin receptor (5-HT1F): a fifth 5-HT1 receptor subtype coupled to the inhibition of adenylate cyclase. Proc Natl Acad Sci USA. 1993;90:408–12.
Labastida-Ramirez A, Rubio-Beltran E, Haanes KA, Danser J, Kovalchin J, Johnson KW, et al. Lasmiditan inhibits dural CGRP release from the rat trigeminovascular system. Cephalalgia. 2018;38(Suppl. 1):45–6.
Edvinsson L, Haanes KA, Warfvinge K, Krause DN. CGRP as the target of new migraine therapies—successful translation from bench to clinic. Nat Rev Neurol. 2018;14:338–50.
Schuster NM, Rapoport AM. Calcitonin gene-related peptide-targeted therapies for migraine and cluster headache: a review. Clin Neuropharmacol. 2017;40:169–74.
Olesen J, Diener HC, Husstedt IW, Goadsby PJ, Hall D, Meier U, et al. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med. 2004;350:1104–10.
Ho TW, Connor KM, Zhang Y, Pearlman E, Koppenhaver J, Fan X, et al. Randomized controlled trial of the CGRP receptor antagonist telcagepant for migraine prevention. Neurology. 2014;83:958–66.
Voss T, Lipton RB, Dodick DW, Dupre N, Ge JY, Bachman R, et al. A phase IIb randomized, double-blind, placebo-controlled trial of ubrogepant for the acute treatment of migraine. Cephalalgia. 2016;36:887–98.
Marcus R, Goadsby PJ, Dodick D, Stock D, Manos G, Fischer TZ. BMS-927711 for the acute treatment of migraine: a double-blind, randomized, placebo controlled, dose-ranging trial. Cephalalgia. 2014;34:114–25.
Tfelt-Hansen P, Loder E. The Emperor’s new gepants: are the effects of the new oral CGRP antagonists clinically meaningful? Headache. 2019;59:113–7.
Allergan. Efficacy, safety, and tolerability of multiple dosing regimens of oral atogepant (AGN-241689) in episodic migraine prevention [ClinicalTrials.gov identifier NCT02848326]. National Institutes of Health, ClinicalTrials.gov. 2019. https://clinicaltrials.gov. Accessed 2 Apr 2019.
Boado RJ, Zhou QH, Lu JZ, Hui EK, Pardridge WM. Pharmacokinetics and brain uptake of a genetically engineered bifunctional fusion antibody targeting the mouse transferrin receptor. Mol Pharm. 2010;7:237–44.
MaassenVanDenBrink A, Meijer J, Villalon CM, Ferrari MD. Wiping out CGRP: potential cardiovascular risks. Trends Pharmacol Sci. 2016;37:779–88.
Deen M, Correnti E, Kamm K, Kelderman T, Papetti L, Rubio-Beltran E, et al. Blocking CGRP in migraine patients—a review of pros and cons. J Headache Pain. 2017;18:96.
Silberstein SD, Dodick DW, Bigal ME, Yeung PP, Goadsby PJ, Blankenbiller T, et al. Fremanezumab for the preventive treatment of chronic migraine. N Engl J Med. 2017;377:2113–22.
Lambru G, Andreou AP, Guglielmetti M, Martelletti P. Emerging drugs for migraine treatment: an update. Expert Opin Emerg Drugs. 2018. https://doi.org/10.1080/14728214.2018.1552939 (Epub 2018 Nov 28).
Zagami AS, Edvinsson L, Goadsby PJ. Pituitary adenylate cyclase activating polypeptide and migraine. Ann Clin Transl Neurol. 2014;1:1036–40.
Tighe AP, Schiavo G. Botulinum neurotoxins: mechanism of action. Toxicon. 2013;67:87–93.
Durham PL, Cady R. Insights into the mechanism of onabotulinumtoxinA in chronic migraine. Headache. 2011;51:1573–7.
Meng J, Wang J, Lawrence G, Dolly JO. Synaptobrevin I mediates exocytosis of CGRP from sensory neurons and inhibition by botulinum toxins reflects their anti-nociceptive potential. J Cell Sci. 2007;120:2864–74.
Aurora SK, Dodick DW, Turkel CC, DeGryse RE, Silberstein SD, Lipton RB, et al. OnabotulinumtoxinA for treatment of chronic migraine: results from the double-blind, randomized, placebo-controlled phase of the PREEMPT 1 trial. Cephalalgia. 2010;30:793–803.
Diener HC, Dodick DW, Aurora SK, Turkel CC, DeGryse RE, Lipton RB, et al. OnabotulinumtoxinA for treatment of chronic migraine: results from the double-blind, randomized, placebo-controlled phase of the PREEMPT 2 trial. Cephalalgia. 2010;30:804–14.
Zhang X, Strassman AM, Novack V, Brin MF, Burstein R. Extracranial injections of botulinum neurotoxin type A inhibit intracranial meningeal nociceptors’ responses to stimulation of TRPV1 and TRPA1 channels: are we getting closer to solving this puzzle? Cephalalgia. 2016;36:875–86.
Dolly JO, Wang J, Zurawski TH, Meng J. Novel therapeutics based on recombinant botulinum neurotoxins to normalize the release of transmitters and pain mediators. FEBS J. 2011;278:4454–66.
Mangione AS, Obara I, Maiaru M, Geranton SM, Tassorelli C, Ferrari E, et al. Nonparalytic botulinum molecules for the control of pain. Pain. 2016;157:1045–55.
Paraskevopoulou M, Perez JT, Miedzik A, Chamberlain J, Lambru G, Davletov B, et al. Non-paralytic botulinum molecules for the control of migraine. Cephalalgia. 2016;36 (Suppl 1):135.
Mustafa G, Anderson EM, Bokrand-Donatelli Y, Neubert JK, Caudle RM. Anti-nociceptive effect of a conjugate of substance P and light chain of botulinum neurotoxin type A. Pain. 2013;154:2547–53.
Maiaru M, Leese C, Certo M, Echeverria-Altuna I, Mangione AS, Arsenault J, et al. Selective neuronal silencing using synthetic botulinum molecules alleviates chronic pain in mice. Sci Transl Med. 2018;10:eaar7384.
Peroutka SJ. Neurogenic inflammation and migraine: implications for the therapeutics. Mol Interv. 2005;5:304–11.
Edvinsson L, Tajti J, Szalardy L, Vecsei L. PACAP and its role in primary headaches. J Headache Pain. 2018;19:21.
Tuka B, Helyes Z, Markovics A, Bagoly T, Szolcsanyi J, Szabo N, et al. Alterations in PACAP-38-like immunoreactivity in the plasma during ictal and interictal periods of migraine patients. Cephalalgia. 2013;33:1085–95.
Harmar AJ, Fahrenkrug J, Gozes I, Laburthe M, May V, Pisegna JR, et al. Pharmacology and functions of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide: IUPHAR review 1. Br J Pharmacol. 2012;166:4–17.
Chan KY, Baun M, de Vries R, van den Bogaerdt AJ, Dirven CM, Danser AH, et al. Pharmacological characterization of VIP and PACAP receptors in the human meningeal and coronary artery. Cephalalgia. 2011;31:181–9.
Amgen. Study to evaluate the efficacy and safety of AMG 301 in migraine prevention [ClinicalTrials.gov identifier NCT03238781]. National Institutes of Health, ClinicalTrials.gov. 2019. https://clinicaltrials.gov. Accessed 2 Apr 2019.
Moldovan LC, Dutzar B, Ojala EW, Hendrix L, Karasek C, Scalley-Kim M, et al. Pharmacologic characterization of ALD1910, a potent humanized monoclonal antibody against the pituitary adenylate cyclase activating peptide. J Pharmacol Exp Ther. 2019;369:26–36.
May V, Buttolph TR, Girard BM, Clason TA, Parsons RL. PACAP-induced ERK activation in HEK cells expressing PAC1 receptors involves both receptor internalization and PKC signaling. Am J Physiol Cell Physiol. 2014;306:C1068–79.
Herbert JM, Savi P. P2Y12, a new platelet ADP receptor, target of clopidogrel. Semin Vasc Med. 2003;3:113–22.
Erb L, Weisman GA. Coupling of P2Y receptors to G proteins and other signaling pathways. Wiley Interdiscip Rev Membr Transp Signal. 2012;1:789–803.
Queiroz G, Talaia C, Goncalves J. ATP modulates noradrenaline release by activation of inhibitory P2Y receptors and facilitatory P2X receptors in the rat vas deferens. J Pharmacol Exp Ther. 2003;307:809–15.
Guarracino JF, Cinalli AR, Fernandez V, Roquel LI, Losavio AS. P2Y13 receptors mediate presynaptic inhibition of acetylcholine release induced by adenine nucleotides at the mouse neuromuscular junction. Neuroscience. 2016;326:31–44.
Gachet C. ADP receptors of platelets and their inhibition. Thromb Haemost. 2001;86:222–32.
Haanes KA, Labastida-Ramirez A, Dirven CM, Danser AHJ, MaassenVanDenBrink A. Purinergic receptors as potential anti-migraine targets. Cephalalgia. 2016;36(Suppl. 1):140.
Wang L, Burmeister BT, Johnson KR, Baillie GS, Karginov AV, Skidgel RA, et al. UCR1C is a novel activator of phosphodiesterase 4 (PDE4) long isoforms and attenuates cardiomyocyte hypertrophy. Cell Signal. 2015;27:908–22.
McNaughton P (2018) HCN2 ion channels- a new target in migraine? MTIS London, Novel Transmitter Systems, 8th Septhember 2018.
Alshammari TM. Ivabradine: do the benefits outweigh the risks? J Cardiovasc Pharmacol Ther. 2017;22:210–8.
Baruscotti M, Bucchi A, Viscomi C, Mandelli G, Consalez G, Gnecchi-Rusconi T, et al. Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene Hcn4. Proc Natl Acad Sci USA. 2011;108:1705–10.
Lee CH, MacKinnon R. Structures of the human HCN1 hyperpolarization-activated channel. Cell. 2017;168:111–20.
Novella RM, Sartiani L, Masi A, Mannaioni G, Manetti D, Mugelli A, et al. HCN channels modulators: the need for selectivity. Curr Top Med Chem. 2016;16:1764–91.
Xiao Y, Richter JA, Hurley JH. Release of glutamate and CGRP from trigeminal ganglion neurons: Role of calcium channels and 5-HT1 receptor signaling. Mol Pain. 2008;4:12.
Schneider SP, Perl ER. Comparison of primary afferent and glutamate excitation of neurons in the mammalian spinal dorsal horn. J Neurosci. 1988;8:2062–73.
Sang CN, Ramadan NM, Wallihan RG, Chappell AS, Freitag FG, Smith TR, et al. LY293558, a novel AMPA/GluR5 antagonist, is efficacious and well-tolerated in acute migraine. Cephalalgia. 2004;24:596–602.
Chan K, MaassenVanDenBrink A. Glutamate receptor antagonists in the management of migraine. Drugs. 2014;74:1165–76.
Waung MW, Akerman S, Wakefield M, Keywood C, Goadsby PJ. Metabotropic glutamate receptor 5: a target for migraine therapy. Ann Clin Transl Neurol. 2016;3:560–71.
Curto M, Lionetto L, Fazio F, Mitsikostas DD, Martelletti P. Fathoming the kynurenine pathway in migraine: why understanding the enzymatic cascades is still critically important. Intern Emerg Med. 2015;10:413–21.
Stone DR, Downs JB, Paul WL, Perkins HM. Adult body temperature and heated humidification of anesthetic gases during general anesthesia. Anesth Analg. 1981;60:736–41.
Fejes-Szabo A, Bohar Z, Vamos E, Nagy-Grocz G, Tar L, Veres G, et al. Pre-treatment with new kynurenic acid amide dose-dependently prevents the nitroglycerine-induced neuronal activation and sensitization in cervical part of trigemino-cervical complex. J Neural Transm (Vienna). 2014;121:725–38.
Lukacs M, Haanes KA, Majlath Z, Tajti J, Vecsei L, Warfvinge K, et al. Dural administration of inflammatory soup or Complete Freund’s Adjuvant induces activation and inflammatory response in the rat trigeminal ganglion. J Headache Pain. 2015;16:564.
Lukacs M, Warfvinge K, Tajti J, Fulop F, Toldi J, Vecsei L, et al. Topical dura mater application of CFA induces enhanced expression of c-fos and glutamate in rat trigeminal nucleus caudalis: attenuated by KYNA derivate (SZR72). J Headache Pain. 2017;18:39.
Cameron C, Kelly S, Hsieh SC, Murphy M, Chen L, Kotb A, et al. Triptans in the acute treatment of migraine: a systematic review and network meta-analysis. Headache. 2015;55(Suppl 4):221–35.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Lars Edvinsson has given lectures on CGRP for Amgen, Novartis, and Teva, and has received minor grant support, though none pertaining to the current manuscript. Kristian Agmund Haanes has no conflicts of interest to report.
Funding
No sources of funding were used to assist with the preparation of this review.
Rights and permissions
About this article

Cite this article
Haanes, K.A., Edvinsson, L. Pathophysiological Mechanisms in Migraine and the Identification of New Therapeutic Targets. CNS Drugs 33, 525–537 (2019). https://doi.org/10.1007/s40263-019-00630-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-019-00630-6