
Abstract
Cranial nerve (CN) disorders are commonly encountered in pediatric practice, since they are related to vision, eye movements, hearing and speech, facial sensation and movements, and swallowing. Localization is an important consideration in the management of these disorders, as well as the knowledge of the various diseases that cause cranial nerve dysfunction. There are 12 pairs of cranial nerves, which are numbered based on the order in which they emerge from the brain (including the brainstem), front to back (i.e., their rostral-caudal [front-back] position when viewing the brain). The first two cranial nerves (olfactory [CN I] and optic [CN II]) arise from the cerebrum, whereas the remaining ten emerge from the brainstem.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Cranial nerve (CN) disorders are commonly encountered in pediatric practice, since they are related to vision, eye movements, hearing and speech, facial sensation and movements, and swallowing. Localization is an important consideration in the management of these disorders, as well as the knowledge of the various diseases that cause cranial nerve dysfunction. There are 12 pairs of cranial nerves, which are numbered based on the order in which they emerge from the brain (including the brainstem), front to back (i.e., their rostral-caudal [front-back] position when viewing the brain). The first two cranial nerves (olfactory [CN I] and optic [CN II]) arise from the cerebrum, whereas the remaining ten emerge from the brainstem.
3.1 Cranial Nerve I (CN I): Olfactory Nerve
The olfactory nerve (CN I), the first and shortest of the cranial nerves, conveys the sense of smell. Passing from its receptors in the nasal mucosa, it reaches the forebrain having entered the skull through the cribriform plate of the ethmoid bone. Thereafter, it sends impulses for interpretation at various regions of the brain including the entorhinal cortex (located in the medial temporal lobe) and the amygdala. The olfactory nerve (CN I) is tested by asking the child to identify different recognizable smells with his or her eyes closed, and specialized forms of olfactory tests have been developed for children [1]. Common causes of smell loss in children are head trauma, allergic rhinitis, and enlarged adenoids. Males with Kallmann syndrome (OMIM # 308700) have anosmia due to agenesis of the olfactory lobes, associated with hypogonadism secondary to deficiency of hypothalamic gonadotropin-releasing hormone (GnRH). Kallmann syndrome is a genetically heterogeneous condition and can be inherited as X-linked, autosomal recessive, or autosomal dominant. There is also a sporadic form of the disease, which is the most common [2]. Males may present early with microphallus and cryptorchidism or later with delayed puberty, whereas females present with primary amenorrhea. Recognition of the syndrome allows proper management of patients with attainment of normal reproductive health and also allows for provision of informed genetic counseling [2].
3.2 Cranial Nerve II (CN II): Optic Nerve
The optic nerve (CN II) is the cranial nerve dedicated for vision and will be discussed in the Chap. 4.
3.3 Cranial Nerve III (CN III): Oculomotor Nerve
The oculomotor nerve (CN III) innervates the extraocular muscles of either eye via motor fibers to levator palpebrae superioris, superior rectus, inferior rectus, medial rectus, and inferior oblique. Via parasympathetic fibers to constrictor pupillae and ciliary muscles, it controls pupillary constriction. Presentations of CN III palsy include diplopia, loss of accommodation, ptosis caused by loss of levator palpebrae superioris function (Fig. 3.1a, b), eye deviation outward, (exotropia) and downward (hypotropia), and pupillary dilation [3].

(a) One-year-old girl who resented with right oculomotor nerve (CN III) palsy manifesting with ptosis and right eye deviation outwards (exotropia) and downwards (hypotropia). (b) Investigations revealed right midbrain atypical teratoid rhabdoid tumor, showing on T2-weighted magnetic resonance image (MRI) as heterogeneous signal intensity (arrow). Inflammatory causes of oculomotor nerve (CN III) palsy include Miller-Fisher syndrome, manifesting in (c) with bilateral ptosis and ophthalmoplegia; whereas the differential diagnosis includes (d) ocular myasthenia gravis which may present as asymmetric ptosis and ocular muscle palsy. (e) 19-month-old boy with synostotic plagiocephaly which manifests with ocular features mimiking congenital trochlear nerve (CN IV) palsy. (f) Cranial 3-dimentional (3D) computed tomography (CT) showed synostosis of the metopic suture and right coronal suture. The left coronal suture was patent (arrow). (g) 4-year-old boy with congenital torticollis resulting from tight sternocleidomastoid muscle complicating sterocleidomastoid tumor of infancy. Congenital torticollis is an important differential diagnosis of congenital trochlear nerve (CN IV) palsy
Dysfunction of CN III can result from lesions occurring along its path between the oculomotor nucleus located in the midbrain, the basilar segment where it courses in the subarachnoid space (alongside the posterior communicating artery) over the base of the skull, intracavernous segment where it pierces the dura mater and enters the cavernous sinus and runs along the lateral wall, and the extraocular muscles within the orbit. Within the intracavernous segment, CN III passes over the trochlear nerve (CN IV) and lays superior to the abducens nerve (CN VI) and medial to the ophthalmic branch of the trigeminal nerve (CN V1).
Third cranial nerve (CN III) palsy may be congenital as in Duane syndrome and disorders formerly known as congenital fibrosis of the extraocular muscles (CFEOM, see below). Stretch injury to the nerve and compression may be caused by trauma leading to herniation of the temporal lobe through the tentorial opening [3]. This can follow birth trauma resulting from forceps injury or compression on the skull as a result of prolonged labor. Traumatic dysfunction of CN III can occur along its course from the nucleus to the orbit as a result of direct of brainstem ischemic injury, direct injury, or diffuse axonal injury.
Of the infectious etiologies, bacterial meningitis is the most common cause [3,4,5]. Other infectious and inflammatory causes include tuberculosis, Tolosa-Hunt syndrome (idiopathic granulomatous inflammation of the cavernous sinus), sarcoidosis, postviral inflammation, and Miller Fisher syndrome (Fig. 3.1c).
Tumors leading to oculomotor nerve (CN III) palsy include pituitary adenoma, craniopharyngioma, astrocytoma, schwannoma, meningioma, peripheral nerve sheath tumor, and teratoid tumor (Fig. 3.1a, b). Cerebral aneurysms are the most common vascular cause of CN III palsy. Ophthalmoplegic migraine, which accounts for 7% of CN III palsies in children, is usually preceded by ipsilateral headache continuing for days [3].
The differential diagnosis of oculomotor nerve (CN III) palsy includes ocular myasthenia gravis which may present as asymmetric ocular muscle palsy (Fig. 3.1d). Diagnostic workup requires neuroimaging in most cases to exclude neoplastic (Fig. 3.1b) or vascular compressive etiology, and the diagnosis of ophthalmoplegic migraine requires fulfillment of its clinical criteria. During or immediately after the migrainous attack, magnetic resonance imaging (MRI) usually detects homogeneous enhancement and enlargement of the CN III at the root exit zone [6]. Ophthalmoplegic migraine is managed by giving systemic steroids and other medications used for migraine prophylaxis, including propranolol, flunarizine, and topiramate.
Children with oculomotor nerve palsy should be promptly referred to an ophthalmologist for the detection and treatment of amblyopia, which may result from ptosis leading to suppression due to ocular misalignment or image deprivation [3]. Management, other than amblyopia, includes surgical ocular alignment and ptosis repair.
3.4 Cranial Nerve IV (CN IV): Trochlear Nerve
Cranial nerve IV (CN IV, trochlear nerve) originates in the pons and passes through the cavernous sinus and superior orbital fissure to provide motor supply to the superior oblique muscle which depresses, internally rotates, and abducts the eye globe. Dysfunction of the trochlear nerve results in inability to move eye downward and laterally, leading to head tilt to the opposite side of the CN IV palsy aiming at decreasing the ocular misalignment. Long-standing head tilt may lead to facial asymmetry with the mouth being slanted upward on the side of the head tilt.
Trochlear nerve palsy can be unilateral or bilateral, congenital or acquired. In children, trauma constitutes the most common cause of acquired CN IV nerve palsy. Other causes include compressive neoplasms, hydrocephalus, and arteriovenous malformations. Diagnostic workup resides on enquiry about history of early onset head tilt, verified by family photographs, and history of head or orbital trauma. Magnetic resonance imaging (MRI) is required in cases of hydrocephalus, suspected aneurysm, or tumor. Differential diagnosis includes synostotic plagiocephaly (Fig. 3.1e, f), which manifests with facial asymmetry, and congenital torticollis resulting from tight sternocleidomastoid muscle, a known complication of sternocleidomastoid tumor of infancy (Fig. 3.1g) [7]. Management of trochlear nerve (CN IV) palsy consists of reducing diplopia, restoring binocularity, and normalizing the anomalous head posture [3].
3.5 Cranial Nerve V (CN V): Trigeminal Nerve
Cranial nerve V (trigeminal nerve) nuclei are located in the brainstem and the upper part of the spinal cord. It has three major branches, the ophthalmic nerve (V1), which passes through the cavernous sinus and exits the skull through the superior orbital fissure, and the maxillary nerve (V2) and the mandibular nerve (V3), which exit via the foramen rotundum and foramen ovale, respectively.
Trigeminal nerve provides sensory supply to face, scalp, and oral cavity (including teeth and tongue) and motor supply to muscles of mastication (the temporal, the masseter, and the medial and lateral pterygoids) and to tensor tympani. Lesions of the ophthalmic nerve (V1) manifest with absent corneal reflex and anesthesia of the forehead, and those of the maxillary nerve (V2) lead to anesthesia of the midface, whereas mandibular nerve (V3) lesions result in paralysis of muscles of mastication, with the jaw deviating toward the side of the lesion. Manifestations of trigeminal nerve nuclei lesions depend on which nuclei are affected. There may be ipsilateral muscles of mastication weakness and/or ipsilateral loss of sensation. Since CN V (trigeminal) and CN VI (abducens) nerves are contiguous in the cavernous sinus, pathology at that site (e.g., cavernous sinus thrombosis) leads to loss of corneal sensation.
A chronic pain condition that affects the trigeminal nerve (CN V) is trigeminal neuralgia, also called tic douloureux. It is a rare condition in children, usually presenting with recurrent sudden severe stabbing pain in the distribution of one or more branches of CN V [8]. Underlying etiology includes vascular compression by blood vessel as CN V exits the brainstem, multiple sclerosis, vascular malformations, and tumors [9, 10]. Neuropathic facial pain can also be due to trigeminal nerve injury following oral surgery, sinus surgery, or facial trauma. Diagnostic investigations require MRI and magnetic resonance angiography (MRA). Carbamazepine is the most frequently used drug for treating trigeminal neuralgia. Other drugs include gabapentin, oxcarbazepine, and phenytoin [8]. On failure of medical therapy to control pain, surgical treatments, including microvascular decompression, may be considered.
Plexiform neurofibroma , which is considered to be pathognomonic feature of neurofibromatosis type 1 (NF1, OMIM # 162200), frequently involves the trigeminal nerves leading to soft tissue hypertrophy and may cause remarkable disfigurement of the face (Fig. 3.2a) [11]. It arises from nerve fascicles and involves multiple nerve branches and plexuses as it grows along the length of the nerve.

(a) Adolescent male with large plexiform neurofibroma along the right trigeminal nerve distribution. (b, c) A child who had mastoiditis leading to ipsilateral right abducens (CN VI) nerve palsy (Gradenigo syndrome). (b) On attempted right gaze, he had no abduction of the right eye, whereas on attempted left gaze (c), he showed full abduction of the left eye
3.6 Cranial Nerve VI (CN VI): Abducens Nerve
Abducens nerve (CN VI) provides motor supply to the lateral rectus muscle, and lesions of CN VI lead to impaired abduction of the affected eye associated with diplopia on attempting lateral gaze. The nucleus of CN VI is located in the pons, and it emerges from the brainstem, to enter the subarachnoid space, at the pontomedullary junction. During its course in the cavernous sinus, CN VI is located in close proximity to the internal carotid artery, and it innervates the lateral rectus muscle after proceeding through the superior orbital fissure.
In children, abducens nerve (CN VI) palsy may be congenital as in congenital cranial dysinnervation disorders (see below), namely, Duane and Moebius syndromes and Bosley-Salih-Alorainy syndrome (BSAS). Nevertheless, intracranial neoplasms constitute the most common cause, including pontine gliomas, cystic cerebellar astrocytomas, ependymoma, and medulloblastoma [3, 12]. The mechanism of injury in intracranial neoplasms is either due to infiltration of the pons or the mass effect of the tumor leading to raised intracranial pressure. Other causes include elevated intracranial pressure secondary to shunt failure, meningitis, venous sinus thrombosis, or idiopathic intracranial hypertension (pseudotumor cerebri) [13, 14]. Elevated intracranial pressure may also follow trauma, and traumatic abducens nerve (CN VI) palsy may result from fracture base of the skull, traumatic carotid cavernous fistulas, or blunt head trauma. Infectious causes include Lyme disease, syphilis, tuberculosis [15], diphtheria [16], and mastoiditis spreading to involve the petrous bone leading to ipsilateral abducens nerve (CN VI) palsy (Gradenigo syndrome, Fig. 3.2b, c). Inflammatory disorders include multiple sclerosis and the Miller Fisher variant of Guillain-Barré syndrome in which bilateral abducens nerve (CN VI) palsy evolves into complete ophthalmoplegia (Fig. 3.1c). Other uncommon causes include infantile botulism, ophthalmoplegic migraine, intracranial hypotension, and following myelography or lumbar puncture.
Antecedent viral illness and immunization were reported to precede a benign idiopathic and recurrent form of abducens nerve (CN VI) palsy. Recurrences are usually seen in females affecting the left eye with high risk of resulting amblyopia [3].
An important differential diagnosis of CN VI palsy in children is accommodative esotropia, in which abduction, unlike cases of abducens palsy, is normal and extraocular muscles movements are intact. Medial wall fracture of the orbit with entrapment of medial rectus muscle leading to limitation of abduction also mimics CN VI palsy. Other diseases which need to be differentiated are myasthenia gravis and thyroid associated orbitopathy with medial rectus involvement.
Workup to diagnose CN VI palsy should include inquiring about history of binocular diplopia (becoming worse at distance or lateral gaze), visual loss, headache, vomiting, facial pain or numbness, otitis media, hearing loss, trauma, recent viral infection, and immunization. Physical examination should document abducens nerve (CN VI) palsy (impaired abduction of the affected eye with esotropia that worsens when looking toward the palsied eye) and the presence of diplopia. It should also include looking for nystagmus (e.g., secondary to pontine glioma or cerebellar tumors), papilledema (heralding increased intracranial pressure), otitis media, and altered sensation in the V1 or V2 distribution of the trigeminal nerve (CN V), which occurs in cavernous sinus lesions. The detection of contralateral hemiparesis associated with brisk tendon jerks will point toward brainstem syndromes that involve CN VI. Magnetic resonance imaging (MRI) is an important diagnostic tool and will help revealing intracranial tumors, venous sinus thrombosis, inflammatory causes (e.g., multiple sclerosis) and infections (e.g., tuberculosis). If the MRI ruled out these conditions, and if there is associated papilledema, lumbar puncture becomes a necessity to assess for meningitis and idiopathic intracranial hypertension (pseudotumor cerebri).
Since abducens nerve (CN VI) palsy is amblyogenic in children, patching of the nonparetic eye is used for the prevention and treatment of amblyopia. However, surgical intervention may be needed to improve ocular alignment [3]. As adjunctive to surgical intervention, the antagonist medial rectus muscle is often injected with botulinum toxin. The treatment of Gradenigo syndrome includes antibiotics and mastoidectomy.
3.7 Cranial Nerve VII (CN VII): Facial Nerve
The facial nerve (CN VII) provides motor supply to muscles of facial expression, and sensation to the tympanic membrane, ear canal, and anterior two-thirds of the tongue. It also provides parasympathetic stimulation to the sublingual, submandibular, and lacrimal glands. Originating in the facial nucleus, located at the caudal pontine area, CN VII travels during its intratemporal course through the petrous temporal bone in a bony canal (facial or fallopian canal). It exits the facial canal via the stylomastoid foramen to pass through the parotid gland, where it divides into temporofacial and cervicofacial trunks. In children, facial nerve (CN VII) palsy can be congenital or acquired.
3.7.1 Perinatal Factors and Other Causes of Congenital Facial Palsy
Due to the relatively superficial course of the extracranial facial nerve (CN VII), it can be damaged during birth. This may follow instrumentation in assisted delivery, and intrapartum compression where the fetal head is compressed against the maternal bony prominences such as the ischial spines, the pubic rami, and sacral prominence.
Congenital nontraumatic facial weakness, known as hereditary congenital facial paresis (HCFP), has recently been classified among the congenital cranial dysinnervation disorders (see below) [17]. Hereditary congenital facial paresis 1 (HCFP1) has been mapped to chromosome 3q and is inherited as autosomal dominant (AD), and the locus for the dominantly inherited HCFP2 has been mapped to chromosome 10q. The latter is associated with variable hearing loss. The third type, HCFP3, is caused by mutation in the HOXB1 gene on chromosome 17q21 and is inherited as autosomal recessive. The condition results from dysfunction of the facial nerve (CN VII) leading to facial palsy, unilateral or bilateral (Fig. 3.3a, b). In HCFP1, there is reduced number of neurons in CN VII, whereas in HCFP3 there is bilateral absence of CN VII [18].

(a, b) An adolescent who has bilateral congenital facial paresis. (b) Attempted closure of the eyes shows upward rolling of both eyes (Bell’s phenomenon). (c) A young girl with Goldenhar syndrome, known to be associated with unilateral facial hypoplasia (occasionally associated with facial palsy), showing mandibular hypoplasia and preauricular tags. (d–f) Familial sclerosteosis, a rare sclerosing bone dysplasia characterized by skeletal overgrowth which may lead to facial palsy, secondary to cranial hyperostosis. (d) Early photograph of a young girl who later developed (e) right facial nerve palsy. (f) X-ray of an affected family member showed cortically dense long tubular bones. (d–f, courtesy of Dr. Ahlam A. Hamed, Faculty of Medicine, University of Khartoum, Sudan)
Syndromic causes of facial palsy include Moebius syndrome, one of the congenital cranial dysinnervation disorders (see below), and Poland sequence (unilateral absence of pectoralis major muscle associated with variable degree of ipsilateral hand and digit anomalies) which can manifest in association with Moebius syndrome [19]. Other syndromes have congenital facial palsy (CFP) as part of their symptoms. These include Goldenhar syndrome (Fig. 3.3c), which is characterized by unilateral facial hypoplasia (occasionally associated with facial palsy), epibulbar dermoid, preauricular skin tags, and cervical vertebral defects [20]. Another syndromic cause is sclerosteosis, a rare sclerosing bone dysplasia characterized by skeletal overgrowth which may lead to facial palsy, secondary to cranial hyperostosis (Fig. 3.3d–f) [21]. Other congenital causes are syringobulbia and Arnold-Chiari malformation. Cayler cardiofacial syndrome (OMIM 125520) can be confused with CFP. However, in this condition, there is isolated weakness of the depressor anguli oris muscle, and the lower corner of the mouth on the involved side fails to move downward on crying (Fig. 3.4). On the same side, the lower lip maybe slightly everted. Accompanying anomalies were reported in 70% of patients involving the head and neck, heart, genitourinary tract, and skeleton [22]. One of the genetic disorders that lead to facial nerve weakness is Charcot-Marie-Tooth disease type 4B1 (Fig. 3.5) [23,24,25]. Overgrowth syndromes involving the face can also mimic CN VII palsy, as in CLOVE syndrome (congenital lipomatous overgrowth, vascular malformations, and epidermal nevi, Fig. 3.6a, b) [26]. Another disease which also mimics CN VII palsy is hemifacial atrophy (Parry-Romberg disease, OMIM 141300) [27], characterized by progressive atrophy of the soft tissues of half the face (Fig. 3.6c), and can be associated with trigeminal neuralgia, migraine, and focal epilepsy. Cancrum oris (Noma), an infectious orofacial gangrenous lesion which destroys soft tissue and bone and progresses to perforate the facial skin, manifests with hemifacial edema which may mimic CN VII palsy (Fig. 3.6d). The disease is associated with extreme poverty, predominantly affects malnourished young children, and is usually triggered by serious eruptive fevers (e.g., measles) [28]. It has been reported throughout history in North America, Europe, Asia, South America, and Africa.

Cayler cardiofacial syndrome (OMIM 125520, characterized by isolated weakness of the depressor anguli oris muscle) in a newborn (a, b), an infant (c, d) and an older child (e). The lower corner of the mouth on the involved side fails to move downward on crying. (e, incorporated from Ref. Salih [19] with permission)

13-Year-old boy with Charcot-Marie-Tooth disease, type 4B1 showing (a) bilateral facial weakness with inability to tightly close his eyes. (b) The hands show wasting of the thenar and hypothenar muscles, associated with (c) distal wasting of the lower limbs and bilateral pes cavus

Disorders which mimc facial nerve palsy. (a, b) Infant with CLOVE syndrome (congenital lipomatous overgrowth, vascular malformations, and epidermal nevi) manifesting epidermal nevi and lipomatous overgrowth involving (a) the left face (leading to facial asymmetry) and (b) the right chest. (c) Hemifacial atrophy (Parry-Romberg disease, OMIM 141300) in a young girl leading to facial asymmetry and mouth deviation to the affected side. (d) A young girl with cancrum oris (Noma), an infectious oro-facial gangrenous lesion which destroys soft tissue and bone. The disease progresses to perforate the facial skin starting with hemifacial oedema which may mimic CN VII palsy. (a, b, courtesy of Dr. Ahlam A. Hamed, Faculty of Medicine, University of Khartoum, Sudan)
3.8 Acquired Facial Paralysis
3.8.1 Bell’s Palsy
3.8.1.1 Definition/Classification
Bell’s palsy is one of the most common neurologic disorders affecting the cranial nerves and is characterized by abrupt unilateral peripheral facial paresis or paralysis with no detectable cause.
3.8.1.2 Epidemiology
The annual incidence of Bell’s palsy is about 25 cases per 100,000 persons in the USA, similar to the rest of the world except for Japan, which has the highest incidence. The incidence of Bell’s palsy is 2.7 per 100,000 in the first decade of life and 10.1 per 100,000 in the second decade. The palsy can occur bilaterally at a rate of less than 1%, and about 1.4% of patients have a family history of the disorder [19].
3.8.1.3 Etiology and Pathophysiology
Bell’s palsy is thought to be caused by inflammation and swelling of the facial nerve (VII) resulting in its compression as it passes through the bony canal (the portion of the temporal bone commonly referred to as the facial canal). Nevertheless, the precise pathophysiology is still unclear, although it is assumed that herpes simplex virus (HSV) is the etiologic agent, being reactivated after remaining latent in the geniculate ganglion and causing, thereafter, local damage to the myelin of the facial nerve.
3.8.2 Clinical Manifestations: Symptoms and Signs
3.8.2.1 Symptoms
The condition may manifest with posterior auricular pain, which precedes the paresis by 2–3 days in a quarter of patients. In the majority, it presents with acute onset of unilateral upper and lower facial paralysis over a period of 48 h. This might be associated with decreased tearing, taste disturbances, and hyperacusis due to paralysis of the stapedius muscle.
3.8.2.2 Signs
On the affected side , weakness and/or paralysis due to involvement of the facial nerve (VII) affects the entire upper and lower part of the face. When the child is asked to raise the eyebrows or look upward, without moving the head, the forehead with the palsy will remain flat. On attempted eye closure, Bell’s phenomenon is observed: the eye on the affected side rolls upward and outward (Fig. 3.7a, b). This phenomenon is a normal response to eye closure. When asked to smile, the face lateralizes to the side opposite to the palsy (Fig. 3.7c). Although the disease can affect both sides, bilateral facial palsy should prompt workup for other causes besides Bell’s palsy.

(a, b) Attempted closure of the eyes showing upward rolling of the left eye (Bell’s phenomenon) in (a) a patient with left facial (VII cranial nerve) palsy, and (b) an adolescent who had traumatic right facial nerve palsy. (c) A child with right facial (VII cranial nerve) palsy. When asked to smile, the face lateralized to the left side, opposite to the palsy
3.8.2.3 Diagnosis
Immediate imaging is not necessary if the history and physical examination lead to a diagnosis of Bell’s palsy. Enhancement of the facial nerve, at or near the geniculate ganglion, may be detected on MRI. When the paralysis progresses over weeks, it is no longer Bell’s palsy, and MRI brain is mandatory to exclude tumors compressing or involving CN VII, such as schwannoma, meningioma, hemangioma, pontine glioma , and rhabdomyosarcoma.
3.8.2.4 Differential Diagnosis
Otitis media and mastoiditis should always be considered as antibiotics and/or surgery may be requested. X-rays and CT of the temporal bone are indicated if the history and examination are suggestive. Hypertension is rarely associated with Bell’s palsy and should systematically be looked for. Herpes zoster of the geniculate ganglion (Ramsay Hunt syndrome) is an uncommon cause of facial palsy in children [29]. Other agents that may cause facial weakness include HIV, poliomyelitis, mumps, chicken pox, and Epstein-Barr viruses.
Bacterial causes of facial palsy include Lyme disease (neuroborreliosis), in which it may be an early sign, bacterial meningitis, tuberculosis, brucellosis, and diphtheria [16, 30]. Facial palsy may be associated with Mycoplasma pneumoniae infections, sometimes in the absence of respiratory symptoms. Traumatic paralysis of the facial nerve is revealed by history (Fig. 3.7b), and CT scan of the temporal bone may be required. Other causes include Guillain-Barré syndrome (bilateral facial palsy), Henoch-Schönlein syndrome, Kawasaki disease, sarcoidosis, leukemia, temporal bone histiocytosis, and tumors of parotid gland , the brainstem, or meninges.
3.8.2.5 Investigations
Laboratory studies should include complete blood count (CBC), erythrocyte sedimentation rate (ESR) measurement, HIV screening, and serological tests for Lyme disease (IgG and IgM titers) and brucellosis (ELISA) in endemic areas. Serum titers for Mycoplasma pneumoniae (IgM) and for HSV may be obtained. Assay of IgM and IgG antibody titers against herpes varicella-zoster is recommended in suspected cases of Ramsay Hunt syndrome. Nerve conduction studies and electromyography (EMG) are useful in severe Bell’s palsy. They are most informative when performed 3–10 days after the onset of paralysis. Comparison to the contralateral (unaffected) side has prognostic implications and helps to determine the extent of nerve injury. Neuroimaging studies are required if there are other neurological manifestations and when suspecting temporal bone fracture, acute mastoiditis, or chronic otitis media [20].
3.8.2.6 Treatment
Impaired eye closure and abnormal tear flow require tear substitutes, lubricants, and eye protection with eye glasses or patches. Significant improvement in outcome was shown in randomized controlled trials, when prednisone was started within 72 h of symptom onset. The recommended pediatric dose is 1 mg/kg/day up to 60 mg/day for 5 days, then tapered over 5 days, for a total of 10 days. Despite evidence to support HSV as a major cause of Bell’s palsy, a recent trial showed no added benefit with the addition of acyclovir to prednisolone. Conversely, children with Ramsay Hunt syndrome should be treated promptly with intravenous steroid in combination with antivirals. The recommended antiviral therapy for children older than 2 years is acyclovir (80 mg/kg per day divided q6 hourly for 5 days) [20].
3.8.2.7 Prognosis
The majority of patients (85%) with Bell’s palsy will achieve complete recovery, 10% have some persistent facial muscle asymmetry, and 5% have severe cosmetic sequelae. Patients showing complete paralysis during the acute phase are at a higher risk for severe sequelae. In comparison, Ramsay Hunt syndrome has a worse prognosis. Bell’s palsy recurs in 10–15% of patients, and recurrences are usually associated with a family history of recurrent Bell’s palsy.
3.8.2.8 Management and Prognosis of Other Causes of Facial Paralysis
Congenital paralysis of CN VII and facial paralysis following perinatal trauma usually have a good prognosis even without treatment. Direct neurorrhaphy and nerve graft have been applied in cases of traumatic neural damage of the facial nerve, and these should be performed within 72 h from the trauma onset [20].
3.8.3 Congenital Cranial Dysinnervation Disorders
Congenital cranial dysinnervation disorders (CCDDs) are a group of neuromuscular diseases characterized by motor unit abnormalities involving ocular motility, eyelid, and/or facial muscles. These disorders result from developmental errors of cranial nerve (CN) innervations [18, 19]. The group includes Duane syndrome, congenital fibrosis of the extraocular muscles (CFEOM), congenital ptosis, horizontal gaze palsy with progressive scoliosis (HGPPS), Bosley-Salih-Alorainy syndrome (BSAS), congenital facial palsy (CFP) , and Moebius syndrome.
3.8.3.1 Duane Syndrome
Duane syndrome (DS) is characterized by congenital limitation of horizontal eye globe movement and some globe retraction on attempted adduction of the eye. It constitutes the most common of the CCDDs with prevalence of 1:10,000 (1–4% of strabismus cases), and 10% of cases are familial (usually autosomal dominant) [19, 31, 32].
The condition results from reduction or absence of the abducens nerve (CN VI) motor neurons associated with aberrant innervations of the lateral rectus by the oculomotor nerve (CN III). In type 1 DS (which constitutes about 80% of cases), abduction is affected with normal or minimally defective adduction, associated with narrowing of the palpebral fissure of the adducting eye. Both abduction and adduction are limited in type 3 DS, whereas in type 2 DS, adduction is limited (Fig. 3.8). Type 1 DS maps to chromosome 8q13, and type 2 DS is caused by mutation in the CHN1 gene on chromosome 2q31, whereas mutation in the MAFB gene (on chromosome 20q12) is causative for type 3 DS.

(a, b) Unilateral Duane syndrome including abduction defect on attempted right gaze, no restriction of adduction in both eyes, and mild narrowing of the palpebral fissure of the left adducting eye. (c, d) Bilateral Duane syndrome in an adolescent who also had Duchenne muscular dystrophy. (c) On attempted left gaze, he had no abduction of the left eye with down shoot of the right eye and retraction of the globe. (d) On attempted right gaze, he had no abduction of the right eye with up shoot of the left eye and retraction of the globe
Duane-radial ray syndrome (also known as Okihiro syndrome, OMIM # 607323) is an autosomal dominant disorder characterized by unilateral or bilateral radial dysplasia, ocular anomalies which usually include Duane anomaly, and in some patients, renal anomalies and sensorineural deafness. The condition is caused by heterozygous mutation in the SALL4 gene on chromosome 20q13 [31, 32]. Other conditions known to be associated with DS include Bosley-Salih-Alorainy syndrome due to HOXA1 gene mutation (OMIM # 601536) [33, 34] and Duchenne muscular dystrophy [35].
3.8.4 Congenital Fibrosis of the Extraocular Muscles
Various forms of congenital fibrosis of the extraocular muscles (CFEOM) result from primary dysinnervation of oculomotor (CN III) and trochlear (CN IV) innervated extraocular muscles. Individuals with CFEOM1 have congenital nonprogressive bilateral external ophthalmoplegia, congenital bilateral ptosis, inability to raise either eye above the horizontal midline, and an infraducted primary position of each eye. The condition is inherited as autosomal dominant and, in most families, results from heterozygous mutations in KIF21A gene leading to axonal stalling and aberrant innervation [18, 31].
Individuals with CFEOM2 are born with bilateral ptosis, with their eyes primarily fixed in an exotropic position and severely limited horizontal and vertical eye movements. Patients also have poorly reactive pupils. The condition results from primary developmental defect of both oculomotor (CN III) and trochlear (CN IV) nuclei, and the only normally functioning extraocular muscle is the abducens (CN VI) innervated lateral rectus that pulls each eye outward. It is inherited as autosomal recessive and results from PHOX2A gene mutations.
In CFEOM3 , at least one affected family member does not meet CFEOM1 criteria, and the phenotype shows variable clinical features. Ptosis may be mild or unilateral, and there may be variable vertical eye movements with the ability of one or both eyes to elevate above the midline [18, 31]. The condition results mainly from a variable defect of the oculomotor (CN III) nucleus development. CFEOM3A, characterized by small orbital nerves (CN II, CN III, CN VI) is caused by mutation in the TUBB3 gene, and CFEOM3B by mutation in the KIF21A gene, whereas CFEOM3C maps to chromosome 13q.
3.8.5 Horizontal Gaze Palsy with Progressive Scoliosis
Individuals affected with horizontal gaze palsy with progressive scoliosis (HGPPS) are born with absent horizontal gaze movements and develop severe progressive scoliosis, starting in infancy or childhood (Fig. 3.9a–c) [36]. In horizontal gaze palsy with progressive scoliosis type 1 (HGPPS1, OMIM # 617542), the nonprogressive horizontal gaze palsy results from hypoplasia of the abducens (CN VI) nucleus with interneuron dysinnervation (medial longitudinal fasciculus and pontine paramedian reticular formation). Unlike other CCDDs, the abducens nerve (CN VI) is present bilaterally, and the extraocular muscles are normal. Inheritance is autosomal recessive, and the condition has been reported in consanguineous pedigrees of several different ethnicities. It results from homozygous or compound heterozygous mutations in ROBO3 gene which encodes a transmembrane receptor required for hindbrain axon midline crossing [37, 38]. Magnetic resonance imaging (MRI) shows normal corpus callosum associated with dysmorphic midbrain and midline cleft in the pons (Fig. 3.10). Electrophysiologic studies and tractography, using MRI diffusion tensor imaging, showed that affected individuals have ipsilateral corticospinal and dorsal column-medial lemniscus tract innervations (Fig. 3.10).

(a, b) Eye movements in a child with horizontal gaze palsy with progressive scoliosis (HGPPS). There is absent abduction of (a) the left eye and (b) the right eye when attempting horizontal eye movements. (c) Early scoliosis in a patient with HGPPS. (d) Bilateral Duane syndrome in an adolescent with Bosley-Salih-Alorainy syndrome (BSAS). There is limitation of horizontal eye globe movement with reduced abduction of both eyes associated with narrowing of the palpebral fissure of the adducting eye. (Courtesy of Prof. Thomas M. Bosley. c and d are incorporated from Ref. Salih [19] with permission)

An adolescent with horizontal gaze palsy with progressive scoliosis revealed by (a) magnetic resonance image (MRI). (b) T2-weighted sagittal MRI image shows pons and medulla oblongata with a reduced volume. (c) T2-weighted axial image of the brainstem demonstrates midline cleft at the level of the pons and tenting of the floor of the fourth ventricle (arrows) with missing facial colliculi. (d) Diffusion tensor imaging shows no crossing over of major fibers at the level of the pons as revealed by the colors which depict the predominant fiber direction. Blue color denotes superior–inferior, green denotes anterior–posterior, and red denotes left–right fiber orientation
Horizontal gaze palsy with progressive scoliosis-2 (HGPPS2) has recently been described (OMIM # 617542) and is caused by homozygous mutation in the DCC gene [39]. Apart from horizontal gaze palsy and progressive scoliosis starting in childhood, the condition is characterized by global developmental delay and intellectual disability. Brain MRI shows corpus callosum agenesis, hypoplasia of the pons and midbrain, and brainstem midline cleft. Diffusion MRI from an affected Saudi girl showed no commissural tracts as well as disorganized white matter tracts [39].
3.8.6 Bosley-Salih-Alorainy (BSAS) and Athabascan Brainstem Dysgenesis Syndromes
Children with Bosley-Salih-Alorainy syndrome (BSAS, OMIM # 601536) have bilateral Duane syndrome (Fig. 3.9d), associated in a subset of them with congenital sensorineural deafness secondary to bilateral absence of the cochlea, semicircular canals and vestibule, malformations of the internal carotid arteries and cardiac outflow tract, mental retardation, and autism in some patients [40, 41]. The phenotype of BSAS overlaps with that of Athabascan brain dysgenesis syndrome (ABDS) , which includes, in addition, central hypoventilation, facial weakness, and vocal cord paralysis. Both syndromes result from mutations in HOXA1 gene [34], a homeobox gene essential to the development of head and neck structures, including hindbrain, ear, and occipital and hyoid bones. Homozygous mutations of HOXA1 have been identified in BSAS consanguineous pedigrees in the Middle East (Saudi Arabia and Turkey) and as a sporadic trait in Native American (Athabascan) children from the American Southwest. In Hoxa1 knockout mice, the abducens nerve (CN VI) is absent. Neuroimaging in BSAS shows the absence of CN VI bilaterally and petrous bone maldevelopment [18].
3.9 Moebius Syndrome
Moebius syndrome (OMIM %157,900) is defined as facial weakness combined with an ocular abduction deficit (Video 3.1). It is a rare sporadic disorder with an estimated prevalence of one case per 50,000 newborns in the USA and four cases per 189,000 newborns in the Netherlands [19]. Necropsy studies in Moebius patients have shown defects ranging from hypoplasia to agenesis of the respective cranial nuclei. Nevertheless, it has not been established yet whether nerve, brainstem, or muscle aplasia is the primary event leading to this phenotype. Cranial nerves (CNs) IX (glossopharyngeal) and X (vagus) may be affected. The hypoglossal nerve (CN XII) is involved in a minority of cases, whereas the oculomotor (CN III) and trochlear (CN IV) can be involved on rare occasions. Moebius syndrome typically occurs sporadically. Nevertheless, familial occurrence and cytogenetic anomalies have rarely been described [42]. The pathogenesis is still unknown, but intrauterine vascular event or drug exposure (misoprostol, cocaine, or thalidomide) during the first trimester have been proposed.
3.9.1 Clinical Manifestations
The condition presents at birth with facial diplegia, incomplete eye closure during sleep, difficulty in sucking, and drooling. Examination reveals masklike immobile facial appearance associated with various gaze palsies in about 80% of patients (Video 3.1). Involvement of the hypoglossal nerve (XII) (in approximately 25% of cases) leads to atrophy and inability to protrude the tongue. Musculoskeletal abnormalities may be present in about a third of patients. These include talipes equinovarus, congenital amputations, arthrogryposis, syndactyly, brachydactyly, and, occasionally (15% of patients), hypoplasia or absence of the pectoralis muscle and breast associated with ipsilateral hand malformation (also called Poland syndrome, OMIM %173,800). Features of autism are known to be associated with some cases of Moebius syndrome [43].
3.9.2 Diagnosis and Differential Diagnosis
Most cases are recognized during infancy , but the diagnosis soon after birth may be difficult because of the rarity of the condition. Moebius syndrome can be confused with facial palsy secondary to birth trauma (especially with the use of forceps in breach deliveries), congenital myotonic dystrophy, congenital myopathies, or congenital muscular dystrophy.
On electromyography (EMG), no features of active denervation will be seen in the facial muscles, which are hypoplastic or aplastic. Conversely, in birth trauma, denervation potentials will be recorded 2–3 weeks (or more) after the facial nuclei or nerves are injured. Cranial computed tomography (CT) may demonstrate bilateral calcifications in the region of the abducens (CN VI) nuclei [44]. Bilateral calcifications of the basal ganglia have also been reported [45]. Brain MRI may show hypoplasia of the brainstem, due to straightened floor of the fourth ventricle depicting absence of the facial colliculus, and exclude other associated cerebral malformations [46].
3.9.3 Treatment
This is generally supportive and symptomatic, depending on the severity of the patient’s deficits, and requires multidisciplinary approach [47]. Attention should be given to check the development of corneal abrasion/ulceration, aspiration pneumonia, dysphagia, and poor nutrition. Physical and occupational therapies are useful for managing associated musculoskeletal problems. Speech therapy is also helpful, as well as psychiatric management when there is an associated autism. Symptomatic and cosmetic surgical care may be required such as tracheostomy, for supporting airway; gastrostomy, for feeding; and correction of foot deformities. Surgery for strabismus is usually delayed because the condition frequently improves with age, and plastic surgery may be required to counteract facial nerve paralysis [47].
3.9.4 Prognosis
Death may occur shortly after birth due to bulbar or respiratory problems. Otherwise, Moebius syndrome is a static neurologic defect with no mortality in its mild form.
3.10 Cranial Nerve VIII (CN VIII): Vestibulocochlear Nerve
The vestibulocochlear nerve (CN VIII) arises from the brainstem slightly posterior to the facial nerve (CN VII). It comprises two nerves, namely, the vestibular nerve, which deals with information related to balance, and the cochlear nerve, which relays information related to hearing. Both of the vestibular and cochlear components exit the brainstem at the pontomedullary junction and enter the internal auditory canal (IAC) within the petrous portion of the temporal bone. In the IAC, the cochlear division courses in the inferior-posterior quadrant, whereas the vestibular division courses in the posterior superior and inferior quadrants. CN VIII then enters the labyrinth which is embedded in the petrous part of the temporal bone.
Sensorineural hearing loss (SNHL) is the major presentation of CN VIII dysfunction and results from abnormality in the cochlea, auditory nerve, neural pathway, or cochlear nerve central connections. In the more developed countries, it constitutes the most common sensory deficit, and two to four children per 1000 have been reported to have SNHL [48]. The limited data from less developed countries indicate a much higher incidence with profound medical, developmental, educational, social, cultural, and economic consequences [49]. Sensorineural hearing loss (SNHL) may be mild, moderate, severe, or profound and can be congenital or acquired. Genetic causes account for more than 50% of congenital cases and are subdivided into syndromic and nonsyndromic forms [50]. Syndromic causes include Waardenburg syndrome (OMIM # 148820), characterized by congenital hearing loss, white forelock, dystopia canthorum (the lateral displacement of the ocular inner canthi), and heterochromic irises. It also includes branchiootorenal syndrome (OMIM # 113650) which is characterized by sensorineural, conductive, or mixed hearing loss, preauricular pits, and branchial, renal, and external ear abnormalities. Alport syndrome (OMIM # 301050) is a genetically heterogeneous disorder of the basement membrane (85% of patients have the X-linked form) resulting in progressive renal failure due to glomerulonephropathy (hematuria and progressive renal impairment), variable sensorineural hearing loss, and variable ocular anomalies (lenticonus, cataract, and retinopathy). Pendred syndrome (OMIM # 274600) is an autosomal recessive (AR) disorder associated with SNHL, developmental abnormalities of the cochlea, and an abnormal perchlorate discharge test or goiter [51]. Another AR disorder associated with SNHL is Usher syndrome which is characterized, as well, by vestibular dysfunction and retinitis pigmentosa (Fig. 3.11a) [48]. Bosley-Salih-Alorainy syndrome (OMIM # 601536) is one of the congenital cranial dysinnervation disorders (see above) which also manifests with SNHL [33, 34, 40]. A small fraction of patients with Wolfram syndrome (also known as DIDMOAD syndrome, OMIM # 222300) present with congenital SNHL, although hearing impairment in this condition is typically progressive and affects mainly the higher frequencies. The disease is characterized by diabetes mellitus, optic atrophy, diabetes insipidus, and deafness (hence the acronym DIDMOAD) [52]. Hearing impairment is universal in mucopolysaccharidosis and may be sensorineural, conductive secondary to otitis media with effusion, or mixed (Fig. 3.11b) [53]. Although patients with microtia (congenital deformity where the pinna is underdeveloped) present with conductive hearing loss, a minority can have SNHL due to associated inner ear anomalies (Fig. 3.11c) [54, 55].

(a) 5-Year-5 months-old girl with congenital sensorineural hearing loss (SNHL) associated with Usher syndrome. Following cochlear implant at the age of 2 years 8 months, she could make two word sentences. (b) 11-Year-old girl with SNHL associated with Sanfilippo syndrome B (mucopolysaccharidosis type IIIB). Note the hearing aid, synophrys and hirsutism. (c) 13-Year-old adolescent with microtia (congenital deformity where the pinna is underdeveloped) following plastic surgery. Although patients with microtia present with conductive hearing loss, yet a minority can have SNHL due to associated inner ear anomalies. (d) A child who had SNHL following kernicterus, wearing hearing aids. (e) As shown in his neonatal chart, his highest total serum bilirubin reached 24.5 mg/dL (419 μmol/L)
A potentially treatable cause of SNHL is Brown-Vialetto-Van-Laere syndrome (OMIM # 211530), a riboflavin transporter deficiency disease caused by homozygous or compound heterozygous mutation in SLC52A3 gene. It is characterized by motor and sensory neuronopathy and cranial neuronopathy (manifesting as optic atrophy, sensorineural deafness, and bulbar palsy) [56]. Oral riboflavin supplementation (10–50 mg/kg/day) is effective (and possibly lifesaving) and should be given as soon as the condition is suspected [56]. Another syndrome that requires to be detected early is Jervell and Lange-Nielsen syndrome (OMIM # 220400), characterized by congenital SNHL, prolongation of the QT interval, ventricular arrhythmias leading to syncopal attacks, and a high risk of sudden death [57].
About 70% of congenital hereditary hearing loss is nonsyndromic, and the inheritance pattern can be autosomal recessive, autosomal dominant, X-linked, or mitochondrial [58]. In many populations, mutations in the gap junction β2 (GJB2) gene, encoding the connexin 26 protein and inherited as autosomal recessive, constitute the predominant cause of congenital severe-to-profound nonsyndromic SNHL [59]. Several other connexin genes (including GJB3) are known to be expressed in the cochlea and are involved in SNHL, and it is noteworthy that deafness-associated variants of GJB2 were reported to be rare in Sudan and Kenya [60, 61].
Acquired SNHL in neonates results from TORCH infections (toxoplasmosis, others [syphilis, varicella-zoster, parvovirus B19], rubella, cytomegalovirus, and herpes simplex viruses) [62, 63], Zika virus [64], prematurity and asphyxiation, hyperbilirubinemia (Fig. 3.11d, e), and mechanical ventilation [48]. Congenital cytomegalovirus (CMV) infection may present with SNHL at birth or throughout the first years of life, and about 50% will have further deterioration or progression of hearing loss during childhood [65]. In infants and children, acquired SNHL is most commonly caused by bacterial meningitis resulting from infections by Hemophilus influenzae, Streptococcus pneumoniae, or Neisseria meningitidis, and for more than a century, meningitis epidemics, caused by serogroup A meningococcus, have regularly recurred across sub-Saharan Africa [4, 5, 66,67,68]. Other infectious pathogens implicated in SNHL in children include Borrelia burgdorferi, Epstein-Barr virus, Lassa virus, measles virus, mumps virus, non-polio enteroviruses, varicella-zoster virus, and Plasmodium falciparum [48]. Craniofacial anomalies with deformation of the pinna and ear canal are also associated with SNHL and ototoxic drugs including aminoglycosides (gentamicin, tobramycin, kanamycin, streptomycin) used for more than 5 days, quinine, platinum-based antineoplastic agents, salicylates, and loop diuretics.
3.11 Diagnosis and Management of Sensorineural Hearing Loss
Screening for hearing loss during the newborn period, adopted by developed countries, has led to the early detection of SNHL as well as to timely intervention [69]. The situation is different in the less developed countries where delay in the diagnosis of deafness leads to speech and language delay and impaired cognitive development with adverse impact on literacy, social skills, academic achievement, and employment opportunities [49, 70, 71].
Newborn screening uses otoacoustic emission testing (OET), which assesses cochlear function and also detects aberrant transmission of sound to and from the cochlea (i.e., conductive hearing loss). Absence of otoacoustic emission is considered diagnostic of cochlear hearing in the presence of relatively normal middle ear function [72]. Another screening test is auditory brainstem response (ABR) [48], which evaluates the auditory pathway from external ear canal to brainstem and detects cochlear origin of hearing loss as well as dysfunction of the auditory nerve. A comprehensive audiologic evaluation should be arranged before the age of 3 months if the newborn fails the screening tests. A third useful physiologic test is impedance audiometry which determines the status of the tympanic membrane and middle ear via tympanometry [48]. Type A tympanogram suggests normal function (Fig. 3.12a), whereas type B tympanogram is found in ear canal occlusion (e.g., with cerumen), otitis media, or perforated tympanic membranes. Auditory steady-state response (ASSR) is another electrophysiological test which uses continuous tonal stimulus for evaluation of hearing ability in young infants. It can estimate the hearing sensitivity in many of those who show no response to ABR testing which uses click stimuli [48].

Audiologic testing of a 5-year-old boy with sensorineural hearing loss (SNHL). (a) Type A tympanogram suggesting normal middle year function in both ears. (b) Pure tone audiogram revealing a high-frequency sloping SNHL in both ears
For infants and young toddlers , subjective behavioral tests of hearing acuity are used. These assess how a child processes auditory information. Visual reinforcement audiometry is used in children aged 6 months to 2.5 years of age, whereas conditioned play audiometry is used for children aged 2.5 to 5 years [48]. Most children can cooperate with pure-tone audiometry (which establishes conduction thresholds in air, bone, or both) after the age of 5 years. It assesses both the peripheral and central auditory systems, with the audiogram showing significant air-bone gaps in conductive hearing loss and high-frequency sloping in SNHL (Fig. 3.12b).
In the less developed countries where universal newborn hearing screening is not applied, most children with SNHL are identified either from parental concerns, known risk factors for hearing loss, or when there is delay in speech and language development. Evaluation of these children should include detailed prenatal, birth, and postnatal history for the affected child. It should also enquire about the vaccination status of the mother, family history of hearing loss, craniofacial anomalies, and consanguinity since the majority of genetic hearing loss is inherited as autosomal recessive [73]. Features of syndromes known to be associated with SNHL should also be ascertained, including renal disorders, progressive visual loss, thyroid disease, and family history of sudden death at a young age. Physical examination should assess the ears and look for dysmorphic features, followed by ophthalmologic evaluation. Laboratory investigations should include complete blood count and differential; creatinine, urea, and electrolytes; thyroid studies (T4 and TSH); serologic tests for syphilis and toxoplasmosis; and urinalysis. If hematuria is detected, renal ultrasound will be required. Electroretinography may be needed to detect coexisting retinitis pigmentosa, and electrocardiography is required to exclude prolongation of the QT interval [74]. Congenital cytomegalovirus, the most common congenital infection worldwide, can be tested by taking a cheek swab from the newborn for culture or real-time PCR assay on saliva swabs [65].
After characterization by determining the type of hearing loss (conductive, sensorineural, or mixed) and the degree of loss (mild, moderate, severe, profound, or anacusic), temporal bone imaging studies will be required. Whereas computed tomography (CT) scan is the modality of choice in identifying abnormalities in the bony ear structures (cochlea, ossicles, and semicircular canals), MRI scanning (Fig. 3.13) can better delineate soft tissue abnormalities (e.g., a mass lesion or cochlear nerve hypoplasia) [75].

(a, b) Axial T2-weighted magnetic resonance image (MRI) sequences for temporal bone of a child with congenital sensorineural hearing loss (SNHL) who received cochlear implant. (a) Shows normal bilateral cochlea (red arrows) and normal bilateral vestibulocochlear nerves (yellow arrows). (b) Shows bilaterally dilated vestibular aqueducts (red arrows). (c) 5-Year-old girl with congenital SNHL who has been managed by auditory brain stem implant, resulting in good sound detection and the development of two word sentences. (d) Her axial T2-weighted MRI sequence for temporal bone revealed small cystic cavity replacing bilateral inner ear structures (red arrows), sever stenosis of right internal auditory canal and absent left, with bilaterally absent cochlear nerves (yellow arrows). (a, b, d, courtesy of Dr. Hamdy H. Hassan, FRCR)
Comprehensive clinical genetic testing is important since it allows evidence-based genetic counseling, provides prognostic information on the possibility of SNHL progression, and guides treatment decisions and cost-effective patient care [59]. Since about 50% of childhood SNHL is due to genetic factors which are heterogeneous, with the majority (75–80%) exhibiting autosomal recessive inheritance, it might be more efficient and cost-effective to apply exome sequencing for exploring a genetic cause, rather than using diagnostic panels with different types and numbers of genes [58].
Management for childhood hearing loss is aimed at correcting disorders that are amenable to treatment. These include congenital syphilis, congenital toxoplasmosis, and congenital CMV infection [76]. The additive use of dexamethasone in Haemophilus influenzae type B meningitis and Streptococcus pneumoniae meningitis, in children aged 2 months and older, showed a significant reduction in the frequency of SNHL [48].
Following confirmation of a hearing loss, the child should be enrolled before the age of 6 months in early intervention, which includes amplification (e.g., hearing aids) and enrollment into programs that facilitate the development of speech [77]. Depending on the type and degree of hearing loss and available facilities, other management options include sign language education, cochlear implantation (which requires the presence of normal cochlea and auditory nerve to have successful outcomes, Fig. 3.13a, b), and auditory brainstem implantation (surgical implantation of an electrode on the cochlear nuclei in brainstem). Auditory brainstem implantation (Fig. 3.13c, d) is the treatment of choice when there is cochlear nerve aplasia or complete labyrinthine aplasia (Michel aplasia) [78].
3.12 Prevention of Sensorineural Hearing Loss
Prevention of SNHL includes vaccination against the causative viral and bacterial diseases (measles, mumps, rubella, and meningitis) via the national immunization programs to ensure widespread coverage [71, 79]. The importance of immunogenic meningococcal conjugate vaccines, which has been highlighted earlier [4], has recently been recognized, and they are now incorporated in national vaccination programs of many countries. Early identification of hearing loss is of vital importance and can be implemented via hearing screening programs for newborns, infants, and young children [71]. An important cornerstone in prevention is to improve maternal and neonatal care and strengthen programs to prevent consequences of congenital CMV infection, preterm deliveries, and low birthweight, birth asphyxia, and neonatal jaundice [71]. Other preventive measures include genetic counseling, strict supervision of ototoxic drug use in pregnant women and children, and noise control (e.g., machinery in neonatal intensive care units , personal audio systems, and fireworks) [71, 79].
In each country, formation of support groups for people with hearing loss and their families should be encouraged, and appropriate technologies (e.g., hearing aid and cochlear implant) should be made accessible; sustainable communication and education of children with hearing loss need to be provided through all available means, and as a prerequisite to implementation, education of individuals, communities, and governments is of vital importance [71, 79].
3.13 The Lower Cranial Nerves
The lower cranial nerves (LCN) include the 9th (glossopharyngeal, CN IX), 10th (vagal, CN X), 11th (accessory, CN XI), and 12th (hypoglossal, CN XII) cranial nerves. They will be highlighted together since isolated lesion of a single nerve is rare and because of their simultaneous involvement in the majority of the cases [80,81,82].
3.14 Cranial Nerve IX (CN IX): Glossopharyngeal Nerve
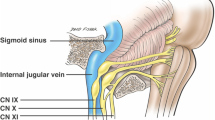
The glossopharyngeal nerve is the ninth cranial nerve (CN IX) , with sensory, motor, and autonomic components. Rostral to the vagus nerve (CN X), it exits the brainstem from the sides of the upper medulla to leave the skull through the pars nervosa of the jugular foramen, passing thereafter between the internal jugular vein and internal carotid artery. Several branches arise at the inferior margin of the stylopharyngeus to provide its motor innervations. It also provides sensation to the carotid sinus and body through the carotid sinus nerve. Passing between the superior and middle pharyngeal constrictors, CN IX enters the pharynx and terminates by dividing into the lingual, tonsillar, and pharyngeal branches. The lingual branch provides general and taste sensation to the posterior one-third of the tongue. The tonsillar branch innervates the palatine tonsils, having formed a network of nerves (the tonsillar plexus), whereas the pharyngeal branch innervates the mucosa of the oropharynx after combining with fibers of CN X to form the pharyngeal plexus. The glossopharyngeal nerve (CN IX) also provides parasympathetic innervation to the parotid gland via fibers which originated in the inferior salivatory nucleus of CN IX.
Glossopharyngeal nerve (CN IX) lesions result in loss of taste sensation in the posterior one-third of the tongue and loss of pain and touch sensations in the same area, pharyngeal walls, and soft palate. Other functions of the CN IX nerve are tested with those of CN X since both nerves travel together.
3.15 Cranial Nerve X (CN X): Vagus Nerve
The vagus nerve (CN X) originates from the medulla and exits the cranium via the jugular foramen accompanied by the glossopharyngeal (CN IX) and accessory (CN XI) nerves and then passes into the carotid sheath down to the neck, chest, and abdomen. The auricular branch of CN X arises in the cranium and provides sensation to the posterior part of the external auditory and canal external ear. Arising in the neck are the pharyngeal branches, which provide motor innervation to the majority of the muscles of the pharynx and soft palate, and the superior laryngeal nerve. The superior laryngeal nerve further splits into internal and external branches, which, respectively, innervate the cricothyroid muscle of the larynx, and provides sensory innervation to the laryngopharynx and superior part of the larynx. Another CN X branch is the recurrent laryngeal nerve (right side only) which innervates the majority of the intrinsic muscles of the larynx.
In the chest, branches from the posterior vagal trunk (formed by the right CN X) and the anterior vagal trunk (formed by the left CN X) innervate the smooth muscle of the esophagus via the esophageal plexus. Other branches are the left recurrent laryngeal nerve which ascends to supply the majority of the intrinsic muscles of the larynx and the cardiac branches which innervate the heart. A lesion to one of the recurrent laryngeal nerves (RLNs) causes dysphonia, whereas aphonia (loss of voice) and a stridor result from paralysis of both RLNs. Entering the abdomen via the esophageal hiatus, the vagal trunks divide into branches that terminate in the esophagus, stomach,, and small and large bowels.
Paralysis of the glossopharyngeal (CN IX) and vagus (CN X) nerves manifests with dysphagia (Fig. 3.14) and leads to abnormal pharyngeal gag reflex, which is elicited by touching the posterior wall of the pharynx, tonsillar area, or base of the tongue, leading to constriction and elevation of the pharyngeal muscles. It also leads to abnormal palatal reflex, elicited by stimulating the soft palate and observing for elevation of the soft palate and ipsilateral deviation of the uvula. There will, as well, be failure of palate elevation upon phonation associated with nasal speech and also nasal regurgitation (especially of liquids) when swallowing. Clinical assessment of CN IX and CN X nerves should start by asking the patient to stick out the tongue and say “Ahh,” and the palate will rise symmetrically when both nerves are intact (Fig. 3.14a). A tongue depressor can be used if the palate could not be seen.

(a) Due to this patient’s right hemi-palate paralysis, the palate deviates to the left side. (b) Multiple cranial nerve palsy in an adolescent with brainstem lesion. There is left abducens (CN VI) and left facial nerve (CN VII) palsies, associated with dysphagia requiring nasogastric tube feeding
3.16 Cranial Nerve XI (CN XI): Accessory Nerve
The accessory nerve (CN XI) is anatomically composed of a spinal and a cranial portion. The spinal portion arises from C1–C5/C6 spinal nerve roots, courses superiorly to enter the cranial cavity through the foramen magnum, and reaches the jugular foramen after traversing the posterior cranial fossa. Briefly meeting the cranial portion of the accessory nerve, it exits the skull through the jugular foramen, accompanied by the glossopharyngeal (CN IX) and vagus (CN X) nerves. The spinal part reaches the sternocleidomastoid muscle, which it innervates, having descended along the internal carotid artery, and then supplies the trapezius muscle having moved across the posterior triangle of the neck.
The cranial portion of the accessory nerve (CN XI) arises from the lateral aspect of the medulla oblongata and briefly meets the spinal part of CN XI before leaving the cranium via the jugular foramen. Combining with the vagus nerve (CN X) at the inferior ganglion of vagus nerve (immediately after emerging from the skull), its fibers then get distributed through CN X.
When acting unilaterally, the action of the sternocleidomastoid is lateral flexion and rotation of the neck, and when acting bilaterally, it extends the neck at the atlanto-occipital joints. Weakness of sternocleidomastoid is revealed by asking the patient to turn the head to one side and then to the other or to anteflect the head against resistance. For testing of the trapezius muscle, the patient is asked to shrug the shoulders with and without resistance. When the trapezius is weak, shrugging of the shoulder is impaired ipsilaterally.
3.17 Cranial Nerve XII (CN XII): Hypoglossal Nerve
The hypoglossal nerve (CN XII) arises from the hypoglossal nucleus in the medulla oblongata and passes laterally, within the subarachnoid space, across the posterior cranial fossa to exit the cranium via the hypoglossal canal. It is responsible for motor innervation of all the extrinsic and intrinsic muscles of the tongue, except for the palatoglossus, and controls the tongue movements required for speech and swallowing. Asking the patient to protrude the tongue will reveal deviation of the tongue toward the damaged side of the hypoglossal nerve (CN XI), as well as possible muscle wasting and fasciculations. Bilateral CN XI lesions cause inability to protrude the tongue associated with dysphagia. Fasciculation of the tongue is an important diagnostic sign of spinal muscular atrophy (SMA) types 1 and 2, and it results from degeneration of the cranial nerve motor nuclei.
3.18 Causes of Lower Cranial Nerve Lesions
In children, causes of lower cranial nerve lesions can be genetic or result from infectious, traumatic, vascular, or neoplastic conditions.
Genetic causes include Brown-Vialetto-Van-Laere syndrome (OMIM # 211530, see above), a riboflavin transporter deficiency disease characterized by bulbar palsy in addition to respiratory compromise, optic atrophy, and SNHL. The condition is potentially treatable by substitution of riboflavin. The autosomal recessively inherited spinal muscular atrophy types 1 (SMA1) and 2 (SMA2), characterized by degeneration of the anterior horn cells leading to death secondary to respiratory failure, present with sucking and swallowing difficulties and fasciculation of the tongue due to involvement of the hypoglossal nucleus. The disease is caused by mutation or deletion in SMN1 gene [83, 84]. Treatment with nusinersen, a modified antisense oligonucleotide, leads to a 50% reduction in deaths or early ventilation in children with SMA1 [85]. Also gene therapy clinical trial, using adeno-associated virus (AAV) vectors encoding SMN protein, revealed positive achievements in survival of affected children as well as in motor milestones [86, 87]. A third disease is the X-linked spinal and bulbar muscular atrophy (Kennedy disease, OMIM # 313200), characterized by speech and swallowing impairment in addition to slowly progressive limb muscle weakness, fasciculations and atrophy, and gynecomastia.
Guillain-Barré syndrome is known to be associated with multiple cranial nerve neuropathy, manifesting with bilateral facial palsy, neck weakness, dysphagia, and dysarthria [88]. Multiple cranial nerve palsies are also known to occur in several types of Charcot-Marie-Tooth disease (CMT) [88, 89]. Vocal cord paralysis has been reported in the X-linked CMTX1 due to GJB1 gene mutations and in congenital hypomyelination neuropathy [81]. The axonal form of CMT4A due to pathogenic variants in GDAP1 is also linked to vocal cord paralysis and diaphragmatic dysfunction [90]. Another recessively inherited peripheral neuropathy is CMT4B1 due to MTMR2 gene mutation in which distal and proximal limb involvements are associated with facial, bulbar, and diaphragmatic weakness [23,24,25]. Hereditary motor and sensory neuropathy Lom (CMT4D) due to pathogenic variants in NDRG1 gene is associated with sensorineural hearing loss and tongue atrophy.
Infectious causes of lower cranial nerves lesions, which are preventable by routine immunizations, include diphtheria and poliomyelitis. Diphtheria can be complicated by systemic radiculoneuropathy and present with palatal and pharyngeal paralysis, tongue weakness, and weakness of the neck muscles, in addition to paralyzed accommodation and abducens nerve (CN VI) and facial nerve (CN VII) palsies [16]. Another disease is bulbar poliomyelitis in which any cranially innervated muscles may be affected [84]. A third preventable disease is tetanus, caused by the endotoxins of Clostridium tetani. Cephalic tetanus presents with trismus in association with paralysis of one or more cranial nerves (usually the facial nerve), accounts for 1–3% of reported cases of tetanus, and has poor prognosis if progressed to generalized tetanus [91].
Herpes zoster of the geniculate ganglion (Ramsay Hunt syndrome), an uncommon cause of facial palsy in children [29], may occasionally involve the vagus nerve (CN X) and manifest with transient dysphagia [81]. Infantile botulism may present with constipation, acute floppiness, ptosis, and bulbar paralysis. The condition is more prevalent in communities adopting the practice of administering honey as a prelacteal feed to newborn babies [92, 93]. Honey is a known dietary reservoir of Clostridium botulinum spores, and fecal excretion of both the organism and its neurotoxin can be detected for weeks to months [92]. Tuberculous meningitis may rarely present with LCN lesions with poor outcome, and craniocervical junction tuberculous spondylitis may present with isolated CN XII palsy [81].
Other causes of LCN palsies include fracture base of the skull, ischemic strokes involving the nuclei or fascicles of any of the four LCN, arterial dissection of the internal carotid or vertebral arteries, and benign or malignant intra and extracranial neoplasms [81].
Miscellaneous conditions involving the LCN include Sandifer syndrome, characterized by reflex torticollis following deglutition in children with gastroesophageal reflux or hiatal hernia, which is considered to be a vagal reflex, with visceral efferents that reach the accessory nerve (CN XI) [81]. Chiari I malformation (>5 mm descent of the caudal tip of cerebellar tonsils past the foramen magnum) is often asymptomatic in childhood. Nevertheless, it may lead to glossopharyngeal neuralgia or compromise the vagal (CN X) or hypoglossal (CN XII) nerves [81].
References
Dalton P, Mennella JA, Cowart BJ, Maute C, Pribitkin EA, Reilly JS. Evaluating the prevalence of olfactory dysfunction in a pediatric population. Ann N Y Acad Sci. 2009;1170:537–42.
Abujbara MA, Hamamy HA, Jarrah NS, Shegem NS, Ajlouni KM. Clinical and inheritance profiles of Kallmann syndrome in Jordan. Reprod Health. 2004;1(1):5.
Rook BS, Phillips PH. Pediatric cranial nerve palsies. J Pediatr Neurol. 2017;15(01):044–52.
Salih MA. Childhood acute bacterial meningitis in the Sudan: an epidemiological, clinical and laboratory study. Scand J Infect Dis Suppl. 1990;66:1–103.
Salih MA, el Hag AI, Sid AH, Bushara M, Yasin I, Omer MI, Hofvander Y, Olcen P. Endemic bacterial meningitis in Sudanese children: aetiology, clinical findings, treatment and short-term outcome. Ann Trop Paediatr. 1990;10(2):203–10.
Ambrosetto P, Nicolini F, Zoli M, Cirillo L, Feraco P, Bacci A. Ophthalmoplegic migraine: from questions to answers. Cephalalgia. 2014;34(11):914–9.
Baisakh MR, Mishra M, Narayanan R, Mohanty R. Cytodiagnosis of sternocleidomastoid tumor of infancy. J Cytol. 2012;29(2):149–51.
Ekici A, Yakut A, Incesulu A, Yimenicioglu S, Kocak O, Carman KB. Trigeminal neuralgia: a rare cause of facial pain in a child. Int J Clin Pediatr. 2013;2(2):74–5.
Childs AM, Meaney JF, Ferrie CD, Holland PC. Neurovascular compression of the trigeminal and glossopharyngeal nerve: three case reports. Arch Dis Child. 2000;82(4):311–5.
Khan M, Nishi SE, Hassan SN, Islam MA, Gan SH. Trigeminal neuralgia, glossopharyngeal neuralgia, and myofascial pain dysfunction syndrome: an update. Pain Res Manag. 2017;2017:7438326.
Misra SR, Maragathavalli G, Baskaran P, Rastogi V. Facial plexiform neurofibroma in a 13-year-old girl with neurofibromatosis-1. J Indian Acad Oral Med Radiol. 2012;24(3):243–9.
Lyons CJ, Godoy F, ALQahtani E. Cranial nerve palsies in childhood. Eye (Lond). 2015;29(2):246–51.
Albakr A, Hamad MH, Alwadei AH, Bashiri FA, Hassan HH, Idris H, et al. Idiopathic intracranial hypertension in children: diagnostic and management approach. Sudan J Paediatr. 2016;16(2):67–76.
Merticariu C, Balta F, Merticariu M, Barac R, Voinea L. Idiopathic intracranial hypertension and associated optic neuropathy in pediatric patients. J Transl Med Res. 2017;22(2):65–70.
Pattnaik L, Sarangi R, Mahapatra S. Opthalmic manifestations of central nervous system tuberculosis—two case reports. Indian J Tuberc. 2011;58:196–8.
Salih MAM, Suliman GI, Hassan HS. Complications of diphtheria seen during the 1978 outbreak in Khartoum. Ann Trop Paediatr. 1981;1:97–101.
Webb BD, Shaaban S, Gaspar H, Cunha LF, Schubert CR, Hao K, Robson CD, Chan W-M, Andrews C, MacKinnon S, Oystreck DT, Hunter DG, Iacovelli AJ, Ye X, Camminady A, Engle EC, Jabs EW. HOXB1 founder mutation in humans recapitulates the phenotype of Hoxb1−/− mice. Am J Hum Genet. 2012;91:171–9.
Bosley TM, Abu-Amero KK, Oystreck DT. Congenital cranial dysinnervation disorders: a concept in evolution. Curr Opin Ophthalmol. 2013;24(5):398–406.
Salih MA. Cranial nerve disorders. In: Textbook of clinical pediatrics. Berlin, Heidelberg: Springer; 2012. p. 3457–62.
Ciorba A, Corazzi V, Conz V, Bianchini C, Aimoni C. Facial nerve paralysis in children. World J Clin Cases. 2015;3(12):973–9.
Appelman-Dijkstra NM, Papapoulos SE. From disease to treatment: from rare skeletal disorders to treatments for osteoporosis. Endocrine. 2016;52(3):414–26.
Lin D-S, Huang F-Y, Lin S-P, Chen M-R, Kao H-A, Hung H-Y, Hsu C-H. Frequency of associated anomalies in congenital hypoplasia of depressor anguli oris muscle: a study of 50 patients. Am J Med Genet. 1997;71:215–8.
Bolino A, Levy ER, Muglia M, Conforti FL, LeGuern E, Salih MA, Georgiou DM, Christodoulou RK, Hausmanowa-Petrusewicz I, Mandich P, Gambardella A. Genetic refinement and physical mapping of the CMT4B gene on chromosome 11q22. Genomics. 2000;63(2):271–8.
Bolino A, Muglia M, Conforti FL, et al. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein 2. Nat Genet. 2000;25:17–9.
Salih MA, Maisonobe T, Kabiraj M, al Rayess M, al-Turaiki MH, Akbar M, Tahan A, Urtizberea JA, Hamadouche T, Guilbot A, Brice A. Autosomal recessive hereditary neuropathy with focally folded myelin sheaths and linked to chromosome 11q23: a distinct and homogeneous entity. Neuromuscul Disord. 2000;10(1):10–5.
Sarici D, Akin MA, Kurtoglu S, Tubas F, Sarici SU. A neonate with CLOVES syndrome. Case Rep Pediatr. 2014;2014:845074. https://doi.org/10.1155/2014/845074.
Deshingkar SA, Barpande SR, Bhavthankar JD, Humbe JG. Progressive hemifacial atrophy (Parry-Romberg Syndrome). Contemp Clin Dent. 2012;3(Suppl1):S78–81.
Enwonwu CO, Falkler WA, Idigbe EO. Oro-facial gangrene (noma/cancrumoris): pathogenetic mechanisms. Crit Rev Oral Biol Med. 1999;11(2):159–71.
Derin S, Derin H, Sahan M, Caksen H. A pediatric case of Ramsay Hunt syndrome. Case Rep Otolaryngol. 2014;2014:469565.
Salih MA, Abdel-Gader AG, Al-Jarallah AA, Kentab AY, Gadelrab MO, Alorainy IA, Hassan HH, Zahraa JN. Infectious and inflammatory disorders of the circulatory system as risk factors for stroke in Saudi children. Saudi Med J. 2006;27:S41–52.
Gutowski NJ, Chilton JK. The congenital cranial dysinnervation disorders. Arch Dis Child. 2015;100(7):678–81.
Oystreck DT, Engle EC, Bosley TM. Recent progress in understanding congenital cranial dysinnervation disorders. J Neuroophthalmol. 2011;31(1):69–77.
Bosley TM, Salih MA, Alorainy IA, et al. Clinical characterization of the HOXA1 syndrome BSAS variant. Neurology. 2007;69:1245–53.
Tischfield MA, Bosley TM, Salih MA, et al. Homozygous HOXA1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nat Genet. 2005;37:1035–7.
Bosley TM, Salih MA, Alkhalidi H, Oystreck DT, El Khashab HY, Kondkar AA, Abu-Amero KK. Duane retraction syndrome in a patient with Duchenne muscular dystrophy. Ophthalmic Genet. 2016;37(3):276–80.
Bosley TM, Salih MA, Jen JC, et al. Neurologic features of horizontal gaze palsy and progressive scoliosis with mutations in ROBO3. Neurology. 2005;59:462–3.
Jen JC, Chan W-M, Bosley TM, Wan J, Carr JR, Rub U, Shattuck D, Salamon G, Kudo LC, Ou J, Lin DDM, Salih MAM, et al. Mutations in a human ROBO gene disrupt hindbrain axon pathway crossing and morphogenesis. Science. 2004;304:1509–13.
Jen J, Coulin CJ, Bosley TM, Salih MAM, Sabatti C, Nelson SF, Baloh RW. Familial horizontal gaze palsy with progressive scoliosis maps to chromosome 11q23-25. Neurology. 2002;59:432–5.
Jamuar SS, Schmitz-Abe K, D’Gama AM, Drottar M, Chan W-M, Peeva M, Servattalab S, Lam A-TN, Delgado MR, Clegg NJ, Al Zayed Z, Dogar MA, et al. Biallelic mutations in human DCC cause developmental split-brain syndrome. Nat Genet. 2017;49:606–12.
Bosley TM, Alorainy IA, Salih MA, Aldhalaan HM, Abu-Amero KK, Oystreck DT, Tischfield MA, Engle EC, Erickson RP. The clinical spectrum of homozygous HOXA1 mutations. Am J Med Genet. 2008;146A:1235–40.
Lintas C, Persico AM. Autistic phenotypes and genetic testing: state-of-the-art for the clinical geneticist. J Med Genet. 2009;46(1):1–8.
Patel RM, Liu D, Gonzaga-Jauregui C, Jhangiani S, Lu JT, Sutton VR, Fernbach SD, Azamian M, White L, Edmond JC, Paysse EA. An exome sequencing study of Moebius syndrome including atypical cases reveals an individual with CFEOM3A and a TUBB3 mutation. Cold Spring Harb Mol Case Stud. 2017;3(2):a000984.
Guillberg C, Steffenburg S. Autistic behavior in Moebius syndrome. Acta Paediatr Scand. 1989;78:314–6.
Kuhn MJ, Clark HB, Morales A, Shekar PC. Group III Möbius syndrome: CT and MR findings. AJNR Am J Neuroradiol. 1990;11(5):903.
Singh B, Shahwan SA, Singh P, al-Deeb SM, Sharif H. Mobius syndrome with basal ganglia calcification. Acta Neurol Scand. 1992;85(6):436–8.
Srinivas MR, Vaishali DM, Vedaraju KS, Nagaraj BR. Mobious syndrome: MR findings. Indian J Radiol Imaging. 2016;26(4):502–5. https://doi.org/10.4103/0971-3026.195790.
Pedersen LK, Maimburg RD, Hertz JM, Gjørup H, Pedersen TK, Møller-Madsen B, Østergaard JR. Moebius sequence—a multidisciplinary clinical approach. Orphanet J Rare Dis. 2017;12:4. https://doi.org/10.1186/s13023-016-0559-z.
Smith RJ, Bale JJ, White KR. Sensorineural hearing loss in children. Lancet. 2005;365(9462):879–90.
Abdalla FM, Omar MA. The role of the health system in the prevention of hearing loss among children in Sub-Saharan Africa. Sudan J Paediatr. 2011;11(1):8–19.
Gürtler N, Gysin C, Schmid N, Pieren C, Vischer M, Schumacher S, Oppermann P, Leuba D, Veraguth D. Bilateral congenital deafness: what investigations should be performed? Swiss Med Wkly. 2017;147:w14416.
Salih MAM, Hashem N. Pendred’s syndrome in a Sudanese family. Sudan J Paediatr. 1984;3:86–94.
Salih MAM, Tuvemo T. Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD syndrome): a clinical study in two Sudanese families. Acta Paediat Scand. 1991;80:567–72.
Soni-Jaiswal A, Jones SA, Callery P, Bruce IA. Hearing loss in mucopolysaccharidosis and its impact on quality of life: a review of the literature. Mol Genet Metab. 2016;117(2):S108.
Tsang WS, Tong MC, Ku PK, Bhatia KS, Joannie KY, Wong TK, van Hasselt CA. Contemporary solutions for patients with microtia and congenital aural atresia–Hong Kong experience. J Otol. 2016;11(4):157–64.
Vrabec JT, Lin JW. Inner ear anomalies in congenital aural atresia. Otol Neurotol. 2010;31(9):1421–6.
Manole A, Houlden H. Riboflavin transporter deficiency neuronopathy. 11 June 2015. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993–2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK299312/.
Al Jarallah AS. Prolonged QT syndrome triggered by swimming: a mimicking cause of seizures not to be missed. J Pediatr Neurol. 2005;3(04):257–60.
Likar T, Hasanhodžić M, Teran N, Maver A, Peterlin B, Writzl K. Diagnostic outcomes of exome sequencing in patients with syndromic or non-syndromic hearing loss. PLoS One. 2018;13(1):e0188578.
Sloan-Heggen CM, Bierer AO, Shearer AE, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet. 2016;135:441–50. https://doi.org/10.1007/s00439-016-1648-8.
Gasmelseed NM, Schmidt M, Magzoub MM, et al. Low frequency of deafness-associated GJB2 variants in Kenya and Sudan and novel GJB2 variants. Hum Mutat. 2004;23(2):206–7.
Salih MA, Dirar AI, Ibrahim NO, Osman MH, Sabeel SM, Yusif NA, Yusif AA, Abdelsamad GH, Elagib AA. Mutation in (GJB3 and GJB4) genes involved in deafness in two Sudanese families using next generation sequencing. Int J Comput Bioinfo In Silico Model. 2014;3(3):388–92.
Brown ED, Chau JK, Atashband S, Westerberg BD, Kozak FK. A systematic review of neonatal toxoplasmosis exposure and sensorineural hearing loss. Int J Pediatr Otorhinolaryngol. 2009;73(5):707–11.
Chau J, Atashband S, Chang E, Westerberg BD, Kozak FK. A systematic review of pediatric sensorineural hearing loss in congenital syphilis. Int J Pediatr Otorhinolaryngol. 2009;73(6):787–92.
Mehrjardi MZ. Is Zika virus an emerging TORCH agent? An invited commentary. Virology (Auckl). 2017;8:1178122X17708993. https://doi.org/10.1177/1178122X17708993.
Marsico C, Kimberlin DW. Congenital cytomegalovirus infection: advances and challenges in diagnosis, prevention and treatment. Ital J Pediatr. 2017;43(1):38.
Mohammed I, Iliyasu G, Habib AG. Emergence and control of epidemic meningococcal meningitis in sub-Saharan Africa. Pathog Glob Health. 2017;111(1):1–6.
Salih MAM, Ahmed HS, Karrar ZA, Kamil I, Osman KA, Palmgren H, Hofvander Y, Olcen O. Features of the major epidemic of group A meningococcal meningitis in Khartoum, Sudan in 1988. Scand J Infect Dis. 1990;22:161–70.
Salih MA, Khaleefa OH, Bushara M, Taha ZB, Musa ZA, Kamil I, Hofvander Y, Olcén P. Long term sequelae of childhood acute bacterial meningitis in a developing country. A study from the Sudan. Scand J Infect Dis. 1991;23(2):175–82.
Dedhia K, Kitsko D, Sabo D, Chi DH. Children with sensorineural hearing loss after passing the newborn hearing screen. JAMA Otolaryngol Head Neck Surg. 2013;139(2):119–23.
Maharani NL, Haksari EL, ArtanaI W. Risk factors for hearing loss in neonates. Paediatr Indones. 2016;55(6):328.
World Health Organization (WHO). Childhood hearing loss: strategies for prevention and care. Geneva, Switzerland: World Health Organization; 2016. http://apps.who.int/iris/bitstream/10665/204632/1/9789241510325_eng.pdf. Retrieved 4 Feb 2018.
Doyle KJ, Burggraaff B, Fujikawa S, et al. Neonatal hearing screening with otoscopy, auditory brain stem response, and otoacoustic emissions. Otolaryngol Head Neck Surg. 1997;116:597–603.
Brookhouser P. Sensorineural hearing loss in children. Pediatr Clin North Am. 1996;43(6):1195–216.
Mehta D, Noon SE, Schwartz E, Wilkens A, Bedoukian EC, Scarano I, Crenshaw EB 3rd, Krantz ID. Outcomes of evaluation and testing of 660 individuals with hearing loss in a pediatric genetics of hearing loss clinic. Am J Med Genet A. 2016;170(10):2523–30.
McClay JE, Booth TN, Parry DA, Johnson R, Roland P. Evaluation of pediatric sensorineural hearing loss with magnetic resonance imaging. Arch Otolaryngol Head Neck Surg. 2008;134(9):945–52.
del Rosal R, Baquero-Artigao F, Blazquez D, et al. Treatment of symptomatic congenital cytomegalovirus infection beyond the neonatal period. J Clin Virol. 2012;55(1):72–4.
Benoit MM. Hearing loss in infants and children. 2017. http://www.medlink.com/article/hearing_loss_in_infants_and_children. Accessed 6 Feb 2018.
Al-Momani MO. Five years audiological outcomes of the first Saudi Auditory Brainstem Implant (ABI). Saudi J Otorhinolaryngol Head Neck Surg. 2017;19(1):32–4.
Alberti PW. The prevention of hearing loss worldwide. Scand Audiol Suppl. 1996;42:15–9.
Erman AB, Kejner AE, Hogikyan ND, Feldman EL. Disorders of cranial nerves IX and X. Semin Neurol. 2009;29(1):85–92. https://doi.org/10.1055/s-0028-1124027.
Finsterer J, Grisold W. Disorders of the lower cranial nerves. J Neurosci Rural Pract. 2015;6(3):377–91.
Walker HK. Chapter 63: Cranial nerves IX and X: the glossopharyngeal and vagus nerves. In: Walker HK, Hall WD, Hurst JW, editors. Clinical methods: the history, physical, and laboratory examinations. 3rd ed. Boston: Butterworths; 1990. Available from: https://www.ncbi.nlm.nih.gov/books/NBK386/.
Al-Jumah M, Majumdar R, Al-Rajeh S, Awada A, Chaves-Carbello E, Salih M, Al-Shahwan S, Al-Subiey K, Al-Uthaim S. Molecular analysis of the spinal muscular atrophy and neuronal apoptosis inhibitory protein genes in Saudi patients with spinal muscular atrophy. Saudi Med J. 2003;24(10):1052–4.
Salih DM. Anterior horn cell diseases. In: Textbook of clinical pediatrics. Berlin, Heidelberg: Springer; 2012. p. 3463–9.
Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377:1723–32.
Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377:1713–22.
Parente V, Corti S. Advances in spinal muscular atrophy therapeutics. Ther Adv Neurol Disord. 2018;11:1756285618754501.
Salih MA. Peripheral nerve disorders. In: Textbook of clinical pediatrics. Berlin, Heidelberg: Springer; 2012. p. 3475–91.
Bird TD. Charcot-Marie-Tooth neuropathy type 4. 24 Sept 1998 [updated 14 Apr 2016]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993–2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1468/.
Sevilla T, Jaijo T, Nauffal D, Collado D, Chumillas MJ, Vilchez JJ, Muelas N, Bataller L, Domenech R, Espinós C, Palau F. Vocal cord paresis and diaphragmatic dysfunction are severe and frequent symptoms of GDAP1-associated neuropathy. Brain. 2008;131:3051–61.
Alhaji MA, Abdulhafiz U, Atuanya CI, Bukar FL. Cephalic tetanus: a case report. Case Rep Infect Dis. 2011;2011:780209.
Abdulla CO, Ayubi A, Zulfiquer F, Santhanam G, Ahmed MAS, Deeb J. Infant botulism following honey ingestion. BMJ Case Rep. 2012;2012:bcr1120115153. https://doi.org/10.1136/bcr.11.2011.5153.
Num SM, Useh NM. Botulism in man and animals. Bulg J Vet Med. 2014;17(4):241–66.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
3.1 Electronic Supplementary Material
An 8-month-old girl with Moebius syndrome showing masklike immobile facial appearance, associated with bilateral horizontal gaze palsy. Note that vertical ocular motility is preserved with full vertical eye movements (AVI 5039 kb)
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Salih, M.A.M. (2020). Cranial Nerve Disorders. In: Salih, M.A. (eds) Clinical Child Neurology. Springer, Cham. https://doi.org/10.1007/978-3-319-43153-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-43153-6_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-43152-9
Online ISBN: 978-3-319-43153-6
eBook Packages: MedicineMedicine (R0)