
Abstract
Poor bioavailability associated with poorly water-soluble compounds remains a challenging issue in drug development. Particle engineering may be used to improve the physicochemical properties of poorly water-soluble compounds, thereby enhancing the bioavailability. Cryogenic technologies, including spray freeze drying (SFD), spray freezing into liquid (SFL), and thin film freezing (TFF), are “bottom-up” precipitation processes to generate amorphous nanostructured aggregates with significantly enlarged surface area, higher dissolution rates, and supersaturation, via rapidly inducing nucleation followed by particle growth arrest through stabilization via polymers and solidification of the solvent. This chapter provides detailed description of each cryogenic process, formulation guidelines, and characterization analyses. Finally, examples of cryogenically engineered drug compositions with improved in vitro and in vivo macroscopic performance are provided to illustrate the potential benefits of cryogenic technologies, especially TFF. The current authors would like to thank and acknowledge the significant contribution of the previous authors of this chapter from the first edition. This current second edition chapter is a revision and update of the original authors’ work.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Particle engineering
- Spray freeze drying (SFD)
- Spray freezing into liquid (SFL)
- Thin film freezing (TFF)
- Amorphous nanostructures
- Dissolution rates
- Supersaturation
- Nucleation
- Particle growth arrest
- Polymers
- Solvents
- Dry powder inhalation
11.1 Introduction
11.1.1 Therapeutic Shortfalls of Poorly Water-Soluble Drugs
In modern drug discovery processes , routine use of high-throughput screening, combinatorial chemistry, and computer-aided drug design appear to result in a higher prevalence of lead compounds of increased molecular weight and lipophilicity, despite the high efficiency of the automated processes. About 60 % of these drug candidates exhibit poor aqueous and nonaqueous solubility (Gao et al. 2008). Among which, for those with high permeability through biomembranes, classified as Biopharmaceutical Classification System (BCS) Class II drugs , the poor dissolution rate limits drug molecules released into biological fluid contacting the absorbing mucosa (Rasenack and Muller 2002). Basically, it is in the form of an aqueous solution that a drug can be absorbed into the systemic circulation and exert its therapeutic effect. Consequently, the poorly water-soluble drugs often result in low and highly variable bioavailability, and sub-optimal therapeutic effects in patients, particularly when delivered via the oral administration (Muller et al. 2001; Patravale et al. 2004).
11.1.2 Solid Dispersion/Solution , Supersaturation
To improve the bioavailability of poorly water-soluble drugs, defined as the rate and extent of the drug that reaches systemic circulation, new technologies and innovative formulations and drug delivery systems were explored to improve dissolution properties of poorly water-soluble drugs throughout the past decade. These include more conventional techniques such as the use of surfactants, cyclodextrin inclusion complexation, emulsification processes, co-solvency, salt formation, powder milling, and spray drying. However, these attempts have been of limited success, and each was found with inherent problems of their efficacy or stability of the final product.
The increased amount of excipients required to formulate the poorly water-soluble drugs may potentially increase side effects, resulting in low patient compliance. Alternatively, invasive dosage forms such as parenteral formulations have to be developed to address the challenges being presented. However, with even less pharmaceutically acceptable excipient options, solubilization of drug is practically limited (Liu 2000).
Salt formation is one of the commonly used means to increase aqueous solubility of poorly water-soluble drugs. Despite the unionized form being much less soluble than its salt, of further interest, therapeutically, it is the unionized form that more readily penetrates biological membranes to exert therapeutic effects (Martin et al. 1993). Salt forms can improve wettability and bioavailability of drugs. For example, albendazole salts exhibited better wettability due to the hydrophilic and ionic nature of their crystals. Microenvironmental pH changes also affect the solubility of the salt forms therefore some salt forms are superior to others. Particle size reduction typically helps increase solubility of the drug. However, in some cases, this is not possible due to the poor wettability (Paulekuhn et al. 2013).
Mechanical milling was generally used to reduce particle size. However, it generates particles with irregular shape and a wide range of size distribution. Moreover, thermo-degradation is another issue associated with milling process. Spray drying is also not an ideal method of choice due to only 50 % dry product recovery (Esclusa-Diaz et al. 1996). Leleux and Williams (2014) recently reviewed mechanical reduction methods regarding particulate systems. There are various bottom-up and top-down techniques utilized to produce crystalline drugs in the micron size range. The pros and cons of both approaches were discussed in terms of their contribution to the pharmaceutical field. However, the top-down methods have demonstrated greater efficiency in the industrial scale. The authors stated that micronization of some extremely low solubility drugs did not have a substantial impact on their solubility. However, size reduction to the nanoscale particle size range was possibly an effective method to enhance solubility of poorly soluble drugs.
To overcome these shortcomings, novel technologies such as hot-melt extrusion, particle engineering by use of supercritical fluid, nanomilling, and solution-based micro-/nanoparticle precipitation (Betageri and Makarla 1995; Mawson et al. 1997; Rogers et al. 2001; Sarkari et al. 2002; Hu et al. 2004b; Matteucci et al. 2007) have been developed to enhance saturation solubility of poorly water-soluble drugs. It may be practical to increase apparent solubility and/or the dissolution rate via alteration of the solid-state form.
Solid dispersion typically refers to systems in which drug particles are homogeneously distributed throughout a solid matrix of excipient(s). This system provides the possibility of reducing the particle size of drugs to nearly a molecular level in order to transform the drug from the crystalline to partially amorphous morphology. A solid solution results when the drug is molecularly dispersed throughout a solid matrix, i.e., complete amorphous morphology (Kapsi and Ayres 2001), in which the particle size of the drug has been reduced to its absolute minimum without any crystalline drug domains (Leuner and Dressman 2000). The amorphous form of a drug has a higher thermodynamic chemical potential than its crystalline counterpart.
Additionally, exposure area of drug to the dissolution media was also greatly enhanced due to significantly increased surface areas of drug compositions obtained in this way. Therefore, poorly water-soluble drugs in the solid solution/dispersion exhibit higher dissolution rates and higher saturation concentration than their intrinsic solubility of crystalline form of drug, i.e., they can produce supersaturated solutions (Betageri and Makarla 1995). Generation of supersaturation provides higher number of free drug molecules in the solution available for absorption, thereby leading to enhanced bioavailability.
11.1.3 Solubility Advantage of Nanoparticles
Production of drug-loaded nanoparticles for the poorly water-soluble drugs is an alternative and promising approach to overcome their low aqueous solubilities and the consequential low bioavailabilities (Muller et al. 2001). Nanoparticles are currently defined as single particles with a diameter less than 100 nm. Agglomerates of nanoparticles can be larger than 100 nm in diameter but may be de-agglomerated either with weak mechanical forces or by dispersing in a solvent.
Although micronization can increase dissolution rate of the poorly water-soluble drugs by reduction of particle size and thereby increased surface area, it does not increase equilibrium solubility. Often, for drugs with very low aqueous solubility, the achieved increase in dissolution rate is limited and insufficient to provide significant enhancement of bioavailability (Muller et al. 2001). However, when the drug particle sizes were deducted in the 100-nm range, they dissolve more quickly and achieve supersaturation versus the micronized drug particles, as described by the Noyes–Whitney and Ostwald–Freundlich equations (Grant and Brittian 1995). Both particle dissolution kinetics and solubility are size dependent. Thus, the dissolution of drug nanoparticles in vivo is usually accompanied by an increase in bioavailability (Hintz and Johnson 1989; Borm et al. 2006).
Furthermore, novel investigation of supersaturation levels of drug nanoparticles in aqueous media demonstrated significantly higher values than the much larger microparticles of drug composition, which have the potential to crystallize during slow dissolution (Matteucci et al. 2007). Amorphous nanostructured formulations of poorly water-soluble drugs have also been developed to enhance therapeutic effectiveness (Yang et al. 2008a).
An emulsion-freeze-drying technique was used to prepare nanoparticles of poorly water-soluble drugs. In situ formation of poorly water-soluble drug nanoparticles within a porous hydrophilic polymer (PVA) scaffold by freeze-drying o/w emulsions has been accomplished. The pore structure maintained the drug nanoparticles and prevented agglomeration. The nanoparticles of the model poorly water-soluble drug loaded in porous polymer rapidly dissolved in water (Grant and Zhang 2011).
He et al. (2013) reported that the dissolution of a model poorly water-soluble drug, indomethacin, was significantly improved due to the resulting nanoparticles prepared by a cryogenic technique. However, preventing nanoparticles from aggregation and agglomeration is also challenging. Nanosuspensions of indomethacin with food proteins as novel stabilizers were prepared by a nanoprecipitation–ultrasonication method following by freeze drying. The nanosuspensions were lyophilized, and the original particle size and particle-size distribution were maintained. The protein stabilizers physically stabilized indomethacin nanosuspensions via a combination of two mechanisms: electrostatic repulsion and steric stabilization.
Published literature reviews have discussed nanoparticle technologies. For example, a review of nanoparticle engineering processes for the enhancement of dissolution rates of poorly water soluble drugs was published by Hu et al. (2004b, c). This overview focused on the commercially viable nanoparticle engineering processes available for increasing the dissolution properties of poorly water-soluble drugs. Cryogenic spray processes and spray freezing into liquid (SFL) were proposed as alternative approaches to prepare nanoparticles of poorly water-soluble drugs. Examples using these techniques were also included. Besides, bottom-up technologies utilized to prepare nanosuspensions of poorly water soluble drugs were recently reviewed by Du et al. (2015). Spray freeze drying and freeze drying were used as the “solidification methods for the nanosuspension prepared by the bottom-up approach”. The article included other solidification techniques, advantages, manufacturing processes, the corresponding characterization methods, drug dosage forms, and limitations of commercial drug products.
11.1.4 Overview of Cryogenic Technologies
Cryogenic particle engineering technologies were developed to improve the solubility and dissolution properties by creating nanostructured amorphous particles with dramatically enlarged surface area at very low temperature conditions, in contrast to micronized crystalline form of poorly water-soluble drugs (Hu et al. 2002, 2004a, b, c; Rogers et al. 2003a; Overhoff et al. 2007a). Cryogenic technologies basically can be categorized into micro-/nanoparticle precipitation technologies or the so-called “bottom-up” particle engineering technologies , with the mechanism of inducing a rapid change in solute solubility to generate solid particles.
Another example of novel bottom-up process using freeze drying to improve the dissolution behavior of poorly water soluble drugs was reported by De Waard et al. (2008). Nanocrystalline particles of fenofibrate produced by the “controlled crystallization during freeze drying” process significantly increased dissolution of the drug compared to tablets containing the physical mixture. The freeze-dried formulation contained fenofibrate, a drug with low Tg, and mannitol as a carrier since it crystallizes. Freezing rate and the ratio of water-to-tertiary butyl alcohol (TBA) affected the nucleation rate that controlled the size of crystals and dissolution performance. The challenge is how to apply the process to high Tg drugs.
There are some recently reported examples of the bottom-up particle engineering based upon cryogenic technologies. For example, Yasmin et al. (2014) successfully utilized lyophilization to prepare silica–lipid hybrid (SLH) microparticles from submicron o/w emulsions stabilized by silica nanoparticles. The authors reported that the performance of microparticles fabricated from this technique were comparable to those produced using spry drying. Hence this technology is another alternative method to manufacture SLH formulations of poorly water-soluble drugs which are thermally labile and also other challenging drugs to process through spray drying.
To the contrary, a comparison of two processing approaches (spray- and freeze-drying) to manufacture solid phospholipid nanoparticles of celecoxib, a BCS Class II drug, was reported. Celecoxib with phospholipid E80 and trehalose was formulated in various drug-to-excipients ratios. Spherically shaped, amorphous nanoparticles (average diameters <1 μm) were produced by spray-drying; while larger particles of the matrix-like structure of solid amorphous phospholipid dispersion were prepared by freeze-drying. Both products significantly improved the dissolution of the drug. The apparent and molecular solubility of the drug from the spray-dried formulation in phosphate buffer (pH 6.5) and in biorelevant fasted state simulated intestinal fluid (pH 6.5) were considerably higher than the freeze-dried powder. This probably resulted from the difference in particle size and surface morphology (Fong et al. 2015).
As well known, solubility is largely determined by the bonding interactions between the solute and solvent on a molecular level; it is also heavily influenced by external factors including temperature, pressure, polarity of the solvent, and pH. Sudden shift of one of these factors can generate strong driving forces to induce nucleation leading to particle formation. Cryogenic technologies utilize cryogens, such as liquid nitrogen, to induce abrupt temperature change of a system containing solubilized poorly water-soluble drug molecules alone or along with excipient molecules. The rapid cooling rates of up to 1.0 × 106 K/s may produce stable amorphous nanostructured particles with significantly enlarged surface areas to facilitate rapid and higher-level dissolution in biological fluids, thereby enhancing bioavailability.
Generally, cryogenic technologies involve the rapid freezing of single solvent or co-solvent-based solution containing drug alone or alone with stabilizer or solubility enhancer. The generated frozen material is then lyophilized to remove the solvent by sublimation like traditional lyophilization and atmospheric freeze drying (ATMFD) (Derle et al. 2010), thus yielding a dry powder of high surface area. Cryogenic processes are defined by the type of injection device (capillary, rotary, pneumatic, and ultrasonic nozzle), location of nozzle (spraying onto or into a cryogenic liquid, or applying the solution onto a cryogenic substrate), and the composition of cryogenic liquid, such as liquid hydrofluoroalkanes, liquid nitrogen, liquid argon, compressed fluid carbon dioxide, and organic solvents.
In this chapter, spray freeze drying (SFD), SFL, and thin film freezing (TFF) cryogenic technologies for pharmaceutical applications will be discussed in detail. Examples of recent studies using the cryogenic technologies with step-by-step procedures to engineer poorly water-soluble drugs for improved in vitro and in vivo performance will be provided and analyzed. It can be readily recognized that applications of these cryogenic technologies may be applied to other freezing processes.
11.1.5 Commonly Used Cryogens
The most commonly used cryogen in cryogenic processes is liquid nitrogen . Liquid nitrogen is a colorless, odorless, and nonflammable liquid that boils at a temperature of approximately −195 °C. Due to its natural abundance in the atmosphere, liquid nitrogen is relatively cheap and is readily available in large quantities throughout the world. Compared to other liquid cryogens, it is relatively safe and is already accepted for use in certain medical applications. The primary disadvantages of liquid nitrogen are that it can pose a safety hazard as an asphyxiant and it also exhibits a behavior known as the Leidenfrost effect, which in some instances may actually result in decreased freezing rates for droplets that come into contact with the liquid nitrogen reservoir but are shielded by a vapor layer for some amount of time before direct contact can be made with the liquid nitrogen.
Liquid argon is the most common alternative to liquid nitrogen for use in SFD. It has many of the same advantages as liquid nitrogen and also suffers from some of the same disadvantages such as being an asphyxiating agent. It boils at nearly the same temperature as liquid nitrogen at −185 °C; however, it is typically more expensive than liquid nitrogen and less widely available.
Another group of alternatives are liquid hydrocarbons such as liquid propane, pentane, and hexane. They have relatively higher boiling point and wider availability than liquid argon. However, these systems are considerably more dangerous to work with due to their extreme combustibility. Additionally, these materials have not been accepted or tested in use with pharmaceutical products and could be a potential source of contamination when brought into direct contact with drug products.
11.2 Cryogenic Technologies
11.2.1 Spray Freeze Drying (SFD)
SFD has been used in pharmaceutical research for over 60 years and is likely the oldest of the cryogenic pharmaceutical processing technologies. One of the first published papers in the literature describing this technique was in 1948, which was used as a means to produce protein powders with varying surface areas for subsequent absorption isothermal analysis (Benson and Ellis 1948). SFD has historically been used as a method to process thermally labile compounds such as proteins and peptides as well as even larger biological molecules in both the pharmaceutical and food industries (Costantino et al. 2000, 2002; Anandharamakrishnan et al. 2010; Ishwarya et al. 2015) because, unlike spray drying, no heat is required to obtain the final powder formulation. In addition to proteins and peptides, SFD has also been applied to different poorly water soluble drugs in order to enhance their solubility and non-pharmaceutical applications such as food processing (Mumenthalera and Leuenberger 1991; Zijlstra et al. 2007; Tong et al. 2011; Niwa et al. 2012, Niwa and Danjo 2013; Wanning et al. 2015)
Spray-freeze drying was applied to fabricate kinetically stable, amorphous solid dispersions. Compared to the commercial tablet, the kinetically stable, amorphous solid dispersions of the BCS Class IV compound, oleanolic acid, with a stabilizer, a wetting agent and a penetration enhancer produced by SFD technique was superior in terms of in vitro dissolution and uniform absorption. Inter-individual variability in oral absorption is common for the BSC Class IV drugs. This SFD-processed formulation not only improved drug absorption but also reduced the large variability of intestinal permeability, which is the critical problem of the compounds in this class (Tong et al. 2011).
A solution of the poorly water soluble drug, cyclosporine, with mannitol was processed through SFD technique to prepare a dry powder inhaler (DPI). Drug was in the amorphous state while mannitol crystallized during the freeze drying process. The hydrophilic property of mannitol promoted dissolution of the drug. Moreover, drug-mannitol co-formulation exhibited better aerosol dispersion since mannitol successfully improved adhesive and cohesive behavior between the drug particles (Niwa et al. 2012).
Niwa and Danjo (2013) also developed a method combining the wet milling and the SFD techniques to generate a novel product for poorly water-soluble drugs. The suspension was prepared by wet milling of drug in an aqueous medium of polymer as a dispersing agent and then sprayed via SFD to yield a dry nanosized powder. The drug in the porous network structure spontaneously released into the nano-scale suspension and rapidly dissolved in both acidic and neutral media. The SFD formulation exhibited a better dipersability and more efficient dispersing into a nanosuspension compared with the product prepared by spray drying (Niwa and Danjo 2013).
The formation of inhalable micronized porous particles containing the poorly water-soluble corticosteroid, budesonide, and mannitol using SFD technique was investigated. The excipients, hydroxypropyl beta-cyclodextrine and/or l-leucine, were used in the formulation. The results demonstrated that both excipients at a suitable ratio influenced the particle shape and morphology as well as improved aerosol performance and dissolution of the particles (Parsian et al. 2014).
Wanning et al. (2015) recently reviewed pharmaceutical SFDs. SFD not only enhanced solubility of poorly water soluble drugs but also exhibited advantages for pulmonary delivery , intradermal ballistic administration and vaccine delivery to the nasal mucosa as well. The authors summarized different studies using SFD technique to prepare both small molecules and biological products for various routes of administration.
11.2.1.1 Process of SFD
SFD is generally described as a three-step process involving the atomization of a drug feed solution or suspension, freezing the atomized droplets, and removal of solvent from the frozen material to obtain a final, typically amorphous, dry powder composition. The equipment and experimental set-up used in SFD are fairly straightforward; however, there are a few choices to consider at each of the three steps of the process.
In the first step of SFD, solutions or suspensions of drug alone or along with excipient(s) are prepared and atomized into small droplets using specialized fluid nozzles or vibrating orifice droplet generators, over a cryogenic vapor to achieve rapid freezing. The primary aspect to consider is the type of nozzle and atomization parameters utilized. To achieve atomization, one of three different types of atomizing nozzles is typically used, including a two-fluid nozzle, an ultrasound or vibration nozzle, and a monodisperse droplet generator. The primary trade-off in these nozzles is particle-size control versus liquid processing rate.
The two-fluid nozzle allows for the highest processing rates up to 15 L/min, but the particle-size distribution created by these nozzles can easily span several orders of magnitude. The particle-size distribution of a given spray created using a two-fluid nozzle is primarily controlled by the properties of the liquid formulation (i.e., the surface tension and viscosity), nozzle geometry, and the flow rate of the liquid and atomizing gas. Two-fluid nozzles are preferred when fast processing is required but droplet-size distribution, hence final particle-size distribution, is not critical. Another disadvantage of the two-fluid nozzle system is that the large volume of atomizing gas utilized by the nozzle can decrease the efficiency and effectiveness of the cryogenic vapor into which the droplets and atomizing gas are being sprayed. This can increase the cost of the overall process and the rate at which cryogen is consumed.
Ultrasonic nozzles allow for relatively high processing rates, potentially up to 100 mL/min, with a better control of particle-size distribution and more importantly do not utilize large volumes of atomizing gas to generate droplets. The particle-size distribution of a spray created using these nozzles is primarily controlled by the properties of the liquid formulation, the properties of nozzle (i.e., orifice size and atomizing surface area), and the frequency of nozzle vibration. Ultrasonic nozzles are preferred for applications where both control of particle size and reasonably high processing rates are needed.
Monodisperse droplet generators are used when extremely precise controls of droplet and particle-size distributions are required. These systems utilize the same technology present in ink jet printing systems to create a controlled monodisperse droplet size; however, the primary disadvantage of these systems is that they have very low processing rates of about 0.1 mL/min. In addition to slow processing rates, these systems are also more prone to clogging and can process only very low viscosity solutions.
The second step in SFD is the freezing of the atomized droplets of drug solution using a cryogenic vapor . In many cases, the cryogenic vapor is created over a cryogenic liquid reservoir. When the atomized droplets fall through the vapor phase, they then encounter the cryogenic liquid reservoir, which can further ensure that the freezing process is completed. The most commonly used cryogen in SFD is liquid nitrogen vapor. When processing poorly water-soluble drugs using SFD, organic solvents are typically used to prepare the drug in solution/suspension. Very low vapor temperatures are therefore required to ensure the freezing point of the solvents reached.
Once the droplets of drug in solution/suspension have been frozen, the final step is removal of the solvents by sublimation to obtain dry powder form of the engineered drug composition. Traditional lyophilization or ATMFD is typically employed to sublime the frozen solvents. In either case, process conditions during sublimation must be controlled precisely to ensure that no melting occurs, which could potentially undo any advantageous physical properties imparted during the rapid freezing of SFD process.
11.2.1.2 Conventional Lyophilization
Conventional lyophilization is conducted at reduced pressure with vacuum level of few hundred millitorr and low shelf temperature to maintain the frozen SFD-processed material. The lyophilization is broken into two stages known as the primary and secondary drying stages. During primary drying, the temperature and pressure inside the lyophilizer are typically kept below that of the triple point of the solvent. This promotes the sublimation of the frozen solvent from the solid phase directly to the gaseous phase, without allowing any melting. After all or the majority of the bulk solvent has been removed, the process is then shifted to the secondary drying stage, where the shelf temperature is typically brought up to room temperature or higher to remove the molecularly bounded solvent. In most cases, convention lyophilization is utilized as it is well studied and accepted for pharmaceutical process.
The enhancement of dissolution rate and oral bioavailability of solid dispersions of valsartan, a poorly water-soluble drug, prepared by a freeze-drying technique was reported. The alkaline solution without organic solvent containing the API, hydrophilic polymers, an alkalizer, and a surfactant was lyophilized. Hydrogen bonds between drug and polymer were detected by FTIR. The resulting amorphous solid dispersions showed greater dissolution rate and significantly higher oral bioavailability as compared with the bulk drug substance (Xu et al. 2014).
11.2.1.3 Atmospheric Freeze Drying (ATMFD)
ATMFD is conducted at atmospheric pressure conditions and thus does not require a vacuum system. In this case, a cold desiccated gas (typically air or nitrogen) is circulated in and around a mass of frozen product in a fluid-bed-style configuration. For these systems to work properly, the circulation gas needs to be colder than the melting point of the solvent to ensure that no melting occurs. Besides low temperature, the circulation gas needs to have a very low partial pressure of the solvent vapor so that a mass transfer driving force is available to allow for sublimation of the frozen solvent. ATMFD has been utilized and well studied in the food industry (Meryman 1959; Boeh-Ocansey 1983; Rahman and Mujumdar 2012) but only recently, in the past 15–20 years, it has begun to gain some traction in the pharmaceutical literature (Mumenthalera and Leuenberger 1991; Rogers et al. 2003a). Its use in the food industry has been entirely focused on the removal of water as a solvent and for this process it has shown great promise. In specific cases, especially with freezing processes that result in more porous or discrete ice chips/particles (e.g., SFD, SFL, and TFF), ATMFD has actually been proven to be a much faster and more energy-efficient sublimation process due to the vastly increased heat and mass transfer afforded by the use of a fluid-bed configuration (Mumenthalera and Leuenberger 1991). However, in the case of poorly soluble compounds, the process is complicated by the use of cosolvent systems. As with traditional lyophilization techniques, cosolvent systems using organic solvents with lower melting points may prove to be problematic due to the low processing temperatures required as well as the large volumes of gas separation that may be required. When removing water as a solvent, standard refrigeration techniques can be used on the large scale to dry and recirculate desiccated air to ensure that the partial pressure of water vapor in the air is low, but when the solvent phase is something other than water it may be more difficult to remove these vapors from the recirculated dry gas. Theoretically, it should be possible to utilize ATMFD for the sublimation and drying of drug products created using cosolvent systems, but currently very little research exists on this topic.)
Rahman and Mujumdar (2012) reported advantages and limitations of the ATMFD process, a comparison between vacuum freeze drying and ATMFD, a novel approach to ATMFD, types of ATMFD and their applications. In addition, mathematical models for ATMFD process optimization were also presented. The models were related to energy, heat and mass transfer of the drying process. Studies of the drying process sought to overcome the limitations of ATMFD.
11.2.1.4 Pros of SFD Process
One potential advantage of SFD over other cryogenic processes is that it typically results in the highest rate of freezing at around 106 K/s when appropriately small droplet sizes are prepared (Engstrom et al. 2007b). This rapid freezing can be useful when working with poorly water-soluble drugs that may have very rapid rates of recrystallization, or when very high surface area powders are needed.
The most common usage of SFD to date has been in the area of the preparation of proteins and peptides for inhalation as a competing and gentler alternative to traditional spray-drying approaches. In contrast to spray drying, SFD process lacks the usage of high and potentially damaging heat to dry powders, and provides very high production yields of more than 95 % versus about 50 % for spray drying (Maa et al. 1999).
Additionally, the SFD process allows for independent control over both the aerodynamic and geometric particle size of prepared powders, which are critical parameters in the development of formulations for inhalation delivery. This enhanced control is due to the fact that geometric particle size is fixed during droplet formation and subsequent freezing so that a specific final particle size can be selected by controlling the size of droplet that is produced during atomization, whereas with spray drying significant droplet shrinkage and deformation occur due to surface-tension forces imparted as the droplet dries. Aerodynamic diameter, on the other hand, is a function of both geometric particle size and density and, in this case, density can also be controlled independently from geometric size by controlling the solids loading and formulation parameters used to prepare the drug solution that is subsequently processed via SFD (Maa et al. 2004).
11.2.1.5 Cons of SFD Process
Compared to other cryogenic techniques such as slow freeze drying in a vial, SFL, and TFF, SFD process can potentially lower biological activity in cases where pure biological compounds without added excipients. SFL and TFF actually result in even higher level of biological activity, because the large droplets used in these processes have relatively less air–water interface. Whereas SFD utilizes atomized fine droplets, a larger air–water interfacial region is created where absorption and denaturation of proteins can occur. However, several recent studies have shown that this effect can be minimized with the incorporation of excipient(s) (stabilizer, complexing agents, cryoprotectants, and surfactants) (Maa et al. 1999; Costantino et al. 2000, 2002; Yu et al. 2006).
11.2.2 Spray Freezing into Liquid (SFL)
SFL, one of the novel cryogenic particle engineering technologies, was developed and patented (Williams et al. 2003) and subsequently commercialized by The Dow Chemical Company.
11.2.2.1 Process of SFL
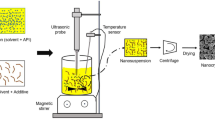
The SFL process involves preparation of a feed liquid, such as an aqueous, organic, or aqueous–organic cosolvent solution, aqueous–organic emulsion, or suspension containing a drug along with pharmaceutical excipient(s) or drug alone: Spray the drug feed liquid under pressure through a small diameter insulating nozzle directly into a liquid cryogen, such as compressed fluid carbon dioxide, helium, propane, ethane, liquid nitrogen, liquid argon, or hydrofluoroethers (Williams et al. 2003). The rapid cooling leads to immediate freezing of the atomized droplets of feed liquid upon contacting the cryogen. A schematic diagram of SFL process is shown in Fig. 11.1. Then the produced frozen materials were collected and subsequently dried by lyophilization or ATMFD as depicted above, to obtain dry and flowable powders of drug compositions.

Schematic diagram of spray freezing into liquid (SFL) process. Reproduced from Rogers et al. (2002b) with permission from Elsevier
The SFL-processed drug powders were generally characterized by micronized structure with amorphous morphology, high surface area, and improved wettability in aqueous media, indicating enhanced dissolution properties of the poorly water-soluble drugs (Hu et al. 2002, 2003; Rogers et al. 2003a, b).
The benefits of SFL process result from intense atomization in conjunction with high freezing rates. Liquid nitrogen has been typically employed as the cryogenic liquid due to nearly instantaneous freezing of the atomized feed liquid resulting from the low boiling point of liquid nitrogen. The nozzle that used to spray feed liquid in SFL process is composed of an insulating material such as poly-ether-ether-ketone (PEEK) tubing to prevent premature freezing of the feed liquid. As the liquid exits the nozzle, liquid–liquid impingement occurs between the pressurized feed liquid exiting the nozzle and the cryogenic liquid, such as liquid nitrogen. The estimated cooling rates are strongly related to droplet particle size of feed liquid, with higher freezing rates observed with smaller droplet sizes due to higher surface area available for heat transfer (Maa and Prestrelski 2000; Engstrom et al. 2007a, b). The cooling rates of SFL process for two different cryogens, iso-pentane and liquid nitrogen, were calculated to be 8.9 × 106 and 1.1 × 105 K/s, respectively (Engstrom et al. 2007a, b). SFL into iso-pentane produced faster cooling rates despite having a higher temperature (−160 °C) compared to liquid nitrogen (−196 °C). This was attributed to the boiling of liquid nitrogen around the inserted spray nozzle and/or sprayed feed fluid formed an insulating layer, known as the Leidenfrost effect (Sitte et al. 1987).
11.2.2.2 Pros of SFL Process
The advantages of the SFL process result from intense atomization of drug feed liquid and the high freezing rates. SFL process used liquid nitrogen nearly exclusively as the cryogen. A very high degree of atomization is achieved by spraying directly into the cryogenic liquid as in contrast to spraying into the vapor phase above the cryogenic liquid, because liquid–liquid impingement occurs between the pressurized feed solution exiting the nozzle and the cryogenic liquid (Rogers et al. 2002b). Thus, high freezing rates can be achieved in SFL process due to the low intrinsic temperature of liquid nitrogen and the high surface area of atomized droplets of the drug feed liquid.
Consequently, amorphous morphology of drug compositions can be formed by SFL process, as a result of the ultra-rapid freezing-induced simultaneous vitrification of the feed solution. The high degree of atomization and ultra-rapid freezing (URF) rates led to the formation of amorphous highly porous nanostructured particles (Hu et al. 2002).
Moreover, it is advantageous to use SFL process to make stable submicron protein drug particles. As discussed previously, SFD has the potential to cause protein aggregation due to the large gas–liquid interface in the spraying step. On the other hand, slow cooling by lyophilization (about 1 K/min) can produce stable protein particles; however, the particle size was found to be a minimum of a few microns in diameter with surface areas less than 1 m2/g. The SFL process can minimize exposure to the gas–liquid interface of droplets containing protein, as the spray nozzle was immersed under the surface of the cryogenic liquid. Thus, the SFL process can reduce protein adsorption, denaturation, and aggregation, and, consequently, lead to higher enzymatic activities than that processed by SFD (Yu et al. 2006; Engstrom et al. 2007a, b). Although the cooling rate in SFL is about 103 K/s, three orders of magnitude less than that in SFD, it is sufficiently fast to arrest the growth of submicron protein particles (Engstrom et al. 2009).
11.2.2.3 Cons of SFL Process
Increase in the drug/excipient(s) concentrations of feed liquid normally leads to increases in viscosity of the feed liquid. The relatively high viscosity of the feed liquid can limit the application of SFL, as it can inhibit liquid jet breakup, resulting in slower cooling rates and larger particle sizes and eventually fibers (Barron et al. 2003).
Moreover, removal of solvent from the collected frozen materials by lyophilization is costly for the equipment (lyophilizers) and is a time- and energy-intensive process that could take days or even weeks to finish (Franks 1992).
11.2.3 Thin Film Freezing (TFF)
TFF, also known as cold metal block freezing, initially was used to cool approximately 100-μm-thick tissue samples at rates between 100 and 10,000 K/s (Gilkey and Staehelin 1986) for nonpharmaceutical application. Impingement and solidification of liquefied droplets onto a cold solid surface have also been used in the electrical and semi-conductor industries to add thin layers of frozen material onto a surface. TFF was also referred as ultra-rapid freezing (URF), spray forming, thermal spray coating, splat cooling, slat quenching solidification, plasma or powder spray deposition, etc. (Overhoff et al. 2009). Recently, TFF has been used as a particle engineering technique to improve the dissolution profiles of poorly water-soluble drugs.
11.2.3.1 TFF Process
In a similar manner to the SFD and SFL processes , the first step in TFF process is preparation of a feed liquid containing a drug along with pharmaceutical excipient(s) or drug alone. Then, droplets of the feed liquid that released from a funnel or a pump/dripper system with a controlled flow rate fall from a given height and impact, spread, and freeze on a cooled surface of a cryogenic substrate, as depicted in a schematic diagram in Fig. 11.2. A cryogenic substrate is selected commonly from materials with a thermal conductivity k between 10 and 20 W/(m K). A rotating cylindrical stainless-steel drum approximately 17 cm in length and 12 cm in diameter with mirror-polished surface was employed to serve as the cryogenic substrate. The drum is hollow with 0.7-cm-thick walls and was filled with cryogen such as dry ice or liquid nitrogen on the inside. As a result of thermal conductivity through the steel, the equilibrium drum surface temperatures were measured to be 223 K or 133 K for dry ice and liquid nitrogen, respectively (Engstrom et al. 2009). The exposure of the cold drum to the atmosphere allowed a thin layer of ice to condense on the drum surface, which may affect the conductivity of the cryogenic substrate, and consequently may affect the freezing rates of the droplets fallen on the drum. To minimize the formation of water-vapor condensation and ice on the steel surface, it is better to place the TFF apparatus in a dry box or humidity-controlled environment with relative humidity less than 15 %. Moreover, a blade made by stainless steel or Teflon is mounted along the rotating drum surface to remove the ice immediately before the droplets of feed liquid impacting the drum.

Schematic diagram of thin film freezing (TFF) process. Reproduced from Overhoff et al. (2007a) with permission from Elsevier
The surface temperature of the drum can be monitored by using a surface moving probe thermocouple attachment. When the temperature on the steel surface reached a proper level, various feed liquids can be applied to the rotating steel drum drop-wise from a height of approximately 10 cm. Upon impacting on the cryogenic surface, the feed liquid droplets (about 2–4 mm in diameter) deformed into thin films (about 100–400 μm in thickness) of disk shape and rapidly cooled until frozen on timescales of 70–1000 ms, which corresponds to a cooling rate of about 102 K/s (Fukai et al. 2000; Pasandideh-Fard et al. 2002), as illustrated in Fig. 11.3 (Engstrom et al. 2009). The frozen disk is scraped off the stainless-steel surface with a blade prior to one full revolution and falls in a collecting pan that filled with dry ice or liquid nitrogen to maintain the frozen disk. After processing a batch, the collecting pan containing the frozen disk is transferred to a lyophilizer where the solvent is removed by sublimation (Overhoff et al. 2007a). The cooling rates in TFF and SFL processes are comparable. Because of rapid conductive heat transfer, resulting in high supersaturation and nucleation rates, TFF process can create powders with high surface area and enhanced dissolution properties, similar to those produced by other rapid freezing technologies. As in other freezing technologies, the rapid freezing of the feed liquid is critical in preventing phase separation of solute and solvent during freezing, allowing for the compositions to molecularly disperse with each other.

Diagram of the thin film freezing process displaying the falling droplet (a), spreading after impact on the stainless-steel surface (b), and during cooling and freezing as a thin film (c) (drawn to scale). Reproduced from Engstrom et al. (2008) with permission from Springer
To tailor the respirable brittle matrix powder, the processing parameters must be taken into consideration. The processing parameters used in the TFF process potentially affected the physicochemical and aerodynamic properties of the resulting matrix powders. Product with improved properties (greater specific surface area, higher porosity and lower density) was prepared at a higher freezing rate by controlling the cryogenic surface temperature. Besides, the lower initial solids content formulation gave the better FPF result possibly due to the increased fragility (Wang et al. (2014a)).
A combination of TFF and another technique (i.e., template emulsion technique) was also used to produce amorphous form of poorly water-soluble drug. The hydrophobic drug was encapsulated into the internal phase of the emulsion through the template emulsion method before rapid freezing through TFF process. Formulation prepared using the TFF-template emulsion technology showed significant improvement of wetting and dissolution properties of the drug (Lang et al. 2014).
11.2.3.2 Advantages of TFF Process
Although the cooling rate in TFF (102 K/s) is lower, compared to those of SFD (106 K/s) and SFL (103 K/s), it is still sufficient to produce rapid nucleation and to prevent significant particle growth during freezing. In TFF process, the size of the unfrozen channels was sufficiently thin and the increase in the viscosity of the unfrozen solution was sufficiently fast to be able to achieve similar particle sizes and morphologies as for the moderately faster process, SFL, and the much faster process, SFD (Engstrom et al. 2008).
TFF on a cold metal surface bypasses the need to maintain aseptic conditions of a liquid cryogen, for example, liquid nitrogen (Gosselin et al. 2003). The cooling rate of the thin films in TFF may be controlled readily by varying the temperature of the metal surface. Also, the surface temperature of the cryogenic substrate may be measured directly. For SFL and SFD, the complex geometry of the turbulent spray in the liquid nitrogen combined with the Leidenfrost effect can be somewhat difficult to control and monitor (Sitte et al. 1987). In TFF, more concentrated and thus more viscous solutions may be processed, as the droplets are not atomized. However, the thickness profile of the film along the radius of the frozen disk may change with viscosity (Overhoff et al. 2009).
TFF process can provide a high yield of products. In TFF, collection of the frozen thin films of the feed liquid droplets leads to nearly 100 % yields, whereas in SFD process yields were about 80 % because of the result of entrainment of uncaptured particles in the atomized aqueous stream, particles attaching to the walls of collection vessels, and inefficient separation of the cryogen from the 10–100-μm frozen particles (Johnson 1997; Overhoff et al. 2009).
TFF process offers flexibility of the amount of drugs to be processed. By using TFF, it is feasible to accommodate either small quantities (<1 mL) of drug feed solution due to the high efficiency in collection of frozen films or large-scale production by adding multiple drippers to make droplets in parallel and increasing the length of the drum. The rotating drum of TFF apparatus offers scale-up advantages over other cryogenic particle engineering technologies by becoming more of a continuous freezing process. Thus, TFF process is not limited by the amount of drug to be processed. It is feasible from the early-stage screening of drug in milligram quantity to commercial product manufacturing at a scale of kilograms to tons.
In addition to the advantage of being a simple, efficient, and robust process for freezing, TFF also renders improvement in the stability of the protein product due to the minimized gas–liquid interface of the feed liquid, in comparison to SFD and SFL. It was found that minimizing gas–liquid interface can improve protein stability by limiting the amount of protein that can absorb to the interface. The surface area to volume ratio of the gas–liquid interface in TFF was 2 orders of magnitude lower than in SFD, leading to much less protein adsorption and aggregation (Engstrom et al. 2008).
11.2.3.3 Disadvantages of TFF Process
First, maintenance of a low humidity for TFF process increases costs for facility design, equipment, and operation, especially for commercial productions.
Second, with all freezing processes, the quantities and quality of cryogen required for manufacturing production-scale batch sizes could also add to production costs. To date, it is not sure which of the aforementioned cryogenic processes is the most cryogenically efficient (Overhoff et al. 2009).
Additionally, similar to the SFL process, removal of solvent from the collected frozen films by lyophilization is costly, for both equipment (lyophilizers) and energy consumption.
11.2.4 Storage of Dried Powders
If the frozen materials were dried by lyophilization, after the lyophilization cycle was complete, the lyophilizer was filled with dried nitrogen gas upon releasing the vacuum to reduce the exposure of the lyophilized powders to moisture in the ambient air before transfer to packaging area, where the humidity is controlled, commonly to less than 15 % RH. The obtained dry powders of the cryogenically processed drug compositions were packaged into hermetically sealed glass containers under dry nitrogen.
11.2.5 Mechanism of Rapid Freezing-Induced Particle Formation
Solubility is heavily influenced by external properties, including temperature, pH, polarity of the solvent, and pressure. Sudden shift of one of these properties can induce nucleation, leading to particle formation. Nanoparticles may be formed by maximizing the supersaturation to induce precipitation instantaneously and then arresting growth (Matteucci et al. 2007). Generally, faster nucleation relative to particle growth leads to a smaller median particle size and more uniform particle-size distribution.
Rapid freezing can be categorized as a precipitation technology, where most of the solvent is separated from the solutes to form ice and the solute phase becomes highly concentrated. Upon initiation of freezing a homogeneous solution, the formation of frozen solvent particles and a drug/polymer-rich phase begin to appear (Tang and Pikal 2004). The rate of cooling in conjunction with other factors, such as solute concentration, plays a key role in determining the final particle size and structure of the solid powders (Overhoff et al. 2007a). The rate of growth and number of solvent crystals in a freezing solution are determined by the degree of supercooling. Higher supercooling results in more/smaller ice crystals and larger ice-specific surface area (Jiang and Nail 1998). As different freezing methods can produce different supercooling effects, freezing with liquid nitrogen basically can provide the highest supercooling, while solutions subjected to slow cooling rates, for example, freezing with the precooled shelf method, give the lowest supercooling. The solvent in the supercooled solution nucleates and forms crystalline solvent particles which grow during freezing. Increased supercooling, in turn, increases the nucleation rate of frozen solvent particles while minimizing the time for frozen solvent particle growth. When the supercooling is extremely high (rapid freezing rate), the formation of a vitrified solution may occur in which the nucleation of crystals may be minimized or fully prevented, leading to an amorphous material (Yu 2001; Overhoff et al. 2009).
During freezing, the supersaturation of the solute in the unfrozen domains as a function of the phase diagram establishes a driving force for precipitation to occur. Particle growth under this condition can occur through condensation of individual molecules onto a growing nucleus, coagulation of two growing particles, or Ostwald ripening, which is the growth of larger structures at the expense of smaller structures (van de Witte et al. 1996). Rapid nucleation at high supersaturation all at one time period will produce uniformly sized particles and lower Ostwald ripening (Overhoff et al. 2009).
As depicted in Fig. 11.4, a greater rapid cooling rate will produce a larger number of nuclei and more solid particles separated by thinner ice domains than in the case of slow cooling and slow nucleation. As the cooling domains vitrify, the high viscosity inhibits the further growth of the particles (Engstrom et al. 2007a, b).

Frozen morphologies of dilute solution with high supercooling (a), concentrated solution with high supercooling (b), dilute solution with low supercooling (c), and concentrated solution with low supercooling (d). Amorphous ice particles are represented as white domains and solute precipitate as solid dots or gray regions. Reproduced from Engstrom et al. (2007b) with permission from Elsevier
11.3 General Guidelines for Cryogenic Technology
11.3.1 Selection of Solvent/Cosolvent Systems for Cryogenic Processes
The first step in the cryogenic processes is to make feed liquids. Despite the diversity of feed liquids (solution, suspension, and emulsion) that can be employed for the cryogenic processes, a complete solution of poorly water-soluble drug alone or along with pharmaceutical excipient(s) was mostly reported.
11.3.1.1 Solubility
Solubility may be defined as the maximum concentration of a substance that may be completely dissolved in a given solvent at a given temperature and pressure. Solute molecules are held together by certain intermolecular forces (dipole–dipole, induced dipole–induced dipole, ion–ion, etc.), as are the molecules of the solvent. In order for dissolution to occur, these cohesive forces of like molecules must be broken and adhesive forces between solute and solvent must be formed. The dissolution process of solids in liquids involves three steps: (1) the removal of a molecule from the solute; (2) creation of a hole in the solvent; and (3) insertion of the solute molecule into the solvent (i.e., solute–solvent interaction) (Hildebrand and Scott 1950). This interaction between the solute and the solvent is obviously dependent on the physical and chemical nature of the participating molecules. The dissolution of hydrophobic materials, which can dissolve readily in nonpolar organic solvents, differs from that of hydrophilic excipient(s) which tend to dissolve in polar aqueous phase.
Generally, hydrophilic excipient(s) would be incorporated into compositions containing poorly water-soluble drug to improve its wettability. To make a feed solution accommodating both hydrophobic API and hydrophilic excipient molecules in a fully dissolved state, it is critical to choose a proper solvent system. One approach is to mix solvents of different polarities to form a solvent system of optimum polarity to dissolve the solutes of different polarities. This method is referred to as solvent blending or cosolvency. When talking about liquids, the term miscibility rather than solubility may be used to describe the affinity between the liquids (Conventional 2000). Liquids that form a homogenous system when mixed in any proportion are said to be miscible (e.g., water and ethanol). Those in which only certain volume ratios produce homogenous mixtures are said to be miscible in certain proportions (e.g., water and chloroform). Immiscible liquids will not produce a homogenous solution in any proportion (e.g., water and olive oil). For the cosolvent used to make feed solution, the solvents must, obviously, be miscible. For example, tetrahydrofuran/water co-solvent was used to form feed solutions for SFL process, because of the ability to dissolve both a poorly water-soluble drug and hydrophilic excipients (Hu et al. 2002; Rogers et al. 2002b). Some poorly water-soluble drugs have relatively low solubility in the tetrahydrofuran/water co-solvent; later on, acetonitrile, acetone, methanol, methylene chloride, 1,3-dioxolane, tert-butanol, and 1,4-dioxane were used to greatly increase the solubility of drugs. However, the percentage of methylene chloride in cosolvent should be less than 1 % to be miscible. Worthy to note is that for the use of low-melting-point solvents, a liquid nitrogen cold trap is necessary to capture the solvent vapor before sucked into vacuum pump during lyophilization.
11.3.1.2 Fluid Dynamics
For SFL process, viscosity of the solvent is an important factor that needs to be considered for preparation of the feed solution to be sprayed through the nozzle into liquid cryogens. Additionally, the melting point of the solvent(s) used in SFL process is better not higher than 0 °C; otherwise, the feed solution may freeze prematurely within the atomizing nozzle before sprayed into cryogens. Examples of case are tert-butanol and 1,4-dioxane, in which many poorly water-soluble drugs show good solubility. However, due to their relatively high viscosity, 3.62 and 1.54 cP, respectively, compared to other organic solvents, and high melting point for liquid, 25 and 12 °C, respectively (see Table 11.1), they are not proper candidates for SFL. Because the TFF technology applies the droplets directly onto the cryogenic substrate, premature freezing is not a concern and solvents with high melting point may be used.
Nevertheless, TFF process also requires consideration of the fluid dynamics for the solvent during spreading and freezing. The ability of an impinging droplet on the cryogenic substrate to spread into a thin large-diameter disk prior to freezing influences the cooling rate of the thin disk. Danazol/polyvinylpyrrolidone (PVP) powders were produced under the same TFF process except for using acetonitrile and tert-butanol, respectively, as the solvent. It was observed that the powder from acetonitrile solution exhibited more uniform nanostructured surface morphology compared to the powder from tert-butanol solution. The morphological difference was attributed to the cooling rate of the two solvents (Overhoff et al. 2007a, 2009). Moreover, solvents of the feed solution with greater thermal diffusivities are more desirable for rapid heat transfer. The rapid heat transfer in TFF process is the result of intimate contact between the solution and cryogenic substrate.
11.3.1.3 Ease of Lyophilization
Freeze drying, i.e., lyophilization , is the commonly used means to obtain dry powder from the cryogenically generated materials by removing the solvent(s). The ideal solvent for freeze drying has the following properties: a high vapor pressure, a melting point either below or slightly above room temperature, a high viscosity, and a low toxicity. It must provide a stable environment for freeze drying and be rapidly and completely removed to produce dry material (Ni et al. 2001).
A variety of different types of organic solvents were used in cryogenic processes, as mentioned in Table 11.1. Most of these water-miscible organic solvents have freezing points below −75 °C. The frozen disk made by the low-melting-point solvents tends to melt during lyophilization and it makes a very challenging task for the lyophilizer condenser to catch the sublimed vapor of these solvents. A cold trap between lyophilizer sample chamber and vacuum pump is necessitated to prevent the solvent vapor from being sucked into the vacuum pump.
Accordingly, organic solvents with higher melting point are of great interest for selecting proper solvent for cryogenic process. Acetonitrile, having a melting point of −45 °C, viscosity of 0.34 cP, good heat transfer, and the unique ability to dissolve both a hydrophobic drug and hydrophilic excipient(s), was widely employed for many compositions containing poorly water-soluble drug in both SFL and TFF processes to increase drug loading with reduced risk of liquid–liquid phase separation. More importantly, the high vapor pressure of 73 mmHg at 25 °C eases the removal of the solvent by lyophilization.
Subsequently, high-melting-point solvents 1,4-dioxane and tert-butanol were often employed in studies of TFF process, which has less concern of premature freezing compared to SFL. Tert-butanol was often used in combination with low-melting-point solvent system for its easy-to-freeze and good lyophilization characteristics. As a result of its high vapor pressure (Table 11.1) and its crystal morphology, the sublimation rate of tert-butanol is greater than 2.5 times that of water. 1,4-dioxane/water co-solvent was extensively used to make feed solutions for TFF process (Overhoff et al. 2007a, b; DiNunzio et al. 2008; Yang et al. 2008a, b), mainly due to its relatively high freezing point and vapor pressure which make the freezing and lyophilization processes easily manageable. These solvents prove beneficial by reducing the lyophilization time (Ni et al. 2001) or eliminating the solvent-removal process altogether as some of these solvents sublime at ambient conditions or higher (Tesconi et al. 1999). Moreover, they have low biological toxicity and basically no harmful impact to the environment.
According to these criteria, a solvent with these ideal properties may not exist for cryogenic processes.
Formation of co-solvent systems can mitigate limitations of certain solvents (solubility) while maintaining some of the ideal fluid dynamics and lyophilization characteristics. Combinations of 1,3-dioxolane (solubility enhancer) and tert-butanol (ideal freezing and lyophilization properties), tetrahydrofuran and water (both are solubility enhancers), acetonitrile (solubility enhancer and freezing and lyophilization properties) and water, and 1,4-dioxane (solubility enhancer and freezing and lyophilization properties) and water have been used to generate nanostructured poorly water-soluble drug compositions using cryogenic technologies.
11.3.2 Selection of Excipients , Concentrations for Drug Formulations Using Cryogenic Processes
The ultimate goal of cryogenic particle engineering technologies is to improve the dissolution properties of poorly water-soluble drug by making an amorphous nanoparticulate form of the drug. However, a primary concern of the formed amorphous material is the inherent instability, due to the higher energy state. Amorphous material may automatically convert back to a low-energy crystalline state. Recrystallization of the amorphous drug may be avoided by inclusion of stabilizing hydrophilic excipients in the composition. Principally, stabilizing excipients combat recrystallization of amorphous drug particles by steric hindrance and/or the formation of hydrogen bonds with drug molecules (Khougaz and Clas 2000; Vasanthavada et al. 2005). These stabilizing excipients must be hydrophilic for improving the wetting properties of drug particles; however, the hydrophilic excipients should not be hygroscopic. Otherwise, they absorb moisture easily when exposed to ambient environment, which can lead to morphological instability of the excipient-stabilized amorphous drug particles by either displacing drug molecules from hydrogen-bonding sites or plasticizing polymeric stabilizers (Forster et al. 2001; Vasanthavada et al. 2004).
As morphological instability and poor wettability are the substantial limitations to particle engineering of poorly water-soluble drugs, concessions need to be made in selecting appropriate stabilizing excipient(s), with respect to rigidity for steric hindrance or hydrophilicity. Many readily soluble and/or wettable excipients do not provide adequate steric hindrance of recrystallization due to low melting points or glass transition temperature (T g). On the contrary, excipients with high melting points or T g’s typically have relatively poor wetting properties. Therefore, polymers with high glass transition temperature (T g) and hydrophilicity, such as PVP and hydroxypropylmethylcellulose (HPMC), are popular candidates to formulate poorly water-soluble drugs for improved aqueous dissolution. To expand the choices of polymers, combinations of rigid and malleable excipients such as surfactants (e.g., poloxamer, sodium dodecyl sulfate, and Tween 80) often can be used to achieve reasonable stability and wettability. Other commonly used polymers include polyvinyl pyrrolidone-covinyl acetate (PVP/VA), hydroxypropylmethylcellulose acetate succinate (HPMCAS), high-molecular-weight polyethylene glycol (PEG), polyvinyl alcohol (PVA), polylactic acids (PLA) and polylactic-co-glycolic acid (PLGA), etc. Catalytic pretreated softwood cellulose (CPSC) isolated from pine wood (Pinus sylvestris) was recently used as a stabilizing carrier polymer of the cryogenic co-ground spray-dried formulation of poorly water-soluble drug (Penkina et al. 2015).
Besides polymers, nonpolymeric natural products (e.g., lecithin) and small-molecule hydrophilic excipients (e.g., sugars) can also be employed to enhance wettability and solubility of poorly water-soluble drugs, with better safety profiles. Sugars such as lactose, sucrose, trehalose, and, recently, fructose oligomer inulin were frequently used as an excipient to stabilize amorphous drugs, peptides, and proteins during drying and subsequent storage (Davies and Feddah 2003; Van Drooge et al. 2004). The addition of sugars has been shown to extend the shelf life of amorphous systems by preventing crystallization (Eriksson et al. 2003).
On the contrary, drugs with low Tg were important for freeze-drying of poorly soluble drugs. To fabricate nanocrystals, fenofibrate, a low Tg, poorly water soluble drug, was mixed with mannitol. Excipient such as mannitol was selected for this process since it was susceptible to crystallize. The produced nanocrystalline particles of poorly water soluble drug also drastically enhanced dissolution of the poorly water-soluble drugs (De Waard et al. 2008).
Worthy to note is that excipient to drug ratio is also an important consideration in formulation design. First, excipient to drug ratio may affect the morphology of the engineered drug composition. The T g of the overall drug composition is a function of the fraction of each component. If there is more amorphous material present in the drug composition, more stabilizing polymer would be needed to be present; otherwise, it may lead to a greater risk of recrystallization. Second, excipient to drug ratio affects hydrophilicity of the engineered drug compositions. It has been demonstrated that as the potency of a poorly water-soluble drug composition increased, the wettability is often decreased, particularly for the matrix system of a drug composition where drug and excipient molecules distribute randomly. Hydrophilic excipients act at the surface of drug composition particles to reduce the solid–liquid interfacial tension between the hydrophobic drug and aqueous media (Sinswat et al. 2005). As potency is increased, a greater proportion of the particle surface area is occupied by drug molecules; hence, the surface is rendered more hydrophobic, i.e., less wettable. Thus, it is often difficult to achieve high-potency drug particles with acceptable wetting properties. However, by carefully selecting excipient(s) and controlling the proportions of drug and excipient(s) in the cryogenically processed drug compositions, it was also possible to achieve high-potency drug compositions with improved wettable surface and high stability against recrystallization. An SFL-processed danazol/PVP K-15 composition with high potency of 91 % was reportedly maintained in its amorphous structure and rapid dissolution characteristics after 1 month of cycled stability conditions (−5 to 40 °C every 3 h) (Hu et al. 2004a, b, c).
In addition to the considerations for characteristics and stability of drug composition, formulation design also needs to comply with regulatory requirements for final dosage forms. It is important to judiciously select excipients that have been accepted/approved by FDA for specific administration routes. Especially for parenteral and pulmonary deliveries, only very limited excipients are approved for use, mainly including biocompatible and biodegradable materials.
Moreover, the total solid loading (drug plus excipients), particularly the excipient to drug ratio in the feed solution for cryogenic processes, affects morphology and surface area of the cryogenically engineered drug compositions. Typically, a higher solid loading infers denser engineered drug composition powder, accordingly, the lower the surface areas of the powder. Itraconazole and hydroxypropylmethyl cellulose phthalate (HP-55) at 2.0 % solid loading demonstrated a porous matrix structure as seen from Fig. 11.5a, b, while decreasing the solid loading to 0.2 % formed discrete nanoparticles, as shown in Fig. 11.5c, d. At 0.2 % solid loading, low polymer loading showed amorphous string-like structures (Fig. 11.5c), whereas increasing the polymer ratio resulted in more spherical nanoparticles (Fig. 11.5d), indicating that the surface morphologies are strongly influenced by the percentage of polymer in the composition. By manipulating the solid loading and polymer portion in the feed solution for cryogenic processes, drug compositions with desired characteristics may be tailored. For example, low-density powders of drug compositions intended for pulmonary delivery can be produced from a feed solution with drug loading commonly in the range of 0.5–1 % (w/v). To make drug composition powders to be further processed into tablets or capsule dosage forms, the feed solution may be made to above 5 % (w/v) if solubility permitted, so that much denser powders can be obtained to press into tablets or fill into capsules.

Scanning electron micrographs of TFF-processed powders containing ITZ and hydroxypropylmethyl cellulose phthalate (HP-55) at various solid loading and drug polymer ratios: (a) 4:1HP55 (2 %), (b) 1:4HP55 (2 %), (c) 4:1HP55 (0.2 %), (d) 1:4HP55 (0.2 %). Reproduced from Overhoff et al. (2007b) with permission from Elsevier
11.3.3 Properties of Pharmaceutical Powders Made by Cryogenic Processes
11.3.3.1 Engineered High Surface Area Powder for Oral Drug Delivery
Based on the extensive studies of the cryogenic particle engineering technologies, drug compositions processed by these technologies have been shown to create nanostructured amorphous particles with dramatically enlarged surface area and improved dissolution properties, in contrast to crystalline bulk drug. Additionally, these processes allow molecular incorporation of hydrophilic pharmaceutical excipient(s) to drug compositions, leading to improvement of the wetting characteristics of the bulk poorly water-soluble drug powder (Hu et al. 2002, 2004a, b; Rogers et al. 2003a, b; ; Overhoff et al. 2007a; Purvis et al. 2007; Yang et al. 2008b). The cryogenic technologies are primarily precipitation, i.e., “bottom-up,” processes, allowing for a reduction in the particle size of drug particles without degradation that induced by heating or mechanical process. As discussed in Sect. 12.2.5 amorphous morphology and uniform small particle size result from the very rapid freezing rates of these processes. Both the spray of drug feed solutions to produce tiny droplets and impinging of the feed solution drop on a solid substrate to form a spreaded very thin layer of liquid enable dramatic enlargement of surface area, which, in turn, enhance the rapid freezing process. According to the Noise–Whitney equation
where dM/dt is the rate of dissolution, A is the surface area available for dissolution, D is the diffusion coefficient of the compound, C s is the solubility of the compound in the dissolution medium, C is the concentration of drug in the medium at time t, and h is the thickness of the diffusion boundary layer. With dramatically increased surface area and decreased particle size, the dissolution rate can be increased. Furthermore, the formation of metastable amorphous form yields higher energy states for the drug and thus a greater thermodynamic driving force for dissolution. Therefore, both dissolution rate and the extent of dissolution are improved, i.e., achieved supersaturation of the drug.
The very rapid freezing of the cryogenic processes enables production of amorphous nanostructured pharmaceutical powders with relatively small amounts of excipient(s) to achieve high drug loadings of commonly 50–86 % drug/total solids, while maintaining high dissolution rates.
Besides the excellent performance of the cryogenically engineered pharmaceutical powders in pulmonary delivery, rapid-release tablet formulation containing SFL-processed danazol micronized compositions by direct compression was also reported (Hu et al. 2004a, b, c). The high surface area, high porosity, and amorphous structure of the SFL-processed danazol compositions contribute to their significantly enhanced dissolution profiles. However, these properties may be changed during the process of incorporation of other tablet excipients and tableting compression. By selection of suitable excipients and direct compression without involving water in the tableting, it was found that over 90 % of danazol released in only 10 min from tablets containing SFL-processed danazol composition (danazol/PVP K-15/SLS = 4:1:1), similar to the dissolution profiles of the SFL composition itself. The amorphous morphology was also maintained. Utilizing high T g excipient in the SFL-processed composition was contributed to prevent recrystallization of the SFL-processed danazol during the tableting process.
An alternative means is to fill the cryogenically engineered pharmaceutical powders into capsules for oral delivery. A solid solution of itraconazole (ITZ) and enteric polymer cellulose acetate phthalate (CAP) (ITZ:CAP = 1:2, w/w) was created by TFF process (DiNunzio, Miller et al. 2008). In vitro supersaturated dissolution results demonstrated significantly lower levels of supersaturation in acidic media and greater extents of supersaturation in neutral media compared to Sporanox pellets, the marketed product of itraconazole. Both the ITZ:CAP = 1:2 and Sporanox pellets were filled into size-9 capsules and dosed to rats. The pharmacokinetics study indicated that ITZ:CAP = 1:2 achieved 2 times higher oral bioavailability and more rapid onset of action versus Sporanox® pellets. The amorphous nature of TFF-engineered ITZ:CAP = 1:2, the intestinal targeting, and the increased durations of supersaturation rendered by enteric polymers were needed to contribute to the improved bioavailability.
A novel way recently patented is utilization of hot-melt extrusion process to prepare extrudates employing relatively low-melting/softening-temperature polymer or a blend of polymers as carrier and cryogenically engineered pharmaceutical powders (Miller et al. 2008). It is critical to conduct the extrusion at a temperature approximating or above the softening or melting temperature of the carrier polymer and below the point of solubilization of drug-containing particles in the carrier system, and below the recrystallization point of amorphous drug in the drug-containing particles. Therefore, high T g excipient(s) need to be employed in the preparation of cryogenically engineered pharmaceutical powders. The obtained extrudate containing the cryogenically engineered pharmaceutical powder particles can be milled for further tableting or capsule filling for oral administration.
Compared to the conventional dosage forms, the lower amount of excipients and drug in cryogenically processed drug compositions to be delivered to patients can provide increased patient compliance, safety, and therapeutic efficacy of the poorly water-soluble drugs (Yang et al. 2008b).
11.3.3.2 Brittle Matrix Powder for Pulmonary Drug Delivery
In addition, cryogenically engineered pharmaceutical powders with the properties described above also offer the versatility of being further assembled into diverse dosage forms for application, such as oral, parenteral and pulmonary delivery. The most extensively explored application is pulmonary delivery, including application of the engineered pharmaceutical powders directly in powder form to various dry powder inhaler (DPI) devices, making aqueous dispersion of the pharmaceutical powder for nebulizers, and dispersing the pharmaceutical powders in propellants such as hydrofluoroalkanes (HFA) for pressurized metered dose inhalers (pMDI).
Pulmonary drug delivery is of great interest due to its targeted delivery of drug to the pathological sites. However, for inhalation delivery, particle-size distribution and morphology of the drug-containing particles have pronounced effects on drug deposition in the respiratory tract and therapeutic effects. There are some drawbacks of nebulization of tacrolimus such as reconstitution, long dosing times, and inefficiently targets the lung. Moreover, drug lost during patient exhalation of more than 50 % resulted in a low efficient therapeutic of drug delivery via vibrating mesh nebulizer and lead to the development of dry powder inhalation formulation (Watts et al. 2013). Unpublished data from our laboratory showed that TFF-engineered dry powders containing tacrolimus and nonpolymer excipient (e.g., sugars) featured with low-density (0.01–0.03 g/mL), high-surface-area amorphous brittle matrices composed of sub-500-nm primary structures. The aerodynamic profiles of the tacrolimus dry powders indicated that they are suitable for deep lung delivery by filling the powders into size-3 capsules and used in Handihaler® and Easyhaler® DPI devices. Shear stress from the Handihaler® DPI device affected the diameter of the respirable brittle powders.
The brittle matrix powders for pulmonary drug delivery of various drugs were prepared using TFF. Formation of the tremendously low-density (<0.01 g/cm3) microparticles of tacrolimus were generated by in situ high shear velocity in a passive commercial pMDI device. The formed particles were delivered to the deep lung.
Lactose and raffinose exhibited better properties than mannitol which showed similar performance to the pure drug formulation. However, both saccharides were hygroscopic materials so, the formulations required a moisture barrier package. On the other hand, mannitol based formulation was less sensitive to humidity (Watts et al. 2013).
The in vitro and in vivo performance the TFF brittle powder and crystalline micronized formulations of tacrolimus were reported by Wang et al. (2014b). Miat® monodose inhaler was used to deliver the low-density particles of tacrolimus to the deep lung. The mass median aerodynamic diameter (MMAD) of 2.26 μm and a fine particle fraction (FPF) of 83.3 % were achieved. Pharmacokinetic results revealed that the brittle powder exhibited higher pulmonary bioavailability but lower systemic concentration than the crystalline dry powder formulation. Moreover, perhaps due to the decreased clearance, the TFF powder retained in the lung longer than the other.
Inhaled rapamycin was also prepared as amorphous brittle matrices using TFF process. The optimized TFF formulation with lactose monohydrate was selected to compare to the milled microparticles of the physical mixture. In terms of aerosolization performance, both formulations were appropriate for pulmonary delivery. The in vitro aerodynamic properties of the brittle matrix powder were superior to the micronized crystalline powder. However, the in vivo study of the single-dose administration showed the lower lung deposition, higher systemic concentration of the fine low-density particles as compared with the crystalline powder. These resulted from the enhancement of drug solubility in the amorphous formulation and the FPF at a more distal part of the lungs (Carvalho et al. 2014).
A fixed-dose combination of two drugs, salmeterol xinafoate and mometasone furoate, were prepared as a brittle matrix powder. The composite particles were studied using a marketed DPI device for inhaled drug delivery. The NGI data indicated that brittle matrix powder enhanced aerodynamic properties as compare to the micronized powder blend. Both drugs co-deposited when they were together in the composite particles. On the other hand, the deposition and dose uniformity of the blend of two micronized drugs were not consistent. Regarding the respirable dose delivered and stability results, the formulation without stabilizing materials was chosen for the in vivo study. Pharmacokinetic study showed that the lung concentration of both drugs from the brittle matrix formulation was greater than the crystalline drugs blend. The systemic drug levels of the TFF processed powder were also higher than the physical mixture due to the rapid absorption and the higher lung tissue exposure (Liu et al. 2015).
Similarly, beclomethasone dipropionate dry powders that were engineered by TFF process demonstrated proper aerodynamic profiles for inhalation when applied to Handihaler® device, suggesting that TFF-generated low-density, nanostructured micron-sized pharmaceutical powders are suitable for application in DPI.
Pulmonary drug delivery targeted to the alveoli for systemic absorption has become an increasingly attractive route of administration for poorly water-soluble drugs (Courrier et al. 2002). The concept of improving bioavailability of poorly water-soluble drugs by pulmonary delivery of nanostructured aggregates was reported in a murine model. An amorphous nanostructured composition containing itraconazole:mannitol:lecithin (1:0.5:0.2, w/w/w) made by TFF process illustrated dramatically enhanced supersaturation dissolution profile and aerodynamic properties suitable for deep lung delivery in vitro. Inhalation of the nebulized colloidal dispersion of this composition by mice for 20 min produced significantly high drug deposition in lung and effective therapeutic concentration in blood. The observed dramatic improvement in bioavailability of the TFF-processed itraconazole was mainly attributed to the amorphous nature, smaller nanoscaled particle size, and the presence of lecithin, which is biocompatible to lung and acts as a permeation enhancer to facilitate ITZ to permeate through the lung epithelium (Yang et al. 2008b).
Another promising application of cryogenically engineered pharmaceutical powders is their incorporation into pMDIs. Suspension stability in pMDIs is a significant challenge because surfactants that have been approved by FDA for inhalation are insoluble in propellants of pMDI. Commercial pMDI products are therefore perplexed by poor suspension stability with settling observed within minutes. Consequently, it requires patients to shake the pMDI device right before inhalation. A novel rod-shaped nanoparticle (nanorods) of bovine serum albumin that was used as a model protein drug, created by TFF process, was found capable of forming stable suspensions in HFA against settling for 1 year and produced optimal aerodynamic properties (Engstrom et al. 2009). The excellent suspension stability of the protein particles in HFA was attributed to their anisotropic geometry. Spherical particles pack together in a more efficient manner than anisotropic particles, such as rods [86]. Therefore, open flocs composed of nanorods take up a larger volume and have lower density, compared to flocs containing spherical particles. TFF process is applicable to a wide variety of drugs to make nanorods for pMDI without the need of stabilizing the primary particles.
11.3.4 Analytical Methods Used to Characterize Pharmaceutical Powders Made by Cryogenic Processes
11.3.4.1 Solid-State Characterization
The solid state of the drug and excipient are important aspects of the physical and chemical stability, as well as pharmaceutical and therapeutic performance of the drug product.
Amorphous state, a disordered phase, having similar mechanical and physical properties of a supercooled liquid existing at temperatures below its thermodynamic crystallization temperature but has not been given sufficient time to anneal and crystallize to its thermodynamically stable ordered phase, inherently has a higher degree of molecular mobility (Hancock et al. 1995). Even a small amount of crystalline form of the drug can significantly affect the in-vivo performance of the amorphous drug (Hancock and Parks 2000). Therefore, it is important to monitor and characterize the extent of crystallinity or disorder during formulation development, manufacturing, and over the intended shelf life of pharmaceutical product to ensure a robust and safe formulation by understanding the behavior of these amorphous systems.
Various analytical techniques have been reported for quantifying amorphous or crystalline phase in solids. The classical methods of evaluating the solid state are powder X-ray diffractometry (Newman and Byrn 2003; Shah et al. 2006) and thermal analyses (Clas et al. 1999).
11.3.4.1.1 Powder X-ray Diffractometry (PXRD)
Diffraction techniques are perhaps the most definitive method of detecting and quantifying molecular order in any system. Conventional, wide-angle and small-angle diffraction techniques have all been used to study order in systems of pharmaceutical relevance (Salekigerhardt et al. 1994). Diffraction is defined as a scattering phenomenon in which the incident X-rays, depending upon the phase difference, are reinforced to form diffracted beams (Suryanarayan 1995).
PXRD is one of the most widely used quantification techniques because of its simplicity and it measures differences in periodicities of atoms/molecules in a powder sample (Stephenson et al. 2001). It provides important insight, based on the degree of long-range order present, into the extent and nature of the crystallinity and microstructure. PXRD patterns of crystalline forms show strong diffraction peaks, whereas amorphous states exhibit diffuse and halo diffraction patterns.
X-ray procedures for the estimation of degree of crystallinity are based upon the measurement of X-ray scattering from the entire sample including the crystalline and amorphous region of the sample. The experimentally measured crystalline and amorphous intensities are proportional to the crystalline and amorphous fraction of the sample. Quantification of amorphous material by PXRD can be achieved by three methods: (1) measuring the characteristic crystalline peak intensities, (2) measuring the integrated peak areas of the principal crystalline peaks, and (3) measuring the intensity of characteristic region of amorphous scattering; of physical mixtures of known crystallinity to yield a calibration curve which is used for further quantification studies (Shah et al. 2006).
Limit of detection (LOD) of crystallinity in amorphous drug compositions of X-ray diffraction is 5–10 %, and the limit of quantitation (LOQ) is 2–5 % (Nagapudi and Jona 2008). The specificity and accurate quantitative nature of this nondestructive technique make it the first line choice for studying crystallinity of pharmaceutical materials.
11.3.4.1.2 Thermal Analysis
Analyses based on thermal energy principle have been widely employed to characterize amorphous pharmaceutical systems.
Crystallization from the amorphous state can be induced by the thermo-analytical techniques to produce an exothermic change whose magnitude is then quantitatively related to the extent of crystallization occurring. This can then be used to determine the crystallinity of a partially amorphous sample provided total crystallization of this sample is known to occur (Salekigerhardt et al. 1994). Differential scanning calorimetry (DSC) in both conventional and modulated modes has been used to quantify the extent of crystallinity.
Amorphous materials can be characterized by their glass transition temperature (T g). By DSC, T g is characterized by a change in heat capacity, which is seen as a change in the baseline. The T g may be important in determining the relative chemical and physical stability of formulations containing amorphous drugs (Yoshioka et al. 1994).
From a pharmaceutical perspective, it was thought that below the T g the molecular mobility was very low, and long-term product stability can be achieved by storing amorphous pharmaceuticals at sub-T g temperatures. In the amorphous form of a hypoglycemic agent for diabetes mellitus, with a glass transition temperature of 71 °C, no recrystallization was found after a 4-month storage at room temperature in the absence of moisture. However, crystallization occurred after storage at 50 °C for 2 months. The extent of recrystallization increased with increasing storage temperature. Some amorphous drugs with high T g can remain stable for extended times. For example, an API with a T g of 125 °C does not crystallize from the solid state in the absence of moisture (Clas et al. 1999). By using scanning calorimetry and thermo-mechanical methods, it was found that in order for the average relaxation time constants to significantly exceed the projected shelf life for a pharmaceutical product (approximately 3 years), it was generally necessary to store the amorphous materials as much as 50 °C below the glass transition region (Hancock et al. 1995; Hancock 2002).
The LOD and LOQ of crystallinity often tend to be better using DSC, with levels of 1–5 % and less than 1 %, respectively (Nagapudi and Jona 2008). DSC also requires much less material than PXRD.
11.3.4.1.3 Spectroscopy
Spectroscopic methods such as Raman, infrared (IR), and near-infrared (NIR) have also been reported for crystallinity quantitation (Head and Rydzak 2003; Brown et al. 2007). They provide chemically resolved information with small amount of material requirements. However, data interpretation from spectroscopic methods can run into problems because of their inability to unambiguously separate peaks from different phases in the sample.
Solid-state nuclear magnetic resonance (ssNMR) is another powerful technique which as yet has not been widely applied (Lefort et al. 2004). The primary advantage of ssNMR lies in its selectivity and ability to probe a variety of nuclei. Among the above-mentioned methods, ssNMR is the only technique that does not require a pure reference standard for phase quantitation. LOQ down to 0.25 % can be achieved when 1H, 31P, or 19F nuclei are used for quantitation, while LOQ of about 3 % can be achieved when using 13C. In spite of the high sensitivity, the usage of ssNMR has been limited due to instrument availability and relatively large amount of material is required (60–200 mg) (Nagapudi and Jona 2008). In contrast, PXRD and DSC have been widely used, as these techniques are usually available in most pharmaceutical labs.
11.3.4.2 Surface Analyses
11.3.4.2.1 Scanning Electron Microscopy (SEM)
SEM is recognized as unique tool in the visual examination of solid-state drug compositions and their surfaces. The resolution is of the order of nanometers (magnifications in the range ×20–×100,000). A fine beam of electrons of medium energy (5–50 keV) scans a gold–palladium-coated sample producing secondary electrons, backscattered electrons, light or cathodoluminescence, and X-rays. The latter allow for X-ray microanalysis for specific elements. SEM is routinely used for imaging particles in the micron and smaller size range and for examining the surfaces of larger particles. The resolution allows identification of specific surface geometric features that are indicative of structural phenomena.)
11.3.4.2.2 Atomic Force Microscopy (AFM)
AFM, a powerful surface and nanoimaging analytical technique, offers a unique opportunity to examine surface structure of a variety of materials with mesoscopic-scale resolution (10–6–10–9 m) and quantify the individual particle and excipient interaction by direct force measurement in a variety of environmental conditions (Jalili and Laxminarayana 2004). Especially for inhalation drug delivery, AFM is very useful to provide tailored investigations of particle–particle interactions within DPIs, particle–DPI wall interactions, and also perform in vivo simulations of inhaled particle–pulmonary surfactant interactions (Sindel and Zimmermann 2001).
AFM can also be used for phase imaging. Surface amorphous domains of sorbitol were reported to be identified and mapped by using both AFM and Raman microscopy (Ward et al. 2005). Also, AFM and Raman microscopy were utilized for screening of miscibility of drug-excipient and stability of solid dispersions. AFM assay technique is able to achieve the characterization of the miscibility and stability of molecularly disperse mixtures in just hours or a couple days instead of weeks or months. which is beneficial for product development (Lauer et al. 2011).
Weiss et al. (2015) recently reviewed the use of AFM as a powerful tool to characterize DPI formulations. This review included theories of adhesion and cohesion forces, summary of the AFM applications for drug particle and formulation characterization, and particularly emphasized its use as a colloidal probe. Assessment of interparticulate forces was also discussed.
11.3.4.2.3 Specific Surface Area Measurement
The surface area of a solid material is the total surface of the sample that is in contact with the external environment. It is expressed as square meters per gram of dry sample. Poorly water-soluble drugs are often rendered more available for absorption by reducing the particle size, i.e., increasing the surface area. Surface area is strongly related to the particle sizes, pore size, and the pore volume. The smaller the particle size and pore size, the higher the surface. The larger is the pore volume, the larger is the surface. The surface area results from the contribution of the internal surface of the pores plus the external surface of the pharmaceutical powders. The pharmaceutical powders generated by cryogenic technologies are generally fluffy and porous from visual and microscopic observations. For such porous systems, the contribution of the external surface to the total is very limited.
Physical and chemical gas adsorption and mercury intrusion porosimetry are the most widely used techniques to characterize powders and solid materials. Gas adsorption porosimetry typically can be performed using the Brunauer–Emmett–Teller (BET) theory (Brunauer et al. 1938) which allows for multilayer adsorption. With nitrogen gas adsorption, depending on the equipment used, pore diameter in the range of 0.3–300 nm, i.e., mesopores and macropores, can be determined.
Mercury intrusion porosimetry is another commonly used method to measure surface area of pharmaceutical powders. The principle, based on the Washburn model, consists of registering the volume of pores penetrated at each intrusion pressure, which can be easily transformed into pore size via the Washburn equation (Washburn 1921) to give a complete pore-size distribution (Carli and Motta 1984).
Both of these techniques can provide reliable information about pore-size/volume distribution, particle-size distribution, and specific surface area for porous solids regardless of their nature and shape.
11.3.4.2.4 Contact Angle
Contact angle is a quantitative measure of the wetting of a solid by a liquid. It is defined geometrically as the angle formed by a liquid at the three-phase boundary where a liquid, gas, and solid intersect. Pharmaceutically, wetting is not an end in itself but is the preliminary step in another process, e.g., dispersion or dissolution, both in vitro and in vivo. The improvement in wettability of a hydrophilic character probably was responsible for the increased dissolution rate of hydrophobic drugs (Lerk et al. 1976). Contact angle is therefore often determined as a measure of the surface energetics of drug substances.
Optical tensiometry (goniometry) is a commonly used method to measure contact angles of drug substances, which are compressed to form smooth and flat-faced tablets. Analysis of the shape of a sessile drop of test liquid placed on the flat-faced tablets is the basis for optical tensiometry. Contact angle can be assessed directly by measuring the angle formed between the solid and the tangent to the drop surface, as shown in Fig. 11.6.

Contact angles formed by a liquid at the liquid, gas, and solid three-phase intersections. Contact angle can be assessed directly by measuring the angle formed between the solid plane and the tangent to the drop surface using optical tensiometry (goniometry)
11.3.4.3 Other Techniques
11.3.4.3.1 Particle Size
Particle size is one of the physicochemical properties influencing the performance of drug product and its manufacturing processability and quality attributes (http://www.ich.org/). The influence includes dissolution rate, drug release profiles, and bioavailability; in vivo particle distribution and deposition, absorption rate, and clearance time; aerosolization behavior and performance of respiratory formulations; content and dose uniformity; and flow and packing properties, mixing and segregation of powders, etc.
There are various principles and techniques used for particle-size measurement. Among the techniques most commonly used in the pharmaceutical research and development, microscopy is often applied as an absolute particle-sizing method because it is the only method where the individual particles can be observed, measured, and their shape determined. Static and dynamic light scattering, Coulter counter (electrical zone sensing), time of flight (TOF), and cascade impactions are also widely used methods (Shekunov et al. 2007).
Pharmaceutical powders generated by cryogenic technologies were found suitable for pulmonary delivery (Vaughn et al. 2006; Yang et al. 2008b). The recent trend of systemic pulmonary drug delivery makes it very important to understand the correlation between the aerodynamic diameter, determined by in vitro measurements or in vivo lung deposition studies, and different geometric diameters measured by a variety of nonaerodynamic techniques. Andersen cascade impactor (ACI), next-generation impactor (NGI), and time-of-flight aerodynamic particle sizer (APS) are commonly employed to measure aerodynamic particle sizes.
11.3.4.3.2 Dissolution
It is well recognized that the low dissolution rate of poorly water-soluble drugs in biological fluids is the rate-limiting step to absorption. Amorphization of poorly water-soluble drugs can increase dissolution rates and achieve supersaturation, thereby the bioavailability (Yamashita et al. 2003). The amorphous forms, due to higher molecular mobility as compared to the corresponding crystalline form, may have enhanced dissolution rate and this difference can then be used to estimate the degree of amorphous content in a given sample. Although the amorphous form will have a higher dissolution rate because of higher free energy, and higher surface area after certain particle engineering processes, there is an inherent risk of recrystallization in the dissolution fluid. Nevertheless, the amount dissolved from a drug composition versus that from the pure crystalline drug, i.e., extent of supersaturation, has been used to quantify crystallinity in drug solid dispersion/solution systems.
11.3.4.3.3 Density Measurement
Solid density is a fundamental physical property of pharmaceutical powders. Generally, crystalline state of materials has a higher density than their amorphous counterparts because the atoms in the crystal lattice are located at a minimum possible distance from each other. An increase in lattice disorder (i.e., increase in amorphous phase) usually results in an increase in volume and therefore a decrease in density (Suryanarayan 1985). Hence, density can also be used as an alternative parameter for investigating the crystallinity of pharmaceutical powders. The cryogenic technologies discussed in this chapter can produce amorphous, low-density pharmaceutical powders. Bulk, tapped and true density measurements, as defined by US Pharmacopeia, are commonly used to characterize these pharmaceutical powders.
11.3.4.3.4 Dynamic Vapor Sorption (DVS)
DVS provides accurate gravimetric data in conjunction with a control of humidity and temperature. It does this by varying the vapor concentration surrounding the sample and measuring the change in mass which this produces in a humidity-controlled microbalance system. Gas and vapor absorption occur into the disordered regions located at the surface, and absorption phenomenon is known to accelerate where disordered surface regions are present. Water vapor is most commonly used, though it is possible to use a wide range of organic solvents. Water vapor sorption is particularly a useful method to assess the presence of amorphous material either as a single component or in combination (Costantino et al. 1998; Stubberud and Forbes 1998).
11.3.4.3.5 Inverse Gas Chromatography (IGC)
IGC is a vapor sorption technique for studying solids using gas chromatography principles, in which the powder is packed into or coated onto a chromatography column and a series of known nonpolar and polar probe gases are eluted. Interactions between the gas molecules and the stationary phase (powder) result in a characteristic net retention volume, which is used in the determination of free energy of adsorption, powder crystallinity, and other thermodynamic surface parameters (Hickey et al. 2007a).
IGC is nondestructive and considers only the outermost layer of the sample (Feeley et al. 1998), i.e., the region of the substance directly involved during interactions. It is a sensitive technique and recently has found its use in pharmaceuticals, such as in characterization of DPI (Sethuraman and Hickey 2002) and pMDI (Traini et al. 2005) formulations, for which adhesional properties are considered crucial for their aerodynamic performance. IGC can be used for the determination of surface energy and surface acid/base properties which directly influence adhesional properties.
11.4 Examples of Poorly Water-Soluble Drug Compositions Made by the Cryogenic Processes with Improved Dissolution Properties
In view of the above description of cryogenic technologies, step-by-step examples of using cryogenic technologies to engineer poorly water-soluble drug compositions to improve their dissolution profiles are provided below.
The majority of published research articles in the field of SFD focus on the preparation of water-soluble biological or proteinaceous materials for inhalation delivery, because of the processing constraints inherent in SFD. It is typically much easier to work with aqueous systems in SFD due to their low viscosity and low freezing point. Here, we provide two examples of poorly water-soluble drugs: cyclosporine A (CsA) and influenza vaccine processed using SFD.
11.4.1 Example 1
CsA, a potent immunosuppressant, is a BCS Class II drug with aqueous solubility of 3.69 μg/mL at 37 °C and log P value of 3.0. Previous research studies have shown that the administration of CsA to the lungs in addition to base therapy can increase the overall survival rate of lung transplant patients (Iacono et al. 1997; Costantino et al. 2004). However, due to its poor solubility, previous studies have utilized nebulized CsA in propylene glycol solution for inhalation to increase its bioavailability (Burkart et al. 2003). The inhalation of propylene glycol resulted in the removal of a number of patients from the study due to extreme irritation of the airways despite the use of local anesthesia. The use of rapidly dissolving, amorphous dry powder of CsA would alleviate the issues associated with the use of propylene glycol while still allowing for improved dissolution and bioavailability. Additionally, the higher deposition efficiency obtained by using a DPI would also allow for lower metered doses and lead to improved patient compliance. SFD-processed amorphous dry powders of CsA and inulin were prepared to examine its viability as a pulmonary delivery formulation.
11.5 Summary
Significant advances have been made in the past few decades toward understanding and tackling the poor bioavailability issues associated with poorly water-soluble drugs. Among various novel methods developed to improve dissolution properties for poorly water-soluble drugs, creation of amorphous pharmaceutical materials holds a lot of promise/demonstrated macroscopic performance advantages versus crystalline counterpart.
Cryogenic particle engineering technologies are “bottom-up” precipitation processes to generate amorphous nanostructured aggregates with significantly enlarged surface area, higher dissolution rates, and supersaturation, by rapidly inducing nucleation followed by particle growth arrest through stabilization via polymers and solidification of the solvent. The improved dissolution properties of poorly water-soluble drugs were achieved by (1) reducing particle size, thereby increasing surface area; (2) creating amorphous morphology; and (3) intimately mixing the drug with hydrophilic excipients. Moreover, without introducing mechanical force and heat, cryogenic processes are specifically suitable for thermal-labile drugs. Compared to SFD and SFL, TFF in particular has been shown to create stable amorphous compositions of poorly water-soluble drugs with significantly improved bioavailability and protein nanoparticles with high activity. Additionally, TFF process is more cost effective and scalable for manufacturing production.
By selecting proper excipient(s) and drug-to-excipient ratio in formulation and appropriate packaging and storage, cryogenic technologies can be used to manufacture stable amorphous drug dosage forms for diverse routes of administration, such as pulmonary, parenteral, and oral, with potentially reduced drug dose and side effects.
Method Capsule 1
Preparation of a Cyclosporine A Solid Dispersion for Inhalation by SFD
Based on the method reported by Zijlstra et al. (2007).
Objective
-
To obtain a dry powder formulation of CsA for pulmonary delivery with enhanced chemical and physical properties, such as wetting, dissolution rate, and aerodynamic performance.
Equipment and Reagents
-
Liquid nitrogen
-
CsA, inulin and tert-butyl alcohol
-
SFD apparatus equipped with a heater two-fluid nozzle (0.5 mm orifice)
-
Christ model Alpha 2–4 stage lyophilizer
Method
-
CsA and inulin were first dissolved into tert-butyl alcohol and water, respectively, and then mixed at a 40/60 volume ratio with a constant 5 % (w/v) solid loading.
-
Formulations with CsA to inulin ratios of 10, 20, 30, 50, and 100 % were prepared.
-
After mixing, solutions were sprayed into liquid nitrogen vapor at a flow rate of 6 mL/min with an atomizing airflow of 500 L/h.
-
After spraying, frozen droplets were then collected and placed into a precooled (−35 °C) stage lyophilizer.
-
Primary lyophilization was conducted with a stage temperature of −35 °C and a vacuum pressure of 165 mTorr for 24 h.
-
Secondary lyophilization was conducted with a stage temperature of −20 °C and a vacuum pressure of 37.5 mTorr for 24 h to obtain the final dry powder formulation.
Results
-
After SFD processing, both pure CsA and CsA/inulin formulations showed high specific surface areas ranging from 145 to 185 m2/g for CsA/inulin formulations and 40 m2/g for pure CsA.
-
Analysis of the secondary structure of the SFD-processed CsA formulations using FTIR confirmed the amorphous nature of these formulations.
-
Cascade impaction studies revealed that SFD-processed CsA formulations had high respirable (>75 %) and fine particle fractions (50 %), making them ideal candidates for inhalation delivery.
-
Dissolution testing showed that SFD-processed CsA formulations had superior wetting and dissolution performance compared to physical mixtures of bulk CsA and inulin.
This study highlighted the use of SFD as a method to prepare an amorphous dry powder formulation of CsA and inulin for pulmonary delivery. By utilizing SFD a high surface, wettable, and rapid dissolving formulation was prepared that had superior aerosol performance and enhanced dissolution. Based on these results, SFD process is a viable alternative to prepare CsA formulations for pulmonary delivery in lung transplant patients, compared to the propylene glycol-based CsA formulation.
11.5.1 Example 2
Influenza affects millions of people each year and has a high mortality for the elderly, infants, and other high-risk populations such as immunocompromised patients (Simonsen et al. 1998). Currently, the influenza vaccine products available on the market are all in liquid form and delivered either as a nasal mist or as an intramuscular injection. To ensure the stability of these liquid-based products prior to administration to the patient, cold chain storage and tracking are required, which add cost and limitations on the shipping and handling of these products. The administration of these vaccines via intramuscular injection is also a major hindrance to patient compliance and acceptance, and even the newer nasal administration forms of these vaccines, which are more acceptable to most patients, also still present a significant barrier to acceptance for a smaller subset of the population. This study utilized the SFD process to prepare a dry powder influenza vaccine to examine its viability as a needle-free epidermal delivery formulation.
Method Capsule 2
Preparation of an Influenza Vaccine Powder for Epidermal Delivery by SFD
Based on the method reported by Maa et al. (2004).
Objective
-
To prepare a stable and viable dry powder formulation of influenza vaccine for epidermal delivery with high density and narrow particle-size distribution.
Equipment and Reagents
-
Liquid nitrogen
-
Trehalose, mannitol, and dextran
-
SFD apparatus equipped with an ultrasonic atomizer nozzle (60 kHz frequency)
-
FTS systems DuraStop lyophilizer
Method
-
Influenza vaccines were first concentrated using tangential flow filtration followed by centrifugal filtration until a desired permeate concentration was collected.
-
Various formulations were then prepared at a final solid loading of 20 % to as high as 35 % by the direct addition of trehalose, mannitol, and dextran to the permeate solutions.
-
All final solutions were prepared at or below a viscosity of 2 poise to allow for effective atomization.
-
Solutions were then sprayed into liquid nitrogen vapor and frozen droplets were then collected and placed into a precooled (−10 °C) stage lyophilizer.
-
Primary lyophilization was conducted with a stage temperature of −10 °C and a vacuum pressure of 100 mTorr for 10 h.
-
Secondary lyophilization was conducted at 15 °C for 5 h and 25 °C for 5 h, both at 100 mTorr, to obtain the final dry powder.
Results
-
After SFD processing, all formulations exhibited a D(50) within the range of 30–60 μm, which is preferred for epidermal skin delivery.
-
Several formulations also exhibited a tapped powder density of greater than 0.5 g/mL, which is required for effective skin penetration in epidermal delivery.
-
Extensive stability and potency testing using sodium dodecyl sulfate–polyacrylamide gel electrophoresis, single radial immunodiffusion assay, and in vivo immunogenicity testing in a mouse model all confirmed that the various formulations remained stable and potent after SFD processing with one formulation exhibiting stability for more than 6 months at 25 °C.
This study highlighted the use of SFD as a method to prepare a dry powder form of the influenza vaccine for epidermal delivery. By utilizing SFD, a stable dry powder with appropriate particle size and density was prepared with excellent long-term stability and no significant loss of antigen potency. Based on these results, it would appear that dry powder forms of influenza vaccine prepared by SFD for epidermal delivery is a viable alternative to the current liquid formulations for intramuscular injection or as a nasal mist.
Early studies of SFL focused on developing drug/excipient(s) combinations to achieve dissolution enhancement, increase drug loading capabilities, and on the effect of solvent systems in achieving higher drug/stabilizer ratios while maintaining enhanced dissolution rates.
11.5.2 Example 3
Carbamazepine was the first poorly water-soluble drug used as a model for SFL process. Carbamazepine is an anticonvulsant and mood-stabilizing drug used primarily in the treatment of epilepsy. Although the drug has been in use for more than 20 years, oral administration of carbamazepine encounters multiple challenges, including low aqueous solubility (17.7 μg/mL at 25 °C, log P value of 2.45) with high dosage required for therapeutic effect (more than 100 mg/day), a narrow therapeutic window, and dissolution-limited bioavailability (Bertilsson and Tomson 1986). Carbamazepine is classified as a BCS Class II drug (Amidon et al. 1995). The low rate of dissolution in aqueous biological media is considered to be a likely cause of the irregular and delayed absorption issues encountered with the oral delivery of carbamazepine (Moneghini et al. 2001). In vivo pharmacokinetic studies revealed a strong correlation between oral bioavailability and the physical form and formulation of carbamazepine (Hickey et al. 2007a, b), suggesting that improvement of the dissolution properties of carbamazepine can result in improved pharmacokinetics and bioavailability. Rogers and co-workers (2002a, b) developed a novel composition of carbamazepine made by SFL process to improve the dissolution profile.
Method Capsule 3
Preparation of Engineered Carbamazepine Compositions by SFL
Based on the method reported by Rogers et al. (2002a, b).
Objective
-
To enhance the dissolution rate of poorly water-soluble drug carbamazepine by using a novel SFL process to engineer the drug composition.
Equipment and Materials
-
SFL apparatus
-
Liquid nitrogen
-
Carbamazepine, sodium lauryl sulfate (SLS), tetrahydrofuran (THF), and purified water
-
Bench-top tray lyophilizer
-
Desiccator and desiccant
Method
-
Prepare carbamazepine feed solution by dissolving the drug in THF, dissolving the hydrophilic excipient SLS in purified water, as listed in the table below, then mix the organic and aqueous solutions to form a one-phase cosolvent solution with a solid loading of 0.44 % (w/v).
Component
Solvent
Organic phase
Carbamazepine, 0.20 g
THF, 29.80 g
Aqueous phase
SLS, 0.20 g
Purified water, 59.6 g
-
Fill a clean large 4-L insulated beaker with liquid nitrogen.
-
Spray and atomize the carbamazepine feed solution beneath liquid nitrogen surface at 5000 psi constant pressure through a 10-cm-long, 63.5-μm ID PEEK nozzle into the beaker by a syringe pump.
-
Monitor liquid nitrogen level in the beaker to ensure that the PEEK nozzle is kept beneath liquid nitrogen level during the spray.
-
Collect the frozen material on a 150-mesh sieve or wait till the liquid nitrogen evaporated after the spray is completed.
-
Transfer the collected frozen material to a precooled shelf in a tray lyophilizer to maintain the frozen material and remove solvents.
-
Store the obtained flowable dry powder of carbamazepine composition at room temperature under vacuum in the desiccator.
Results
-
SEM images demonstrated that porous micron-sized particles consisting of carbamazepine and SLS matrix system were created by SFL process.
-
XRD indicated that the highly crystalline carbamazepine bulk powder was transferred to complete amorphous state by the SFL process.
-
SFL-processed carbamazepine composition was wetted and dissolved immediately in purified water; almost 100 % carbamazepine was released within 10 min, significantly greater than carbamazepine bulk powder and the physical mixture of carbamazepine and SLS.
This study was a proof of concept that SFL process can significantly improve the dissolution properties of poorly water-soluble drug.
11.5.3 Example 4
Danazol is a synthetic steroid derived from ethisterone that has low aqueous solubility (0.5 μg/mL at 37 °C) with high permeability across biological membranes (log P value of 4.53) (Bakatselou et al. 1991; Badawya et al. 1996). Therefore, danazol is classified as BCS Class II drug. The oral bioavailability of danazol is dissolution rate-limited. Many techniques have been utilized to enhance the dissolution of danazol. However, because of the practically insoluble nature of crystalline danazol, the improvement of aqueous solubility was limited.
The several different danazol formulations engineered by SFL process were reported (Hu et al. 2002; Rogers et al. 2002a, b). After initial proof-of-concept studies, formulations with high potency and enhanced stability of amorphous state (i.e., achieved high T g), besides high dissolution rates, were designed.
Method Capsule 4
Preparation of Engineered High-Potency Danazol Compositions by SFL
Based on the study reported by Hu et al. (2004a, b, c).
Objective
-
To investigate the use of organic solvents in the SFL particle engineering process to make rapid-dissolving high-potency danazol powders and to examine their particle size, surface area, and dissolution rate.
Equipment and Materials
-
SFL apparatus
-
Liquid nitrogen
-
Danazol micronized powder, polyvinylpyrrolidone (PVP) K-15, sodium lauryl sulfate (SLS), acetonitrile, dichloromethane (DCM), and purified water
-
Bench-top tray lyophilizer
-
Desiccator and desiccant
Method
-
Prepare danazol feed solutions by dissolving the drug and PVP K-15 in various weight ratios in acetonitrile or acetonitrile/dichloromethane mixtures to form solutions with total solid loading up to 1.6 % (w/v), as listed in the table below.
Danazol/PVP K-15 ratio (w/w)
Danazol (g)
PVP K-15 (g)
Acetonitrile (mL)
DCM (mL)
Potency (%)
Solid loading (%)
1:2
0.2
0.4
70
–
33
0.86
1:1
0.2
0.2
70
–
50
0.57
2:1
0.4
0.2
70
–
66
0.86
3:1
0.6
0.2
65
5
75
1.14
10:1
1
0.1
55
15
91
1.57
-
Fill a clean insulated container with liquid nitrogen.
-
Spray and atomize the danazol feed solutions beneath liquid nitrogen surface, with a constant pressure of 2000 psi to provide a flow rate of 50 mL/min for the feed solution to spray through PEEK tubing of 127 μm ID into liquid nitrogen using a syringe pump.
-
Monitor liquid nitrogen level in the beaker to ensure that the PEEK nozzle is kept beneath liquid nitrogen level during the spray.
-
Collect the frozen material on a 150-mesh sieve or wait till the liquid nitrogen evaporated after the spray is completed.
-
Transfer the collected frozen material to a precooled shelf in a tray lyophilizer to maintain the frozen material and remove solvents. A cold trap is connected to the lyophilizer for the compositions prepared with DCM.
-
Store the obtained dried powders in glass vials at room temperature under vacuum in the desiccator.
Results
-
XRD indicated that the SFL-processed micronized danazol/PVP K-15 compositions with potencies of 50–91 % were all amorphous.
-
Surface areas of these SFL-processed danazol/PVP K-15 powders were in the range of 28–115 m2/g, in a reversed order to the increasing solid loading of the SFL feed solutions.
-
Contact angles of these SFL-processed danazol/PVP K-15 powders were in the range of 22–35°, much reduced compared to 57° of the unprocessed bulk danazol; among the SFL-processed compositions, contact angles increased with increasing potencies of danazol in the compositions.
-
SFL-processed danazol compositions exhibited significantly enhanced dissolution rates with 95 % of danazol dissolved in only 2 min for the high-potency composition, whereas the micronized bulk danazol dissolved slowly with only 30 % of the danazol released in the same time frame.
The rapid freezing of SFL process produced porous, nanostructured aggregates of the danazol/PVP K-15 compositions as seen in Fig. 11.7b, with smooth primary particle size of about 100 nm in diameter as shown in Fig. 11.7c, d, in contrast to the micron-sized crystalline bulk danazol in Fig. 11.7a. Because SFL-processed powders have high surface areas and contain amorphous danazol, enhanced dissolution of the poorly water-soluble drug in aqueous media was achieved.

Representative SEM images of SFL-processed danazol/PVP K-15 compositions: bulk micronized danazol (a); SFL danazol/PVP K-15 (50 % potency) powder at magnification of 20k (b); at magnification of 60k (c); at magnification of 200k (d). Adapted from Hu et al. (2004a, b, c) with permission from Elsevier
A subsequent stability study was conducted for the SFL-processed danazol/PVP K-15 powder (75 % potency) at cycle conditions (−5 to 40 °C every 3 h). It was found that the amorphous structure and rapid dissolution characteristics were maintained after 1 month of cycled stability conditions (Hu et al. 2004a, b, c). The high stability of amorphous SFL powders was partially attributed to the selection of PVP K-15 as stabilizer in the composition. PVP K-15 has a T g of 146 °C; the interaction of danazol with PVP K-15 may lead to a reduction in the molecular mobility of danazol in the formed solid solution/dispersion of danazol/PVP K-15 powder.
11.5.4 Example 5
Because of the innate hydrophobicity of the poorly water-soluble drugs, there was an upper limit for the drug concentrations that could be dissolved in solvent/cosolvent system. Low drug concentrations in the feed solution for SFL process resulted. To further increase drug loading in feed solution, o/w emulsions with higher danazol and excipient concentrations were formulated for SFL process (Rogers et al. 2003b), as the total concentration of drug in the o/w emulsions could be much larger than in the cosolvent because of the high solubility of hydrophobic drug in the internal organic phase of the emulsion.
Method Capsule 5
Preparation of Amorphous High-Potency Danazol Compositions Using SFL Processed o/w Emulsions
Based on the study reported by Rogers et al. (2003a, b).
Objective
-
To further increase drug loading in feed solution, o/w emulsions with higher danazol and excipient concentrations were formulated for SFL process.
Equipment and Materials
-
SFL apparatus
-
Liquid nitrogen
-
Danazol micronized powder, poly(vinyl alcohol (PVA, MW 22 000), Poloxamer 407, PVP K-15, THF, ethyl acetate dichloromethane (DCM), and purified water
-
Rotor-and-stator homogenizer, high-pressure homogenizer
-
Bench-top tray lyophilizer
-
Desiccator and desiccant
Method
-
Prepare danazol SFL solution by dissolving the drug and excipients in THF and water, respectively, then mix the organic and aqueous phases to form feed solution with a potency of 40 %, as listed in the table below.
Formulations
Component
Wt. ratio
Solvent
Potency (%)
Solution
Danazol
2
THF/water
40
PVA (MW 22 000)
1
Poloxamer 407
1
PVP K-15
1
Emulsion
Danazol
20
Ethyl acetate/water, or DCM/water
87
PVA (MW 22 000)
1
Poloxamer 407
1
PVP K-15
1
-
Prepare oil-in-water (o/w) emulsions of danazol for SFL process by high-pressure homogenization. Dissolve danazol and excipients in organic solvents and water, respectively. Slowly pour the organic phase into the aqueous phase under constant mixing, then blend for 1 min using a high-speed rotor-and-stator homogenizer. Further homogenize the emulsion for 10 cycles at 20,000 PSI (138 MPa) using a high-pressure homogenizer to reduce the oil droplets to <1 μm in diameter.
-
Fill clean insulated containers with liquid nitrogen for SFL process.
-
Spray and atomize the danazol feed solution and emulsions beneath liquid nitrogen surface at 5000 psi (34.5 MPa) at a flow rate of 20 mL/min through a 127 μm ID PEEK nozzle measuring 15 cm in length by a syringe pump.
-
Monitor and top up liquid nitrogen level in the container to ensure that the PEEK nozzle merged under liquid nitrogen level all the time.
-
Collect the frozen material on a 150-mesh sieve or wait till the liquid nitrogen evaporated after the spray is completed.
-
Lyophilize the collected frozen material using a tray lyophilizer equipped with a liquid nitrogen cold trap to condense DCM or ethyl acetate, of which the low melting points exceed the capture capacity of condenser. Maintain vacuum of 100 mTorr throughout the lyophilization cycle.
-
Store the obtained dried powders under vacuum in the desiccator at room temperature.
Results
-
Total drug/excipients concentrations of 5.75–7.5 % were achieved in the emulsion formulations, compared to that of 0.55 % in the cosolvent solution composition.
-
XRD indicated that the lyophilized danazol SFL-solution composition and SFL-emulsion compositions were amorphous, even for SFL-emulsion compositions with high API-to-excipients ratio of 20:3.
-
Surface areas increased with increasing drug and excipient concentrations, ranging from 8.9 m2/g of the SFL-solution composition to 83.1 m2/g of the SFL-emulsion compositions.
-
Both danazol SFL-solution composition and SFL-emulsion compositions wetted and dissolved rapidly (100 % in 2 min) than the slowly frozen counterpart and bulk danazol (50 % in 2 min). Even for the SFL-emulsion compositions of high API-to-excipients ratios, >90 % of the danazol dissolved within 5 min. The SFL-emulsion compositions retained the high dissolution rates that achieved from SFL-solution composition.
High-potency formulations with high drug-to-excipient ratios and rapid dissolution rates would be advantageous in increasing dosages and in ameliorating side effects attributed to less excipients needed.
11.5.5 Example 6
The SFL and TFF particle engineering technologies were not only utilized to enhance dissolution rate of poorly water-soluble compounds but also used to enhance the stability of potentially labile compounds, such as proteins and peptides such as lactate dehydrogenase (LDH), and influenza vaccine (Yu et al. 2004; Engstrom et al. 2007a, b).
Due to the instability of proteins in solution, it is often necessary to produce a solid protein composition to achieve an acceptable shelf life. Ideally, the formulation should achieve high protein loadings with minimum burst release, high surface area, and submicron protein particles uniformly incorporated into 10–50 μm microspheres. However, it is challenging to produce stable submicron protein particles with surface area exceeding 10 m2/g, relative to less than 1 m2/g for lyophilized formulation (Engstrom et al. 2007a, b). It has been shown that the SFL process leads to less protein denaturation and subsequent aggregation relative to SFD since the destabilizing gas–liquid interfacial area is lower (Yu et al. 2006).
LDH is a fragile protein extensively studied. Producing stable high surface area submicron particles of LDH is of practical interest in protein storage and in various applications in controlled release. TFF process can also be used to produce stable submicron protein particle without loss of protein activity. Engstrom et al. reported LDH and lysozyme particles engineered by TFF process (Engstrom et al. 2008), with the detailed procedures described as following.
Method Capsule 6
Production of Highly Stable, Submicron Protein Particles by TFF
Based on the study reported by Engstrom et al. (2008).
Objective
-
To produce highly stable, submicron LDH and lysozyme particles by TFF processing the aqueous solutions followed by lyophilization.
Equipment and Materials
-
TFF apparatus
-
Liquid nitrogen
-
LDH, lysozyme, trehalose, and purified water
-
Bench top tray lyophilizer
-
Desiccator and desiccant
-
Particle-size analyzer by laser light scattering
-
BET apparatus
Method
-
Prepare 0.25 mg/mL LDH in both 30 and 100 mg/mL trehalose solutions in 10 mM K3PO4 buffer with pH 7.5. Prepare 5 and 50 mg/mL aqueous solutions of lysozyme.
-
Fill dry ice inside the hollow cylindrical stainless-steel drum. An equilibrium drum surface temperature of −50 °C was achieved.
-
TFF process of feed solutions: pass the protein feed solutions through a 17-gauge stainless-steel syringe needle at 4 mL/min to produce individual liquid droplets. The droplets fell from a height of 10 cm above a rotating stainless-steel cryogenic drum. Drum surface temperature was monitored by a moving surface temperature probe. Upon impacting, the droplets freeze simultaneously into round thin film disks.
-
Collect the frozen thin disks into a 400-mL glass beaker filled with liquid nitrogen.
-
Transfer the glass beakers containing the frozen disks of proteins to a −80 °C freezer to evaporate excess liquid nitrogen.
-
Cover the beakers with a single layer Kim-wipe to prevent particles exiting during drying, and transfer into a tray lyophilizer with precooled shelf temperature of −40 °C. Primary drying was carried out at −40 °C for 36 h at 300 mTorr and secondary drying at 25 °C for 24 h at 100 mTorr.
-
Upon completion of the lyophilization, purge the lyophilizer with nitrogen to release vacuum to minimize exposure of the protein powders to moisture in the ambient air.
-
Rapidly transfer the dried powders to a dry box with humidity less than 15 % RH, and package the powders into 20-mL scintillation vials. Purge vials with dry nitrogen gas for 2 min via a needle through the septa and an additional needle for the gas effluent.
-
Store the protein powders in glass vials at room temperature under vacuum in desiccator.
Results
-
Protein particles with an average diameter of about 300 nm and 100 % enzyme activity upon reconstitution (for LDH) were engineered by TFF process.
-
The surface areas were significantly enlarged by TFF process, in the range of 30–75 m2/g. The higher the concentration of protein loading in the feed solution, the lesser the surface area of protein particles.
In TFF, the exposure of protein to the gas–liquid interface is minimized. The TFF-processed LDH composition was determined very high enzyme activities upon reconstitution, similar to that processed by direct lyophilization. TFF process has intermediate cooling rate (102 K/s), relative to that of the ultra-rapid cooling process SFD (106 K/s) and the slow process lyophilization (1 K/min), as shown in Fig. 11.8. Although the cooling rate of TFF is slower than that of SFL (103 K/s), it was sufficiently fast to arrest particle growth, whereas the relatively minimized liquid–gas interfacial surface area in TFF process can improve protein stability by limiting the amount of protein adsorption to the interface, unfolding, and aggregation.

SEM of particles from 5 mg/mL lysozyme solutions processed by thin film freezing (TFF) at surface temperatures of −50 °C (a) and processed by spray freeze into liquid nitrogen (SFL) (b). SEM of particles from 50 mg/mL lysozyme solution processed by TFF at surface temperatures of −50 °C (c) and processed by SFL (d). Adapted from Engstrom et al. 2008 with permission from Springer
Later studies mainly focus on TFF-technology-engineered solid solutions/dispersions of poorly water-soluble drugs, such as tacrolimus, sirolimus, itraconazole, and repaglinide, to achieve supersaturated dissolution properties. Correlations between the enhanced in vitro properties of engineered drug compositions to improved in vivo performance in animal models were sought, as described in the following examples.
11.5.6 Example 7
Tacrolimus is a hydrophobic macrolide antibiotics used as a potent immunosuppressive agent, and has superior immunosuppressive effect compared to cyclosporine A. However, the erratic oral absorption profiles of tacrolimus have limited its therapeutic efficiency. It was reported that the oral bioavailability of tacrolimus ranges from 4 to 93 % with a mean value of 25 % (Wallemacq and Verbeeck 2001). Various innovative formulations and technologies have been used to improve the bioavailability of tacrolimus, with the theory that increasing the solubility greater than twofold through polymorphism can have a significant increase in biopharmaceutical activity. TFF technology was also employed to make engineered tacrolimus compositions with hydrophilic stabilizers (Overhoff et al. 2008), as described below.
Method Capsule 7
Preparation of Tacrolimus Solid Dispersions for Oral Delivery Using TFF
Based on the study reported by Overhoff et al. (2008).
Objective
-
To investigate tacrolimus solid dispersions containing various stabilizers prepared by TFF process, and to determine the effect on their ability to form supersaturated solutions in aqueous media and on enhancing transport across biological membranes.
Equipment and Materials
-
TFF apparatus
-
Liquid nitrogen
-
Tacrolimus (TAC), poly(vinyl alcohol (PVA), poloxamer 407 (P407), and sodium dodecyl sulfate (SDS)
-
Bench-top tray lyophilizer
-
Desiccator and desiccant
-
Dissolution tester
Method
-
Prepare feed solutions of TAC compositions by dissolving TAC and excipient(s) at 1:1 (w/w) ratio and 1.0 % solid loading in a cosolvent acetontrile/water (60/40, v/v), as listed in the table below.
TAC/excipient(s) ratio (w/w)
Excipient(s)
Acetonitrile/water
Potency (%)
Solid loading (%)
1:1
SDS
60/40
50
1.0
1:1
PVA/P407 (1/1)
60/40
50
1.0
1:1
P407
60/40
50
1.0
-
Apply the feed solutions to the rotating stainless-steel drum that precooled to −150 °C drop-wisely from a glass funnel tip 10 cm above the drum. The feed solution drops spread and form thin layer frozen disk simultaneously upon impacting on the drum.
-
Place a metal pan filled with liquid nitrogen under the drum to collect and maintain the generated frozen material.
-
Transfer immediately the frozen material into a bench-top tray lyophilizer with shelf precooled to −60 °C after evaporation of excessive liquid nitrogen in the collecting pan, and start lyophilization.
-
Store the obtained dried powders of TAC compositions at room temperature under vacuum in the desiccator until characterization.
Results
-
XRD indicated that the TFF-engineered TAC compositions were all amorphous.
-
SEM displayed highly porous network of nanostructured aggregates of the TFF-engineered TAC compositions, in contrast to the plate-shaped crystalline bulk TAC with particle sizes ranging from a few microns to over 120 μm in diameter.
-
Supersaturation dissolution testing demonstrated the three TFF-engineered TAC compositions, as well as the commercial product Prograf® showed rapid dissolution reaching their maximum supersaturation within 2 h and achieved supersaturation relative to the solubility of bulk crystalline TAC. However, only the TFF-processed TAC/SDS = 1/1 composition had higher supersaturation than Prograf®.
In this study, the selected stabilizers including partially hydrolyzed PVA which has been shown to increase drug concentration in vivo (Suzuki and Sunada 1998), P407 which has been shown to alter surface properties of crystals, and SDS which is a nonpolymeric anionic surfactant. SDS may be used to facilitate wetting and dissolution rates. TAC has good solubility in organic solvent and is readily dissolvable in the cosolvent.
Single-dose pharmacokinetic study of orally administered TAC compositions in a rat model was conducted to determine the in vivo performance of the TFF-processed compositions. The results suggested that TFF-processed TAC/P407 = 1/1 achieved the greatest absorption with a 1.5-fold increase in AUC and higher C max compared to Prograf®. All the TFF-engineered tacrolimus compositions had a shorter T max compared to Prograf®, as seen from Fig. 11.9. It is therefore concluded that the enhanced physico-chemical properties of TFF-engineered TAC compositions led to enhanced in vivo absorption over the current commercial product of TAC.

Mean whole blood absorption levels of tacrolimus compositions produced using the TFF process compared to Prograf®. Powders were given in gelatin capsule containing 1.5 mg equivalent tacrolimus (5 mg/kg) dosed via oral gavage to a rat model: TFF-processed tacrolimus/SDS (filled square), TFF-processed tacrolimus/PVA:P407 (filled triangle), TFF-processed tacrolimus/P407 (filled circle), Prograf® capsule powder (filled diamond). Reproduced from Overhoff et al. (2008) with permission from Springer
11.5.7 Example 8
Itraconazole is a broad-spectrum antimycotic triazole used for both prophylaxis and treatment of invasive fungal diseases for the last two decades. Itraconazole has pH-dependent solubility with extremely low value of approximately 1 ng/mL at neutral pH and approximately 4 μg/mL at pH 1 (Peeters et al. 2002). Given the high log P value of 6.2, itraconazole is classified as a BCS class II drug (Amidon et al. 1995). Sporanox® oral capsule is a currently available marketed oral dosage form of itraconazole. However, the oral absorption of ITZ in a subset of immunocompromised patients was not optimal, and the pharmacokinetics varied considerably among patients (Poirier et al. 1997). To treat invasive fungal infection, especially Aspergillus spp. infections, itraconazole levels of greater than 0.5 μg/g of lung tissue, or 0.5 μg/mL of blood, are required (Sobel 2000).
A novel composition containing itraconazole:mannitol:lecithin (1:0.5:0.2, w/w/w) for pulmonary delivery was made by TFF technology (Yang et al. 2008b). Here, we provide a step-by-step procedure for a standard batch of the itraconazole composition.
Method Capsule 8
Preparation of Amorphous Nanoparticulate Itraconazole Composition for Pulmonary Delivery Using TFF
Based on the study reported by Yang et al. (2008a, b).
Objective
-
To develop amorphous nanoparticulate formulations of itraconazole for improved bioavailability following pulmonary administration.
Equipment and Materials
-
TFF apparatus
-
Liquid nitrogen
-
Itraconazole, mannitol, lecithin, 1,4-dioxane, and purified water
-
Bench-top tray lyophilizer
-
Desiccator and desiccant
Method
-
Prepare itraconazole feed solution: dissolve lecithin (118 mg) in 200 mL co-solvent of 1,4-dioxane and purified water (65/35, v/v); Subsequently dissolve itraconazole (588 mg) and mannitol (294 mg) to form a solution of 0.5 % (w/v) solid loading with itraconazole: mannitol: lecithin = 1:0.5:0.2 weight ratio.
-
Precool the stainless-steel drum of TFF apparatus to −70 °C.
-
Apply the feed solution to the cold drum of TFF apparatus from a glass funnel set 10 cm above the top surface of the rotating stainless-steel drum.
-
Collect the generated frozen material in a container filled with liquid nitrogen.
-
Transfer immediately the frozen material into a bench-top tray lyophilizer with shelf precooled to −20 °C after evaporation of excessive liquid nitrogen, and start lyophilization.
-
Store the lyophilized dry powder at room temperature in the desiccator under vacuum.
Results
-
XRD and DSC confirmed that TFF-processed itraconazole:mannitol:lecithin = 1:0.5:0.2 was amorphous.
-
SEM images showed the TFF-processed itraconazole composition has a highly porous structure with more regularly round-shaped particles in aggregated network.
-
Aqueous colloidal dispersion of the TFF-processed itraconazole composition has a mean particle size of 230 nm.
-
Dissolution testing revealed TFF-processed itraconazole composition achieved about five-times higher supersaturation than a crystalline Wet-milled itraconazole.
For comparison, a crystalline itraconazole nanoparticle composition was made by wet ball milling process (named Wet-milled itraconazole). SEM images showed that Wet-milled itraconazole was composed of fractured, irregular-shaped particles with various sizes, ranging from about 150 to 600 nm in length, as shown in Fig. 11.10a. In contrast, TFF-processed itraconazole composition exhibited a highly porous structure with more regularly round-shaped particles in aggregated network, as shown in Fig. 11.10b. Both the milling and TFF process dramatically reduced itraconazole particles to nanosize range compared to bulk micron-sized itraconazole particles shown in Fig. 11.10c.

SEM images of (a) wet-milled itraconazole (wet milling–processed pure ITZ) powder at a magnification of 20k, (b) URF–ITZ (URF-processed ITZ/mannitol/lecithin = 1:0.5:0.2, weight ratio) powder at a magnification of 20k, and (c) bulk ITZ as received at a magnification of 10k. Reproduced from Yang et al. (2010) with permission from Elsevier
Their corresponding mean particle size in aqueous dispersion was 230 and 570 nm, respectively. Dissolution testing revealed TFF-processed itraconazole composition achieved about 27-times higher supersaturation versus itraconazole equilibrium solubility, and five times higher dissolved itraconazole than the Wet-milled itraconazole, as seen from Fig. 11.11 (Yang et al. 2010).

Dissolution profiles of Wet-milled itraconazole colloidal dispersion (ITZ/mannitol/lecithin = 1:0.5:0.2) and TFF-processed itraconazole/mannitol/lecithin = 1:0.5:0.2 colloidal dispersion in simulated lung fluid (pH 7.4) at supersaturation conditions (i.e., 100-times equilibrium solubility of micronized crystalline itraconazole was added) and 37 °C. Reproduced from Yang et al. (2010) with permission from Elsevier
A subsequent in vivo single-dose 24 h pharmacokinetic study of inhaled nebulized colloidal dispersions of the TFF-processed amorphous itraconazole composition and the crystalline Wet-milled itraconazole (equivalent to 20 mg itraconazole/mL) were conducted in a rat model. The results demonstrated a significantly higher systemic absorption and Cmax of itraconazole in blood of rats inhaled TFF-processed amorphous itraconazole composition than those inhaled crystalline Wet-milled itraconazole (Fig. 11.12a), although the lung depositions of itraconazole were comparable for both inhaled compositions (Fig. 11.12b). It is concluded from this study that using TFF technology to make amorphous nanoparticulate composition of poorly water-soluble drugs is an alternative and promising approach to overcome the low aqueous solubility issues by providing higher dissolution rate and apparent solubility, and subsequently higher bioavailability.

(a) Plasma concentration of itraconazole in rats, (b) lung deposition of itraconazole in rats at 0 and 24 h post inhalation of a single-dose nebulized aqueous Wet-milled itraconazole colloidal dispersion (ITZ/mannitol/lecithin = 1:0.5:0.2) and TFF-processed itraconazole/mannitol/lecithin = 1:0.5:0.2 colloidal dispersion. Data are presented as mean ± SD. *p < 0.05, **p < 0.01. Adapted from Yang et al. (2010) with permission from Elsevier
These examples are merely few representatives of the many poorly water-soluble drugs that contemplated by the cryogenic particle engineering technologies for enhanced dissolution properties and hence improved bioavailability.
TFF technology has been successfully used for cGMP manufacturing of stable amorphous drug products for clinical trial with improved patient compliance.
Method Capsule 9
Preparation of Amorphous Fenofibrate Solid Disp ersions by TFF
Based on the study reported by Zhang et al. (2012).
Objective
-
To prepare amorphous fenofibrate solid dispersions using the TFF method and to incorporate the solid dispersions into pharmaceutically acceptable dosage forms for improving the bioavailability.
Equipment and Materials
-
TFF apparatus
-
Liquid nitrogen
-
Fenofibrate (FB), fenofibric acid, Methocel® E5, Soluplus®, Hydroxypropyl methylcellulose phthalate NF (HP55), Hydroxypropyl methylcellulose acetate succinate LF (HPMCAS-LF), 1,4-dioxane and water
-
Bench top tray freeze dryer
-
Particle-size analyzer by laser light scattering
-
BET apparatus
-
Differential scanning calorimeter (DSC)
-
Powder X-ray diffraction (XRD)
-
Fourier-transform infrared spectroscopy-attenuated total reflectance (FTIR-ATR)
-
Dissolution tester
Method
-
Prepare 1 % w/v solids content of FB:polymer at designed ratio of 1:4, 1:6 or 1:8 in 1,4-dioxane or its mixture with water at ratio of 8:2 v/v as shown in the table below
Formulation
FB:polymer ratio (%w/w)
Solvent
FB:Soluplus
1:4, 1:6, 1:8
1,4-Dioxane
FB:HPMC E5
1:4, 1:6, 1:8
1,4-Dioxane:water (8:2, v/v)
FB:HPMCAS
1:4, 1:6, 1:8
1,4-Dioxane
FB:HP55
1:4, 1:6, 1:8
1,4-Dioxane
-
Apply solutions of FB-excipient onto the pre-cooled cryogenic substrate (−45 °C)
-
Collect frozen solids and lyophilize using freeze dryer in which the temperature increased from −40 to 25 °C over 48 h
-
Store the dry powders in desiccator at room temperature under vacuum condition
Results
-
FB-polymer solid dispersions prepared by TFF technique provided extremely high surface area, microstructure, and wettability.
-
Morphology of amorphous aggregates of samples using SEM illustrated a sponge-like structure resulting in the creation of high surface area and the formation of loose connection between particles.
-
Both PXRD and MDSC results revealed the drug was molecularly dispersed in polymers.
-
There were no crystals of FB observed in TFF samples.
-
Supersaturation dissolution of FB solid dispersions prepared by TFF method showed success in enhancing drug release that exceeding the equilibrium solubility and maintaining FB in solution.
-
In vivo study demonstrated that levels of plasma drug concentration of the amorphous FB prepared using TFF processing were drastically higher than the bulk FB.
The morphology of the porous aggregates of amorphous FB solid dispersion from the SEM images agreed with those surface area measurements using BET. The specific surface area of the TFF formulations increased as compared with the bulk FB. Preparation of homogeneous solutions prior to the rapid freezing procedure resulted in no crystalline structure observed in TFF formulations. Polymers possibly prevented crystal growth of FB due to the fact that they were adsorbed on the drug crystal interface and the crystal nucleation was inhibited.
Dissolution of amorphous FB significantly increased after TFF processing. FB-HP-55 formulation had a largest surface area as compared to the other formulations. Although, it unexpectedly exhibited the worst dissolution results under supersaturation conditions and in the dissolution media the recrystallization rate of this formulation was fastest. This resulted from the decrease of FB solubility in acidic solution. HP-55 contains a larger number of acidic groups compared to other polymers, which results in the poorest dissolution performance of FB. After the amorphous TFF powders were compressed into slug tablets, compression triggered the recrystallization of FB, which resulted in lower dissolution.
Figure 11.13 showed the success of the enhancement of drug absorption of FB solid dispersion. This significant improvement of oral bioavailability was achieved in these non-surfactant, non-porous excipients processed through the TFF approach.

Pharmacokinetic profile of drug plasma concentration of fenofibric acid after single dose oral administration of different formulations to rats. Reproduced from Zhang et al. (2012) with permission
Method Capsule 10
Development of Voriconazole Dry Powder Inhalation Produced by TFF
Based on the method reported by Beinborn et al. (2012).
Objective
-
To manufacture voriconazole formulations using TFF to enhance properties for dry powder inhalation and to study the process parameters which impact the morphology and aerodynamic properties of the resulting formulations.
Equipment and Reagents
-
Liquid nitrogen
-
Voriconazole (VRC), lactose monohydrate, polyvinylpyrrolidone K30 (Povidone K-30, USP), polyvinylpyrrolidone K12 (Plasdone® K12), hydroxypropyl methylcellulose (HPMC) K3 (Methocel™ K3), 1,4-dioxane and methanol.
-
TFF apparatus
-
Bench top tray freeze dryer
Method
-
Prepare VRC TFF formulations in various process parameters including the effect of stabilizing excipient, drug to excipient ratio, percentage of solids content, and solvent composition
-
Dissolve VRC and excipients in 1,4-dioxane or deionized water
-
Drop the solutions onto a precooled rotating cryogenic, steel surface (approximately −40 °C) to produce frozen thin films
-
Remove thin films from the steel surface using a scraper and keep in frozen mass in liquid nitrogen
-
Sublimate the solvents by lyophilization to dry powders (perform the lyophilization over 48 h at pressures below 200 mTorr while the shelf temperature was gradually ramped from −40 to 25 °C)
-
Transfer and store the dried powders in vacuum desiccators at room temperature
Results
-
Microstructured, crystalline low-density aggregate particles with the specific surface area of approximately 10 m2/g of VCR were obtained from the TFF formulation without stabilizing excipients.
-
Nanostructured, amorphous low density aggregate particles were obtained from TFF VRC–PVP solutions. The specific surface area of formulations (ranging from 15 to 180 m2/g) depended on the solvent system compositions, grade of polymer, and drug-polymer ratio.
-
The lowest specific surface area for VRC formulations were obtained from formulations manufactured with 1,4-dioxane, with and without PVP K12 however, exhibited the best aerodynamic properties.
-
Microstructured crystalline TFF–VRC and nanostructured amorphous TFF–VRC–PVP K12 (1:2) demonstrated total emitted fractions of 80.6 % and 96.5 %, fine particle fractions of 43.1 % and 42.4 %, and mass median aerodynamic diameters of 3.5 and 4.5 μm, respectively when delivery using a Handihaler® dry powder inhaler (DPI).
The engineered particles were successfully generated using the TFF process to enhance powder properties. VRC TFF formulation without stabilizing excipients produced microstructured, crystalline aggregate particles. There was no notable influence of solvent composition or percentage of solids content on the solid-state properties and the aerodynamic performance of the formulation. Drug-excipient ratio had an effect on preparation of the nanostructured of amorphous solid dispersions formulations using the TFF technique.
All investigated parameters including type or grade of stabilizing excipient, drug-excipient ratio, solvent composition, and percentage of solids content in the liquid feed solution significantly affected physicochemical properties and morphology of the resulting formulations. Solvent composition in the TFF liquid feed solution also influenced the spreading of the droplets on the cryogenic surface which appeared in the specific surface area and macroscopic powder of the TFF formulations. Two solid-state properties were the most significant factors affecting the aerodynamic properties of VRC TFF powders.
Method Capsule 11
Manufacture of Amorphous Rapamycin Solid Dispersions for Dry Powder Inhalation Using TFF
Based on the study reported by Carvalho et al. (2014).
Objective
-
To study the in vivo behavior and pharmacokinetic profiles of crystalline and amorphous rapamycin when administered through dry powder inhalation to the lungs of rats
Equipment and Materials
-
TFF apparatus
-
Liquid nitrogen
-
Rapamycin (Sirolimus), lactose monohydrate (Lactohale® LH 200), acetonitrile, and water
-
Bench top tray freeze dryer
-
Wet ball milling
-
Particle-size analyzer by laser light scattering
-
BET apparatus
-
Differential scanning calorimeter (DSC)
-
Powder X-ray diffraction (XRD)
-
Next generation cascade impactor (NGI)
Method
-
Dissolve rapamycin:lactose in a ratio of 1:1 by weight in a co-solvent mixture of acetonitrile and water (3:2) with different final solids content 0.40 and 0.75 % (w/v)
-
Freeze the solution rapidly onto the cryogenically cooled stainless steel surface (−80 °C)
-
Collect frozen films in a container filled with liquid nitrogen
-
Transfer the frozen formulation to a −70 °C freezer until liquid nitrogen is completely evaporated
-
Lyophilize the frozen material in the lyophilizer over 24 h at −40 °C at pressure 400 mTorr and increase the temperature to 25 °C over 24 h with a pressure below 200 mTorr, and hold at 25 °C for 24 h
-
Prepare micronized crystalline rapamycin and lactose at 1:1 weight ratio using wet ball milling and lyophilize at −80 °C in the lyophilizer for 48 h
-
Keep all dry powder products in a desiccator under vacuum condition at room temperature
Results
-
An absence of crystalline peaks from the XRD diffractogram indicated the amorphous state of both TFF formulations of rapamycin:lactose with solids content 0.40 % and 0.75 %. Also, modulated DSC indicated that both formulations existed in the amorphous form.
-
A resulting TGA profile showed the TFF formulations initially decomposed at a temperature of about 180 °C. Minimal solvent evaporation of both formulations was only 3.2 % through the range of temperature from 45 to 110 °C.
-
The specific surface area of TFF powder formulations dramatically increased compared to the bulk and wet milling materials (see table below).
Formulation
Specific surface area ± SD (m2/g)
Lactose LH200
0.34 ± 0.02
Rapamycin
0.57 ± 0.08
Wet ball milled lactose
10.76 ± 0.01
Wet ball milled repamycin
14.29 ± 0.25
RapaLac_0.75 %
69.57 ± 6.25
RapaLac_0.40 %
85.72 ± 19.86
-
Diluted solid loading solution (0.40 %) resulted in a less dense TFF powder with greater specific surface area. Therefore, this TFF powder exhibited better performance using the dry powder inhaler device by generating greater FPF values with smaller MMAD and GSD values than the least porous powder.
-
In vitro aerosol performance was demonstrated using NGI. The results are presented as follows:
Formulation
Total emitted dose—TED (%)
Fine particle fraction—FPF (%)
Mass media aerodynamic diameter—MMAD (μm)
Geometric standard deviation—GSD
RapaLac physical mixture
78.92
36.79
1.81
4.26
RapaLac_0.40 %
97.14
72.11
2.1
2.25
RapaLac_0.75 %
94.71
61.29
2.43
2.76
-
Resulting GSD indicated that the physical mixture had a wider aerodynamic particle size distribution than the TFF formulations.
-
In vivo systemic bioavailability of rapamycin from TFF formulation was greater than its crystalline counterpart. The dissolution rate of the crystalline formulation was low and this resulted in poor absorption from the lung fluid and low systemic bioavailability. The level of drug in the lung from 12 to 24 h indicated that TTF powder and crystalline powder stayed in the lung for the same period of time. However, the TFF formulation exhibited a higher level of systemic bioavailability. The percentage of FPF at a more distal part of the lung also increased.
The wet ball milling reduced the particle size of bulk rapamycin and lactose to sub-micron range. The irregular to round-shaped particles of the drug adhered to the lactose surface were confirmed by SEM images. The improvement of aerosolization during inhalation of the milled samples was expected. Nonetheless, both TFF powders gave better aerosolization characteristics compared to the milled formulation. This study further investigated the influence of solids loading concentration on aerosolization properties of the rapamycin TFF formulation. The specific surface area of TFF powder formulations was more increased for the formulation containing a solids content of 0.40 % than for the 0.75 % formulation. SEM pictures also showed the lower density of the lower solids content formulation.
The TFF powder formulation showed superior in vitro aerosol performance to the physical mixture because the brittle powder matrix of TFF samples is easy to disintegrate to low density aerosolized particles. The reduction of electrostatic and van der Waals forces is likely to result in low adhesion between particles.
Rapamycin in the TFF formulation was an amorphous material, which basically enhances solubility and dissolution rate of its crystalline form. In vivo study exhibited a 24-h pharmacokinetic profile determined in BAL, lung tissue and blood. The amount of drug per gram of lung tissue detected in the lung was more than that detected in broncho-alveolar lavage (BAL). The rate of absorption of rapamycin from the TFF formulation into the systemic circulation is drastically greater than the crystalline physical mixture since dissolution is the rate-limiting step for the crystalline sample. This resulted in high drug levels in BAL, low absorption from lung fluid and low systemic bioavailability.
Amorphous rapamycin from the TFF process appeared to be eliminated from the lung more rapidly than the crystalline physical mixture. This is possibly due to the hygroscopic property of the TFF formulation and the hydrophilic characteristics of the lactose, which facilitates the water sorption of the formulation. Hygroscopic TFF processed rapamycin particles may deposit more in the conducting zone and be eliminated by the mucociliary clearance. Amorphous rapamycin dissolved in the lining fluid may generate a supersaturation environment, which precipitated and increased susceptibility of clearance mechanisms.
Method Capsule 12
Dry Powder Inhalation Formulations of Tacrolimus Prepared by TFF
Based on the study reported by Wang et al. (2014b).
Objective
To compare performance of dry powder inhalation formulations of tacrolimus (TAC) prepared using TFF to that of produced by micronization in the in vitro and in vivo studies
Equipment and Materials
-
TFF apparatus
-
Liquid nitrogen
-
Tacrolimus monohydrate, mannitol (MAN), Polysorbate 80, acetonitrile
-
Bench-top tray lyophilizer
-
Desiccator and desiccant
-
Next generation pharmaceutical impactor (NGI)
Method
-
Dissolve MAN in purified water and dissolve TAC in acetonitrile
-
Prepare a co-solvent mixture of ACN and water (60:40 v/v) containing TAC and MAN (1:1 w/w) with total solids content of 0.75 % w/v
-
Freeze the co-solvent solution rapidly on a cryogenically cooled rotating stainless steel surface (−50 ± 3 °C).
-
Remove resulting thin film from the surface using a scraper
-
Keep frozen samples in liquid nitrogen
-
Sublimate solvents in lyophilizer over 48 h at pressures below 200 mTorr
-
Ramp the shelf temperature up from −40 to 25 °C
-
Purge dry nitrogen into the chamber to equilibrate to atmospheric pressure before remove sample from the lyophilizer
-
Store final product in a vacuum desiccator at room temperature
-
Prepare a physical mixture of micronized TAC and micronized MAN (1:1 by weight, micronized TACMAN) by blending the two powders in a tubular mixer
Results
-
DSC and XRD results indicated that TAC in the TFF TACMAN was amorphous while MAN in the TFF TACMAN (1:1) exhibited crystallinity.
-
TAC in the TFF TACMAN formulation remained in an amorphous form for up to 6 months.
-
SEM images showed a highly porous structure of the TFF formulations.
-
Figures 11.14 and 11.15 presented a highly porous structure and a relatively small aerodynamic size distribution (GSD = 2.08 μm) of the TFF TACMAN formulation.
Fig. 11.14 
The SEM images of the micronized (a) and the TFF (b) of the TACMAN at low (×2.70k, top) and high (×25.00k, bottom) magnifications. Reproduced from Wang et al. (2014b) with permission from Springer.
Fig. 11.15 
NGI results of aerodynamic diameter distribution of TFF TACMAN formulation released from a Miat® monodose inhaler at the flow rate of 90 L/min. Reproduced from Wang et al. (2014b) with permission from Springer


-
Almost 80 % of the TFF formulation was delivered to the lung using Miat® Monodose inhaler.
-
In vivo study presented that the AUC of the TFF formulation was higher than the micronized TAC while the lung clearance rate of the micronized particles was a lot faster than the aerosolized TFF particles.
-
The TFF TACMAN formulation could avoid clearance mechanisms of the lung and deliver TAC on pulmonary epithelium, while minimizing systemic concentration.
According to the particle size distribution, the TFF formulation of TAC is appropriate for deep lung delivery when incorporated into a commercially available DPI. ATR-FTIR results showed that there was no interaction between TAC and MAN molecules. As a result, drug molecules did not have an influence from seeding of crystalline mannitol. Regarding in vivo study, the TFF formulation exhibited higher AUC and slower clearance rate in the lung tissue compared to the micronized formulation due to the faster dissolution rate and the less susceptible of the TFF formulation to the clearance mechanisms of the lung.
Method Capsule 13
Pulmonary Delivery of A Fixed-Dose Combination Drug Formulation Prepared Using TFF
Based on the study reported by Liu et al. (2015).
Objective
To develop a fixed dose combination formulation and evaluate its suitability as an inhaled product by in vitro and in vivo studies
Equipment and Materials
-
TFF apparatus
-
Liquid nitrogen
-
Salmeterol xinafoate (SX), mometasone furoate (MF), alpha-lactose monohydrate, mannitol, d-Trehalose anhydrous, glycine, acetonitrile and methanol
-
Bench-top tray lyophilizer
-
Desiccator and desiccant
-
NGI
-
Dynamic vapor sorption (DVS)
Method
-
Prepare a combination of SX and MF (ratio of 50:220 by weight) with or without other pharmaceutical excipients (lactose, mannitol, trehalose, or glycine) (1:1 molar ratio) in a co-solvent mixture of tertiary butanol, 1,4-dioxane, acetonitrile and purified water (2:1:3:3, v/v)
-
Apply the solution to a rotating cryogenically cooled steel surface of the thin film apparatus (−90 ± 3 °C)
-
Remove thin frozen films using a scraper
-
Collect and maintain the frozen samples in a container filled with liquid nitrogen
-
Lyophilize samples in a freeze dryer
-
Collect dry powders and store in a vacuum desiccator at room temperature
-
Prepare the micronized physical blends of SX, MF and excipients, in the same ratio as used in the TFF formulations for comparison
Results
-
The TFF product of neat SX crystallized during the lyophilization process.
-
The Brittle matrix powder (BMP) of co-deposition was successfully formulated using the TFF technology.
-
DSC thermograms and PXRD patterns agreed in the presence of the co-amorphous state of the BMP combination formulations.
-
BMP fixed dose combinations exhibited a sponge-like matrix consisting of large porous, homogeneous and brittle particles. In contrast, the micronized drug physical mixtures were not homogeneous.
-
The specific surface area of the BMP was approximately 30-fold greater than that of the micronized formulations.
-
Figure 11.16 showed the sorption and desorption isotherms of the BMP fixed-dose combinations using DVS. The BPM of lactose and trehalose based formulations were hygroscopic matrix. On the other hand, the mannitol and glycine based formulations were non-hygroscopic.
Fig. 11.16 
Sorption (solid line) and desorption (dashed line) isotherms of BMP formulations; (square) BMP SXMF, (diamond) BMP SXMFLac, (triangle) BMP SXMFMan, (cross symbol) BMP SXMFGly and (circle) SXMFTre after the full cycle of 0 % RH to 90 % RH. Reproduced from Liu et al. (2015) with permission from Elsevier
-
Aerosolized BMP combination formulations exhibited the co-deposition of each drug and the uniformity of the delivered dose.
-
About 50 % of the loaded dose from the excipient-free BMP was delivered in the respirable range.
-
This formulation was stable in an amorphous state at ambient conditions up to 6 months.
-
After the single dose administration, the pharmacokinetic study in the lung exhibited that the concentration of drugs from the BMP formulation was much higher than that of the crystalline physical mixture (Fig. 11.17).
Fig. 11.17 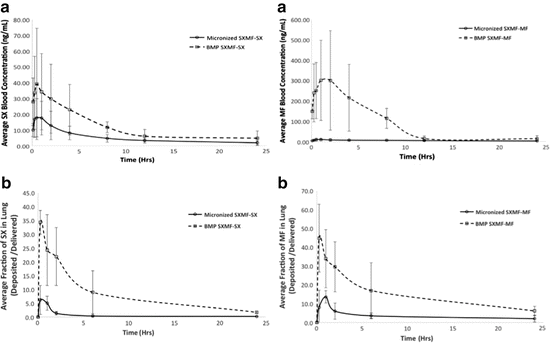
In vivo pharmacokinetic study of SX and MF in (a) plasma, (b) lung tissue following dry powder insufflation; (square) BMP SXMF formulation and (circle) micronized SXMF formulation (n = 3). The plot for lung tissue was normalized with respect to the target delivered SX and MF dose of 450 μg/kg and 1980 μg/kg, respectively. Reproduced from Liu et al. 2015 with permission from Elsevier


The TFF formulation of neat SX was not able to maintain its amorphous morphology during lyophilization as it had a low Tg. The absence of melting peaks of drugs, the single Tg(s) and the re-crystallization peak in the BMP formulations confirmed the formation of the drug-drug homogeneous amorphous phase. One drug was dissolved in the other drug (or the excipients). The co-amorphous solids improved solubility, stability and drug concentration. Hence, to retain these improvements, the formulations must withstand a propensity for recrystallization. Stability study exhibited no changes in amorphous state of the most BMP samples after 6 months at ambient condition. The formulations with higher Tg(s) had lower molecular movement which can reduce crystallization. Likewise, the hygroscopic formulations supposed to be less stable in co-amorphous BMP formulations. FPFs of the formulations with non-hygroscopic excipients or without excipient were higher than the hygroscopic powders. Owing to the stability and aerodynamic performance, the formulation without excipient was selected for the in vivo study. Pharmacokinetic data, the absorption rate and plasma AUC0–24h, of both drugs from TFF formulations were significantly higher than those of the micronized powders due to the higher dissolution and absorption. However, the enhancement of absorption resulted in the higher drug concentration. Hence, the adverse effect should be taken into account.
References
Amidon GL, Lennernas H et al (1995) A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res 12(3):413–420
Anandharamakrishnan C, Rielly CD et al (2010) Spray-freeze-drying of whey proteins at sub-atmospheric pressures. Dairy Sci Technol 90(2–3):321–334
Badawya SIF, Ghorabb MM et al (1996) Characterization and bioavailability of danazol-hydroxypropyl β-cyclodextrin coprecipitates. Int J Pharm 128(1–2):45–54
Bakatselou V, Oppenheim RC et al (1991) Solubilization and wetting effects of bile salts on the dissolution of steroids. Pharm Res 8(12):1461–1469
Barron MK, Young TJ et al (2003) Investigation of processing parameters of spray freezing into liquid to prepare polyethylene glycol polymeric particles for drug delivery. AAPS PharmSciTech 4:1–13
Beinborn NA, Lirola HL et al (2012) Effect of process variables on morphology and aerodynamic properties of voriconazole formulations produced by thin film freezing. Int J Pharm 429(1):46–57
Benson SW, Ellis DA (1948) Surface areas of proteins; surface areas and heats of absorption. J Am Chem Soc 70(11):3563–3569
Bertilsson L, Tomson T (1986) Clinical pharmacokinetics and pharmacological effects of carbamazepine and carbamazepine-10,11-epoxide. An update. Clin Pharmacokinet 11(3):177–198
Betageri GV, Makarla KR (1995) Enhancement of dissolution of glyburide by solid dispersion and lyophilization techniques. Int J Pharm 126(1–2):155–160
Boeh-Ocansey O (1983) A study of the freeze drying of some liquid foods in vacuo and at atmospheric pressure. Drying Technol 2:389–405
Borm P, Klaessig FC et al (2006) Research strategies for safety evaluation of nanomaterials, Part V: role of dissolution in biological fate and effects of nanoscale particles. Toxicol Sci 90(1):23–32
Brown SC, Claybourn M et al (2007) Optimizing raman spectroscopy to quantify polymorphic forms of a drug molecule. Am Pharm Rev 10(58):60–67
Brunauer S, Emmett P et al (1938) Adsorption of gases in multimolecular layer. J Am Chem Soc 60:309–319
Burkart GJ, Smaldone GC et al (2003) Lung deposition and pharmacokinetics of cyclosporine after aerosolization in lung transplant patients. Pharm Res 20(2):252–256
Carli F, Motta A (1984) Particle size and surface area distributions of pharmaceutical powders by microcomputerized mercury porosimetry. J Pharm Sci 73(2):197–203
Carvalho SR, Watts AB et al (2014) Characterization and pharmacokinetic analysis of crystalline versus amorphous rapamycin dry powder via pulmonary administration in rats. Eur J Pharm Biopharm 88(1):136–147
Clas SD, Dalton CR et al (1999) Differential scanning calorimetry: applications in drug development. Pharm Sci Technol Today 2(8):311–320
Conventional, U.P. (2000) The United States pharmacopoeia. The United States Pharmacopoeia Conventional, Rockville, p 8
Costantino HR, Curley JG et al (1998) Water sorption behaviour of lyophilised protein-sugar systems and implications for solid-state interactions. Int J Pharm 166:211–221
Costantino HR, Firouzabadian L et al (2000) Protein spray-freeze drying. Effect of atomization conditions on particle size and stability. Pharm Res 17(11):1374–1383
Costantino HR, Firouzabadian L et al (2002) Protein spray freeze drying. 2. Effect of formulation variables on particle size and stability. J Pharm Sci 91(2):388–395
Costantino HR, Johnson OL et al (2004) Relationship between encapsulated drug particle size and initial release of recombinant human growth hormone from biodegradable microspheres. J Pharm Sci 93(10):2624–2634
Courrier HM, Butz N et al (2002) Pulmonary drug delivery systems: recent developments and prospects. Crit Rev Ther Drug Carrier Syst 19(4–5):425–498
Davies NM, Feddah MR (2003) A novel method for assessing dissolution of aerosol inhaler products. Int J Pharm 255(1–2):175–187
De Waard H, Hinrichs WLJ et al (2008) A novel bottom–up process to produce drug nanocrystals: controlled crystallization during freeze-drying. J Control Release 128(2):179–183
Derle D, Patel J et al (2010) Particle engineering techniques to enhance dissolution of poorly water soluble drugs. Int J Curr Pharm Res 2(1):10–15
DiNunzio JC, Miller DA et al (2008) Amorphous compositions using concentration enhancing polymers for improved bioavailability of itraconazole. Mol Pharm 5(6):968–980
Du J, Li X et al (2015) Nanosuspensions of poorly water-soluble drugs prepared by bottom-up technologies. Int J Pharm 495(2):738–749
Engstrom JD, Simpson DT et al (2007a) Stable high surface area lactate dehydrogenase particles produced by spray freezing into liquid nitrogen. Eur J Pharm Biopharm 65(2):163–174
Engstrom JD, Simpson DT et al (2007b) Morphology of protein particles produced by spray freezing of concentrated solutions. Eur J Pharm Biopharm 65(2):149–162
Engstrom JD, Lai ES et al (2008) Formation of stable submicron protein particles by thin film freezing. Pharm Res 25(6):1334–1346
Engstrom J, Tam J et al (2009) Templated open flocs of nanorods for enhanced pulmonary delivery with pressurized metered dose inhalers. Pharm Res 26(1):101–117
Eriksson JHC, Hinrichs WLJ et al (2003) Investigations into the stabilization of drugs by sugar glasses: III. The influence of various high-pH buffers. Pharm Res 20:1437–1443
Esclusa-Diaz MT, Guimaraens-Mendez M et al (1996) Characterization and in vitro dissolution behaviour of ketoconazole/β- and 2-hydroxypropyl-β-cyclodextrin inclusion compounds. Int J Pharm 143:203–210
Feeley JC, York P et al (1998) Determination of surface properties and flow characteristics of salbutamol sulphate, before and after micronisation. Int J Pharm 172:89–96
Fong SYK, Ibisogly A et al (2015) Solubility enhancement of BCS Class II drug by solid phospholipid dispersions: Spray drying versus freeze-drying. Int J Pharm 496(2):382–391
Forster A, Hempenstall J et al (2001) Characterization of glass solutions of poorly water-soluble drugs produced by melt extrusion with hydrophilic amorphous polymers. J Pharm Pharmacol 53(3):303–315
Franks F (1992) Freeze-drying: from empiricism to predictability. The significance of glass transitions. Dev Biol Stand 74:9–18, discussion 19
Fukai J, Ozaki T et al (2000) Numerical simulation of liquid droplet solidification on substrates. J Chem Eng Jpn 33:630–637
Gao L, Zhang D et al (2008) Drug nanocrystals for the formulation of poorly soluble drugs and its application as a potential drug delivery system. J Nanopart Res 10:845–862
Gilkey JC, Staehelin LA (1986) Advances in ultrarapid freezing for the preservation of cellular ultrastructure. J Electron Microsc Tech 3:177–210
Gosselin PM, Thibert R et al (2003) Polymorphic properties of micronized carbamazepine produced by RESS. Int J Pharm 252(1–2):225–233
Grant DJW, Brittian HG (1995) Physical characterisation of pharmaceutical solids. Marcel Dekker, New York
Grant N, Zhang H (2011) Poorly water-soluble drug nanoparticles via an emulsion-freeze-drying approach. J Colloid Interface Sci 356(2):573–578
Hancock BC (2002) Disordered drug delivery: destiny, dynamics and the Deborah number. J Pharm Pharmacol 54(6):737–746
Hancock BC, Parks M (2000) What is the true solubility advantage for amorphous pharmaceuticals? Pharm Res 17(4):397–404
Hancock BC, Shamblin SL et al (1995) Molecular mobility of amorphous pharmaceutical solids below their glass transition temperatures. Pharm Res 12(6):799–806
He W, Lu Y et al (2013) Food proteins as novel nanosuspension stabilizers for poorly water-soluble drugs. Int J Pharm 441(1):269–278
Head T, Rydzak J (2003) Chemometric models using diamond attenuated total reflectance IR and Raman spectroscopy to characterize and quantitate polymorphs in pharmaceuticals. Am Pharm Rev 6:78–84
Hickey AJ, Mansour HM et al (2007a) Physical characterization of component particles included in dry powder inhalers. I. Strategy review and static characteristics. J Pharm Sci 96(5):1282–1301
Hickey MB, Peterson ML et al (2007b) Performance comparison of a co-crystal of carbamazepine with marketed product. Eur J Pharm Biopharm 67(1):112–119
Hildebrand JH, Scott RL (1950) Solubility of nonelectrolytes. Reinhold, New York, pp 11–13, 47, 160, 175–197
Hintz RJ, Johnson KC (1989) The effect of particle-size distribution on dissolution rate and oral absorption. Int J Pharm 51(1):9–17
Hu J, Rogers TL et al (2002) Improvement of dissolution rates of poorly water soluble APIs using novel spray freezing into liquid technology. Pharm Res 19(9):1278–1284
Hu J, Johnston KP et al (2003) Spray freezing into liquid (SFL) particle engineering technology to enhance dissolution of poorly water soluble drugs: organic solvent versus organic/aqueous co-solvent systems. Eur J Pharm Sci 20(3):295–303
Hu J, Johnston K et al (2004a) Rapid release tablet formation of micronized danazol powder produced by spray freezing into liquid (SFL). J Drug Deliv Sci Technol 14(4):305–311
Hu J, Johnston KP et al (2004b) Nanoparticle engineering processes for enhancing the dissolution rates of poorly water soluble drugs. Drug Dev Ind Pharm 30(3):233–245
Hu J, Johnston KP et al (2004c) Rapid dissolving high potency danazol powders produced by spray freezing into liquid process. Int J Pharm 271(1–2):145–154
Iacono AT, Smaldone GC et al (1997) Dose-related reversal of acute lung rejection by aerosolized cyclosporine. Am J Respir Crit Care Med 155(5):1690–1698
Ishwarya SP, Anandharamakrishnan C et al (2015) Spray-freeze-drying: a novel process for the drying of foods and bioproducts. Trends Food Sci Technol 41(2):161–181
Jalili N, Laxminarayana K (2004) A review of atomic force microscopy imaging systems: application to molecular metrology and biological sciences. Mechatronics 14(8):907–945
Jiang S, Nail SL (1998) Effect of process conditions on recovery of protein activity after freezing and freeze-drying. Eur J Pharm Biopharm 45(3):249–257
Johnson KA (1997) Preparation of peptide and protein powders for inhalation. Adv Drug Deliv Rev 26(1):3–15
Kapsi SG, Ayres JW (2001) Processing factors in development of solid solution formulation of itraconazole for enhancement of drug dissolution and bioavailability. Int J Pharm 229(1–2):193–203
Khougaz K, Clas SD (2000) Crystallization inhibition in solid dispersions of MK-0591 and poly(vinylpyrrolidone) polymers. J Pharm Sci 89(10):1325–1334
Lang B, Chow KT et al (2014) Thin film freezing-template emulsion of itraconazole to improve the dissolution properties of poorly water-soluble drugs. J Drug Deliv Sci Technol 24(2):205–211
Lauer ME, Grassmann O et al (2011) Atomic force microscopy-based screening of drug-excipient miscibility and stability of solid dispersions. Pharm Res 28(3):572–584
Lefort R, De Gusseme A et al (2004) Solid state NMR and DSC methods for quantifying the amorphous content in solid dosage forms: an application to ball-milling of trehalose. Int J Pharm 280(1–2):209–219
Leleux J, Williams RO (2014) Recent advancements in mechanical reduction methods: particulate systems. Drug Dev Ind Pharm 40(3):289–300
Lerk CF, Schoonen AJ et al (1976) Contact angles and wetting of pharmaceutical powders. J Pharm Sci 65(6):843–847
Leuner C, Dressman J (2000) Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm 50(1):47–60
Liu R (2000) Water-insoluble drug formulation. Interpharm Press, Englewood
Liu S, Watts AB et al (2015) Formulation of a novel fixed dose combination of salmeterol xinafoate and mometasone furoate for inhaled drug delivery. Eur J Pharm Biopharm 96:132–142
Maa YF, Prestrelski SJ (2000) Biopharmaceutical powders: particle formation and formulation considerations. Curr Pharm Biotechnol 1(3):283–302
Maa YF, Nguyen PA et al (1999) Protein inhalation powders: spray drying vs spray freeze drying. Pharm Res 16(2):249–254
Maa YF, Ameri M et al (2004) Influenza vaccine powder formulation development: spray-freeze-drying and stability evaluation. J Pharm Sci 93(7):1912–1923
Martin A, Swarbrick J et al (1993) Physical pharmacy: physical chemical principles in the pharmaceutical sciences. Lippincott Williams & Wilkins, Philadelphia, PA, pp 125–142, 212–250, 329–334
Matteucci ME, Brettmann BK et al (2007) Design of potent amorphous drug nanoparticles for rapid generation of highly supersaturated media. Mol Pharm 4(5):782–793
Mawson S, Yates MZ et al (1997) Stabilized polymer microparticles by precipitation with a compressed fluid antisolvent. 2. Poly(propylene oxide)- and poly(butylene oxide)-based copolymers. Langmuir 13(6):1519–1528
Meryman H (1959) Sublimation freeze drying without vacuum. Science 130:628–629
Miller DA, McConville JT, et al (2008) Stabilized HME composition with small drug particles. USPTO. USA, Board of the regents, The University of Texas at Austin System. US 2008/0274194 Al
Moneghini M, Kikic I et al (2001) Processing of carbamazepine-PEG 4000 solid dispersions with supercritical carbon dioxide: preparation, characterisation, and in vitro dissolution. Int J Pharm 222(1):129–138
Muller RH, Jacobs C et al (2001) Nanosuspensions as particulate drug formulations in therapy Rationale for development and what we can expect for the future. Adv Drug Deliv Rev 47(1):3–19
Mumenthalera M, Leuenberger H (1991) Atmospheric spray-freeze drying: a suitable alternative in freeze-drying technology. Int J Pharm 72(2):97–110
Nagapudi K, Jona J (2008) Amorphous active pharmaceutical ingredients in preclinical studies: preparation, characterization, and formulation. Curr Bioact Compd 4:213–224
Newman AW, Byrn SR (2003) Solid-state analysis of the active pharmaceutical ingredient in drug products. Drug Discov Today 8(19):898–905
Ni N, Tesconi M et al (2001) Use of pure t-butanol as a solvent for freeze-drying: a case study. Int J Pharm 226(1–2):39–46
Niwa T, Danjo K (2013) Design of self-dispersible dry nanosuspension through wet milling and spray freeze-drying for poorly water-soluble drugs. Eur J Pharm Sci 50(3–4):272–281
Niwa T, Mizutani D et al (2012) Spray freeze-dried porous microparticles of a poorly water-soluble drug for respiratory delivery. Chem Pharm Bull (Tokyo) 60(7):870–876
Overhoff KA, Engstrom JD et al (2007a) Novel ultra-rapid freezing particle engineering process for enhancement of dissolution rates of poorly water-soluble drugs. Eur J Pharm Biopharm 65(1):57–67
Overhoff KA, Moreno A et al (2007b) Solid dispersions of itraconazole and enteric polymers made by ultra-rapid freezing. Int J Pharm 336(1):122–132
Overhoff KA, McConville JT et al (2008) Effect of stabilizer on the maximum degree and extent of supersaturation and oral absorption of tacrolimus made by ultra-rapid freezing. Pharm Res 25(1):167–175
Overhoff KA, Johnston KP et al (2009) Use of thin film freezing to enable drug delivery: a review. J Drug Deliv Sci Technol 19(2):89–98
Parsian AR, Vatanara A et al (2014) Inhalable budesonide porous microparticles tailored by spray freeze drying technique. Powder Technol 260:36–41
Pasandideh-Fard M, Chandra S et al (2002) A three dimensional model of droplet impact and solidification. Int J Heat Mass Transfer 45(11):2229–2242
Patravale VB, Date AA et al (2004) Nanosuspensions: a promising drug delivery strategy. J Pharm Pharmacol 56(7):827–840
Paulekuhn GS, Dressman JB et al (2013) Salt screening and characterization for poorly soluble, weak basic compounds: case study albendazole. Int J Pharm Pharm Sci 68(7):555–564
Peeters J, Neeskens P et al (2002) Characterization of the interaction of 2-hydroxypropyl-beta-cyclodextrin with itraconazole at pH 2, 4, and 7. J Pharm Sci 91(6):1414–1422
Penkina A, Semjonov K et al (2016) Towards improved solubility of poorly water-soluble drugs: cryogenic co-grinding of piroxicam with carrier polymers. Drug Dev Ind Pharm 42(3):378–388
Poirier JM, Hardy S et al (1997) Plasma itraconazole concentrations in patients with neutropenia: advantages of a divided daily dosage regimen. Ther Drug Monit 19(5):525–529
Purvis T, Mattucci ME et al (2007) Rapidly dissolving repaglinide powders produced by the ultra-rapid freezing process. AAPS PharmSciTech 8(3):E58
Rahman SMA, Mujumdar AS (2012) 7 Atmospheric freeze drying. Prog Food Preserv 143
Rasenack N, Muller BW (2002) Dissolution rate enhancement by in situ micronization of poorly water-soluble drugs. Pharm Res 19(12):1894–1900
Rogers TL, Johnston KP et al (2001) Solution-based particle formation of pharmaceutical powders by supercritical or compressed fluid CO2 and cryogenic spray-freezing technologies. Drug Dev Ind Pharm 27(10):1003–1015
Rogers TL, Hu J et al (2002a) A novel particle engineering technology: spray-freezing into liquid. Int J Pharm 242(1–2):93–100
Rogers TL, Nelsen AC et al (2002b) A novel particle engineering technology to enhance dissolution of poorly water soluble drugs: spray-freezing into liquid. Eur J Pharm Biopharm 54(3):271–280
Rogers TL, Nelsen AC et al (2003a) Enhanced aqueous dissolution of a poorly water soluble drug by novel particle engineering technology: spray-freezing into liquid with atmospheric freeze-drying. Pharm Res 20(3):485–493
Rogers TL, Overhoff KA et al (2003b) Micronized powders of a poorly water soluble drug produced by a spray-freezing into liquid-emulsion process. Eur J Pharm Biopharm 55(2):161–172
Salekigerhardt A, Ahlneck C et al (1994) Assessment of disorder in crystalline solids. Int J Pharm 101(3):237–247
Sarkari M, Brown J et al (2002) Enhanced drug dissolution using evaporative precipitation into aqueous solution. Int J Pharm 243(1–2):17–31
Sethuraman V, Hickey A (2002) Powder properties and their influence on dry powder inhaler delivery of an antitubercular drug. AAPS PharmSciTech 3:E28
Shah B, Kakumanu VK et al (2006) Analytical techniques for quantification of amorphous/crystalline phases in pharmaceutical solids. J Pharm Sci 95(8):1641–1665
Shekunov BY, Chattopadhyay P et al (2007) Particle size analysis in pharmaceutics: principles, methods and applications. Pharm Res 24(2):203–227
Simonsen L, Clarke MJ et al (1998) Pandemic versus epidemic influenza mortality: a pattern of changing age distribution. J Infect Dis 178(1):53–60
Sindel U, Zimmermann I (2001) Measurement of interaction forces between individual powder particles using an atomic force microscope. Powder Technol 117:247–254
Sinswat P, Gao X et al (2005) Stabilizer choice for rapid dissolving high potency itraconazole particles formed by evaporative precipitation into aqueous solution. Int J Pharm 302(1–2):113–124
Sitte H, Edelmann L et al (1987) Cryofixation without pretreatment at ambient pressure. In: Steinbrecht RA, Zierold K (eds) Cryotechniques in biological electron microscopy. Springer, Berlin, pp 87–113
Sobel JD (2000) Practice guidelines for the treatment of fungal infections. For the Mycoses Study Group. Infectious Diseases Society of America. Clin Infect Dis 30(4):652
Stephenson GA, Forbes RA et al (2001) Characterization of the solid state: quantitative issues. Adv Drug Deliv Rev 48(1):67–90
Stubberud L, Forbes RT (1998) The use of gravimetry for the study of the effect of additives on the moisture induced recrystallization of amorphous lactose. Int J Pharm 163:145–156
Suryanarayan R (1985) Evaluation of two concepts of crystallinity using calcium gluceptate as a model compound. Int J Pharm 24:1–17
Suryanarayan R (1995) X-ray powder diffractometry. In: Brittain H (ed) Physical characterization of pharmaceutical solids. Marcel Dekker, New York, pp 187–221
Suzuki H, Sunada H (1998) Influence of water-soluble polymers on the dissolution of nifedipine solid dispersions with combined carriers. Chem Pharm Bull (Tokyo) 46(3):482–487
Tang X, Pikal MJ (2004) Design of freeze-drying processes for pharmaceuticals: practical advice. Pharm Res 21(2):191–200
Tesconi MS, Sepassi K et al (1999) Freeze-drying above room temperature. J Pharm Sci 88(5):501–506
Tong HHY, Du Z et al (2011) Spray freeze drying with polyvinylpyrrolidone and sodium caprate for improved dissolution and oral bioavailability of oleanolic acid, a BCS Class IV compound. Int J Pharm 404(1–2):148–158
Traini D, Rogueda P et al (2005) Surface energy and interparticle forces correlations in model pMDI formulations. Pharm Res 22(5):816–825
van de Witte P, Dijkstra PJ et al (1996) Phase separation processes in polymer solutions in relation to membrane formation. J Membrane Sci 117:1–31
Van Drooge DJ, Hinrichs WLJ et al (2004) Incorporation of lipophilic drugs in sugar glasses by lyophilization using a mixture of water and tertiary butyl alcohol as solvent. J Pharm Sci 93(3):713–725
Vasanthavada M, Tong WQ et al (2004) Phase behavior of amorphous molecular dispersions I: determination of the degree and mechanism of solid solubility. Pharm Res 21(9):1598–1606
Vasanthavada M, Tong WQ et al (2005) Phase behavior of amorphous molecular dispersions II: role of hydrogen bonding in solid solubility and phase separation kinetics. Pharm Res 22(3):440–448
Vaughn JM, McConville JT et al (2006) Single dose and multiple dose studies of itraconazole nanoparticles. Eur J Pharm Biopharm 63(2):95–102
Wallemacq PE, Verbeeck RK (2001) Comparative clinical pharmacokinetics of tacrolimus in paediatric and adult patients. Clin Pharmacokinet 40(4):283–295
Wang Y-B, Watts AB et al (2014a) Effect of processing parameters on the physicochemical and aerodynamic properties of respirable brittle matrix powders. J Drug Deliv Sci Technol 24(4):390–396
Wang Y-B, Watts AB et al (2014b) In vitro and in vivo performance of dry powder inhalation formulations: comparison of particles prepared by thin film freezing and micronization. AAPS PharmSciTech 15(4):981–993
Wanning S, Süverkrüp R et al (2015) Pharmaceutical spray freeze drying. Int J Pharm 488(1–2):136–153
Ward S, Perkins M et al (2005) Identifying and mapping surface amorphous domains. Pharm Res 22(7):1195–1202
Washburn EW (1921) The dynamics of capillary flow. Phys Rev 17:273–283
Watts AB, Wang Y-B et al (2013) Respirable low-density microparticles formed in situ from aerosolized brittle matrices. Pharm Res 30(3):813–825
Weiss C, McLoughlin P et al (2015) Characterisation of dry powder inhaler formulations using atomic force microscopy. Int J Pharm 494(1):393–407
Williams RO, Hu J et al (2003) Process for production of nanoparticles and microparticles by spray freezing into liquid. US Patent 20030041602
Xu WJ, Xie HJ et al (2016) Enhanced dissolution and oral bioavailability of valsartan solid dispersions prepared by a freeze-drying technique using hydrophilic polymers. Drug Deliv 23:41–48
Yamashita K, Nakate T et al (2003) Establishment of new preparation method for solid dispersion formulation of tacrolimus. Int J Pharm 267(1–2):79–91
Yang W, Peters JI et al (2008a) Inhaled nanoparticles—a current review. Int J Pharm 356(1–2):239–247
Yang W, Tam J et al (2008b) High bioavailability from nebulized itraconazole nanoparticle dispersions with biocompatible stabilizers. Int J Pharm 361(1–2):177–188
Yang W, Johnston KP et al (2010) Comparison of bioavailability of amorphous versus crystalline itraconazole nanoparticles via pulmonary administration in rats. Eur J Pharm Biopharm 75(1):33–41
Yasmin R, Tan A et al (2014) Lyophilized silica lipid hybrid (SLH) carriers for poorly water‐soluble drugs: physicochemical and in vitro pharmaceutical investigations. J Pharm Sci 103(9):2950–2959
Yoshioka M, Hancock BC et al (1994) Crystallization of indomethacin from the amorphous state below and above its glass transition temperature. J Pharm Sci 83(12):1700–1705
Yu L (2001) Amorphous pharmaceutical solids: preparation, characterization and stabilization. Adv Drug Deliv Rev 48(1):27–42
Yu Z, Garcia AS et al (2004) Spray freezing into liquid nitrogen for highly stable protein nanostructured microparticles. Eur J Pharm Biopharm 58(3):529–537
Yu Z, Johnston KP et al (2006) Spray freezing into liquid versus spray-freeze drying: influence of atomization on protein aggregation and biological activity. Eur J Pharm Sci 27(1):9–18
Zhang M, Li H et al (2012) Formulation and delivery of improved amorphous fenofibrate solid dispersions prepared by thin film freezing. Eur J Pharm Biopharm 82(3):534–544
Zijlstra GS, Rijkeboer M et al (2007) Characterization of a cyclosporine solid dispersion for inhalation. AAPS J 9(2):E190–E199
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 American Association of Pharmaceutical Scientists
About this chapter
Cite this chapter
Surasarang, S.H., Williams, R.O. (2016). Pharmaceutical Cryogenic Technologies. In: Williams III, R., Watts, A., Miller, D. (eds) Formulating Poorly Water Soluble Drugs. AAPS Advances in the Pharmaceutical Sciences Series, vol 22. Springer, Cham. https://doi.org/10.1007/978-3-319-42609-9_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-42609-9_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-42607-5
Online ISBN: 978-3-319-42609-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)