
Abstract
C-type lectin-like receptor 2 (CLEC-2) is a glycoprotein expressed on the surface of platelets that induces platelet activation including aggregation. Binding of the endogenous ligand podoplanin or the exogenous ligand rhodocytin to CLEC-2 induces tyrosine phosphorylation of the hemITAM motif expressed in its cytoplasmic tail. This hemITAM phosphorylation induces subsequent downstream signalling leading to calcium mobilisation and platelet activation. Although CLEC-2 stimulation induces powerful platelet activation, the interaction of CLEC-2 with podoplanin is not crucial for haemostasis. During development, CLEC-2 interacts with podoplanin on lymphatic endothelial cells (LECs) and is crucial for blood/lymphatic vessels separation, as well as development of the cerebrovasculature. Moreover, CLEC-2 critically maintains the integrity of high-endothelial venules within lymph nodes post-development through its interaction with podoplanin on fibroblastic reticular cells.
Under pathologic conditions, blockade of the CLEC-2/podoplanin interaction has been shown to elicit both detrimental and beneficial effects in a disease-dependent manner. The interaction has been shown to be detrimental in the case of tumour metastasis, HIV propagation and salmonella-mediated liver thrombosis and inflammation. In contrast, upregulation of podoplanin on TH17 cells impairs T-cell expansion and survival in multiple sclerosis and results in enhanced resolution of the disease. This book chapter will discuss the structure and function of the novel platelet receptor CLEC-2 from its discovery to the latest studies carried out on the physiological and pathological role of CLEC-2. This chapter is divided into four sections that cover the identification of CLEC-2 on platelets, the key signalling pathways involved, the role of CLEC-2 in embryonic development and its function in and beyond the vasculature.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
- Lymphatic Endothelial Cell
- Podoplanin Expression
- Lymphatic Vasculature
- Fibroblastic Reticular Cell
- Tail Bleeding Time
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
In 2000, Colonna et al. performed a bioinformatics study and identified the CLEC-2 gene Clec1B on chromosome 12 in the Dectin-1 gene cluster. The CLEC-2 gene is located near the genes for six other C-type lectin receptors and was identified as a novel member of the C-type lectin-like superfamily (Colonna et al. 2000). Originally, CLEC-2 mRNA was found in the liver and on some haematopoietic cells, in particular dendritic cells (DCs) and natural killer cells in human and mice (Colonna et al. 2000). Later, in 2006, CLEC-2 mRNA and CLEC-2 protein expression was found to be abundant in the megakaryocyte/platelet lineage, liver sinusoidal endothelial cells and liver Kupffer cells, although expression in liver has yet to be independently confirmed (Chaipan et al. 2006; Suzuki-Inoue et al. 2006; Senis et al. 2007). In 2009, CLEC-2 was further shown to be expressed on subsets of murine myeloid cells and dendritic cells under basal conditions as well as on a variety of leukocyte subsets in response to inflammatory stimuli (Kerrigan et al. 2009; Acton et al. 2012). However, a recent study identified a much more restrictive expression profile, demonstrating CLEC-2 expression to be localised on platelets and on a subset of circulating inflammatory dendritic cells, which was lost upon entry into secondary lymphoid organs (Lowe et al. 2015a, b). The discrepancy in regard to expression to haematopoietic subsets is explained by off-target actions of the available antibodies. Investigation of the function of CLEC-2 in platelets was facilitated by its identification as a receptor for the snake venom rhodocytin using affinity chromatography and mass spectrometry (Suzuki-Inoue et al. 2006). CLEC-2 is a type II membrane protein with an extracellular carbohydrate-like recognition domain, a transmembrane domain and a cytoplasmic tail that is essential for downstream signalling. The CLEC-2 cytosolic tail has a hemi (or half) immunoreceptor tyrosine-based activation motif (ITAM) which consists of a single YXXL sequence which is phosphorylated by Src and Syk tyrosine kinases. CLEC-2 has two well-characterised ligands: an exogenous ligand, rhodocytin, and an endogenous ligand, podoplanin. In addition, it has been shown to be activated by the sulphated sugar, fucoidan, which also activates the platelet collagen receptor GPVI.
Podoplanin is a transmembrane glycoprotein that is widely expressed outside of the vasculature under physiological conditions including on kidney podocytes, alveolar type 1 epithelial cells, lymphatic endothelial cells (LECs), cerebral choroid plexus and many epithelial beds as well as being induced in subsets of haematopoietic cells. The expression of podoplanin on LECs is crucial for prevention of mixing of the blood and lymphatic vasculatures during development through its interaction with platelet CLEC-2 (Bertozzi et al. 2010; Herzog et al. 2013; Lowe et al. 2015a, b). On kidney podocytes, the absence of podoplanin was suggested to impair blood filtration, resulting in the presence of proteins in the urine (Koop et al. 2008). However, with the generation of transgenic mice with targeted deletion of podoplanin on kidney podocytes, the function of podoplanin in the kidney, both physiologically and under inflammatory conditions, is now questionable and requires further investigation (Rayes and Watson, unpublished). Podoplanin has been shown to be upregulated under pathologic conditions, such as inflammation, cancer and autoimmune disease on inflammatory macrophages, tumour cells and T-helper 17 (Th17) cells, respectively, and in development.
The role of podoplanin and CLEC-2 has been studied in a variety of disorders. It is important to note that the beneficial or detrimental effect of the CLEC-2-podoplanin interaction seems to be disease-dependent. Upregulation of podoplanin on macrophages and tumour cells induces platelet aggregation and thrombosis, whereas the expression of podoplanin on TH17 cells alters TH17 cell function and survival and improves recovery in a mouse model of multiple sclerosis (spontaneous experimental autoimmune encephalomyelitis) on a susceptible background.
In this chapter, CLEC-2 signalling pathways and the role of CLEC-2 in developmental pathways and in both thrombotic and non-thrombotic disorders will be discussed.
CLEC-2 Signalling
(Hem)ITAM Receptors in Platelets
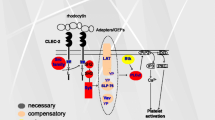
As mentioned above, the C-type lectin receptor CLEC-2 is a glycoprotein receptor and a member of the (hem)ITAM family of receptors, which includes other receptors such as the B-cell antigen receptor, the low affinity immune receptor FcγRIIa and the major platelet signalling collagen receptor, GPVI/FcRγ chain (referred to as GPVI). An ITAM consists of two YXXL sequences (single amino acid code) separated by 6–12 amino acids. HemITAM receptors such as CLEC-2 differ as they contain only one YXXL sequence in their intracellular tails (Fuller et al. 2007). CLEC-2 signalling is additionally dependent on a conserved triacidic amino acid sequence immediately upstream of the hemITAM motif, which is also a feature of all but one of the other five known hemITAM-containing receptors (Hughes et al. 2013; Ruckrich and Steinle 2013). CLEC-2 is expressed as a dimer, thus providing two YXXL sequences (Fig. 1).

Graphical representation of the proximal events in CLEC-2 mediated platelet activation. Upon receptor engagement, the hemITAM contained within the intracellular tail of CLEC-2 becomes phosphorylated by Syk and possibly also by Src family kinases. HemITAM phosphorylation allows recruitment of Syk via its SH2 domains, allowing downstream phosphorylation of LAT and the formation of the LAT signalosome. The result of this signalosome formation is the mobilisation of intracellular Ca2+ via recruitment and activation of PLCγ2
(Hem)ITAM Signalling
Upon receptor engagement via endogenous or exogenous ligands, the tyrosine residues contained within ITAM domains become targets of phosphorylation via Src family kinases (SFKs) such as Src and Lyn (Senis et al. 2014). A key difference between CLEC-2 and traditional ITAM signalling is that the hemITAM motif appears to be more reliant upon spleen tyrosine kinase (Syk) than SFKs for initial phosphorylation, with SFKs being important for activation of Syk and for signalling further downstream (Spalton et al. 2009; Severin et al. 2011). Upon platelet activation by agonists such as rhodocytin or podoplanin, the hemITAM sequence is phosphorylated, allowing recruitment and docking of the tandem SH2 domain-containing kinase Syk. Once recruited to the phosphorylated hemITAM via its SH2 domains, Syk undergoes a conformational change, moving from an auto-inhibited to an active conformation. In this active conformation, Syk undergoes further auto- and trans-phosphorylation. The latter is mediated by SFKs, allowing for full activation of Syk and phosphorylation of several downstream proteins. One of the most important of these is LAT, an adapter protein containing several tyrosine residues that undergo phosphorylation. LAT has no intrinsic activity and serves as the core of the ‘LAT signalosome’, allowing the docking and activation of many of the effectors of this pathway including Grb-2, Gads, SLP-76, PI3-K, Btk and PLCγ2 (Suzuki-Inoue et al. 2006; Fuller et al. 2007). PLCγ2 is generally considered to be the most important, giving rise to formation of inositol 1,4,5-trisphosphate (IP3) and 1,2-dacylglycerol (DAG) which mobilise Ca2+ and activate protein kinase C, respectively. These changes culminate in the exocytosis of intracellular granules, such as α- and dense granules, as well as inside-out activation of platelet integrins including αIIbβ3 (Hughes et al. 2015; Zheng et al. 2015). The release of ADP from dense granules and formation of thromboxane A2, made de novo from membrane phospholipids, play critical positive feedback roles in activation of CLEC-2 in human platelets (Pollitt et al. 2010). CLEC-2 activation in human is also dependent on the Rho GTPase, Rac (Pollitt et al. 2010). This contrasts with activation by the collagen ITAM receptor GPVI which shows a reduced rather than complete dependency on the role of the two feedback messengers and Rac.
Functional Roles of CLEC-2 Signalling
Platelet activation and aggregation is a multistep process that involves different receptors and signalling pathways with the aim of prevention of excessive blood loss following vessel injury. CLEC-2 is expressed on platelets with an average copy number of 2000 receptor per platelet. Several studies have investigated a possible role of CLEC-2 in haemostasis and thrombus formation using various mouse models. CLEC-2-deficient platelets aggregate normally in response to classical platelet agonists but not to rhodocytin. Moreover, CLEC-2-deficient platelets have normal adhesion and spreading on extracellular matrices including collagen, laminin, von Willebrand factor and fibrinogen (Suzuki-Inoue et al. 2010). Several groups have investigated the consequence of CLEC-2 deficiency in a tail bleeding assay (haemostasis) or laser, photochemical or FeCl3 thrombosis model with contrasting results. Using an anti-CLEC-2 antibody to deplete CLEC-2 from the platelet surface in vivo, May et al. (2009) showed a prolonged bleeding time and profound defect in arterial thrombus formation following FeCl3 injury (May et al. 2009). Using irradiated mice reconstituted with Clec-2−/− foetal liver, Suzuki-Inoue et al. (2010) showed a reduction in thrombus formation following photochemical injury but no significant change in tail bleeding times (Suzuki-Inoue et al. 2010). In contrast, Hughes et al. (2010) did not observe a significant change in tail bleeding time or aggregation on collagen at high shear using radiation chimeric mice reconstituted with CLEC-2-deficient bone marrow, and additionally were unable to demonstrate phosphorylation of CLEC-2 under flow conditions, providing indirect evidence that it is not activated (Hughes et al. 2010). The reason for the discrepancy between the various studies remains unclear. A recent study showed a profound defect in thrombus formation in mice double deficient in GPVI and CLEC-2 suggesting possible compensatory roles of the two (hem)ITAM receptors (Bender et al. 2013), although this which must involve adhesion and signalling as a similar result is not seen in mice deficient in Syk. Overall the consensus is that CLEC-2 has a minor role (at best) in haemostasis, consistent with the absence of an identified ligand in the vasculature, and that its primary role is to support many of the non-canonical roles for platelets that are beginning to emerge.
CLEC-2 in Development
Blood-Lymphatic Vascular Separation Defect in CLEC-2-Deficient Mice
Two decades ago, mice deficient in the downstream CLEC-2 signalling molecule, Syk, were shown to develop subcutaneous haemorrhages in the skin and oedema at mid-gestation (Cheng et al. 1995; Turner et al. 1995). A similar phenotype was observed in mice deficient in the adaptor protein SLP-76 (Clements et al. 1998; Pivniouk et al. 1998; Clements et al. 1999) or PLC-γ2 (Wang et al. 2000). The development of haemorrhages and oedema was later shown to result from aberrant connections between the blood and lymphatic vasculatures causing the appearance of blood-filled lymphatic vessels and impaired lymphatic drainage, respectively (Abtahian et al. 2003; Ichise et al. 2009). Support for the involvement of CLEC-2 in this phenotype came following the characterisation of mice deficient in its endogenous ligand, podoplanin, which were also shown to harbour blood-filled lymphatic vessels in the skin and gut at birth (Schacht et al. 2003), a phenotype later identified in CLEC-2-deficient embryos in mid-gestation (Bertozzi et al. 2010; Suzuki-Inoue et al. 2010; Finney et al. 2012). Both CLEC-2- and podoplanin-deficient mice experience almost 100 % perinatal mortality that is thought to be caused by defective lymphatic function and fluid retention leading to a failure to inflate the lungs (Schacht et al. 2003; Finney et al. 2012). Furthermore, the mice that survived for sufficient time to suckle developed chylous ascites, further indicative of impaired lymphatic function (Abtahian et al. 2003; Finney et al. 2012; Hess et al. 2014).
Identifying the Causative CLEC-2-Expressing Cell That Underlies the Defects in Lymphatic Development
The mechanism by which podoplanin and CLEC-2 prevent the mixing of the blood and lymphatic vasculatures during development remains controversial. A role for podoplanin specifically on the lymphatic endothelium has been confirmed by the presence of blood-filled lymphatic vessels in mice with a Tie-2-Cre-mediated deletion of podoplanin or the glycosyltransferase enzyme, T-synthase, which is important for the glycosylation of the podoplanin extracellular domain (Fu et al. 2008; Herzog et al. 2013). The interacting CLEC-2-expressing cell type has been investigated through a series of lineage tracing studies that initially pinpointed Syk/SLP-76 activity in a circulating endothelial progenitor (Abtahian et al. 2003; Sebzda et al. 2006) but which was later shown to be a circulating cell of the myeloid lineage (Bohmer et al. 2010). Using a series of transgenic mice, a role for Syk in macrophages, T- and B-lymphocytes, was eliminated, while the loss of CLEC-2, Syk or SLP-76 in the megakaryocyte/platelet lineage (generated by crossing floxed homozygotes to a PF4-Cre transgenic) was shown to be indispensable for blood-lymphatic vascular separation (Bertozzi et al. 2010; Finney et al. 2012).
A role for CLEC-2 on platelets/megakaryocytes is supported by the presence of blood-filled lymphatic vessels following the selective ablation of this lineage using diphtheria toxin (again achieved using a PF4-Cre transgenic), through treatment with aspirin in utero or in mice that lack functional megakaryocytes due to a deficiency in the transcription factor Meis-1 (Carramolino et al. 2010; Uhrin et al. 2010). Furthermore, the contribution of platelet-specific effects over megakaryocytes is supported by the development of blood-filled intestinal lymphatic vessels in irradiated mice reconstituted with Syk-, SLP-76-, PLCγ2- or CLEC-2-deficient bone marrow, where the number of circulating megakaryocytes is negligible (Ichise et al. 2009; Bertozzi et al. 2010; Finney et al. 2012).
Unravelling the Molecular Mechanism of the Impairment in Lymphatic Development
Initially it was proposed that a direct interaction between platelets and LECs at the cardinal vein was critical to ‘clot off’ the primary budding lymphatic structures and prevent the formation of blood-lymphatic vascular connections (Bertozzi et al. 2010; Uhrin et al. 2010; Hess et al. 2014). However, high-resolution microscopy studies of developing embryos have failed to identify any connection between the blood and lymphatic vasculatures at this stage (Hagerling et al. 2013). Furthermore, blood-filled lymphatic vessels can be seen in irradiated adult mice reconstituted with CLEC-2- or Syk-deficient bone marrow where the lymphatic system is already established (Bertozzi et al. 2010; Finney et al. 2012). It was recently proposed that a thrombus, formed through podoplanin-induced CLEC-2 activation on platelets, forms at the joining of the thoracic duct and the subclavian vein to prevent backflow of blood into the lymphatic system during development and throughout adulthood (Hess et al. 2014). However, an important role for thrombus formation is disputed by the absence of a blood-lymphatic vascular separation defect in mice lacking the major platelet integrin αIIbβ3 (Uhrin et al. 2010).
It has also been proposed that one or more of the bioactive molecules or growth factors that are abundant in platelet dense, α- and lysosomal secretory granules and released upon their activation could contribute towards influencing lymphatic endothelial cell behaviour. So far, there has been little evidence to support this mechanism since patients or mice with defective dense- or α-granule secretion syndromes known as Hermansky-Pudlak syndrome or grey platelet syndrome, respectively, have not been described to exhibit lymphatic defects (e.g. Deppermann et al. 2013). Platelets additionally store large concentrations of the bioactive lipid sphingosine-1-phosphate (S1-P), which have been shown to have potent effects on endothelial cell migration, but while the loss of S1-P in mice is associated with widespread haemorrhaging and embryonic lethality, no phenotype of blood-lymphatic mixing was observed (Liu et al. 2000).
Other potential mechanisms include the direct interaction between platelet-CLEC-2 and podoplanin on LECs, which has been shown to inhibit LEC migration (Finney et al. 2012). It is proposed that these inhibitory signals are critical to arrest the growth of budding lymphatic vessels and prevent the formation of aberrant connections (anastomoses) with the blood vascular endothelium (Finney et al. 2012). The ability of CLEC-2 to cluster its receptor podoplanin on the surface of LECs was later demonstrated. This interaction supports platelet adhesion and is also proposed to inhibit signalling by podoplanin in LECs as shown in other cell types (Pollitt et al. 2014). The presence of anastomoses between veins and lymphatic veins has been described by many groups, including direct visualisation of mixing of the two vasculatures in mice with a platelet-specific loss of functional Syk (Hughes et al. 2015).
Other Developmental Functions of CLEC-2
Podoplanin was identified on fibroblastic reticular cells (FRCs) of the spleen over two decades ago and has since been shown to have several important roles in the organisation and function of lymphoid compartments (Farr et al. 1992; Yu et al. 2007). Recently, a number of these functions have been linked to CLEC-2 including trafficking of CLEC-2-expressing DCs to lymph nodes which is dependent on podoplanin expression on lymphatic endothelium and on FRCs (Acton et al. 2012). In addition, CLEC-2 has been shown to play a role in maintaining the integrity of high-endothelial venules within lymph nodes through its interaction with podoplanin on FRCs (Herzog et al. 2013). In this study, mice with a platelet/megakaryocyte-specific loss of CLEC-2 (i.e. PF4-Cre transgenic CLEC-2fl/fl) developed blood-filled lymph nodes, and while this does not influence the primary immune response, it impairs the regulation of acquired immune responses (Herzog et al. 2013; Benezech et al. 2014). A much more severe phenotype is observed following constitutive loss of CLEC-2 or podoplanin where pups are born with a complete absence of mesenteric and inguinal lymph nodes due to a defect in lymph node maturation during late embryogenesis (Peters et al. 2011; Benezech et al. 2014). Significantly, lymph nodes are present in PF4-CLEC-2-deficient mice indicating that the developmental defect is not due to loss of platelet activation. This suggests a potential role for CLEC-2 outside of the platelet lineage during development and a second critical role for platelet-derived CLEC-2 in maintaining lymph node vascular integrity post-development.
A role for CLEC-2 in maintaining vascular integrity has been described in the developing brain. Podoplanin expression was shown to be restricted to the choroid plexus and ependymal lining of the ventricles at E16.5, but has since been seen to be widely expressed throughout the neuroepithelium between E10.5 and E12.5 (Williams et al. 1996; Schacht et al. 2003; Lowe et al. 2015a, b). Haemorrhages were first reported in the midbrain parenchyma of CLEC-2-deficient embryos at E12.0 (Tang et al. 2010). While severe cerebral haemorrhaging had been described in T-synthase-deficient embryos at E12.0, only recently were podoplanin-deficient embryos characterised and shown to exhibit extensive cerebral haemorrhaging between E11.5 and E12.5 (Lowe et al. 2015). Developing cerebral blood vessels in CLEC-2- and podoplanin-deficient mice were visibly tortuous and prone to haemorrhage at E10.5 caused by aberrant associations between the endothelium and the surrounding mural cells. CLEC-2-induced platelet aggregation is implicated in the haemorrhaging as bleeding is also seen in αIIb-deficient embryos. Thus, activation of platelets by podoplanin-CLEC-2 leads to platelet aggregation and sealing of gaps in developing vessels. It remains unclear whether this also contributes to the altered patterning of vessels that is seen before the onset of haemorrhage.
CLEC-2- and podoplanin-deficient mice experience respiratory failure at birth due to the inability to inflate their lungs (Ramirez et al. 2003; Finney et al. 2012). This is likely secondary to impairment in lymphatic function leading to fluid retention. In addition, it has been speculated that this may also be due to loss of podoplanin on alveolar type I cells, although as yet there is no direct evidence for this (Millien et al. 2006).
Podoplanin-deficient mice have been reported to exhibit myocardial pathology, where hypoplasia in the myocardium is proposed to result from impaired development of the epicardium (Mahtab et al. 2008). However, no such phenotype has been reported in CLEC-2-deficient mice, suggesting podoplanin plays a functional role independent of CLEC-2 in cardiac development.
CLEC-2 Beyond Haemostasis: The Beneficial and Detrimental Role of the Interaction of CLEC-2 and Podoplanin
In the last few years, increasing evidence for a role of platelets in the regulation of inflammation, infection and cancer has emerged in human disease and mouse models. Platelets are now recognised as having multiple roles in inflammation and infection, including the ability to regulate leukocyte functions, such as secretion of granular contents and production of reactive oxygen species (ROS) from neutrophils (Gros et al. 2015), while the formation of a thrombus inside a blood vessel supports innate immunity through pathogen recognition and containment (Engelmann and Massberg 2013). Many of these new roles for platelets have been shown to involve CLEC-2 and podoplanin (Fig. 2).

Possible beneficial and detrimental roles of platelet CLEC-2/podoplanin interactions in normal physiology and in disease
The Role of CLEC-2-Podoplanin in Inflammatory Diseases
Multiple studies have shown the selective role of CLEC-2-podoplanin interaction in inflammatory and non-inflammatory conditions. The beneficial or detrimental effect depends largely on the inflammatory state and the nature of the platelet-leukocyte interaction. Indeed, in a mouse model of multiple sclerosis (MS), an autoimmune inflammatory disorder of the central nervous system (CNS), podoplanin expression on a subset of CD4+ cells, TH17 cells, alters TH17 expansion and survival resulting in an increased rate of resolution of the inflammation (Peters et al. 2015). This beneficial effect of podoplanin was abrogated by a global deletion of CLEC-2 showing that the interaction between CLEC-2 and podoplanin mediates the resolution of the inflammation. However, whether CLEC-2 on platelets, DCs or possibly B-cells (where expression is controversial) contributes to this effect remains unclear. Significantly, upregulation of podoplanin was also observed in human patients with active MS plaque but not in glioblastoma multiforme (GBM), an aggressive malignant primary brain tumour (Nylander et al. 2015) indicating this upregulation is disease-specific. Thus, podoplanin-CLEC-2 interaction seems to modulate the inflammatory response in MS infiltrates, and this effect depends on the presence of a specific microenvironment.
It has been suggested that platelets contribute to several stages of atherosclerosis, including initiation, lesion growth and thrombus formation upon plaque disruption. Interestingly, the level of expression of podoplanin on smooth muscle cells and macrophages in atherosclerotic lesions from human patients correlates with the severity of the lesion, although the significance of this remains to be determined (Hatakeyama et al. 2012).
CLEC-2 expression has been reported in synovium of patients with rheumatoid arthritis (RA), a chronic systemic inflammatory disease leading to progressive destruction of the articular cartilage and bone (Del Rey et al. 2014). Importantly, the interaction of synovial fibroblasts with platelet CLEC-2 has been shown to lead to an increase in the proinflammatory cytokines IL6 and IL8 (Del Rey et al. 2014). CLEC-2 expression on microvesicles is also increased in RA patients (Gitz et al. 2014). It has been speculated that the abundant levels of microvesicles in RA may induce potent pro-inflammatory effects on the synovial fluid (Boilard et al. 2010; Del Rey et al. 2014). Moreover, podoplanin expression is increased in areas of inflammation, and synovial fibroblast activation and synovial transformation (Ekwall et al. 2011; Miyamoto et al. 2013; Del Rey et al. 2014).
The Role of CLEC-2 and Podoplanin in Infection
There is increasing recognition of the importance of platelets in bacterial and viral infections (Cox et al. 2011; Chabert et al. 2015). Severe and sometimes life-threatening bleeding can occur in certain viral infections, with thrombocytopenia often arising from viral-induced platelet destruction, consumption or sequestration. Platelet-viral interactions are complex and will depend on the species of the virus and/or the platelet environment. One of the most important viral agents that CLEC-2 associates with is the human immunodeficiency virus (HIV-type 1). Zucker-Franklin et al. (1990) were the first group to visualise HIV internalisation by megakaryocytes and platelets (Zucker-Franklin et al. 1990). HIV-1-infected cells release HIV-1 virions expressing podoplanin on their capsules that allow HIV-1 to interact with CLEC-2 and DC-SIGN on platelets. This interaction mediates HIV-1 capture and facilitates the spread of infection (Chaipan et al. 2006; Chaipan et al. 2010). Encouragingly, a CLEC-2-specific antiserum reduced HIV-1 transmission by platelets by approximately 50 % (Chaipan et al. 2006). Podoplanin depletion only diminished CLEC-2-dependent HIV transmission in B-THP cells, a Raji B-cell line, suggesting that podoplanin incorporation for HIV spread is dependent on podoplanin expression in the targeted cell for HIV infection (Chaipan et al. 2010). Therefore, incorporation of a second, as yet unrecognised, CLEC-2 ligand may be responsible for CLEC-2-dependent capture of T-cell-derived and peripheral blood mononuclear cell-derived viruses, facilitating CLEC-2-driven HIV transmission (Ozaki et al. 2009; Chaipan et al. 2010; Lowe et al. 2015a, b).
In a mouse model of salmonella infection, Hitchcock et al. (2015) have recently reported that upregulation of podoplanin in the liver leads to an inflammation-driven occlusive thrombosis. Significantly, the venous thrombosis was substantially abrogated in the absence of CLEC-2 on platelets or in the absence of macrophages without affecting the time course of the bacteraemia. The upregulation of podoplanin in the liver is driven by a TLR4- and INFγ-dependent inflammation. Given that CLEC-2 does not have a major role in haemostasis, this highlights the C-type lectin-like receptor as a novel target in some forms of thromboinflammatory disease.
The Role of CLEC-2 and Podoplanin in Cancer
A potential role of podoplanin in cancer metastasis has also emerged. Podoplanin is upregulated in a wide range of cancer types such as colorectal adenocarcinoma (Kato et al. 2003), testicular germ cell tumours (Kato et al. 2004), squamous cell carcinomas of the lung, cervix, oral cavity, larynx and mesothelioma and in tumours of the central nervous system (Kato et al. 2005; Kimura and Kimura 2005; Schacht et al. 2005; Shibahara et al. 2006; Rodrigo et al. 2010). Tumour cells can interact with platelets to form heterogeneous aggregates which protect the invasive cells from shear stress and immunological assault (Gay and Felding-Habermann 2011). It is speculated that the coating of platelets allows the tumour cells to evade the immune system as well as supporting adherence to the vessel wall. The antihuman podoplanin antibody (MS-1 mAb) has been shown to reduce platelet aggregation in vitro and pulmonary metastasis in vivo (Takagi et al. 2013). This highlights podoplanin as a promising target for developing novel antitumour and anti-metastatic agents.
Take-Home Messages
-
CLEC-2 interaction with podoplanin induces platelet activation and aggregation.
-
During development, CLEC-2-podopanin interaction is crucial for blood/lymphatic vessel separation.
-
Post-development, CLEC-2-podoplanin interaction maintains the integrity of high endothelial venules in lymph nodes.
-
CLEC-2-podoplanin is not crucial for haemostasis.
-
Detrimental role of CLEC-2-podoplanin: HIV propagation, salmonella-induced liver thrombosis and cancer metastasis.
-
Beneficial role of CLEC-2-podoplanin inhibition of T-cell expansion and survival in multiple sclerosis.
Conclusion
Although CLEC-2 and podoplanin have been intensively studied in the last decade, many questions concerning their roles remain unanswered, most notably the roles outside of the vasculature. Anti-CLEC-2 or anti-podoplanin drugs may have a beneficial effect in many diseases, in particular in inflammatory disorders. Additional studies are required to fully understand the mechanism underlying the role of CLEC-2 and podoplanin in physiology and pathology.
References
Abtahian F, Guerriero A, Sebzda E, Lu MM, Zhou R, Mocsai A, Myers EE, Huang B, Jackson DG, Ferrari VA, Tybulewicz V, Lowell CA, Lepore JJ, Koretzky GA, Kahn ML (2003) Regulation of blood and lymphatic vascular separation by signaling proteins SLP-76 and Syk. Science 299(5604):247–251
Acton SE, Astarita JL, Malhotra D, Lukacs-Kornek V, Franz B, Hess PR, Jakus Z, Kuligowski M, Fletcher AL, Elpek KG, Bellemare-Pelletier A, Sceats L, Reynoso ED, Gonzalez SF, Graham DB, Chang J, Peters A, Woodruff M, Kim YA, Swat W, Morita T, Kuchroo V, Carroll MC, Kahn ML, Wucherpfennig KW, Turley SJ (2012) Podoplanin-rich stromal networks induce dendritic cell motility via activation of the C-type lectin receptor CLEC-2. Immunity 37(2):276–289
Bender M, May F, Lorenz V, Thielmann I, Hagedorn I, Finney BA, Vogtle T, Remer K, Braun A, Bosl M, Watson SP, Nieswandt B (2013) Combined in vivo depletion of glycoprotein VI and C-type lectin-like receptor 2 severely compromises hemostasis and abrogates arterial thrombosis in mice. Arterioscler Thromb Vasc Biol 33(5):926–934
Benezech C, Nayar S, Finney BA, Withers DR, Lowe K, Desanti GE, Marriott CL, Watson SP, Caamano JH, Buckley CD, Barone F (2014) CLEC-2 is required for development and maintenance of lymph nodes. Blood 123(20):3200–3207
Bertozzi CC, Schmaier AA, Mericko P, Hess PR, Zou Z, Chen M, Chen CY, Xu B, Lu MM, Zhou D, Sebzda E, Santore MT, Merianos DJ, Stadtfeld M, Flake AW, Graf T, Skoda R, Maltzman JS, Koretzky GA, Kahn ML (2010) Platelets regulate lymphatic vascular development through CLEC-2-SLP-76 signaling. Blood 116(4):661–670
Bohmer R, Neuhaus B, Buhren S, Zhang D, Stehling M, Bock B, Kiefer F (2010) Regulation of developmental lymphangiogenesis by Syk(+) leukocytes. Dev Cell 18(3):437–449
Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, Massarotti EM, Remold-O’Donnell E, Farndale RW, Ware J, Lee DM (2010) Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 327(5965):580–583
Carramolino L, Fuentes J, Garcia-Andres C, Azcoitia V, Riethmacher D, Torres M (2010) Platelets play an essential role in separating the blood and lymphatic vasculatures during embryonic angiogenesis. Circ Res 106(7):1197–1201
Chabert A, Hamzeh-Cognasse H, Pozzetto B, Cognasse F, Schattner M, Gomez RM, Garraud O (2015) Human platelets and their capacity of binding viruses: meaning and challenges? BMC Immunol 16:26
Chaipan C, Soilleux EJ, Simpson P, Hofmann H, Gramberg T, Marzi A, Geier M, Stewart EA, Eisemann J, Steinkasserer A, Suzuki-Inoue K, Fuller GL, Pearce AC, Watson SP, Hoxie JA, Baribaud F, Pohlmann S (2006) DC-SIGN and CLEC-2 mediate human immunodeficiency virus type 1 capture by platelets. J Virol 80(18):8951–8960
Chaipan C, Steffen I, Tsegaye TS, Bertram S, Glowacka I, Kato Y, Schmokel J, Munch J, Simmons G, Gerardy-Schahn R, Pohlmann S (2010) Incorporation of podoplanin into HIV released from HEK-293T cells, but not PBMC, is required for efficient binding to the attachment factor CLEC-2. Retrovirology 7:47
Cheng AM, Rowley B, Pao W, Hayday A, Bolen JB, Pawson T (1995) Syk tyrosine kinase required for mouse viability and B-cell development. Nature 378(6554):303–306
Clements JL, Yang B, Ross-Barta SE, Eliason SL, Hrstka RF, Williamson RA, Koretzky GA (1998) Requirement for the leukocyte-specific adapter protein SLP-76 for normal T cell development. Science 281(5375):416–419
Clements JL, Lee JR, Gross B, Yang B, Olson JD, Sandra A, Watson SP, Lentz SR, Koretzky GA (1999) Fetal hemorrhage and platelet dysfunction in SLP-76-deficient mice. J Clin Invest 103(1):19–25
Colonna M, Samaridis J, Angman L (2000) Molecular characterization of two novel C-type lectin-like receptors, one of which is selectively expressed in human dendritic cells. Eur J Immunol 30(2):697–704
Cox D, Kerrigan SW, Watson SP (2011) Platelets and the innate immune system: mechanisms of bacterial-induced platelet activation. J Thromb Haemost 9(6):1097–1107
Del Rey MJ, Fare R, Izquierdo E, Usategui A, Rodriguez-Fernandez JL, Suarez-Fueyo A, Canete JD, Pablos JL (2014) Clinicopathological correlations of podoplanin (gp38) expression in rheumatoid synovium and its potential contribution to fibroblast platelet crosstalk. PLoS One 9(6):e99607
Deppermann C, Cherpokova D, Nurden P, Schulz JN, Thielmann I, Kraft P, Vogtle T, Kleinschnitz C, Dutting S, Krohne G, Eming SA, Nurden AT, Eckes B, Stoll G, Stegner D, Nieswandt B (2013) Gray platelet syndrome and defective thrombo-inflammation in Nbeal2-deficient mice. J Clin Invest 123(8):3331–3342
Ekwall AK, Eisler T, Anderberg C, Jin C, Karlsson N, Brisslert M, Bokarewa MI (2011) The tumour-associated glycoprotein podoplanin is expressed in fibroblast-like synoviocytes of the hyperplastic synovial lining layer in rheumatoid arthritis. Arthritis Res Ther 13(2):R40
Engelmann B, Massberg S (2013) Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol 13(1):34–45
Farr AG, Berry ML, Kim A, Nelson AJ, Welch MP, Aruffo A (1992) Characterization and cloning of a novel glycoprotein expressed by stromal cells in T-dependent areas of peripheral lymphoid tissues. J Exp Med 176(5):1477–1482
Finney BA, Schweighoffer E, Navarro-Nunez L, Benezech C, Barone F, Hughes CE, Langan SA, Lowe KL, Pollitt AY, Mourao-Sa D, Sheardown S, Nash GB, Smithers N, Reis e Sousa C, Tybulewicz VL, Watson SP (2012) CLEC-2 and Syk in the megakaryocytic/platelet lineage are essential for development. Blood 119(7):1747–1756
Fu J, Gerhardt H, McDaniel JM, Xia B, Liu X, Ivanciu L, Ny A, Hermans K, Silasi-Mansat R, McGee S, Nye E, Ju T, Ramirez MI, Carmeliet P, Cummings RD, Lupu F, Xia L (2008) Endothelial cell O-glycan deficiency causes blood/lymphatic misconnections and consequent fatty liver disease in mice. J Clin Invest 118(11):3725–3737
Fuller GL, Williams JA, Tomlinson MG, Eble JA, Hanna SL, Pohlmann S, Suzuki-Inoue K, Ozaki Y, Watson SP, Pearce AC (2007) The C-type lectin receptors CLEC-2 and Dectin-1, but not DC-SIGN, signal via a novel YXXL-dependent signaling cascade. J Biol Chem 282(17):12397–12409
Gay LJ, Felding-Habermann B (2011) Contribution of platelets to tumour metastasis. Nat Rev Cancer 11(2):123–134
Gitz E, Pollitt AY, Gitz-Francois JJ, Alshehri O, Mori J, Montague S, Nash GB, Douglas MR, Gardiner EE, Andrews RK, Buckley CD, Harrison P, Watson SP (2014) CLEC-2 expression is maintained on activated platelets and on platelet microparticles. Blood 124(14):2262–2270
Gros A, Syvannarath V, Lamrani L, Ollivier V, Loyau S, Goerge T, Nieswandt B, Jandrot-Perrus M, Ho-Tin-Noe B (2015) Single platelets seal neutrophil-induced vascular breaches via GPVI during immune-complex-mediated inflammation in mice. Blood 126(8):1017–1026
Hagerling R, Pollmann C, Andreas M, Schmidt C, Nurmi H, Adams RH, Alitalo K, Andresen V, Schulte-Merker S, Kiefer F (2013) A novel multistep mechanism for initial lymphangiogenesis in mouse embryos based on ultramicroscopy. EMBO J 32(5):629–644
Hatakeyama K, Kaneko MK, Kato Y, Ishikawa T, Nishihira K, Tsujimoto Y, Shibata Y, Ozaki Y, Asada Y (2012) Podoplanin expression in advanced atherosclerotic lesions of human aortas. Thromb Res 129(4):e70–e76
Herzog BH, Fu J, Wilson SJ, Hess PR, Sen A, McDaniel JM, Pan Y, Sheng M, Yago T, Silasi-Mansat R, McGee S, May F, Nieswandt B, Morris AJ, Lupu F, Coughlin SR, McEver RP, Chen H, Kahn ML, Xia L (2013) Podoplanin maintains high endothelial venule integrity by interacting with platelet CLEC-2. Nature 502(7469):105–109
Hess PR, Rawnsley DR, Jakus Z, Yang Y, Sweet DT, Fu J, Herzog B, Lu M, Nieswandt B, Oliver G, Makinen T, Xia L, Kahn ML (2014) Platelets mediate lymphovenous hemostasis to maintain blood-lymphatic separation throughout life. J Clin Invest 124(1):273–284
Hitchcock JR, Cook CN, Bobat S, Ross EA, Flores-Langarica A, Lowe KL, Khan M, Dominguez-Medina CC, Lax S, Carvalho-Gaspar M, Hubscher S, Rainger GE, Cobbold M, Buckley CD, Mitchell TJ, Mitchell A, Jones ND, Van Rooijen N, Kirchhofer D, Henderson IR, Adams DH, Watson SP, Cunningham AF (2015) Inflammation drives thrombosis after Salmonella infection via CLEC-2 on platelets. J Clin Investig 125(12):4429–4446
Hughes CE, Navarro-Nunez L, Finney BA, Mourao-Sa D, Pollitt AY, Watson SP (2010) CLEC-2 is not required for platelet aggregation at arteriolar shear. J Thromb Haemost 8(10):2328–2332
Hughes CE, Sinha U, Pandey A, Eble JA, O’Callaghan CA, Watson SP (2013) Critical Role for an acidic amino acid region in platelet signaling by the HemITAM (hemi-immunoreceptor tyrosine-based activation motif) containing receptor CLEC-2 (C-type lectin receptor-2). J Biol Chem 288(7):5127–5135
Hughes CE, Finney BA, Koentgen F, Lowe KL, Watson SP (2015) The N-terminal SH2 domain of Syk is required for (hem)ITAM, but not integrin, signaling in mouse platelets. Blood 125(1):144–154
Ichise H, Ichise T, Ohtani O, Yoshida N (2009) Phospholipase Cgamma2 is necessary for separation of blood and lymphatic vasculature in mice. Development 136(2):191–195
Kato Y, Fujita N, Kunita A, Sato S, Kaneko M, Osawa M, Tsuruo T (2003) Molecular identification of Aggrus/T1alpha as a platelet aggregation-inducing factor expressed in colorectal tumors. J Biol Chem 278(51):51599–51605
Kato Y, Sasagawa I, Kaneko M, Osawa M, Fujita N, Tsuruo T (2004) Aggrus: a diagnostic marker that distinguishes seminoma from embryonal carcinoma in testicular germ cell tumors. Oncogene 23(52):8552–8556
Kato Y, Kaneko M, Sata M, Fujita N, Tsuruo T, Osawa M (2005) Enhanced expression of Aggrus (T1alpha/podoplanin), a platelet-aggregation-inducing factor in lung squamous cell carcinoma. Tumour Biol 26(4):195–200
Kerrigan AM, Dennehy KM, Mourao-Sa D, Faro-Trindade I, Willment JA, Taylor PR, Eble JA, Reis e Sousa C, Brown GD (2009) CLEC-2 is a phagocytic activation receptor expressed on murine peripheral blood neutrophils. J Immunol 182(7):4150–4157
Kimura N, Kimura I (2005) Podoplanin as a marker for mesothelioma. Pathol Int 55(2):83–86
Koop K, Eikmans M, Wehland M, Baelde H, Ijpelaar D, Kreutz R, Kawachi H, Kerjaschki D, de Heer E, Bruijn JA (2008) Selective loss of podoplanin protein expression accompanies proteinuria and precedes alterations in podocyte morphology in a spontaneous proteinuric rat model. Am J Pathol 173(2):315–326
Liu Y, Wada R, Yamashita T, Mi Y, Deng CX, Hobson JP, Rosenfeldt HM, Nava VE, Chae SS, Lee MJ, Liu CH, Hla T, Spiegel S, Proia RL (2000) Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest 106(8):951–961
Lowe KL, Finney BA, Deppermann C, Hagerling R, Gazit SL, Frampton J, Buckley C, Camerer E, Nieswandt B, Kiefer F, Watson SP (2015a) Podoplanin and CLEC-2 drive cerebrovascular patterning and integrity during development. Blood 125(24):3769–3777
Lowe KL, Navarro-Nunez L, Benezech C, Nayar S, Kingston BL, Nieswandt B, Barone F, Watson SP, Buckley CD, Desanti GE (2015b) The expression of mouse CLEC-2 on leucocyte subsets varies according to their anatomical location and inflammatory state. Eur J Immunol 45(9):2484–2493
Mahtab EA, Wijffels MC, Van Den Akker NM, Hahurij ND, Lie-Venema H, Wisse LJ, Deruiter MC, Uhrin P, Zaujec J, Binder BR, Schalij MJ, Poelmann RE, Gittenberger-De Groot AC (2008) Cardiac malformations and myocardial abnormalities in podoplanin knockout mouse embryos: correlation with abnormal epicardial development. Dev Dyn 237(3):847–857
May F, Hagedorn I, Pleines I, Bender M, Vogtle T, Eble J, Elvers M, Nieswandt B (2009) CLEC-2 is an essential platelet-activating receptor in hemostasis and thrombosis. Blood 114(16):3464–3472
Millien G, Spira A, Hinds A, Wang J, Williams MC, Ramirez MI (2006) Alterations in gene expression in T1 alpha null lung: a model of deficient alveolar sac development. BMC Dev Biol 6:35
Miyamoto Y, Uga H, Tanaka S, Kadowaki M, Ikeda M, Saegusa J, Morinobu A, Kumagai S, Kurata H (2013) Podoplanin is an inflammatory protein upregulated in Th17 cells in SKG arthritic joints. Mol Immunol 54(2):199–207
Nylander A, Ramanan S, Debartolo D, Park C, Hafler D, Pitt D (2015) Immune infiltrates expressing podoplanin (PDPN) are present in multiple sclerosis but not glioblastoma (S12.002). Neurology 84(14 Supplement): S12.002.
Ozaki Y, Suzuki-Inoue K, Inoue O (2009) Novel interactions in platelet biology: CLEC-2/podoplanin and laminin/GPVI. J Thromb Haemost 7(Suppl 1):191–194
Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, Wucherpfennig K, Turley S, Carroll MC, Sobel RA, Bettelli E, Kuchroo VK (2011) Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity 35(6):986–996
Peters A, Burkett PR, Sobel RA, Buckley CD, Watson SP, Bettelli E, Kuchroo VK (2015) Podoplanin negatively regulates CD4+ effector T cell responses. J Clin Invest 125(1):129–140
Pivniouk V, Tsitsikov E, Swinton P, Rathbun G, Alt FW, Geha RS (1998) Impaired viability and profound block in thymocyte development in mice lacking the adaptor protein SLP-76. Cell 94(2):229–238
Pollitt AY, Grygielska B, Leblond B, Desire L, Eble JA, Watson SP (2010) Phosphorylation of CLEC-2 is dependent on lipid rafts, actin polymerization, secondary mediators, and Rac. Blood 115(14):2938–2946
Pollitt AY, Poulter NS, Gitz E, Navarro-Nunez L, Wang YJ, Hughes CE, Thomas SG, Nieswandt B, Douglas MR, Owen DM, Jackson DG, Dustin ML, Watson SP (2014) Syk and Src family kinases regulate C-type lectin receptor 2 (CLEC-2)-mediated clustering of podoplanin and platelet adhesion to lymphatic endothelial cells. J Biol Chem 289(52):35695–35710
Ramirez MI, Millien G, Hinds A, Cao Y, Seldin DC, Williams MC (2003) T1alpha, a lung type I cell differentiation gene, is required for normal lung cell proliferation and alveolus formation at birth. Dev Biol 256(1):61–72
Rodrigo JP, Garcia-Carracedo D, Gonzalez MV, Mancebo G, Fresno MF, Garcia-Pedrero J (2010) Podoplanin expression in the development and progression of laryngeal squamous cell carcinomas. Mol Cancer 9:48
Ruckrich T, Steinle A (2013) Attenuated natural killer (NK) cell activation through C-type lectin-like receptor NKp80 is due to an anomalous hemi-immunoreceptor tyrosine-based activation motif (HemITAM) with impaired Syk kinase recruitment capacity. J Biol Chem 288(24):17725–17733
Schacht V, Ramirez MI, Hong YK, Hirakawa S, Feng D, Harvey N, Williams M, Dvorak AM, Dvorak HF, Oliver G, Detmar M (2003) T1alpha/podoplanin deficiency disrupts normal lymphatic vasculature formation and causes lymphedema. EMBO J 22(14):3546–3556
Schacht V, Dadras SS, Johnson LA, Jackson DG, Hong YK, Detmar M (2005) Up-regulation of the lymphatic marker podoplanin, a mucin-type transmembrane glycoprotein, in human squamous cell carcinomas and germ cell tumors. Am J Pathol 166(3):913–921
Sebzda E, Hibbard C, Sweeney S, Abtahian F, Bezman N, Clemens G, Maltzman JS, Cheng L, Liu F, Turner M, Tybulewicz V, Koretzky GA, Kahn ML (2006) Syk and Slp-76 mutant mice reveal a cell-autonomous hematopoietic cell contribution to vascular development. Dev Cell 11(3):349–361
Senis YA, Tomlinson MG, Garcia A, Dumon S, Heath VL, Herbert J, Cobbold SP, Spalton JC, Ayman S, Antrobus R, Zitzmann N, Bicknell R, Frampton J, Authi KS, Martin A, Wakelam MJ, Watson SP (2007) A comprehensive proteomics and genomics analysis reveals novel transmembrane proteins in human platelets and mouse megakaryocytes including G6b-B, a novel immunoreceptor tyrosine-based inhibitory motif protein. Mol Cell Proteomics 6(3):548–564
Senis YA, Mazharian A, Mori J (2014) Src family kinases: at the forefront of platelet activation. Blood 124(13):2013–2024
Severin S, Pollitt AY, Navarro-Nunez L, Nash CA, Mourao-Sa D, Eble JA, Senis YA, Watson SP (2011) Syk-dependent phosphorylation of CLEC-2: a novel mechanism of hem-immunoreceptor tyrosine-based activation motif signaling. J Biol Chem 286(6):4107–4116
Shibahara J, Kashima T, Kikuchi Y, Kunita A, Fukayama M (2006) Podoplanin is expressed in subsets of tumors of the central nervous system. Virchows Arch 448(4):493–499
Spalton JC, Mori J, Pollitt AY, Hughes CE, Eble JA, Watson SP (2009) The novel Syk inhibitor R406 reveals mechanistic differences in the initiation of GPVI and CLEC-2 signaling in platelets. J Thromb Haemost 7(7):1192–1199
Suzuki-Inoue K, Fuller GL, Garcia A, Eble JA, Pohlmann S, Inoue O, Gartner TK, Hughan SC, Pearce AC, Laing GD, Theakston RD, Schweighoffer E, Zitzmann N, Morita T, Tybulewicz VL, Ozaki Y, Watson SP (2006) A novel Syk-dependent mechanism of platelet activation by the C-type lectin receptor CLEC-2. Blood 107(2):542–549
Suzuki-Inoue K, Inoue O, Ding G, Nishimura S, Hokamura K, Eto K, Kashiwagi H, Tomiyama Y, Yatomi Y, Umemura K, Shin Y, Hirashima M, Ozaki Y (2010) Essential in vivo roles of the C-type lectin receptor CLEC-2: embryonic/neonatal lethality of CLEC-2-deficient mice by blood/lymphatic misconnections and impaired thrombus formation of CLEC-2-deficient platelets. J Biol Chem 285(32):24494–24507
Takagi S, Sato S, Oh-hara T, Takami M, Koike S, Mishima Y, Hatake K, Fujita N (2013) Platelets promote tumor growth and metastasis via direct interaction between Aggrus/podoplanin and CLEC-2. PLoS One 8(8):e73609
Tang T, Li L, Tang J, Li Y, Lin WY, Martin F, Grant D, Solloway M, Parker L, Ye W, Forrest W, Ghilardi N, Oravecz T, Platt KA, Rice DS, Hansen GM, Abuin A, Eberhart DE, Godowski P, Holt KH, Peterson A, Zambrowicz BP, de Sauvage FJ (2010) A mouse knockout library for secreted and transmembrane proteins. Nat Biotechnol 28(7):749–755
Turner M, Mee PJ, Costello PS, Williams O, Price AA, Duddy LP, Furlong MT, Geahlen RL, Tybulewicz VL (1995) Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk. Nature 378(6554):298–302
Uhrin P, Zaujec J, Breuss JM, Olcaydu D, Chrenek P, Stockinger H, Fuertbauer E, Moser M, Haiko P, Fassler R, Alitalo K, Binder BR, Kerjaschki D (2010) Novel function for blood platelets and podoplanin in developmental separation of blood and lymphatic circulation. Blood 115(19):3997–4005
Wang D, Feng J, Wen R, Marine JC, Sangster MY, Parganas E, Hoffmeyer A, Jackson CW, Cleveland JL, Murray PJ, Ihle JN (2000) Phospholipase Cgamma2 is essential in the functions of B cell and several Fc receptors. Immunity 13(1):25–35
Williams MC, Cao Y, Hinds A, Rishi AK, Wetterwald A (1996) T1 alpha protein is developmentally regulated and expressed by alveolar type I cells, choroid plexus, and ciliary epithelia of adult rats. Am J Respir Cell Mol Biol 14(6):577–585
Yu H, Gibson JA, Pinkus GS, Hornick JL (2007) Podoplanin (D2-40) is a novel marker for follicular dendritic cell tumors. Am J Clin Pathol 128(5):776–782
Zheng Y, Adams T, Zhi H, Yu M, Wen R, Newman PJ, Wang D, Newman DK (2015) Restoration of responsiveness of phospholipase Cgamma2-deficient platelets by enforced expression of phospholipase Cgamma1. PLoS One 10(3):e0119739
Zucker-Franklin D, Seremetis S, Zheng ZY (1990) Internalization of human immunodeficiency virus type I and other retroviruses by megakaryocytes and platelets. Blood 75(10):1920–1923
Acknowledgements
This work was supported by the British Heart Foundation, the Wellcome Trust, and medical research council.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Rayes, J., Hardy, A.T., Lombard, S.E., Montague, S.J., Watson, S.P., Lowe, K.L. (2017). The Role of CLEC-2 in and Beyond the Vasculature. In: Gresele, P., Kleiman, N., Lopez, J., Page, C. (eds) Platelets in Thrombotic and Non-Thrombotic Disorders. Springer, Cham. https://doi.org/10.1007/978-3-319-47462-5_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-47462-5_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-47460-1
Online ISBN: 978-3-319-47462-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)