
Abstract
Corneal and conjunctival epithelia, along with the tear film, serve as biological barriers to protect the eye from the entrance of potentially harmful substances. These barriers create constraint effective drug medication of ocular diseases with topical ocular formulations. Designing an effective therapy for ocular diseases, especially for the anterior segment, has been considered a challenging task. Nano-drug carriers giving us an array of hope for ocular drug therapy, owing to its potential to improve the ocular retention, controlled release, trans-corneal permeation and thus intra-ocular drug availability. Nanotechnology-based formulation design is important in ocular pharmaceuticals yet knowledge of anatomy and physiology of eyes are critical along with the understanding of nanoparticles design. Here, we discussed the ocular transport of topically applied drug, different barriers in its path and how the nanoparticles as drug carriers can improve the drug delivery to the eyes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Ocular drug delivery
- Topical administration
- Drug transport barriers
- Controlled drug release
- Nanotechnology
- Targeting
Introduction
The development of drug delivery approach for the transportation of drug in a bioavailable and safe manner to the target site is now becoming an exceedingly important area of biopharmaceutical research. To be sure, a large number of novel drug delivery technologies surface every year and every segment of the body part has been attempted as a potential target for the site of action.
Ophthalmic drug delivery is one of the most interesting and challenging endeavors facing the pharmaceutical scientist. The anatomy, physiology, and biochemistry of the eye render this organ highly impervious to foreign substances. This appears to be quite challenging for drug delivery scientist to circumvent the protective barriers of the eye without causing permanent tissue damage. Development of newer, more sensitive diagnostic techniques and novel therapeutic agents continued to provide ocular delivery systems with high therapeutic efficacy.
The goal of pharmacotherapeutics is to treat a disease in a consistent and predictable fashion. An assumption is made that a correlation exists between the concentration of a drug at its intended site of action and the resulting pharmacological effect. The specific aim of designing a therapeutic system is to achieve an optimal concentration of a drug at the active site for the appropriate duration. Ocular disposition and elimination of a therapeutic agent are dependent upon its physicochemical properties as well as the relevant ocular anatomy and physiology (Hanrahan et al. 2012) for treating anterior ocular disorders, topical administration into the conjunctival cul-de-sac is usually the preferred route of delivery. A successful design of a drug delivery system, therefore, requires an integrated knowledge of the drug molecule and the constraints offered by the ocular route of administration.
Conventional Approach for Ocular Drug Delivery
Medication is applied to the surface of the eye for two purposes: to treat the outside of the eye for such infections as conjunctivitis, blepharitis, keratitis sicca, or to provide intraocular treatment through the cornea for diseases such as glaucoma or uveitis. Most ocular diseases are treated with topical application of solutions administered as eye drops. These conventional dosage forms account for nearly 90 % of the currently accessible marketed formulations. Eye drops used for soluble drug, require frequent instillations of highly concentrated solutions. The practical reasons for selecting solutions are the generally favorable cost advantage, the greater simplicity of formulation development and production and the good acceptance by patients despite a little blurring (Fitzgerald and Wilson 1994; Araujo et al. 2009; Singh et al. 2011). In that, most of the drugs are washed from the eye by various mechanisms like lacrimation, tear dilution, and tear turnover. Moreover, the relatively impermeable corneal barrier restricts the entry of foreign substances. As a result, less than 5 % of administered drug penetrates the cornea and reaches intraocular tissues. Many researchers have made significant efforts to improve the ocular bioavailability by increasing the corneal residence/contact time by using novel delivery system such as aqueous eye drop with increased viscosity (Sechoy et al. 2000; Tissie et al. 2002), colloidal particles (De Campos et al. 2004; Sahoo et al. 2008; Gupta et al. 2010; Wadhwa et al. 2010), in situ gel system, dendrimers (Vandamme and Brobeck 2005), Ocular inserts (Ding 1998), collagen shields (Hill et al. 1993) and ion pair (Higashiyama et al. 2006).
Drug Transport Mechanism and Barriers in Ocular Drug Delivery
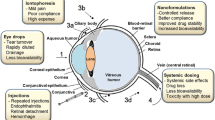
The eyes are among the most readily accessible organs in terms of location in the body and topical delivery into the cul-de-sac is, by far, the most common route of ocular drug delivery. Absorption from this site may be corneal or non-corneal. The so-called non-corneal route of absorption involves penetration across the sclera and conjunctiva into the intraocular tissues. This mechanism of absorption is usually non-productive, as drug penetrating the surface of the eye beyond the corneal-scleral limbus is taken up by the local capillary beds and removed to the general circulation. This non-corneal absorption, in general, precludes the entry into the aqueous humor. But drug delivery to eye tissues is particularly problematic due to the presence of ocular barriers like tear film, cornea, conjunctiva and sclera (Fig. 9.1a, d).

Anatomical structure of human eye and barriers in ocular drug delivery. a Human eye. b Cornea. c Tear film. d Conjunctiva
Tear Film
The tear (precorneal film) film is the liquid layer bathing the cornea and conjunctiva. The film thickness is reported to be about 3–10 μm (Robinson 1993; King-Smith et al. 2000; Szczesna et al. 2006, 2007; Szczesna-Iskander and Iskander 2012; Dhubhghaill et al. 2012). The tear film consists of a three-layered structure comprising a lipid (oil) layer, an aqueous (water) layer and a mucous layer over the corneal epithelium as shown in Fig. 9.1c. The lipid layer is the outermost surface and polishes the corneal surface. It is produced by the meibomian glands and provides a smooth tear surface and retarding the rate of tear evaporation from the cornea. Mechanically traps and flushes out foreign bodies and chemicals contain bacteriostatic substances that inhibit the growth of microorganisms. The aqueous layer produced by the lachrymal gland is composed of water, proteins, and other substances, such as lipocalin, lactoferrin, lysozyme, and lacritin. This layer is responsible for control of infection, osmotic balance and water promoting spreading of the tear film. The inner-most layer of the tear film is mucus layer. It is produced by goblet cells of the conjunctiva and acts as a hydrophilic layer (water soluble) and serves as an anchor for the tear film and helps it adhere to the eye. The tear film creates a smooth surface for light to pass through the eye, nourishes the front of the eye, and provides protection from infection (Wolff 1946, 1954; King-Smith et al. 2000; Rathore and Nema 2009; Montes et al. 2010; Stahl et al. 2012). Tear film which reduces the effective concentration of the administrated drugs due to dilution by the tear turnover (approximately 1 µL/min), accelerated clearance, and binding of the drug molecule to the tear proteins (Schoenwald et al. 1998; King-Smith et al. 2000; Her et al. 2013). The Osmolarity of the tear film equals 310–350 mOsm/kg in normal eyes and is adjusted by the principal inorganic ions Na+, K+, Cl−, HCO3−, and proteins. The mean pH value of normal tears is about 7.4. Depending on age and diseases, values between 5.2 and 9.3 have been measured. Diurnal patterns of pH changes exist, with a general shift from acid to alkaline during the day. The buffer capacity of the tears is determined by bicarbonate ions, proteins, and mucin. Tears exhibit a non-Newtonian rheological behavior. The viscosity is about 3 mPas (Greaves et al. 1993; Pandit et al. 1999). The surface tension depends on the presence of soluble mucin, lipocalins, and lipids. The mean surface tension value is about 44 mN/m (Nagyova and Tiffany 1999; Tiffany 2003).
Cornea
The cornea is the transparent, dome-shaped window covering the front of the eye and one of the most densely innervated tissues on the surface of the body (Marfurt et al. 1989; Muller et al. 2003). It is a powerful refracting surface, providing 2/3 of the eye’s focusing power. Because there are no blood vessels in the cornea, it is normally clear and has a shiny surface. It’s approximately 11.7 mm in diameter, 500 μm thick in the center with around 700 μm at the periphery and radius of curvature of anterior surface about 7.7 mm while the radius of curvature of the globe is approximately 12 mm (Fig. 9.1b) (Maurice and Mishima 1984; Hitzenberger et al. 1994). The cornea consists of three layers; epithelium, stroma, and endothelium and passes the mechanical barrier of foreign substances. Each layer possesses a different polarity and a rate-limiting structure for drug permeation. The corneal epithelium is of lipophilic nature, and tight junctions among cells are formed to restrict paracellular drug permeation from the tear film and permeated lipophilic drugs (transcellular pathway). Hydrophilic charged cationic compounds permeate more easily through the cornea than anionic forms, because the corneal epithelium is negatively charged above its isoelectric point (Rojanasakul et al. 1992; Bourlais et al. 1998; Gipson and Argueso 2003; Baspinar et al. 2008; Araujo et al. 2009). The highly hydrated structure of the stroma acts as a barrier to permeation of lipophilic drug molecules. Corneal endothelium is the innermost monolayer of hexagonal-shaped cells and acts as a separating barrier between the stroma and aqueous humor. The endothelial junctions are leaky and facilitate the passage of macromolecules between the aqueous humor and stroma (Bourlais et al. 1998; Fischbarg 2006; Araujo et al. 2009).
Conjunctiva
The conjunctiva is the thin, vascularized mucous membrane, transparent tissue that covers the outer surface of the eye. Histologically the conjunctiva consists of non-keratinized, stratified squamous epithelium, with interspersed goblet cells. It is involved in the formation and maintenance of the tear film, In addition, conjunctiva or episclera has a rich supply of capillaries and lymphatics as shown in Fig. 9.1d (Sugar et al. 1957; Raviola 1983; Robinson 1993; Gausas et al. 1999; Singh 2003; Hosoya et al. 2005) therefore, drugs administered in the conjunctival or episcleral space may be cleared through blood and lymph. The conjunctival blood vessels do not form a tight junction barrier (King-Smith et al. 2000), which means drug molecules can enter into the blood circulation by pinocytosis and/or convective transport through paracellular pores in the vascular endothelial layer. The conjunctival lymphatics act as an efflux system for the efficient elimination of the conjunctival space. Recently, it has been reported that at least 10 % of a small molecular weight hydrophilic model compound (sodium fluorescein), administered in the subconjunctival space, was eliminated via the lymphatics within the first hour in rat eyes (Lee et al. 2010). Therefore, drugs transported by lymphatics in conjunction with the elimination by blood circulation can contribute to systemic exposure, since the interstitial fluid is returned to the systemic circulation after filtration through lymph nodes.
Sclera
The sclera (white of the eye) is white, fibrous collagen tissues, and has three layers (anterior to posterior): episclera, scleral stroma, and lamina fusca and continued to cornea anteriorly. Sclera provides the structural integrity that defines the shape and length of the eye (Newell 1993; Ludwig 2005; Pescina et al. 2011). Scleral permeability has been shown to have a strong dependence on the molecular radius. Scleral permeability decreases roughly exponentially with molecular radius. Additionally, the posterior sclera is composed of a looser weave of collagen fibers than the anterior sclera (Miao et al. 2013), and the human sclera is relatively thick near the limbus (0.53 ± 0.14 mm), thin at the equator (0.39 ± 0.17 mm) and much thicker near the optic nerve (0.9–1.0 mm). Thus, the ideal location for transscleral drug delivery is near the equator at 12–17 mm posterior to the corneoscleral limbus (Myles et al. 2005; Qi et al. 2013). Hydrophobicity of drugs affects scleral permeability; an increase of lipophilicity shows lower permeability; and hydrophilic drugs may diffuse through the aqueous medium of proteoglycans in the fiber matrix pores more easily than lipophilic drugs (Maurice and Mishima 1984; Cruysberg et al. 2002; Wen et al. 2013).
Pharmacokinetic Consideration in Ocular Drug Delivery
The most significant reason for not conducting ocular pharmacokinetic studies in the human eye is the inability to sample tissues or fluids from the intact eye without risking pain and/or injury. Although the rabbit eye is useful in predicting human ocular toxicities (McDonald and Shadduck 1977), the eyes of each species are dissimilar in anatomy and physiology (Table 9.1) such that predicting human ocular pharmacokinetics from rabbit data may not be very precise for certain drugs.
After topical administration of an ophthalmic drug solution, the drug is firstly mixed with the lachrymal fluid. The contact time of drug with ocular tissues is relatively short (1–2 min) because of the permanent production of lachrymal fluid (0.5–2.2/µl/min). Then, approximately half of the drug flows through the upper canaliculus and the other half, through the lower canaliculus into the lachrymal sac, which opens into the nasolacrimal duct. Drainage of lachrymal fluid during blinking (every 12 s) towards the nasolacrimal duct induces a rapid elimination of conventional dosage forms (Ahmed and Patton 1985, 1987). The drug is absorbed into the retina-choroid via a corneal or scleroconjunctival route; the iris and ciliary body are presumably supplied via both the transcorneal and the extracorneal pathways.
Drugs penetrate across the corneal epithelium via the transcellular or paracellular pathway. Lipophilic drugs prefer the transcellular route, while hydrophilic drugs penetrate primarily through the paracellular pathway, which involves passive or altered diffusion through intercellular spaces. The transcorneal penetration appears to be hindered by the binding of the drug to the corneal tissues. The cornea may act as a drug reservoir, slowly releasing the drug into the aqueous humor, where levels decrease very slowly.
Then, drugs are distributed from the aqueous humor to the intraocular tissues, i.e., iris-ciliary body, lens, vitreous and choroid-retina and eliminated mainly via aqueous humor turnover and venous blood flow in the anterior uvea (Fig. 9.2). It is suggested that ocular penetration via the scleroconjunctival route is more rapid (for a hydrophilic drug) than via the transcorneal route (Worakul and Robinson 1997). Both transconjunctival absorption and transnasal absorption after drainage via the nasolacrimal duct are generally undesirable, not only because of the loss of active ingredient into the systemic circulation but also because of possible side-effects, for instance the effects on the heart when beta-blockers are administered for the treatment of wide-angle glaucoma (Schoenwald 1990; Jtirvinen et al. 1995; Meseguer et al. 1996).

Systemic representation of drug distribution after topical instillation in human eye
Formulation Approach for Ocular Drug Delivery
Mainly two types of approaches i.e., conventional and controlled drug delivery approaches. Conventional drug delivery approaches such as solutions, suspensions, and ointments, account for almost 90 % of the currently accessible ophthalmic formulations on the market (Lang 1995; Bourlais et al. 1998). They offer some advantages such as their ease of administration by the patient, ease of preparation and low production costs. However, there are also significant disadvantages, especially with the use of conventional solutions, including the very short contact time with the ocular surface and the fast nasolacrimal drainage, both leading to poor bioavailability of the drug. Nevertheless, conventional eye drops remain the most commonly used dosage forms in ocular delivery. The various drawbacks of conventional ocular drug delivery are described in Table 9.2.
Advantages of Modified Ocular Drug Delivery (Wilson 2004; Vyas and Khar 2008)
-
To circumvent the protective barriers like drainage, lacrimation and diversion of exogenous chemicals into the systemic circulation by the conjunctiva
-
To increase the ocular bioavailability of the drug by increasing corneal contact time. This can be achieved by effective coating or adherence to corneal surface so that the released drug effectively reaches the anterior chamber.
-
To overcome the side effects of pulsed dosing produced by conventional systems.
-
To provide comfort and compliance to the patient and yet improve the therapeutic performance of the drug over conventional systems.
-
To provide sustained and controlled drug delivery.
-
To provide targeting within the ocular globe, so as to prevent the loss to other organs.
Colloidal Carrier Exploited for Ocular Drug Delivery
Colloidal delivery systems, particularly nanoparticulate system are widely employed for the treatment of ocular disease. It would prolong the ocular retention of the drug, allowing the drug to remain in contact with the cornea for a longer duration, sustained release and thus increasing bioavailability. These delivery systems include liposomes, microemulsions/nanoemulsions, and nanoparticulate system etc.
Microemulsions/Nanoemulsions
Microemulsions/nanoemulsion (ME/NE) are of interest to the pharmaceutical scientists as promising drug delivery vehicles due to their, small size, simple and inexpensive preparation, and their sterilization easily by filtration (Soukharev and Wojciechowska 2005), low viscosity, greater ability as drug carrier, absorption promoter, and low surface tension of microemulsions, show spreading on the cornea mixing with the precorneal film constituents (Fialho 2004), long term stability, low toxicity and irritancy, considerable capacity for solubilisation of a variety of drug molecules (lipophilic and hydrophilic) and great potential in bioavailability improvement (Fialho 2004; Soukharev and Wojciechowska 2005; Gupta and Moulik 2008; Djekic et al. 2008; Kesavan et al. 2013). It consists of water, oil and a mixture of surfactants and co-surfactant, making a homogeneous, optically isotropic and thermodynamically stable and a small droplet size in the dispersed phase (<1.0 mm) (Soukharev and Wojciechowska 2005; Djekic et al. 2008).
The penetration enhancing the property of surfactant and co-surfactant of microemulsions increases the drug permeability (drug uptake) and facilitates the passage of drug by the corneal membrane. Moreover, microemulsion/nanoemulsion achieve sustained release of a drug applied to the cornea and higher penetration into the deeper layers of the ocular structure and the aqueous humor than the eye drops. Due to prolonged release of drug from microemulsions, they find an important role in ocular delivery which can decrease the frequency of application.
Kesavan and coworkers developed microemulsions of dexamethasone and found that small stable globule size, acceptable physicochemical behavior, good mucoadhesive properties, and ability to enhance bioavailability through its longer precorneal residence time sustained the release of the drug (Kesavan et al. 2013).
Ammar and co-workers described dilutable nanoemulsions of dorzolamide. They developed nanoemulsion of dorzolamide hydrochloride using different oils, surfactants, and co-surfactants by applying pseudo ternary-phase diagrams. These nanoemulsions showed acceptable physicochemical properties and exhibited slow drug release and no sign of inflammation on Draize rabbit. Biological evaluation of dorzolamide hydrochloride nanoemulsions on normotensive albino rabbits indicated that these products had higher therapeutic efficacy, faster onset of action, and prolonged effect relative to either drug solution or the market product (Ammar et al. 2009).
The high-level indomethacin in inner ocular structure, aqueous humor in rabbit eyes was achieved upon topical instillation of chitosan nanoemulsion as compared with indomethacin drug solution. The chitosan nanoemulsion prepared were able to make contact intimately with the cornea thus giving a slow continuing drug release with long-term drug intensity, thus enhancing delivery to both internal and external ophthalmic tissues (Badawi et al. 2008; Addo et al. 2010, 2015). The microemulsions system exploited for ocular delivery of various drugs is shown in Table 9.3
Tayel and co-workers developed controlled-release in situ ocular drug-loaded nanoemulsion using pseudo ternary phase diagrams technique (NE) gels. The formulation contained isopropyl myristate/Miglyol 812), surfactants (Tween 80/Cremophor EL), co-surfactant (polyethylene glycol 400) and water and gellan gum solution (0.2 %, w/w). The optimized formulation was evaluated in vitro and in vivo and showed transparency, rheological behavior, mucoadhesive force, sustained drug release. On instillation in rabbit eye, it showed good consistency, thermodynamically stable and no irritation and histopathological assessment of ocular irritation. The gel had significantly (P < 0.01) higher Cmax, delayed Tmax, prolonged mean residence time and increased bioavailability as compared to terbinafine hydrochloride eye drops and maintained effective aqueous humor concentrations (Tayel et al. 2013).
Nanoparticles (Nps)
Nanoparticles (NPs) are one of the most studied colloidal systems with the object of improving targeting of the drug to organs and increasing drug bioavailability across biological membranes. NPs are sub-microscopic, a colloidal system consisting of macromolecular substances that vary in size from 1 to 1000 nm. In NPs the drug may be dissolved, entrapped, adsorbed, attached or encapsulated into the polymer matrix.
Depending on the method of preparation, it can be classified into two groups: NPs (nanospheres) and nanocapsules and have different release profile of drug (Sahoo and Labhasetwar 2003; Omidi et al. 2012).
Nanospheres are small solid spheres constituting of a dense solid polymeric network having a large surface area (Omidi et al. 2012). Drugs can either be incorporated into the matrix system or adsorbed on the surface of the nanospheres. On the other hand, nanocapsules have a small central cavity (oily droplet) surrounded by a polymeric membrane and drug can either be incorporated into the matrix system or adsorbed on the surface (Fig. 9.3).

Different type of NPs exploited in ocular drug delivery
NPs represent promising drug carriers for ophthalmic applications. After optimal binding of these particles, the drug absorption in the eye is enhanced significantly in comparison to eye drop solutions owing to the much slower ocular elimination rate of particles. Smaller particles are better tolerated by the patients than larger particles; therefore NPs may comfortably be used for prolonged action ophthalmic delivery systems.
Different polymers can be used to fabricate NPs such as biodegradable polymers like polylactide (PLAs), poly (D, L-lactides), polylactic-co-glycolic acid, E-caprolactone, (Fessi et al. 1989; Kumari et al. 2010; Aksungur et al. 2011; Gupta et al. 2011), polyacrylamide, poly cyanoacrylate and polymethylmethacrylate (Zimmer et al. 1991; Wenger et al. 2011) and natural polymers like CS (Jain et al. 2014), gelatin (Vandervoort and Ludwig 2004), sodium alginate (Zhu et al. 2012), albumin (Zimmer et al. 1994; Merodio et al. 2002) and tamarind kernel polysaccharide (Kaur et al. 2012) can be used effectively for efficient drug delivery to the ocular tissues. Many researchers conducted the study on nanoparticulate drug delivery system and found that it prevents the degradation of the drug in an ocular environment and release of drug over an extended period of time which gives the desired effect (Gupta et al. 2010; Javadzadeh et al. 2010) (Table 9.4).
Başaran and coworkers showed in an in vivo study that CS-NPs of cyclosporine prolonged release of active agent due to the positive charge of CS. This may be attributed to enhanced residence time at the corneal and conjunctival surfaces (Başaran et al. 2014).
Sabzevari and coworkers studied the biodegradable poly β-amino ester-NPs of triamcinolone acetonide of anti-inflammatory effects in the rabbit eye. It was concluded that polymeric NPs of triamcinolone acetonide will provide good anti-inflammatory effects as subconjunctival injection method and are better compared to other drug delivery systems (Sabzevari et al. 2013).
Wadhwa and coworkers reported that hyaluronic acid (HA) gave the synergistic effect for mucoadhesion in association with dorzolamide hydrochloride or timolol maleate loaded CS-NPs and revealed that CS-HA-NPs show a higher reduction in intraocular pressure level as compared to pure drug solution (Wadhwa et al. 2010).
Agnihotri and Vavia evaluated diclofenac sodium–loaded PLGA-NPs for ocular use and found good biocompatibility with the eye (Agnihotri and Vavia 2009).
Jwala and coworkers studied the PLGA-NPs delivery of acyclovir prodrug for the treatment of ocular herpes keratitis. It was found that NPs exhibited a biphasic release behavior i.e., initial burst phase followed by sustained release because PLGA-NPs may retard the degradation of the prodrug in the precorneal area and further sustain the release in deep corneal tissues. Dispersion of NPs in thermosensitive gels completely eliminated the burst release phase (Jwala et al. 2011).
Basaran and co-workers developed cyclosporine NPs for ocular delivery by spray drying technique by using different grade (molecular weight) of CS and characterized in vitro and in vivo. In vitro study showed sustained release. The in vivo study was done using sheep and sample was analyzed by enzyme immune assay. It showed that prolonged release of active agent from positively charged CS formulations after 72 h of study in both aqueous and vitreous humor samples. This was attributed to increasing in the corneal and conjunctival surfaces residence, so it was concluded that CS possess bioadhesive and permeation enhancing property (Başaran et al. 2014).
Tayel and coworkers formulated terbinafine hydrochloride NPs by positively charged Eudragit® RS 100 and CS polymer in a different ratio by nanoprecipitation technique using Soybean lecithin (1 %, w/v) and Pluronic® F68 as surfactant and stabilizer. The NPs have small particles size, zeta potential (73.29–320.15 nm and +20.51 to +40.32 mV respectively) and sustained release profile in simulated tear fluid (pH 7.4). The NPs were physically stable at 4 and 25 °C. NP suspension showed significantly (P < 0.05) increased drug mean residence time and improved its ocular bioavailability; 1.657-fold in rabbits aqueous humor as compared to terbinafine hydrochloride eye drops (0.25 %, w/v). To enhance the permeation and residence time of the formulation, by overcoming the limitations associated with protective ocular barriers (Tayel et al. 2013).
Pathak and co-workers evaluated novel pH-triggered nano-emulsified in situ gel (NE-ISG) for ophthalmic delivery of fluconazole. Pseudoternary phase diagrams were constructed using capmul MCM (oil phase), tween 80 (surfactant) and transcutol P (cosurfactant) to identify the nanoemulsion region and formation of spontaneous emulsification. The formulations were characterized by permeation, corneal irritation, and corneal toxicity. The optimized nanoemulsion in situ gels showed 3-fold (337.67 µg/cm2) higher Ex vivo transcorneal permeation than commercial eye drops (112.92 µg/cm2). Formulation did not show any visual signs of tissue damage. Hence it was concluded that nanoemulsion gel may offer a more intensive treatment of ocular fungal infections due to higher permeation, prolonged precorneal residence time and sustained drug release along with higher in vitro efficacy, safety and greater patient compliance (Pathak et al. 2013).
Mohammed and coworkers developed and studied the fluconazole loaded chitin nanogels (Flu-CNGs) for the treatment of corneal fungal infections. Flu-CNGs have controlled release pattern at a prolonged period of time. Flu-CNGs are hemocompatible, cytocompatible and also showed very good cell uptake in human dermal fibroblast cells and penetration to the deeper sections of the porcine cornea with no signs of destruction or inflammation to corneal cells (Mohammed et al. 2013).
Du Toit and coworkers developed and compared two specific embodiments of an ocular nanosystem (NS) i.e., CS-poly (ε-caprolactone), and the composite lipoidal-polymeric NS for intelligent treatment of inflammatory disorders (specifically ocular afflictions). The anti-inflammatory efficacy, demonstrated by a decrease in 4-chloro-7-nitrobenzo-2-oxa-1, 3-diazole complex formation (0.0031 Vs 0.0023 mmol/L) and decrease in NFκB formation (decrease in relative optical density of 0.2027 vs. 0.2420). It was found that indomethacin lipoidal-polymeric NS has enhanced efficacy in terms of tissue permeation, cell uptake, and anti-inflammatory activity (Du Toit et al. 2013).
Zhou and co-worker developed a self-aggregated nanoparticulate vehicle using an amphiphilic poly (lactic acid)-grafted-CS (PLA-g-CS) copolymer and to evaluate its potential for ocular delivery of amphotericin B. The self-aggregated PLA-g-CS NPs had a core-shell structure with an average particle size of approximately 200 nm and zeta potentials higher than +30 mV with high encapsulation efficiency. The formulation showed sustained release profile and no sign of irritation after instillation in rabbit eye was scored. The significant antifungal activity was observed when compared with amphotericin B against Candida albicans. The in vivo ocular pharmacokinetic study suggested that the PLA-g-CS-NPs have the advantage of prolonging residence time at the ocular surface. The corneal penetration study showed that the PLA-g-CS NPs could penetrate into the cornea (Zhou et al. 2013).
Kaur and co-workers developed the spanlastics which consisted of spans and an edge activator prepared by the ether injection method and characterized by size, shape, and the number of vesicles/ml by optical microscopy, Entrapment efficiency, ex vivo corneal permeability study and MTT assay. Spanlastics formulation is smaller in size (3 times) and showed a better permeation in comparison to a corresponding niosomal formulation and 3-fold increase compared to the marketed formulation due to its elastic nature. The developed system is novel and provides an effective and safe formulation of fluconazole (Kaur et al. 2012).
Başaran and co-workers developed cyclosporine A NPs using cationic Eudragit RS for ocular application. The formulation was characterized in vitro and in vivo. The NPs showed the extended release of incorporated drug. In vivo study showed prolonged residence time off, cyclosporine A in the deeper layers (vitreous humor) of the eye due the positively charged nature of Eudragit RS (Başaran et al. 2011).
Singh and coworkers developed CS-NPs of brimonidine tartrate (BT) for ocular delivery and characterized by TEM, SEM, particle size, and polydispersity index (PI), DSC, IR, and entrapment efficiency which gave an insight of physicochemical interaction that influenced the CS-NPs formation and entrapment of BT. In vitro release showed sustained release. In vivo studies confirmed a significant sustained effect BT-NPs were compared with conventional eye drops. It was concluded that BT loaded CS-NPs could help to reduce dosing frequency by sustained drug release in the treatment of glaucoma (Singh et al. 2011).
Liposomes
Liposomes were first introduced as drug delivery carriers by Bangham et al. (1965). They bilayer lipid vesicles composed of altering aqueous compartments enclosed by lipid bilayers (mainly phospholipids and cholesterol) of natural and synthetic origin, and usually within the size range of 10 nm to 1 μm or greater (Meisner and Mezei 1995; Mishra et al. 2011; Honda et al. 2013). Liposomes have advantages like, improved bioavailability of ophthalmic drugs after topical administration (Monem et al. 2000; Kaur and Kanwar 2002; Wadhwa et al. 2009), no toxicity and low antigenicity (Rooijen and Nieuwmegen 1980) stable, biocompatible, biodegradable and metabolize in vivo encapsulating both lipophilic, hydrophilic and amphiphilic molecules (Klibanov et al.1990; Abrishami et al. 2009) hydrophilic drugs are entrapped in the aqueous layer, while hydrophobic drugs are stuck in the lipid bilayers. Depending on their size and number of bilayers, liposomes can be classified as small unilamellar vesicles (SUVs, 20 nm to ~200 nm), giant unilamellar vesicles (GUVs, >1 μm), and large unilamellar vesicles (LUVs, 200 nm to ~1 μm) and, Multilamellar vesicles (MLV, >0.5 μm) (Jesorka and Orwar 2008). Unilamellar vesicles are composed of single layer of lipid such as lecithin or phosphatidylglycerol encapsulating aqueous interior core whereas Multilamellar vesicle is composed of various layers of lipid bilayers (Kaur et al. 2004; Mainardes et al. 2005; Shen and Tu 2007; Elbayoumi and Torchilin 2010; Dai et al. 2013). Liposome can also be divided into three categories i.e., negatively charged, positively charged and neutral liposomes. The charge present on the liposome is due to charged ingredient used in formulation development. Many researchers investigated that positively charged liposomes exhibited prolonged precorneal retention than negative and neutral charge liposome, due to electrostatic interaction with the negatively charged corneal epithelium, thus enhance drug absorption (Monem et al. 2000; Abuzaid et al. 2003; Danion et al. 2007; Dai et al. 2013).
Hitoshi Sasaki and co-workers reported the surface modification of liposome with poly-L-lysine to enhance the efficiency of coumarin-6 as a model drug and fluorescent marker to the retina (delivery to the posterior segment of the eye). It was found that surface modification with low molecular weight poly-L-lysine significantly increase the delivery as compared to high molecular weight poly-L-lysine, because aggregation of surface-modified liposomes, increased particle size hampered distribution to inner ocular tissue (Sasaki et al. 2013).
Hathout and co-workers reported acetazolamide liposome showed that positively charged and neutral liposomes exhibited greater lowering in IOP and a more prolonged effect than the negatively charged ones. The findings suggest that liposomes enhance corneal penetration of drug by being adsorbed onto the corneal surface, with direct transfer of drug from liposomal to epithelial cell membranes (Hathout et al. 2007).
Drug loading capacity and entrapment efficiency of liposomes depend on many factors such as the size of liposomes, concentration and types of lipid used, and physicochemical properties of therapeutic agent itself. Loading capacity and entrapping efficiency are poor for SUVs in comparison to MLVs. However, LUVs provide a balance between size, loading capacity and entrapment efficiency (Jesorka and Orwar 2008; Ding et al. 2005).
Bochot and coworkers reported the phosphodiester oligonucleotide encapsulated in liposome showed sustained release in the vitreous humor, retina and choroid (37 % even after 15 days) compared with the release from the solution and in a reduced distribution to the non-targeted tissues (sclera, lens) (Bochot et al. 1998a, b).
Szulc and coworkers reported that the liposomal encapsulation of the hydrophilic drug, pilocarpine, enhanced its pharmacological effect in rabbits when using positively charged vesicles (Szulc et al. 1988).
Chetoni and coworkers compared Acyclovir containing positively charged unilamellar liposomes compared with pure acyclovir in solution and marketed acyclovir ointment containing same acyclovir concentrations (0.12 %). It was observed that the liposomal formulation have the highest drug concentration in the aqueous humor of rabbits. The in vitro release profile of liposomal formulation showed sustained release. It was concluded that positively charged liposome bind to negatively charge corneal epithelium enhances the efficacy of acyclovir. These results indicated a significant advantage of acyclovir liposome as an alternative to acyclovir ointment (Chetoni et al. 2004).
Similarly many antiviral drugs like iododeoxyuridine, ganciclovir and acyclovir-loaded in a liposome (immuno-liposome) have been reported for the treatment of ocular herpes infection (herpes simplex virus) (Norley et al. 1986).
Niosomes
Niosome is a novel nonionic surfactant, a bilayered vesicular system formed by self-assembly of hydrated surfactant. Like liposome, they deliver a drug in a controlled manner to enhance bioavailability and get therapeutic effect over a longer period of time (Mujoriya and Bodla 2011; Karim et al. 2012). But the liposome has some drawbacks like high cost and limited shelf life, and these are overcome by developing the new vesicular system niosome (Mahale et al. 2012). The vesicle suspension is a water based vehicle; this offers high patient compliance in comparison with an oily delivery system. They possess an infrastructure consisting of hydrophilic, amphiphilic and lipophilic moieties together and can entrap both hydrophilic and lipophilic drug with a wide range of solubilities. The bilayers of the niosomes protect the enclosed active pharmaceutical ingredient from the heterogeneous factors present both inside and outside the body so can be used for labile and sensitive drugs (Khan et al. 2011). Mainly nonionic surfactant preferred due to their nontoxic, stable, ability to maintain pH up to physiological pH, act as solubilizer, wetting agents, and permeability enhancer, improve the bioavailability, controlled release of drug, nonimmunogenic biodegradable and biocompatible properties. The order of toxicity potential of various surfactant are cationic > anionic > ampholytic > nonionic. It comprised of both polar and nonpolar segments and has high interfacial activity. The formation of bilayered vesicles instead of micelles is dependent on the hydrophilic–lipophilic balance (HLB), chemical structure, critical packing parameter (CPP) of the surfactant. The chain length and size of the hydrophilic head group of the nonionic surfactant affect the entrapment efficiency of the drug. Nonionic surfactants with stearyl (C18) chains show higher entrapment efficiency than those with lauryl (C12) chains. The Tween series of surfactants bearing a long alkyl chain and a large hydrophilic moiety in combination with cholesterol in a 1:1 ratio have the highest entrapment efficiency (Arunothayanun et al. 2000). HLB value in the range 14–17 is not suitable to produce niosomes whereas one with an HLB value of 8.6 gives niosomes with the highest entrapment efficiency. Entrapment efficiency decreases as the HLB value decreases from 8.6 to 1.7 (Lawrence et al. 1996; Biswal et al. 2008; Shahiwala et al. 2002; Kumbhar et al. 2013). For HLB > 6, cholesterol must be added to the surfactant in order to form a bilayered vesicle and for lower HLB values, cholesterol enhances the stability of vesicles. It is also seen that the addition of cholesterol enables more hydrophobic surfactants to form vesicles, suppresses the tendency of the surfactant to form aggregates, and provides greater stability to the lipid bilayers by promoting the gel-liquid transition temperature of the vesicle of water soluble drugs (Khazaeli et al. 2007). Different non-ionic surfactant used in the manufacturing of niosomes like alkyl ethers and alkyl glyceryl ethers. The component of niosomes are surfactant, ether linked surfactants i.e., polyoxyethylene 4 lauryl ether (Brij 30), polyoxyethylene acetyl ethers (Brij 58) (Carafa et al. 2002; Manosroi et al. 2003; Raymzond et al. 2006), Polyoxyethylene fatty acid esters (Brij 72) (Pardakhti et al. 2007; Balakrishnan et al. 2009), Sorbitan fatty acid esters (Guinedi et al. 2005); Cholesterol; Charge Inducers: increase the stability of the vesicles by induction of charge on the surface preventing the fusion of vesicles due to repulsive forces of the same charge and provide higher values of zeta potential, negative charge inducers are diacetyl phosphate, dihexadecyl phosphate and lipoamine acid and positive charge inducers are sterylamine and cetyl pyridinium chloride (Shan et al. 2008). Gemini and Bola surfactants also used for preparation of niosome have, non-toxic, more stable, non-irritating non-haemolytic higher solubility, lower aggregation number and have no drawback like other surfactants such as Polysorbate (Harmful to persons with Crohn’s disease) (Hait and Moulik 2002; Ikeda 2003; Yan et al. 2009; Carol and Keita 2010).
Different methods are used for the preparation of niosomes: such as lipid layer hydration, reversed phase evaporation, micro-fluidization, ether injection, the bubbling of nitrogen, Handjani–Vila method, enzymatic method, single pass technique involving evaporation to produce a lipid film followed by hydration with the hydration medium.
Basha and co-workers developed span 60 based nanovesicles with edged activator Tween 80 (TW80), sodium cholate (SC) or sodium deoxycholate (SDC) of clotrimazole. They found that spherical unilamellar vesicle with 87.92 % entrapment efficiency and displayed sustained antifungal effect over 12 h against Candida albicans. The AUC of the optimized formulation was 3.09 times more than that of drug suspension with no sign of irritation after testing for ocular tolerance (Basha et al. 2013).
Hamdy and coworkers reported the Span 60-based niosomes for ocular delivery of naltrexone HCl (NTX) and found that 5-fold increase in NTX entrapment efficiency (EE %). Ex vivo transcorneal permeation studies conducted using excised cow corneas showed that niosomes were capable of controlling NTX permeation, enhanced its corneal permeability and practically nonirritant (Hamdy et al. 2011).
Akhter and coworker developed ganciclovir mucoadhesive niosomal dispersion. It was revealed from the results that the developed formulations were nonirritant and nontoxic in nature. It was further concluded that ganciclovir nanoformulation could be utilized as a potential delivery system for treatment of ocular infections by topical instillation (Akhter et al. 2011).
Kapadia and coworkers reported niosomes incorporated in situ gel hydrogel for ocular delivery are found effective in controlling the drainage of the formulation from the ocular site and were effective in improving the formulation performance (Kapadia et al. 2009).
Aggarwal and coworkers reported that CS-coated timolol maleate (0.25 %) niosomal formulation exhibits more effect on intraocular pressure reduction with less chance of cardiovascular side effects when compared to a marketed formulation timolol solution (TMS; 0.25 %) (Aggarwal and Kaur 2005).
Ahmed and coworkers developed the non-ionic surfactant niosome delivery of acetazolamide. The results of this study showed the effect of cholesterol content, the type of surfactant and the method of preparation on the entrapment efficiency and in vitro drug release. The higher entrapment efficiency was obtained with multilamellar niosomes prepared with span 60 and cholesterol in a 7:6 molar ratio. The in vivo evaluation of acetazolamide niosomes in comparison to acetazolamide solution and niosomes showed that acetazolamide multilamellar niosomes most effectively prolonged the decrease in the IOP. Slight irritation could be observed in corneal tissues after long treatment with these niosomes which is reversible and abolished by time (Ahmed et al. 2005).
Dendrimers
Dendrimers are particularly auspicious potentially usable in numerous applications. It is a tree like highly branched synthetic nanostructured polymers with a three-dimensional structure. These very monodisperse molecules are composed of an initiator core, interior layers of repeating units, and multitudinous terminal groups. Their branched layered architectures displayed a high number of controlled terminal groups which favored biomedical applications (Bai et al. 2006; Newkome and Shreiner 2008; Kambhampati and Kannan 2013). They are classified by the number of branches and terminal groups. Various cores and units can be used, which can change the properties and shape of the dendrimer. The drug delivery can be achieved by coupling a drug through one of two approaches. Hydrophobic drugs can be complexed within the hydrophobic dendrimer interior to make them water-soluble or drugs can be covalently coupled to the surface of the dendrimer. Dendrimers can either attach to a therapeutic agent by a permanent or separable bond to the end groups or be enclosed within the dendrimer itself. Because of its multiple terminal groups and its polymer backbone, dendrimers can have multiple functionalities. In addition, dendrimers have been shown to be capable of bypassing efflux transporters and enable the efficient transport of drugs across cellular barriers (Yang and Kao 2006; Samad et al. 2009; Abdelkader et al. 2011).
Dendrimeric structures are of particular interest in the field of drug delivery due to their peculiar structural properties including controllable internal cavities bearing specific species for the encapsulation of guest drugs and external periphery with 3D multiple functional moieties for solubilisation, conjugation of bioactive compounds and targeting molecules, and recognition purposes The main successes of dendrimers resulted in their appropriate, reproducible and optimized design parameters addressing physicochemical limitations of classical drugs (e.g., solubility, specificity, stability, biodistribution and therapeutic efficiency) (Jain and Gupta 2008) and their ability to overcome biological issues to reach the right target(s) (e.g., first-pass effect, immune clearance, cell penetration, off-target interactions, etc.). Improvement of pharmacokinetic (PK) and pharmacodynamic (PD) behaviors of both drug-dendrimer conjugates and drug–dendrimer encapsulates versus drugs demonstrates their strong potentials in medicine as nano-carriers (Gajbhiye et al. 2009; Mintzer and Grinstaff 2011). The most commonly used dendrimers in nanomedicine are polyamidoamines (PAMAM), poly(L-lysine), scaffold dendrimers (PLL), polyesters (PGLSA-OH), polypropylimines (PPI), poly(2,2-bis (hydroxymethyl) propionic acid scaffold dendrimers (bis-MPA) and aminobis (methylene phosphonic acid) scaffold dendrimer.
Contemporary View: Paradigm Shift Towards Mucoadhesive Nano-System
From last 20 years, efforts have been directed in the rational design of ocular drug delivery consisting of mucoadhesive nanocarriers. Thereafter, the plan was directed to generate nanocarriers with a hydrophilic coating with the idea of improving their stability and their interaction with the mucosa (Calvo et al. 1997; Schipper et al. 1997; De Campos et al. 2003; Ludwig 2005). It was proficient via optimization of nanocarrier’s ocular drug delivery to obtain long-lasting bioadhesive/residence time by the so-called ‘mucoadhesive property’ based on entrapment of particles in the ocular mucous layer and interaction of bioadhesive polymer chain with mucin (Henriksen et al. 1996; Du Toit et al. 2013). Maintenance of the designed nanocarriers in the ocular delivery following topical application is thus decisive to accomplish unremitting drug release and prolonged therapeutic activity. The concept of enhanced ocular resident time and bioavailability of drug through mucoadhesive nanocarriers and positively charged mucoadhesive nano-system and its interaction with the ocular surface are illustrated in Fig. 9.4.

Proposed mechanism of mucoadhesion over the corneal surface exploiting mucoadhesive and positively charged nano-system
Therefore, a design of nanocarriers from mucoadhesive materials is crucial for improved retention in the ocular cul-de-sac (Du Toit et al. 2013). Ocular mucoadhesion, exclusively, refers to the capability of certain polymers to hold on to the mucous layer casing the conjunctival and corneal surfaces of the eye by noncovalent bonds (Round et al. 2002). Washout time for the mucoadhesive polymeric system is reduced since this depends on mucous turnover rate rather than lachrymal discharge turnover rate. A mucoadhesive polymer with plentiful hydrophilic functional group viz sulfate, hydroxyl, carboxyl, amide has found a fundamental role in ocular drug delivery system owing to their adhesion property with precorneal/conjunctival mucin layer via non-covalent bonds, and remaining in place for as long as the mucin is available there. Using this concept, various investigators planned the cationic polymer CS and CMKP as a polymer of choice because of its unique properties, including good enough biodegradability and biocompatibility permeability enhancer by affecting both paracellular and intracellular pathways of epithelial cells in a reversible manner without affecting cell viability or causing membrane wounds (Hirano et al. 1990; Takeuchi et al. 1996; Calvo et al. 1997; Felt et al. 1999; Motwani et al. 2008).
For the selection of bioadhesive polymer intended for ophthalmic drug delivery, the viscosity and wetting properties of the polymer are considered. Viscosity measures the resistance to flow which depends on upon its molecular mass, concentration, temperature and shear stress. Polymer showing non-Newtonian behaviour, when incorporated in formulation possessing pseudoplastic behaviour in which viscosity decreases with increasing shear rate (due to blinking and ocular movement), this results is significantly less resistance to blinking and demonstrates greater acceptance as compared to formulation possessing polymer exhibiting Newtonian flow, no real improvement of bioavailability in polymer showing Newtonian flow system (Schoenwald et al. 1978; Greaves et al. 1993; Ludwig and Ooteghem 1992; Van Ooteghem 1995). The mucoadhesive properties of polyacrylic acid hydrogel and their ability to penetrate the mucin at the surface of the eye have been investigated extensively (Ponchel et al. 1987; Slovin and Robinson 1993; Duchfine et al. 1988; Meng et al. 2011). There are many mucoadhesive polymers used for designing of a colloidal carrier (Greaves et al. 1993; Govender et al. 2005) in ocular drug delivery. Various mucoadhesive polymers and their mucoadhesive strength are graded in Table 9.5.
Several other synthetic polymers have been examined for the fabrication of mucoadhesive nanocarriers for ocular delivery, for example, Pignatello and coworkers reported the formulation and evaluation of nanocarriers composed of Eudragit-RL100 with good ocular tolerance, and no inflammation or discomfort in the rabbit eye (Pignatello et al. 2002). Similarly, Barbault-Foucher and co-workers reported the hyaluronic acid (HA) colloidal system as a natural, non-irritating carrier system that showed pseudoplastic behavior with desirable ocular mucoadhesive properties (Barbault-Foucher et al. 2002).
References
Abdelkader H, Ismail S, Kamal A, Alany RG (2011) Design and evaluation of controlled release niosomes and discomes for naltrexone hydrochloride ocular delivery. J Pharm Sci 100:1833–1846
Abrishami M, Zarei-Ganavati S, Soroush D, Rouhbakhsh M, Jaafari MR, Malaekeh Nikouei B (2009) Preparation, characterization, and in vivo evaluation of nanoliposomes-encapsulated bevacizumab (avastin) for intravitreal adminis-tration. Retina 29:699–703
Abuzaid SS, El-Ghamry HA, Hammad M (2003) Liposomes as ocular drug delivery system for atenolol. Egypt J Pharm Sci 44:227–245
Addo RT, Siddig A, Patel NJ, Siwale R, Akande J, Uddin AU, D’Souza MJ (2010) Formulation, characterization, and testing of tetracaine hydrochloride-loaded albumin-chitosan microparticles for ocular drug delivery. J Microencapsul 27(2):95–104
Addo RT, Yeboah KG, Siwale RC, Siddig A, Jones A, Ubale RV, Akande J, Nettey H, Patel NJ, Addo E, D’Souza MJ (2015) Formulation and characterization of atropine sulfate in albumin-chitosan microparticles for in vivo ocular drug delivery. J Pharm Sci 104(5):1677–1690
Aggarwal D, Kaur IP (2005) Improved pharmacodynamics of timolol maleate from mucoadhesive niosomal ophthalmic drug delivery system. Int J Pharm 16:155–159
Aggarwal D, Pal D, Mitra AK, Kaur IP (2007) Study of the extent of ocular absorption of acetazolamide from a developed niosomal formulation, by micro dialysis sampling of aqueous humor. Int J Pharm 29:21–26
Agnihotri SM, Vavia PR (2009) Diclofenac-loaded biopolymeric nanosuspensions for ophthalmic application. Nanomed Nanotech Boil Med 5:90–95
Ahmed I, Patton TF (1985) Importance of the noncorneal absorption route in topical ophthalmic drug delivery. Invest Ophthalmol Vis Sci 26(4):584–587
Ahmed I, Patton TF (1987) Disposition of timolol and inulin in the rabbit eye following corneal versus non-corneal absorption. Int J Pharm 38:9–21
Ahmed S Guinedi, Nahed DM, Samar M, Rania MH (2005) Preparation and evaluation of reverse-phase evaporation and multilamellar niosomes as ophthalmic carriers of acetazolamide. Int J Pharm 306:71–82
Akhter S, Talegaonkar S, Khan ZI, Jain GK, Khar RK, Ahmad FJ (2011) Assessment of ocular pharmacokinetics and safety of ganciclovir loaded nanoformulation. Biomed Nanotechnol 7:144–145
Aksungur P, Demirbilek M, Denkbas EB, Vandervoort J, Ludwig A, Unlu N (2011) Development & characterization of cyclosporine A loaded nanoparticles for ocular drug delivery: cellular toxicity, uptake and kinetic studies. J Control Rel 151:286–294
Ameeduzzafar Ali J, Bhatnagar A, Kumar N, Ali A (2014) Chitosan nanoparticles amplify the ocular hypotensive effect of cateolol in rabbits. Int J Biol Macromol 65:479–491
Ammar HO, Salama HA, Ghorab M, Mahmoud AA (2009) Nanoemulsion as a potential ophthalmic delivery system for dorzolamide hydrochloride. AAPS PharmSciTech 10(3):808
Araujo J, Gonzalez E, Egea MA, Garcia ML, Souto EB (2009) Nanomedicines for ocular NSAIDs: safety on drug delivery. Nanomedicine 5:394–401
Arunothayanun P, Bernard MS, Craig DQ, Uchegbu IF, Florence AT (2000) The effect of processing variables on the physical characteristics of non-ionic surfactant vesicles (niosomes) formed from hexadecyl diglycerol ether. Int J Pharm 201:7–14
Badawi AA, El-Laithy HM, El Qidra RK, El Mofty H, El dally M (2008) Chitosan based nanocarriers for indomethacin ocular delivery. Arch Pharm Res 31(8):1040-1049
Bai S, Thomas C, Rawat A, Ahsan F (2006) Recent progress in dendrimer-based nanocarriers. Crit Rev Ther Drug Carrier Syst 23:437–495
Balakrishnan P, Shanmugam S, Lee WS, Lee WM (2009) Formulation and in vitro assessment of minoxidil niosomes for enhanced skin delivery. Int J Pharm 377:1–8
Bangham D, Standish MM, Watkins JC (1965) Diffusion of univalent ions across the lamellae of swollen phospholipids. J Mol Biol 13:238–252
Barbault-Foucher S, Gref R, Russo P, Guechot J, Bochot A (2002) Design of poly--caprolactone nanospheres coated with bioadhesive hyaluronic acid for ocular delivery. J Control Release 83(3):365–375
Başaran, E, Demirel, M, Sırmagül, B, Yazan, Y (2011) Polymeric cyclosporine-A nanoparticles for ocular application. J Biomed Nanotechnol 7(5):714–723
Başaran E, Yenilmez E, Berkman MS, Büyükköroğlu G, Yazan Y (2014) Chitosan nanoparticles for ocular delivery of cyclosporine A. J Microencapsul 31(1):49–57
Basha M, Hosam , El-Alim A, Shamma RN, Awad GEA (2013) Design and optimization of surfactant based nanovesicles for ocular delivery of clotrimazole
Baspinar Y, Bertelmann E, Pleyer U, Buech G, Siebenbrodt I, Borchert HH (2008) Corneal permeation studies of everolimus microemulsion. J Ocul Pharmacol Ther 24(4):399–402
Biswal S, Murthy PN, Sahu J, Sahoo P, Amir F (2008) Vesicles of non-ionic surfactants (niosomes) and drug delivery potential. Int J Pharm Sci Nanotechnol 1:1–8
Bochot A, Fattal E, Grossiord JL, Puisieux F, Couvreur P (1998a) Characterization of a new drug delivery system based on dispersion of liposomes in a thermosensitive gel. Int J Pharm 162:119–127
Bochot A, Gulik FA, Couarraze G, Couvreur P (1998b) Liposomes dispersed within a thermosensitive gel: a new dosage form for ocular delivery of oligonucleotides. Pharm Res 15:1364–1369
Bourlais CL, Acar L, Zia H, Sado PA, Needham T, Leverge R (1998) Ophthalmic drug delivery systems: recent advances. Prog Retin Eye Res 17:33–58
Calvo P, Remunan-Lopez C, Vila-Jato JL, Alonso MJ (1997) Novel hydrophilic chitosan-polyethylene oxide nanoparticles as protein carriers. J Appl Polym Sci 63:125–132
Carafa M, Santucci E, Lucania G (2002) Lidocaine loaded non ionic surfactant vesicles: characterization and in vitro permeation studies. Int J Pharm 231:21–32
Carol L, Keita AV (2010) Translocation of Crohn’s disease Escherichia coli across M cells: contrasting effects of soluble plant fibers and emulsifiers. Gut 59:1331–1339
Chen Y, Lu Y, Zhong Y, Wang Q, Wu W, Gao S (2012) Ocular delivery of cyclosporine A based on glyceryl monooleate/poloxamer 407 liquid crystallinenanoparticles: preparation, characterization, in vitro corneal penetration and ocular irritation. J Drug Target 20(10):856–863
Chetoni P, Rossi S, Burgalassi S, Monti D, Mariotti S, Saettone MF (2004) Comparison of liposome-encapsulated acyclovir with acyclovir ointment: ocular pharmacokinetics in rabbits. J Ocul Pharmacol Ther 20:169–177
Cruysberg LP, Nuijts RM, Geroski DH, Koole LH, Hendrikse F, Edelhauser HF (2002) In vitro human scleral permeability of fluorescein, dexamethasone-fluorescein, methotrexate-fluorescein and rhodamine 6G and the use of a coated coil as a new drug delivery system. J Ocul Pharmacol Ther 18:559–569
Dai Y, Zhou R, Liu L, Lu Y, Qi J, Wu W (2013) Liposomes containing bile salts as novel ocular delivery systems for tacrolimus (FK506): in vitro characterization and improved corneal permeation. Int J Nanomed 8:1921–1933
Danion A, Arsenault I, Vermette P (2007) Antibacterial activity of contact lenses bearing surface-immobilized layers of intact liposomes loaded with levofloxacine. J Pharm Sci 96:2350–2363
De Campos AM, Sanchez A, Gref R, Calvo P, Alonso MJ (2003) The effect of a PEG versus a chitosan coating on the interaction of drug colloidal carriers with the ocular mucosa. Eur J Pharm Sci 20:73–81
De Campos AM, Diebold Y, Carvaiho ELS, Sanchez A, Alonso MJ (2004) Chitosan nanoparticles as new ocular drug delivery system: in vitro stability, in vivo fate, and cellular toxicity. Pharm Res 21:803–810
Dhubhghaill SN, Humphries MM, Kenna PF (2012) Further development of barrier modulation as a technique for systemic ocular drug delivery. Adv Exp Med Biol 723:155–159
Ding S (1998) Recent developments in ophthalmic drug delivery. Pharm Sci Technol Today 1:328–335
Ding X, Alani WG, Robinson JR (2005) Extended-release and targeted drug delivery systems. In: Troy DB (ed) Remington: the science and practice of pharmacy. Lippincott Williams and Wilkins, Philadelphia, PA, USA
Djekic L, Ibric S, Primorac M (2008) The application of artificial neural networks in the prediction of microemulsion phase boundaries in PEG-8 caprylic/capric glycerides based systems. Int J Pharm 361:41–46
Du Toit LC, Govender T, Carmichael T, Kumar P, Choonara YE, Pillay V (2013) Design of an anti-inflammatory composite nanosystem and evaluation of its potential for ocular drug delivery. J Pharm Sci 102(8):2780–2805
Duchfine D, Touchard F, Peppas NA (1988) Pharmaceutical and medical aspects of bioadhesive systems of drug administration. Drug Dev Ind Pharm 14:283–318
Elbayoumi TA, Torchilin VP (2010) Current trends in liposome research. Methods Mol Biol 605:1–27
Felt O, Furrer P, Mayer JM, Plazonnet B, Buri P, Gurny R (1999) Topical use of chitosan in ophthalmology: tolerance assessment and evaluation of pre-corneal retention. Int J Pharm 180:185–193
Fessi H, Puisieux F, Devissaguet JP, Ammoury N, Benitas S (1989) Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int J Pharm 55:R1–R4
Fialho SL (2004) New vehicle based on a microemulsion for topical ocular administration of dexamethasone. Clin Exp Ophthalmol 32:626–632
Fischbarg J (2006) The corneal endothelium. In: Fischbarg J (ed) The Biology of Eye. Academic Press, New York, NY, USA, pp 113–125
Fitzgerald P, Wilson C (1994) Polymeric systems for ophthalmic drug delivery. In: Severian D (ed) Polymeric systems for ophthalmic drug delivery. Marcel Dekker, New York. 373–398
Gajbhiye V, Palanirajan VK, Tekade RK, Jain NK (2009) Dendrimers as therapeutic agents: a systematic review. J Pharm Pharmacol 61:989–1003
Garty N, Lusky M (1994) Pilocarpine in submicron emulsion formulation for treatment of ocular hypertension: a phase II clinical trial. Invest Ophthalmol Vis Sci 35:2175–2185
Gasco MR, Gallarate M, Trotta M, Bauchiero L, Gremmo E and Chiappero O (1989) Microemulsions as topical delivery vehicles: Ocular administration of timolol. J Pharm Biomed Analysis 7(4):433–439
Gausas RE, Gonnering RS, Lemke BN, Dortzbach RK, Sherman DD (1999) Identification of human orbital lymphatics. Ophthal Plast Reconstr Surg 15:252–259
Gipson IK, Argueso P (2003) Role of mucin in the function of the corneal and conjunctival epithelia. Int Rev Cytol 231:1–49
Govender S, Pillay V, Chetty DJ, Essack SY, Dangor CM, Govender T (2005) Optimization and characterization of bioadhesive controlled release tetracycline microspheres. Int J Pharm 306:24–40
Greaves JL, Wilson CG, Birmingham AT (1993) Assessment of the precorneal residence of an ophthalmic ointment in healthy subjects. Br J Clin Pharmacol 35:188–192
Guinedi AS, Mortada ND, Mansour S, Hathout RM (2005) Preparation and evaluation of reverse-phase evaporation and multilamellar niosomes as ophthalmic carriers of acetazolamide. Int J Pharm 306:71–82
Gupta S, Moulik SP (2008) Biocompatible Microemulsions and their prospective uses in drug delivery. J Pharm Sci 97:22–45
Gupta H, Aqil M, Khar RK, Ali A, Bhatnagar A, Mittal G (2010) Sparfloxacin-loaded PLGA nanoparticles for sustained ocular drug delivery. Nanomed Nanotech Biol Med 6:324–333
Gupta H, Aqil M, Khar RK, Ali A, Bhatnagar A, Mittal G (2011) Biodegradable levofloxacine nanoparticles for sustained ocular drug delivery. J Drug Target 19(6):409–417
Hanrahan F, Campbell M, Nguyen AT, Suzuki M, Kiang AS, Tam LC, Gobbo OL, Dhubhghaill SN, Humphries MM, Kenna PF (2012) Further development of barrier modulation as a technique for systemic ocular drug delivery. Adv Exp Med Biol 723:155–159
Hait SK and Moulik SP (2002) Gemini surfactants: A distinct class of self-assembling molecules. Curr Sci 82:1101–1111
Hamdy A, Sayed I, Amal K, Raid GA (2011) Design and evaluation of controlled-release niosomes and discomes for naltrexone hydrochloride ocular delivery. J Pharm Sci 100(5):1833–1846
Hathout RM, Mansour S, Mortada ND (2007) Liposomes as an ocular delivery system for acetazolamide: in vitro and in vivo studies. AAPS Pharm Sci Tech 8:1–12
Henriksen I, Green KL, Smart JD, Smistad G, Karlsen J (1996) Bioadhesion of hydrated chitosans: an in vitro and in vivo study. Int J Pharm 145:231–240
Her Y, Lim JW, Han SH (2013) Dry eye and tear film functions in patients with psoriasis. Jpn J Ophthalmol 57(4):341–346
Higashiyama M, Tajika T, Inada K, Ohtori A (2006) Improvement of the ocular bioavailability of carteolol by ion pair. J Ocular Pharmacol Ther 22:333–339
Hill JM, O’Callaghan RJ, Hobden JA, Kaufman E (1993) Corneal collagen shields for ocular drug delivery. Ophthalmic drug delivery systems. Marcel Dekker, New York, pp 261–275
Hirano S, Seino H, Akiyama I, Nonaka I (1990) Chitosan: a biocompatible material for oral and intravenous administration. In: Gebelein CG, Dunn RL (eds) Progress in biomedical polymers. Plenum Press, New York, pp 283–289
Hitzenberger CK, Baumgartner A, Drexler W et al (1994) Interferometric measurement of corneal thickness with micrometer precision. Am J Ophthalmol 118:468–476
Honda M, Asai T, Oku N, Araki Y, Tanaka M, Ebihara N (2013) Liposomes and nanotechnology in drug development: focus on ocular targets. Int J Nanomed 8:495–503
Hosoya K, Vincent HL, Kim LKJ (2005) Roles of the conjunctiva in ocular drug delivery: a review of conjunctival transport mechanisms and their regulation. Eur J Biopharm 60:227–240
Ikeda I (2003) Synthesis of gemini and elated substances. In: Zana R, Xia J (eds) Gemini surfactants synthesis, interfacial and solution phase-behavior, and applications, vol 1. Marcel Dekker, New York, pp 26–30
Irache JM, Merodio M, Arnedo A, Camapanero MA, Mirshahi M, Espuelas S (2005) Albumin nanoparticles for the intravitreal delivery of anticytomegaloviral drugs. Mini Rev Med Chem 5:293–305
Jain NK, Gupta U (2008) Application of dendrimer-drug complexation in the enhancement of drug solubility and bioavailability. Expert Opin Drug Discov 4:1035–1051
Jain K, Kumar RS, Sood S, Dhyanandhan G (2013) Betaxolol hydrochloride loaded chitosan nanoparticles for ocular delivery and their anti-glaucoma efficacy. Curr Drug Deliv 10:493–499
Jain S, Thompson JR, Foot B, Tatham A, Eke T (2014) Severe intraocular pressure rise following intravitreal triamcinolone: a national survey to estimate incidence and describe case profiles. Eye (Lond) 28(4):399–401. doi:10.1038/eye.2013.306
Javadzadeh Y, Ahadi F, Davaran S, Mohammadi G, Sabzevari A, Adibkia K (2010) Preparation and physicochemical characterization of naproxen-PLGA nanoparticles. Colloids Surf B Biointerfaces 81:498–502
Jesorka A, Orwar O (2008) Liposomes: technologies and analytical applications. Ann Rev Anal Chem 1:801–832
Jóhannesson G, Moya-Ortega MD, Asgrímsdottir GM, Agnarsson BA, Lund SH, Loftsson T, Stefansson E (2014) Dorzolamide cyclodextrin nanoparticle suspension eye drops and Trusopt in rabbit. J Ocul Pharmacol Ther 30(6):464–467
Jtirvinen K, Tomi J, Arto Urttia S (1995) Ocular absorption following topical delivery. Adv Drug Deliv Rev 16:3–19
Jwala J, Boddu SHS, Shah S, Sirimulla S, Pal D, Mitra AK (2011) Ocular sustained release nanoparticles containing stereoisomeric dipeptide prodrugs of acyclovir. J Ocul Pharmacol Ther 27(2):163–172
Kalam MA (2016) The potential application of hyaluronic acid coated chitosan nanoparticles in ocular delivery of dexamethasone. Int J Biol Macromol 6(89):559–568
Kambhampati SP, Kannan RM (2013) Dendrimer nanoparticles for ocular drug delivery. J Ocul Pharmacol Ther 29(2):151–165
Kapadia R, Khambete H, Katara R (2009) Novel approach for ocular delivery of acyclovir via niosomes entrapped in situ hydrogel system. J Pharm Res 2:745–751
Karim KM, Mandal AS, Biswas N, Guha A, Chatterjee S, Behera M, Kuotsu K (2012) Niosome: a future of targeted drug delivery systems. J Adv Pharm Tech Res 1:374–380
Katiyar S, Pandit J, Mondal RS, Mishra AK, Chuttani K, Aqil M, Ali A, Sultana Y (2014) In situ gelling dorzolamide loaded chitosan nanoparticles for the treatment of glaucoma. Carbohydr Polym 15(102):117–124
Kaur IP, Kanwar M (2002) Ocular preparations: the formulation approach. Drug Dev Ind Pharm 28:473–493
Kaur IP, Garg A, Singla AK, Aggarwal D (2004) Vesicular systems in ocular drug delivery: an overview. Int J Pharm 269:1–14
Kaur IP, Aggarwal D, Singh H, Kakkar S (2010) Improved ocular absorption kinetics of timolol maleate loaded into a bioadhesive niosomal delivery system. Graefes Arch Clin Exp Ophthalmol 248:1467–1472
Kaur H, Ahuja M, Kumar S, Dilbaghi N (2012) Carboxymethyl tamarind kernel polysaccharide nanoparticles for ophthalmic drug delivery. Int J Biol Macromol 50:833–839
Kesavan K, Kant S, Singh PN, Pandit JK (2013) Mucoadhesive chitosan-coated cationic microemulsion of dexamethasone for ocular delivery: in vitro and in vivo evaluation. Curr Eye Res 38:342–352
Khan A, Sharma PK, Visht S, Malviya R (2011) Niosomes as colloidal drug delivery system: a review. J Chronotherapy Drug Deliv 2:15–21
Khazaeli P, Pardakhty A, Shoorabi H (2007) Caffeine-loaded niosomes: characterization and in vitro release studies. Drug Deliv 14:447–452
King-Smith PE, Fink BA, Fogt N et al (2000) The thickness of the human precorneal tear film: evidence from reflection spectra. Invest Ophthalmol Visual Sci 41:3348–3359
Klibanov AL, Maruyama K, Torchilin VP, Huang L (1990) Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett 268:235–237
Kumari A, Yadav SK, Yadav SC (2010) Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf A 72(1):1–18
Kumbhar D, Wavikar P, Vavia P (2013) Niosomal gel of lornoxicam for topical delivery: in vitro assessment and pharmacodynamic activity. AAPS Pharm Sci Tech 14(3):1072–1082
Lang JC (1995) Ocular drug delivery conventional ocular formulations. Advanced Drug Delivery Reviews 16:39–43
Lawrence MJ, Chauhan S, Lawrence SM, Barlow DJ (1996) The formation, characterization and stability of non-ionic surfactant vesicles. STP Pharm Sci 1:49–60
Lee SJ, He W, Robinson SB, Robinson MR, Csaky KG, Kim H (2010) Evaluation of clearance mechanisms with trans-scleral drug delivery. Invest Ophthalmol Vis Sci 51:5205–5212
Li J, Wu L, Wu W, Wang B, Wang Z, Xin H, Xu Q (2013) A potential carrier based on liquid crystal nanoparticles for ophthalmic delivery of pilocarpine nitrate. Int J Pharm 15 455(1–2):75–84
Lin J, Wu H, Wang Y, Lin J, Chen Q, Zhu X (2016) Preparation and ocular pharmacokinetics of hyaluronan acid-modified mucoadhesive liposomes. Drug Deliv 23(4):1144–1151
Liu R, Wang S, Fang S, Wang J, Chen J, Huang X, He X, Liu C (2016) Liquid crystalline nanoparticles as an ophthalmic delivery system for tetrandrine: development, characterization, and in vitro and in vivo. Nanoscale Res Lett 11(1):254. doi:10.1186/s11671-016-1471-0 Cited 2016 May 17
Ludwig A (2005) The use of mucoadhesive polymers in ocular drug delivery. Adv Drug Delivery Rev 57:1595–1639
Ludwig A, Ooteghem VM (1992) Influence of, viscoslysers on the residence of ophthalmic solutions evaluated by slip lamp fluorophotometry. STP Pharm Sci 2(1):81–87
Lv FF, Zheng LQ, Tung CH (2005) Phase behavior of the microemulsions and the stability of the chloramphenicol in the microemulsion-based ocular drug delivery system. Int J Pharm 301(1–2):237–246
Ma SW, Gan Y, Gan L, Zhu CL, Zhu JB (2008) Preparation and in vitro corneal retention behaviour of novel cationic microemulsion/in situ gel system. Yao Xue Xue Bao 43:749–755
Mahale NB, Thakkar PD, Mali RG, Walunj DR, Chaudhari SR (2012) Niosomes: novel sustained release nonionic stable vesicular systems—an overview. Adv Colloid Interface Sci 15:46–54
Mainardes RM, Urban MCC, Cinto PO (2005) Colloidal carriers for ophthalmic drug delivery. Curr Drug Targets 6:363–371
Manosroi A, Wongtrakul P, Manosroi J, Sakai H, Sugawara F, Yuasa N (2003) Characterization of vesicles prepared with various nonionic surfactants mixed with cholesterol. Colloids Surf B Biointerfaces 30:129–138
Marfurt CF, Kingsley RE, Echtenkamp SE (1989) Sensory and sympathetic innervation of the mammalian cornea. A retrograde tracing study. Invest Ophthalmol Vis Sci 30:461–472
Maurice DM, Mishima S (1984) Pharmacology of the eye. Handb Exp Pharmacol 69:109–116
McDonald TO, Shadduck JA (1977) Eye irritation. Adv Mod Toxicol 4:139–191
Meisner D, Mezei M (1995) Liposome ocular delivery systems. Adv Drug Deliv Rev 16:75–93
Meng J, Sturgis TF, Youan BC (2011) Engineering tenofovir loaded chitosan nanoparticles to maximize microbicide mucoadhesion. Eur J Pharm Sci 44:57–67
Merodio M, Irache JM, Valamanesh F, Mirshahi M (2002) Ocular disposition and tolerance of ganciclovir-loaded albumin nanoparticles after intravitreal injection in rats. Biomaterials 23:1587–1594
Meseguer G, Buri P, Plazonnet B, Rozier A, Gurny R (1996) Gamma scintigraphic comparison of eyedrops containing pilocarpine in healthy volunteers. J Ocul Pharmacol Ther 12:481–488
Miao H, Wu B, Tao Y, Li X. (2013). Diffusion of macromolecules through sclera. Acta Ophthamologica 91(1):e1–e6
Mintzer MA, Grinstaff MW (2011) Biomedical applications of dendrimers: a tutorial. Chem Soc Rev 40(1):173–190
Mishra GP, Bagui M, Tamboli V, Mitra AK (2011) Recent applications of liposomes in ophthalmic drug delivery. J Drug Deliv 14:205–2018
Mohammed N, Rejinold NS, Mangalathillam S, Biswas R, Nair SV, Jayakumar R (2013) Fluconazole loaded chitin nanogels as a topical ocular drug delivery agent for corneal fungal infections. J Biomed Nanotechnol 9(9):1521–1531
Monem AS, Ali FM, Ismail MW (2000) Prolonged effect of liposomes encapsulating pilocarpine HCl in normal and glaucomatous rabbits. Int J Pharmaceutics 198:29–38
Montes-Mico R, Cervino A, Ferrer-Blasco T, Garcia-Lazaro S, Madrid-Costa D (2010) The tear film and the optical quality of the eye. Ocul-Surf 8:185–192
Motwani SK, Chopra S, Talegaonkar S, Kohli K, Ahmad FJ, Khar RK (2008) Chitosan–sodium alginate nanoparticles as submicroscopic reservoirs for ocular Delivery: formulation, optimization and in-vitro characterization. Eur J Pharm Biopharm 68:513–525
Mujoriya RZ, Bodla RB (2011) Niosomes—challenge in preparation for pharmaceutical scientist. Int J App Pharm 3:11–15
Muller LJ, Marfurt CF, Kruse F, Tervo TM (2003) Corneal nerves: structure, contents and function. Exp Eye Res 76:521–542
Myles ME, Neumann DM, Hill JM (2005) Recent progress in ocular drug delivery for posterior segment disease: emphasis on transscleral iontophoresis. Adv Drug Del Rev 57:2063–2079
Nagarwal RC, Kumar R, Pandit JK (2012) Chitosan coated sodium alginate-chitosan nanoparticles loaded with 5-FU for ocular delivery: in vitro characterization and in vivo study in rabbit eye. Eur J Pharm Sci 47(4):678–685
Nagyova B, Tiffany JM (1999) Components responsible for the surface tension of human tears. Curr Eye Res 19:4–11
Newell FW (1993) Ophthalmology: principles and concepts. 7th edn. Mosby Year Book, St. Louis
Newkome GR, Shreiner CD (2008) Amidoamine, polypropylenimine, and related dendrimers and dendrons possessing different 1 → 2 branching motifs: An overview of the divergent procedures. Polymer 49:1–173
Norley SG, Huang L, Rouse BT (1986) Targeting of drug loaded immunoliposomes to herpes simplex virus infected corneal cells: an effective means of inhibiting virus replication in vitro. J Immunol 136(2):681–685
Omidi, Y., Barar, J., Hamzeiy, H., 2012. Nanomedicines Impacts in Ocular Delivery and Targeting. Nanotechnol Health Care, 43–106
Pandit JC, Nagyova B, Bron AJ, Tiffany JM (1999) Physical properties of stimulated and unstimulated tears. Exp Eye Res 68:247–253
Pardakhti A, Moshefi MH, Moteshafi H (2007) Preparation of niosomes containing chloramphenicol sodium succinate and evaluation of their physicochemical and antimicrobial properties. Pharm Sci Spr 1:11–21
Parra A, Mallandrich M, Clares B, Egea MA, Espina M, García ML, Calpena AC (2015) Design and elaboration of freeze-dried PLGA nanoparticles for the transcorneal permeation of carprofen:Ocular anti-inflammatory applications. Colloids Surf B Biointerfaces 136:935–943
Pathak MK, Chhabra G, Pathak K (2013) Design and development of a novel pH triggered nanoemulsified in-situ ophthalmic gel of fluconazole: ex-vivo transcorneal permeation, corneal toxicity and irritation testing. Drug Dev Ind Pharm 39:780–790
Pescina S, Santi P, Ferrari G, Nicoli S (2011) Trans-scleral delivery of macromolecules. Ther Deliv 2:1331–1349
Pignatello R, Bucolo C, Spedalieri G, Maltese A, Puglisi G (2002) Flurbiprofen-loaded acrylate polymer nanosuspensions for ophthalmic application. Biomaterials 23:3247–3255
Ponchel G, Touchard F, Duchfine D, Peppas NA (1987) Bioadhesive analysis of controlled-release systems. I. Fracture and interpenetration analysis in poly (acrylic acid)-containing systems. J Control Release 5:129–141
Qi HP, Gao XC, Zhang LQ, Wei SQ, Bi S, Yang ZC, Cui H (2013) In vitro evaluation of enhancing effect of borneol on transcorneal permeation of compounds with different hydrophilicities and molecular sizes. Eur J Pharmacol 705:20–25
Rathore KS, Nema PK (2009) An insight into ophthalmic drug delivery system. Int J Pharm Sci Drug Res 1:1–5
Raviola G (1983) Conjunctival and episcleral blood vessels are permeable to blood-borne horseradish peroxidase. Invest Ophthalmol Vis Sci 24:725–736
Raymzond CR, Paul JS, Sian CO (2006) Polyoxyethylenealkyl ethers. Handbook of pharmaceutical excipients, 5th edn. Pharmaceutical Press, London, pp 564–571
Robinson JC (1993) Ocular anatomy and physiology relevant to ocular drug delivery. In: Mitra AK (ed) Ophthalmic drug delivery systems. Marcel Dekker, New York, pp 29–57
Rojanasakul Y, Wang LY, Bhat M (1992) The transport barrier of epithelia: a comparative study on membrane permeability and charge selectivity in the rabbit. Pharm Res 9:1029–1934
Rooijen VN, Nieuwmegen VR (1980) Liposomes in immunology: multilamellar phosphatidylcholine liposomes as a simple, biodegradable and harmless adjuvant without any immunogenic activity of its own. Immunol Commun 9:243–256
Round A, Berry M, McMaster T, Stoll S, Gowers D, Corfield A, Miles M (2002) Heterogeneity and persistence length in human ocular mucins. Biophys J 83:1661–1670
Sabzevari A, Adibkia K, Hashemi H, De-Geest BG, Mohsenzadeh N, Atyabi F, Ghahremani MH, Khoshayand MR, Dinarvand R (2013) Improved anti-inflammatory effects in rabbit eye model using biodegradable poly beta-amino ester nanoparticles of triamcinolone acetonide. Invest Ophthalmol Vis Sci 54:5520–5526
Sahoo SK, Labhasetwar V (2003) Nanotech approaches to drug delivery and imaging. Drug Discov Today 8:1112–1120
Sahoo SK, Dilnawaz F, Krishnakumar S (2008) Nanotechnology in ocular drug delivery. Drug Discov Today 13:144–151
Salama AH, Mahmoud AA, Kamel R (2015) A novel method for preparing surface-modified fluocinolone acetonide loaded PLGA nanoparticles for ocular use: in vitro and in vivo evaluations. AAPS Pharm Sci Tech, 1–14
Samad A, Alam MI, Saxena K (2009) Dendrimers: a class of polymers in the nanotechnology for the delivery of active pharmaceuticals. Curr Pharm 15(25):2958–2969
Sasaki H, Karasawa K, Hironaka K, Tahara K, Tozuka Y, Takeuchi H (2013) Retinal drug delivery using eyedrop preparations of poly-L-lysine-modifiedliposomes. Eur J Biopharm 83:364–369
Schipper NG, Olsson S, Hoogstraate JA, deBoer AG, Varum KM, Artursson P (1997) Chitosans as absorption enhancers for poorly absorbable drugs 2: mechanism of absorption enhancement. Pharm Res 14:923–929
Schoenwald RD (1990) Ocular drug delivery: pharmacokinetic considerations. Clin Pharmacokinet 18:255–269
Schoenwald RD, Ward RL, Desantis LM, Roehrs RE (1978) Influence of high viscosity vehicles on miotic effect of pilocarpine. J Pharm Sci 67:1280–1283
Schoenwald RD, Vidvauns S, Wurster DE, Barfknecht CF (1998) The role of tear proteins in tear film stability in the dry eye patient and in the rabbit. In: Sullivan DA, Dartt DA and Menerary MA (eds) Lacrimal gland, Tear Film and Dry Eye Sydromes. 2. Plenum Press: New York, pp 391–400
Sechoy O, Tissiiee G, Sebastian C, Maurin F, Driot JY, Trinquand C (2000) A new long acting ophthalmic formulation of Carteolol containing alginic acid. Int J Pharm 207:109–116
Shahiwala A, Misra A (2002) Studies in topical application of niosomally entrapped nimesulide. J Pharm Sci 5:220–225
Shan W, Liu H, Shi J, Yang L, Hu N (2008) Self-assembly of electroactive layer-by-layer films of heme proteins with anionic surfactant dihexadecyl phosphate. Biophys Chem 134:101–109
Shen Y, Tu J (2007) Preparation and ocular pharmacokinetics of ganciclovir liposomes. AAPS Pharm Sci Tech 9:371–377
Singh D (2003) Conjunctival lymphatic system. J Cataract Refract Surg 29:632–633
Singh V, Ahmad R, Heming T (2011) The challenges of ophthalmic drug delivery: a review. Int J Drug Discov 3:56–62
Slovin EM, Robinson JR (1993) Bioadhesive in ocular drug delivery. In: Edman P (ed) Biopharmaceutics of ocular drug delivery. CRC Press, Boca Raton, Florida, pp 145–157
Soukharev AR, Wojciechowska J (2005) Microemulsions as potential ocular drug delivery systems: phase diagrams and physical depending on ingredients. Acta Pol Pharm Drug Res 62:465–471
Stahl U, Willcox M, Stapleton F (2012) Osmolality and tear film dynamics. Clin Exp Optom 95(1):3–11
Sugar HS, Riazi A, Schaffner R (1957) The bulbar conjunctival lymphatics and their clinical significance. Trans Am Acad Ophthalmol Otolaryngol 61:212–223
Sun KX, Wang AP, Huang LJ, Liang RC, Liu K (2006) [Preparation of diclofenac sodium liposomes and its ocular pharmacokinetics]. Yao Xue Xue Bao. 41(11):1094–1098
Szczesna DH, Jaronski J, Kasprzak HT, Stenevi U (2006) Interferometric measurements of dynamic changes of tear film. J Biomed Opt 11(3):340–352
Szczesna DH, Kasprzak HT, Jaronski J, Rydz A, Stenevi U (2007) An interferometric method for the dynamic evaluation of the tear film. Acta Ophthalmol Scand 85:202–208
Szczesna-Iskander DH, Iskander DR (2012) Future directions in non-invasive measurements of tear film surface kinetics. Optom Vis Sci 89(5):749–759
Szulc J, Woyczikowski B, De Laval W (1988) Effect of pilocarpine hydrochloride liposomes on the intraocular pressure of the rabbit eye pupil. Farm Pol 44:462–465
Takeuchi H, Yamamoto H, Niwa T, Hino T, Kawashima Y (1996) Enteral absorption of insulin in rats from mucoadhesive chitosan coated liposomes. Pharm Res 13:896–901
Tayel SA, El-Nabarawi MA, Tadros MI, Abd-Elsalam WH (2013) Positively charged polymeric nanoparticle reservoirs of terbinafine hydrochloride: preclinical implications for controlled drug delivery in the aqueous humor of rabbits. AAPS Pharm Sci Tech 14:782–793
Tiffany JM (2003) Tears in health and disease. Eye 17:1–4
Tissie G, Sebastian C, Elena PP, Driot JY, Trinquand C (2002) Alginic acid effect on carteolol ocular pharmacokinetic in the pigmented rabbit. J Ocul Pharmacol Ther 18:65–73
Van Ooteghem M (1995) Preparations ophtalmiques. In: Galenica (ed) Technique and documentation. Lavoisier, Paris
Vandamme TF, Brobeck L (2005) Poly (amidoamine) dendrimers as ophthalmic vehicles for ocular delivery of pilocarpine nitrate and tropicamide. J Control Release 102(1):23–38
Vandervoort J, Ludwig A (2004) Preparation and evaluation of drug-loaded gelatin nanoparticles for topical ophthalmic use. Eur J Biopharm 57:251–261
Vyas, S.P., Khar, R.K., 2008. Ocular drug delivery, in controlled drug delivery. 384–385
Wadhwa S, Paliwal R, Paliwal SR, Vyas SP (2009) Nanocarriers in ocular drug delivery: an update review. Curr Pharm Des 15:2724–2750
Wadhwa S, Paliwal R, Paliwal SR, Vyas SP (2010) Hyaluronic acid modified chitosan nanoparticles for effective management of glaucoma: development, characterization, and evaluation. J Drug Target 18(4):292–302
Warsi MH, Anwar M, Garg V, Jain GK, Talegaonkar S, Ahmad FJ, Khar RK (2014) Dorzolamide-loaded PLGA/vitamin E TPGS nanoparticles for glaucoma therapy: pharmacoscintigraphy study and evaluation of extended ocular hypotensive effect in rabbits. Colloids Surf B Biointerfaces 122:423–431
Wen H, Hao J, Li SK (2013) Characterization of human sclera barrier properties for transscleral delivery of bevacizumab and ranibizumab. J Pharm Sci 102:892–903
Wenger Y, Schneider RJ, Reddy GR, Kopelman R, Jolliet O, Philbert MA (2011) Tissue distribution and pharmacokinetics of stable polyacrylamide nanoparticles following intravenous injection in the rat. Toxicol Appl Pharmacol 251:181–190
Wilson CG (2004) Topical drug delivery in the eye. Exp Eye Res 78(3):737–743
Wolff E (1946) The muco-cutaneous junction of the lid margin and the distribution of the tear fluid. Trans Ophthamol Soc UK 66:291–308
Wolff E (1954) The Anatomy of the Eye and Orbit, fourth ed. H.K Lewis and Co, London. 49
Worakul N, Robinson JR (1997) Ocular pharmacokinetics-pharmacodynamics. Eur J Pharm Biopharm 44(1627):71–83
Yan Y, Ting L, Huang J (2009) Recent advances in bolaamphiphiles and oppositely charged conventional surfactants. J Colloid Interface Sci 337:1–10
Yang H, Kao WJ (2006) Dendrimers for pharmaceutical and biomedical applications. J Biomater Sci Polym Ed 17:13–19
Zeng W, Li Q, Wan T, Liu C, Pan W, Wu Z, Zhang G, Pan J, Qin M, Lin Y, Wu C, Xu Y (2016) Hyaluronic acid-coated niosomes facilitate tacrolimus ocular delivery: mucoadhesion, precorneal retention, aqueous humor pharmacokinetics, and transcorneal permeability. Colloids Surf B Biointerfaces 1(141):28–35
Zhou W, Wang Y, Jian J, Song S (2013) Self-aggregated nanoparticles based on amphiphilic poly (lactic acid)-grafted-chitosan copolymer for ocular delivery of amphotericin B. Int J Nanomed 8:3715–3728
Zhu X, Su M, Tang S, Wang L, Liang X, Meng F, Hong Y, Xu Z (2012) Synthesis of thiolated chitosan and preparation nanoparticles with sodium alginate for ocular drug delivery. Mol Vis 18:1973–1982
Zimmer AK, Kreuter J, Robinson JR (1991) Studies on the transport pathway of PBCA nanoparticles in ocular tissues. J Microencapsul. 8(4):497–504
Zimmer AK, Zerbe H, Kreuter J (1994) Evaluation of pilocarpine-loaded albumin particles as drug delivery systems for controlled delivery in the eye. In vitro and in vivo characterisation. J Control Release 32:57–70
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing AG
About this chapter
Cite this chapter
Zafar, A., Ahmad, J., Addo, R.T., Akhter, S. (2016). Progress of Controlled Drug Delivery Systems in Topical Ophthalmology: Focus on Nano and Micro Drug Carriers. In: Addo, R. (eds) Ocular Drug Delivery: Advances, Challenges and Applications. Springer, Cham. https://doi.org/10.1007/978-3-319-47691-9_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-47691-9_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-47689-6
Online ISBN: 978-3-319-47691-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)