
Abstract
The innate immune system plays an essential role in initiating the early response against microbial infection, as well as instructing and shaping subsequent responses. Microbial pathogens are enormously diverse in terms of the niches they occupy, their metabolic properties and requirements, and the cellular pathways that they target. Nevertheless, innate sensing of pathogens triggers a relatively stereotyped set of responses that involve transcriptional induction of key inflammatory mediators, as well as post-translational assembly and activation of a multiprotein inflammatory complex termed ‘the inflammasome.’ Along with classical Pattern Recognition Receptors, the inflammasome activation pathway has emerged as a key regulator of tissue homeostasis and immune defense. Components of the inflammasome generally exist within the cell in a soluble, monomeric state, and oligomerize in response to diverse enzymatic activities associated with infection or cellular stress. Inflammasome assembly triggers activation of the pro-enzyme caspase-1, resulting in the cleavage of caspase-1 targets. The most extensively studied targets are the cytokines of the IL-1 family, but the recent discovery of Gasdermin D as a novel target of caspase-1 and the related inflammatory caspase, caspase-11, has begun to mechanistically define the links between caspase-1 activation and cell death. Cell death is a hallmark of macrophage infection by many pathogens, including the gram-negative bacterial pathogens of the genus Yersinia. Intriguingly, the activities of the Yersinia-secreted effector proteins and the type III secretion system (T3SS) itself have been linked to both inflammasome activation and evasion during infection. The balance between these activating and inhibitory activities shapes the outcome of Yersinia infection. Here, we describe the current state of knowledge on interactions between Yersinia and the inflammasome system, with the goal of integrating these findings within the general framework of inflammasome responses to microbial pathogens.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
1.1 Evolutionary Relationships Among the Pathogenic Yersinia
Cell death is an evolutionarily conserved immune response to microbial infection, as it prevents pathogen replication and can provide pro-inflammatory signals necessary for an effective immune response (Campisi et al. 2014; Munoz-Pinedo 2012). Extensive death of immune cells has been a well-appreciated hallmark of infection by pathogenic bacteria of the Yersinia species. The three pathogenic Yersiniae, Y. pestis (Yp), Y. pseudotuberculosis (Yptb), and Y. enterocolitica (Ye), all harbor a 70 kB virulence plasmid encoding a conserved type III secretion system (T3SS) and virulence factors, termed the Yersinia outer proteins (Yops) (Viboud and Bliska 2005; Wren 2003). While environmental Yersinia species that do not possess the T3SS are capable of causing disease in fish, for example, Y. ruckeri, all mammalian pathogenic Yersinia require the T3SS and associated Yops to cause disease. The T3SS provides multiple functions that enable Yersinia to evade or modulate host responses, including blockade of phagocytosis, interference with oxidative burst in neutrophils, and disruption of host signaling pathways to suppress innate and adaptive immune responses (Cornelis 2006). The mammalian pathogenic Yersinia have therefore long been thought to replicate extracellularly in infected lymphoid organs, although it is possible that an intracellular stage exists during the Yersinia life cycle in vivo (Grabenstein et al. 2004). In this review, we focus on the interactions between Yersinia virulence factors and host cell death machinery. While a great deal of work has been done in this area, recent advances in understanding distinct pathways and mechanisms of cell death in the context of infection and inflammation have enabled further advances in understanding how Yersinia interference with these pathways might alter the balance between pathogenesis and host defense.
Yp was identified as the causative agent of plague in 1894 independently by Alexander Yersin and Shibasaburo Kitasato, who came from the Pasteur and Koch schools of microbiology, respectively (Rosenberg 1968). Transmission typically occurs either through the bite of an infected flea or the inhalation of Yp containing droplets from an individual infected with the pneumonic form of plague (Wren 2003). Currently, wild animal populations in the western USA provide a reservoir for Yp in the USA, and a number of cases have occurred recently in National Parks (Ben Ari et al. 2008; Kwit et al. 2015). The appearance of multidrug resistance and the potential use of Yp as a bioterrorism agent continue to draw interest in understanding the biology of Yersinia infection. Inside the mammalian host, Yp has a preference for leukocytes (Balada-Llasat and Mecsas 2006; Maldonado-Arocho et al. 2013; Marketon et al. 2005). During the bubonic form of the plague, bacteria traffic to and replicate in the lymph node, resulting in inflammation and swelling of the lymph node. Yp can also spread to the lungs (pneumonic plague) or systemically (septicemic plague). Yp evolved directly from Yptb through a combination of gene acquisition and gene loss that allowed for transmission by fleas, a change in virulence, and restriction of lifestyle (Achtman et al. 1999; Chain et al. 2004; Hinnebusch et al. 2002; Zimbler et al. 2015).
While Yp is a vector-borne disease that often spreads to systemic tissues and sites, both Yptb and Ye are transmitted through the oral–fecal route (Wren 2003). All pathogenic Yersinia share a tropism for lymph nodes and leukocytes (Durand et al. 2010; Koberle et al. 2009). In contrast to pestis, the diseases caused by the gastro-intestinal Yersinia species are generally self-limiting in immunocompetent individuals. Interestingly, Ye is found as a commensal in over 80 % of commercially farmed pigs and is the most common cause of food-borne Yersinia infection (Bhaduri et al. 2005; Wesley et al. 2008).
Yptb in contrast, is a natural pathogen of sylvatic rodents, and like Ye, causes systemic disease in rodent models of infection. Spread of Yptb occurs from peripheral sites in the intestine and Peyer’s patches to mesenteric lymph nodes, and the systemic tissues of the reticuloendothelial system, the spleen, and liver. Interestingly, this is not a linear process and involves at least some fraction of bacteria that bypass the lymph nodes to go directly to the systemic tissues (Barnes et al. 2006). This may involve spread via the circulation either as free bacteria or potentially subsequent to uptake by intestinal phagocytic cells that traffic the bacteria to systemic sites, which has been found to occur in Salmonella infection (Vazquez-Torres et al. 1999). Thus, both Ye and Yptb have been used extensively as models of systemic Yersinia infection in mice. Below we discuss key aspects of the Yersinia–host interaction from the standpoint of how the innate immune system detects Yersinia infection, and the mechanisms by which Yersinia modulates these inflammatory responses.
1.2 Features of Yersinia-Induced Cell Death
The YopJ protein of Yp and Yptb (termed YopP in Y. enterocolitica) has both deubiquitinase and acyltransferase activities (Mittal et al. 2006; Mukherjee et al. 2006; Zhou et al. 2005), which potently blocks nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) signaling (Palmer et al. 1998; Schesser et al. 1998). Blockade of NF-κB and MAPK signaling in the context of TLR engagement results in both inhibition of cytokine production and death of Yersinia-infected cells (Mills et al. 1997; Monack et al. 1997; Ruckdeschel et al. 1998). Among the sequenced strains of pathogenic Yersiniae, YopJ and YopP share 95–98 % identity across the full length of the protein sequence; however, key polymorphisms impact both YopJ/P enzymatic activity and translocation, which tunes the precise outcome of Yersinia infection (Brodsky and Medzhitov 2008; Ruckdeschel et al. 2001; Zauberman et al. 2006; Zheng et al. 2011). In particular, while YopJ is an important virulence factor that facilitates the systemic spread of Yptb to systemic sites following oral infection (Monack et al. 1998), elevated YopJ activity in the context of Yptb or Yp attenuates the infection and induces a more robust adaptive immune response (Brodsky and Medzhitov 2008; Zauberman et al. 2009).
Yersinia-infected cells have been observed to exhibit features of apoptosis, pyroptosis, or necrosis, depending on the state of the cells and the cell type involved (Bergsbaken and Cookson 2007; Erfurth et al. 2004; Grobner et al. 2006; Philip et al. 2014; Zheng et al. 2012). In particular, priming of cells in vitro via TLR stimulation results in protection from Yersinia-induced cell death relative to cells that encounter Yersinia prior to such stimulation (Bergsbaken and Cookson 2007; Brodsky et al. 2010). Furthermore, dendritic cells are significantly more resistant to YopJ-induced cell death than macrophages in vitro (Brodsky and Medzhitov 2008). A number of studies have revealed key players in cell death pathways during Yersinia infection, providing insight into mechanisms of Yersinia-induced cell death, but little is currently known about the precise nature and role of Yersinia death in vivo. Yersinia is thought to primarily replicate as an extracellular pathogen that evades phagocytosis by neutrophils and monocytic cells in lymphoid tissues. Cell death has therefore been viewed as a strategy for Yersinia to eliminate host phagocytes (Monack et al. 1998). However, several studies suggest that host cell death during Yersinia infection promotes anti-Yersinia immunity, although the downstream consequences of Yersinia-induced cell death on induction of immune responses in vivo are not entirely clear (Bergman et al. 2009; Brodsky and Medzhitov 2008; Zauberman et al. 2009). Interestingly, recent studies in Y. pestis models of pneumonic plague have revealed that the Plasminogen activator protease Pla targets Fas Ligand for degradation, which limits apoptosis in vivo and promotes pulmonary bacterial replication (Caulfield et al. 2014). If Yersiniae transit through an intracellular stage during in vivo infection (Grabenstein et al. 2004), excessive death of the infected cell due to the activity of YopJ may eliminate its intracellular replicative niche. Since cell death is a characteristic feature of infection by many pathogens, including Yersinia, Yersinia has served as a genetically tractable and powerful system to investigate the mechanisms and consequences of cell death in immune defense.
2 Mechanisms of Yersinia-Induced Cell Death
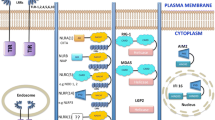
The original reports of Yersinia-induced death of macrophages identified a role for YopJ and described this death as apoptosis (Mills et al. 1997; Monack et al. 1997). Interestingly, in contrast to other forms of apoptosis, Yersinia infection also induced activation of caspase-1 (Bergsbaken and Cookson 2007; Lilo et al. 2008) via mechanisms that are distinct from other known activators of caspase-1 (Brodsky et al. 2010; Philip et al. 2014). In contrast to other pathogens, Yersinia infection appears to both activate and inhibit multiple pathways of cell death, depending on the availability of different host and bacterial inflammatory signals (Fig. 1). Below, we discuss the different pathways of cell death that are triggered during Yersinia infection, and the way in which they are induced.

Activation and inhibition of cell death signaling during Yersinia infection. TLR signaling induces upregulation of NF-κB- and MAPK-dependent gene expression, including proinflammatory mediators and pro-survival factors. These prosurvival factors limit induction of caspase-8-dependent apoptosis. Blockade of IKK and MKK function by YopJ de-represses caspase-8 activation, which triggers activation of multiple caspases, including caspase-1. This caspase-1 activation pathway is distinct from both canonical and non-canonical inflammasomes as it requires blockade of NF-κB and MAPK signaling by YopJ and does not engage other inflammasome sensors. Yersinia injection of YopK and YopM limits the activation of caspase-1 and pyroptosis in LPS-primed macrophages, thereby potentially allowing Yersinia to evade these responses. These responses are induced in murine macrophages by both Y. pestis and Y. pseudotuberculosis indicating that either murine cells sense Y. pestis hepta-acylated LPS enough to trigger death signaling or that alternative TLRs are also linked to these pathways
2.1 YopJ-Induced Death via the Extrinsic Apoptosis Pathway
Macrophages stimulated with LPS in the presence of protein synthesis inhibitors or inhibitors of NF-κB signaling undergo cell death (Philip et al. 2014; Ruckdeschel et al. 2004; Zhang et al. 2005). Consistent with this, Tlr4 −/− macrophages are resistant to YopJ-dependent apoptosis, as are cells deficient in the TLR3/4 adaptor TRIF, but not MyD88 (Haase et al. 2003; Philip et al. 2014; Ruckdeschel et al. 2004; Zhang et al. 2005). Importantly, infection of dendritic cells with Yersinia leads to the formation of a Fadd/caspase-8/Ripk1 complex and caspase-8 activation (Grobner et al. 2007). In macrophage-like cell lines, activation of caspase-8 is accompanied by Yersinia-induced cleavage of the pro-apoptotic protein Bid, leading to the classical hallmarks of apoptosis including cytochrome-c release, and caspase-3 and caspase-7 cleavage (Denecker et al. 2001). While early studies observed that treatment with broad-spectrum caspase inhibitors reduced the number of TUNEL+ cells during Yersinia infection (Denecker et al. 2001; Monack et al. 1998), Yersinia-infected dendritic cells treated with a pan-caspase inhibitor undergo substantial cell death that exhibits morphological features of necrosis (Grobner et al. 2006).
While the mechanisms responsible for this alternative form of cell death were not clear initially, more recent studies uncovered a caspase-independent pathway of cell death driven by Receptor Interacting Protein Kinases 1 and 3 (Ripk1 and Ripk3), termed programmed necrosis (Degterev et al. 2008; He et al. 2011). Intriguingly, treatment of Yersinia-infected cells with caspase inhibitors induces cell death that is blocked either by Ripk3 deficiency or pharmacological inhibitors of Ripk1 or Ripk3, indicating that this cell death is programmed necrosis (Philip et al. 2014; Weng et al. 2014). Importantly however, Ripk3 deficiency or treatment with necrostatin has no impact on the levels of Yersinia-induced cell death on its own, suggesting that the primary mode of cell death that takes place in response to the activity of YopJ is caspase-8-mediated apoptosis (Philip et al. 2014).
Interestingly, YopJ-dependent apoptosis is also associated with caspase-1 activation (Brodsky et al. 2010; Lilo et al. 2008; Zheng et al. 2011), and the extent of YopJ-mediated NF-κB inhibition correlates with the degree of caspase-1 activation (Zheng et al. 2011) (Fig. 1). This is consistent with the finding that deletion of IKKβ in macrophages induces spontaneous inflammasome activation (Greten et al. 2007). Although Nlrp3 and the inflammasome adaptor Asc are required for YopJ-dependent secretion of IL-1β and IL-18 (Zheng et al. 2011), the mechanism by which YopJ activates caspase-1 is unclear, as caspase-1 processing and YopJ-dependent cell death still occur equivalently to wild-type cells in the absence of Asc, Nlrc4, or Nlrp3 (Brodsky et al. 2010; Philip et al. 2014). Intriguingly, during Yersinia infection, YopJ triggers a unique pathway of caspase-1 activation that requires activation of caspase-8 (Philip et al. 2014; Weng et al. 2014). Caspase-8 was also found to play a role in inflammasome priming in the context of canonical inflammasome stimuli (Gurung et al. 2014; Man et al. 2013). However, the caspase-8-dependent activation of caspase-1 during Yersinia infection occurs when priming itself is inhibited by the NF-κB-blocking activity of YopJ. Moreover, both the presence and enzymatic activity of caspase-8 were necessary for activation of caspase-1 by Yersinia infection (Philip et al. 2014; Weng et al. 2014).
Distinct inflammasome complexes with different functions have been identified and could potentially account for these observations. Caspase-1 could be recruited to an Nlrp3/Asc complex that regulates IL-1β and IL-18 production and to a separate caspase-8-containing complex that activates cell death. Indeed, during Salmonella infection, a complex containing catalytically active caspase-1, but not Asc triggers cell death but not cytokine secretion, while a distinct Asc-containing focus mediates caspase-1 processing and cytokine secretion (Broz et al. 2010).
2.2 Inflammasome Sensing of Yersinia Infection and Its Evasion by YopK
Nlrp12 was also recently found to induce inflammasome activation in response to Y. pestis infection, and both Nlrp3 and Nlrp12 contributed to host defense against Yersinia, presumably via induction of caspase-1-dependent IL-1β and IL-18 (Brodsky et al. 2010; Vladimer et al. 2013). Nlrp12 inflammasome activation occurs under conditions when Yp LPS is altered to provide increased stimulatory activity to TLR4, as Y. pestis normally evades TLR4 by producing a tetra-acylated LPS molecule when grown at 37 °C. YopJ may also activate an Nlrp12 inflammasome, although this remains to formally be demonstrated. Whether Nlrp12 also plays a cryptic role in other instances of TLR4 priming, or whether NLRP12 activation depends on the interaction between increased TLR stimulation and an activity of the Yersinia T3SS (potentially YopJ) remains to be determined. NLRP12 protein levels are upregulated by LPS, similarly to Nlrp3 raising the possibility that Nlrp12 activation in this context may be due to enhanced upregulation of Nlrp12 in response to elevated levels of Yp hexa-acylated LPS. Interestingly, in the absence of Nlrp12, there is no difference in levels of IL-1β secretion in response to parental Yp and Yp expressing hexa-acylated LPS (Vladimer et al. 2013), suggesting that NLRP12 responds preferentially to this LPS modification. Whether Nlrp12 also responds to other hexa-acylated forms of LPS, or whether the Nlrp12 inflammasome responds to a combination of signals, such as YopJ together with hexa-acylated LPS, remains to be established.
In addition to Nlrp12, the other primary NLR protein that responds to Yersinia infection is Nlrp3, which is triggered in response to the Yersinia T3SS in the absence of all known secreted effectors, particularly upon deletion of YopK (Brodsky et al. 2010). The T3SS of bacterial pathogens could be viewed as a ‘Pattern of Pathogenesis,’ analogous to pore-forming toxins of gram-positive pathogens that are essential to the virulence of the organism, but which also disrupt host membrane integrity and modulate signaling networks, resulting in increased host sensing (Vance et al. 2009). Interestingly, the pore-forming activity of the Yersinia T3SS is not required for its ability to induce inflammasome activation (Kwuan et al. 2013; Zwack et al. 2014). Rather, the ability to inject components of the T3SS translocon was particularly important to activate the T3SS-induced inflammasome. Notably, increased translocation of the T3SS pore-forming translocon proteins is responsible for inflammasome activation in response to Yptb infection (Zwack et al. 2014).
The precise mechanism by which the Yersinia T3SS triggers inflammasome activation remains unclear; however, a secreted effector protein, termed YopK, specifically prevents inflammasome activation in response to the Yersinia translocon proteins (Brodsky et al. 2010). YopK is a general regulator of Yersinia translocation, as yopK mutant Yersinia strains exhibit significantly greater level of translocation than their wild-type counterparts (Holmstrom et al. 1995, 1997). Interestingly, YopK-deficient Yersinia inject increased levels of the translocon proteins YopB and YopD into host cells, and limiting this translocation prevents inflammasome activation by the Yersinia T3SS (Zwack et al. 2014). How YopK prevents hyper-injection of translocon components into the cell remains unclear. While YopK interacts biochemically with translocon components (Brodsky et al. 2010; Thorslund et al. 2011), it is also translocated into the cytosol of infected cells and has been proposed to regulate translocation from within the cytosol (Dewoody et al. 2011). Interactions between YopK and the scaffolding protein Rack1 are important for the ability of YopK to regulate translocation, but whether these interactions are also important for YopK to limit inflammasome activation is not known (Thorslund et al. 2011).
In contrast to other gram-negative pathogens with T3SSs, such as Salmonella and Shigella, Yersinia does not trigger large amounts of NLRC4 inflammasome activation, and the contribution of Nlrc4 to anti-Yersinia inflammasome responses is only revealed in the absence of NLRP3 (Brodsky et al. 2010). This is likely due to the counter-regulation between the Yersinia T3SS and flagellin, which is downregulated at 37 °C (Minnich and Rohde 2007). Like the inner rod protein of the Salmonella pathogenicity island I T3SS, the Yersinia homolog YscI may also trigger the Naip/Nlrc4 inflammasome at low levels, as YscI possesses the conserved C-terminal sequence required to trigger this inflammasome (Miao et al. 2010). Interestingly, the Yersinia T3SS activates both the canonical Nlrp3 inflammasome, and the more recently described non-canonical caspase-11 inflammasome, which is activated in response to direct binding of cytosolic LPS (Casson et al. 2013). This non-canonical inflammasome requires priming by LPS and signaling through a TRIF-IFN-IFNAR feedback loop to upregulate caspase-11, which directly senses the presence of cytosolic LPS (Broz et al. 2012; Gurung et al. 2012; Kayagaki et al. 2011; Rathinam et al. 2012; Shi et al. 2014). It is possible that the canonical and non-canonical inflammasomes are triggered by distinct patterns of pathogenesis present within the cytosol during Yersinia infection (injection of translocon Yops vs. LPS). An alternative possibility, which remains to be tested, is that Yersinia lacking YopK trigger both canonical and non-canonical pathways via a common trigger independently of cytosolic LPS.
2.3 Evasion of Inflammasome by YopM
YopM is a T3SS effector that was originally found to play a role in the suppression of innate immune responses during Yp infection (Nemeth and Straley 1997). Interestingly, YopM interacted with two host kinases, Rsk1 and Prk2, and was found to play a role in systemic induction of the immuno-suppressive cytokine IL-10 during systemic mouse infection with Yptb (McPhee et al. 2010, 2012). YopM is a leucine-rich repeat (LRR)-containing effector protein, whose LRR comprises the majority of the protein length (Evdokimov et al. 2001). Surprisingly, YopM is highly polymorphic among the different pathogenic Yersinia species, and even within strains of the same species. The length of the LRR domain is highly variable, containing between 13 and 21 repeats in the different Yersinia strains, and is followed by an unstructured tail that is conserved in different YopM isoforms. Consistent with other LRR-containing proteins, the YopM LRR domain adopts an overall horseshoe-like structure, and this fold is predicted to serve as a binding platform for host cell proteins (Evdokimov et al. 2001). In the host cytosol, YopM binds to and activates two protein kinases, Rsk1 and Prk2 (Hentschke et al. 2010; McDonald et al. 2003). The C-terminal tail of YopM is required for both Rsk1 binding in macrophages and virulence of Yersinia in mice (McCoy et al. 2010; McPhee et al. 2010). However, it is currently not clear whether the interaction between YopM and Rsk1 or Prk2 is specifically responsible for the contribution of YopM to Yersinia virulence, or whether an alternative function of YopM is affected by ablation of the C terminus.
Notably, a particular YopM isoform from the YPIII strain of Yp and some Yptb strains binds to and inhibits caspase-1 in macrophages (LaRock and Cookson 2012). This inhibitory effect of YopM on inflammasome activation is revealed in LPS-primed macrophages, in the presence of YopK (Chung et al. 2014; LaRock and Cookson 2012). During infection with YopK-deficient Yersinia, both primed and unprimed macrophages undergo rapid inflammasome activation as a consequence of the activity of the T3SS (described above). These findings suggest an interaction between the macrophage activation state and the activity of various Yersinia effector proteins that determines the cellular response to Yersinia infection (Ruckdeschel and Richter 2002). Interestingly, recent findings indicate that in primed macrophages, YopJ, which normally induces inflammasome activation (as described above), actually functions together with YopM to prevent inflammasome activation (Ratner et al. 2016; Schoberle et al. 2016). While YopM can bind either to caspase-1 directly, thereby acting as a decoy substrate, or interact with the small GTPase IQGAP1, which also limits inflammasome activation (Chung et al. 2014; LaRock and Cookson 2012), the mechanism for how YopJ limits inflammasome activation in primed cells is not clear.
As might be expected, in naïve or unstimulated innate immune cells, YopJ limits inflammasome priming due to its ability to interfere with induction of both MyD88- and Trif-dependent responses (Rosadini et al. 2015), thereby likely interfering with upregulation of inflammasome pathway constituents in innate cells (Ratner et al. 2016; Rosadini et al. 2015). YopM interactions with caspase-1 prevent both recruitment of caspase-1 to inflammasomes and caspase-1 auto-cleavage (LaRock and Cookson 2012). Initial studies observed that translocon insertion by wild-type Yptb triggers caspase-1 in LPS-activated macrophages (Bergsbaken and Cookson 2007), but with delayed kinetics compared with a yopM mutant. Purified YopM bound to cleaved caspase-1 and inhibited caspase-1 activity in vitro. Binding to and inhibition of caspase-1 required Asp residue 271 located in the 10th LRR of YopM. This Asp residue is located in a consensus caspase-1 cleavage motif (YLTD); however, caspase-1 did not cleave YopM, and not all YopM isoforms have this motif. These data suggest that the YLTD motif in YopM acts as a pseudosubstrate inhibitor of caspase-1, similar to endogenous inhibitors such as Flightless-1 (LaRock and Cookson 2012).
Direct evidence that inhibition of caspase-1 by YopM is functionally linked to bacterial virulence was demonstrated by the findings that the virulence deficiency of YopM deletion is overcome by infection of caspase-1-deficient mice (LaRock and Cookson 2012; Chung et al. 2014). The above data suggest a mechanism for how YopM inhibits pro-inflammatory responses, and also raise a number of important questions. For example, how do YopM isoforms that lack the YLTD motif also inhibit caspase-1? Is YopM a bifunctional effector, with the LRR region acting as a caspase-1 inhibitor and the C-terminal tail working in conjunction with RSK1? It will also be interesting to determine if inhibition of caspase-1 is linked to the remarkable demonstration that purified Y. enterocolitica YopM can penetrate into cultured cells and inhibit transcription of pro-inflammatory cytokine genes (Ruter et al. 2014).
3 Interactions Between Yersinia and Cell Death Pathways In Vivo
3.1 Impact of Yersinia-Induced Cell Death on Pathogenesis and Host Defense
Early studies observed that macrophages and dendritic cells infected by Yersinia exhibit characteristics of apoptosis, specifically membrane blebbing, nuclear condensation, DNA fragmentation, and formation of large cytoplasmic vacuoles (Monack et al. 1997; Ruckdeschel et al. 1997). Apoptosis has classically been viewed as immunologically silent, but growing evidence suggests that during infection or in the context of tissue stress, apoptosis may in fact promote inflammatory responses (Green et al. 2009; Torchinsky et al. 2009). Furthermore, apoptotic cells can be phagocytosed, and their associated microbial antigens used to prime CD8+ T-cell responses (Heath and Carbone 2001). Therefore, while cell death during Yersinia infection is thought to be apoptotic, it may not be immunologically silent. Below, we discuss the current state of understanding how Yersinia-induced cell death contributes to bacterial virulence or host defense in vivo.
3.2 Activation of Cell Death by Yersinia Virulence Factors In Vivo
A number of studies indicate that YopJ/P promotes Yersinia virulence in vivo. Oral infection with Yptb and Ye demonstrate that YopJ/P contributes to systemic disease and barrier dysfunction (Jung et al. 2012; Meinzer et al. 2012; Monack et al. 1998). YopJ is dispensable for colonization of the Peyer’s patches (PPs) and mesenteric lymph nodes (mLNs), especially at higher infectious doses; however, YopJ-deficient Yersinia had significantly reduced levels of spleen colonization after oral infection (Monack et al. 1998). Spleens and mLNs from mice infected with YopJ-sufficient bacteria had a higher percentage of Mac1+ TUNEL+ and total TUNEL+ cells compared to YopJ-deficient bacteria, consistent with the role of YopJ in apoptosis in vivo. Furthermore, in competitive index experiments, YopJ-deficient Yersinia showed colonization defects in PPs, mLNs, and spleen. YopJ-deficient Yersinia were not defective for splenic replication following intraperitoneal infection, indicating that YopJ primarily regulates dissemination from mucosal tissues, rather than replication at systemic sites (Monack et al. 1998). Consistently, YopJ-deficient Yp are still able to cause systemic infection in a rat model of bubonic plague, despite a defect in induction of apoptosis and cytokine inhibition (Lemaitre et al. 2006). These findings imply that apoptosis may be utilized by Yersinia to eliminate immune cells at mucosal surfaces resulting in barrier dysfunction and dissemination. An alternative possibility is that YopJ-mediated blockade of cytokine production during early stages of infection limits the ability of the host to control bacterial dissemination.
Paradoxically, ectopic expression of a hypercytotoxic YopP from Ye in Yptb results in its attenuation in oral mouse infection (Brodsky and Medzhitov 2008). While both Yptb and Ye cause cell death in cultured macrophages and infected tissues, infection with YopP-expressing Ye induced a significant increase in TUNEL+ CD11b+, CD11c+, and B220+ cells in mLNs relative to mice infected with the YopP-expressing strain (Brodsky and Medzhitov 2008). Similarly, Yp strains expressing YopP had higher cytotoxic potency than strains expressing YopJ, both in vitro and in tissues of infected mice; furthermore, expression of YopP in Yp also resulted in lower virulence following subcutaneous, but not intranasal or intravenous routes of infection (Zauberman et al. 2009). Interestingly, subcutaneous administration of Yp expressing YopP protected against infection with virulent Y. pestis, regardless of the route of challenge. These observations suggest that YopJ contributes to dissemination of Yersinia from barrier surfaces, but may be less important once bacteria have spread to systemic sites. Whether YopJ or additional immunosuppressive virulence mechanisms play a role in dampening the early inflammatory response to Yersinia infection in pneumonic plague (Lathem et al. 2007) also remains to be determined. Notably, pathogenesis of pneumonic plague is influenced by levels of apoptosis, as degradation of Fas Ligand by Yp Pla protease results in reduced apoptosis, increased bacterial burdens, and reduced levels of inflammatory cytokines in lung homogenates (Caulfield et al. 2014). This implies a common role for apoptosis in the induction of inflammatory responses in multiple organ systems during Yersinia infection.
Consistent with observations that YopJ promotes systemic dissemination following oral infection, YopJ contributes to gut barrier disruption (Jung et al. 2012; Meinzer et al. 2012). Specifically, YopJ can induce TLR2-dependent IL-1β secretion in PPs, which was associated with increased barrier permeability, suggesting that TLR2 signaling mediates YopJ-dependent gut disruption (Jung et al. 2012). Conversely, TLR2-deficient mice have been reported to be more susceptible to oral infection by Yptb, due to a loss of TLR2-dependent Reg3β expression in the gut epithelium (Dessein et al. 2009). Thus, the precise role of TLR2 in Yersinia infection remains to be further dissected. Notably, IL-1α production during intestinal infection is associated with pathological intestinal inflammation and increased dissemination of Ye (Dube et al. 2001), but the role of YopP or TLR2 in this context has not been examined.
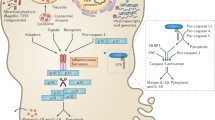
In addition to cell death induced by the activity of a bacterial virulence factor, CD8+ cytotoxic T cells also induce death of Yersinia-infected cells and are important for control of Yersinia infection, as demonstrated by the more severe disease that occurs in infected β2m −/−, anti-CD8α-treated, or perforin-deficient mice (Bergman et al. 2009). CD8+ T-cell-mediated killing of bacteria-associated cells targeted them for phagocytosis by uninfected macrophages and could bypass the anti-phagocytic activity of Yersinia Yops (Bergman et al. 2009). Interestingly however, CD8+ T cells were not responsible for increased resistance to YopP-expressing Y. pestis, suggesting that the increased cytotoxicity of these bacteria induces immune clearance through an alternative pathway (Zauberman et al. 2009). Indeed, the more cytotoxic YopP may bypass the requirement for CD8+ T-cell-mediated killing due to the elevated cytotoxicity induced by the bacteria. These studies collectively suggest that regulation of cytotoxicity during Yersinia infection impacts virulence and that a balance between the cytokine-blocking and death-inducing functions of YopJ is required for optimal virulence. Specifically, absence of YopJ results in failure of Yersinia to suppress cytokine production or induce cell death and causes a defect in dissemination (Fig. 2). However, Yptb and Y. pestis expressing YopP, which enables stronger inhibition of cytokine production and elevated levels of cell death, are also significantly attenuated in vivo. Thus, while the relative contributions of bacteria-induced and T-cell-induced cell death during Yersinia infection in vivo are not yet defined, activation of cell death in vivo either in response to YopJ activity, or as a consequence of T-cell-mediated cytotoxicity plays an important role in immune control of Yersinia infection.

Interplay of cell death signaling in virulence and host defense during intestinal Yersinia infection in vivo. Intestinal infection with Yersinia leads to dissemination of the bacteria to systemic tissues from a replicating pool in the intestine as well as spread from lamina propria to mesenteric lymph nodes (MLN). Infection of monocyte/macrophage (Mf/Mo) and DC populations and injection of YopJ results in activation of cell death pathways through caspase-8 and caspase-1. Depending on the activation state of these cells however, the combined activities of YopK, YopM, and YopJ may also limit the induction of these pathways, thus potentially limiting the activation of downstream inflammatory responses, for example, release of inflammatory mediators and T-cell responses that control anti-Yersinia immune defense
3.3 In Vivo Interactions Between Yersinia and Inflammasome Responses
Given the interconnected nature of death signaling pathways, a current challenge remains defining the degree to which inflammasome activation and pyroptosis occur in vivo, and their precise contribution to host defense. Interestingly, in the absence of YopK, Yptb infection is significantly attenuated, and this attenuation is reversed in caspase-1/11-deficient mice, implying that YopK promotes virulence at least in part by enabling Yersinia to evade inflammasome-mediated responses (Brodsky et al. 2010). Similarly, YopM deficiency results in significant attenuation that is reversed in a Casp1/11 −/− genetic background deficient, thus also implicating YopM in inflammasome evasion in vivo (LaRock and Cookson 2012). In both cases, whether the role of these virulence factors is to limit inflammasome-induced pyroptosis, or whether it is to limit the production of inflammasome-dependent cytokine responses, remains to be defined. Analysis of dual IL-1R/IL-18R-deficient animals would help address this, although other IL-1 family cytokines and inflammatory mediators are still released in the context of inflammasome activation and pyroptosis.
It is notable that Yersinia employs two distinct non-overlapping strategies to target the inflammasome. These strategies may function in a context- specific way in vivo, as the bacteria will encounter diverse cell types in distinct activation states during infection of different tissues over the course of the infection, whose responses will likely differ depending on their activation state and balance of injected effectors that they encounter. Interestingly, in primed bone marrow derived macrophages, YopJ functions together with YopM to limit inflammasome activation, which is very distinct from the function of YopJ in other contexts (Ratner et al. 2016; Schoberle et al. 2016). Furthermore, in vivo during intravenous infection, there was an additive effect of yopJ and yopM deficiency, providing further support for an in vivo role of YopJ activity in virulence. Again the specific nature of how YopJ activity promotes virulence in vivo—i.e., whether it is through blockade of cytokine production, inducing apoptosis, or preventing pyroptosis, remains to be determined.
During pneumonic plague infection in murine models, Y. pestis induces minimal inflammatory responses during the initial stages of infection within the first 24–36 h (Lathem et al. 2007). This may be due to the combination of both the non-stimulatory, tetra-acylated LPS that Y. pestis produces within mammalian hosts as well as the blockade of NF-κB and MAPK signaling by YopJ discussed above (Palmer et al. 1998; Rebeil et al. 2006; Ruckdeschel et al. 1997). Interestingly, as with Yptb intestinal infections, YopK is also an important Yp virulence factor that modulates cell death pathways in the context of pneumonic plague and may therefore also contribute to Yersinia immune evasion in this context (Peters et al. 2013). Notably, while caspase-1 activation does occur in the lung, accompanied by production of IL-1β and IL-18, Y. pestis also induces expression of the IL-1 receptor antagonist (IL-1RA), thereby limiting the ability of the host to respond to the presence of these cytokines (Pechous et al. 2016; Sivaraman et al. 2015). This pre-inflammatory phase may contribute to dissemination of the bacteria throughout the tissue and to systemic sites, resulting in a profound subsequent inflammatory response that is accompanied by neutrophilic infiltrate and immunopathologic tissue destruction (Lathem et al. 2007; Sivaraman et al. 2015). Whether YopJ-induced apoptosis of innate immune cells contributes to host defense or to bacterial dissemination in this context has not been teased apart, in large part because it has not been possible to separate blockade of cytokine production and induction of apoptosis during Yersinia infection (Philip and Brodsky 2012; Philip et al. 2014).
4 Concluding Remarks
Like all pathogens, Yersinia employs multiple virulence factors and strategies to balance the need to disable host defenses with the need to evade recognition by innate immune responses. The T3SS confers the ability to subvert host signaling pathways but also exposes Yersinia to detection by cytosolic sensing systems. Notably, Yersinia has solved this problem by means of multiple virulence factors that limit detection—in particular, two secreted effector proteins, YopK and YopM, prevent inflammasome activation by limiting the translocation of molecules that trigger inflammasome responses, and by direct blocking of caspase-1 activation, respectively. Whether other pathogens with T3SS utilize similar strategies to evade inflammasome activation remains undefined. While no sequence homologs of YopK exist, other pathogens also modulate inflammasome activation during infection, and understanding how pathogens interfere with inflammasome activation provides insight into inflammasome functioning (Shin and Brodsky 2015; Wynosky-Dolfi et al. 2014). Interestingly, YopJ, classically viewed as an activator of cell death responses, was recently found to act as a redundant factor with YopM in preventing inflammasome activation (Schoberle et al. 2016). How macrophage priming alters their responsiveness, and how macrophage priming switches the response to YopJ from activating to inhibiting inflammasome activation remains to be defined. It is possible that priming of macrophages upregulates a novel set of transcriptional responses that provide a target for YopJ activity that is absent from unprimed cells. Treatment of cells with LPS and pharmacological inhibitors of both MAPK and IKK results in robust cell death that in many ways phenocopies the activity of YopJ. Whether it is possible to similarly pharmacologically mimic the inflammasome inhibition function of YopJ that has been observed in primed macrophages remains to be determined.
The mechanisms and signaling pathways engaged by the host to conduct various cell death programs during Yersinia infection have been characterized extensively in vitro. Future studies will need to define how modulation of different cell death pathways impacts replication, dissemination, and host cytokine production in vivo. Addressing this question is likely to shed significant new light on our understanding of infection dynamics and may provide new targets for intervention in the context of many types of infections that modulate cell death pathways. An important question remains whether some commensal bacteria might also utilize T3SSs for purposes of long-term colonization, and consequently, whether such bacteria might modulate key host signaling components and pathways in order to limit activation of inflammasomes or other inflammatory cell death pathways. Indeed, a ‘pathobiont’ E. coli that expresses a T3SS was recently identified as being expanded in a genetically susceptible mouse strain lacking Nlrc4 and results in increased susceptibility to a mouse model of colitis (Ayres et al. 2012). How this strain avoids inducing overt disease in the absence of a colitis-inducing stimulus is unclear, but may involve limiting induction of inflammasome or other inflammatory death signaling pathways in a manner conceptually similar to those described here. Despite the nearly 20 years since the initial discovery that Yersinia induces cell death in infected macrophages, a great deal likely remains to be discovered. The availability of CRISPR-based approaches to rapidly make knockouts and point mutations in cellular signaling components that interact with bacterial virulence factors is likely to facilitate rapid progress in this area.
References
Achtman M, Zurth K, Morelli G, Torrea G, Guiyoule A, Carniel E (1999) Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc Natl Acad Sci USA 96(24):14043–14048
Ayres JS, Trinidad NJ, Vance RE (2012) Lethal inflammasome activation by a multidrug-resistant pathobiont upon antibiotic disruption of the microbiota. Nat Med 18(5):799–806. doi:10.1038/nm.2729
Balada-Llasat JM, Mecsas J (2006) Yersinia has a tropism for B and T cell zones of lymph nodes that is independent of the type III secretion system. PLoS Pathog 2(9):e86
Barnes PD, Bergman MA, Mecsas J, Isberg RR (2006) Yersinia pseudotuberculosis disseminates directly from a replicating bacterial pool in the intestine. J Exp Med 203(6):1591–1601
Ben Ari T, Gershunov A, Gage KL, Snall T, Ettestad P, Kausrud KL, Stenseth NC (2008) Human plague in the USA: the importance of regional and local climate. Biol Lett 4(6):737–740. doi:10.1098/rsbl.2008.0363
Bergman MA, Loomis WP, Mecsas J, Starnbach MN, Isberg RR (2009) CD8(+) T cells restrict Yersinia pseudotuberculosis infection: bypass of anti-phagocytosis by targeting antigen-presenting cells. PLoS Pathog 5(9):e1000573. doi:10.1371/journal.ppat.1000573
Bergsbaken T, Cookson BT (2007) Macrophage activation redirects Yersinia-infected host cell death from apoptosis to caspase-1-dependent pyroptosis. PLoS Pathog 3(11):e161
Bhaduri S, Wesley IV, Bush EJ (2005) Prevalence of pathogenic Yersinia enterocolitica strains in pigs in the United States. Appl Environ Microbiol 71(11):7117–7121. doi:10.1128/AEM.71.11.7117-7121.2005
Brodsky IE, Medzhitov R (2008) Reduced secretion of YopJ by Yersinia limits in vivo cell death but enhances bacterial virulence. PLoS Pathog 4(5):e1000067. doi:10.1371/journal.ppat.1000067
Brodsky IE, Palm NW, Sadanand S, Ryndak MB, Sutterwala FS, Flavell RA, Bliska JB, Medzhitov R (2010) A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe 7(5):376–387
Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM (2010) Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med 207(8):1745–1755. doi:10.1084/jem.20100257
Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, Monack DM (2012) Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490(7419):288–291. doi:10.1038/nature11419
Campisi L, Cummings RJ, Blander JM (2014) Death-defining immune responses after apoptosis. Am J Transplant 14(7):1488–1498. doi:10.1111/ajt.12736
Casson CN, Copenhaver AM, Zwack EE, Nguyen HT, Strowig T, Javdan B, Bradley WP, Fung TC, Flavell RA, Brodsky IE, Shin S (2013) Caspase-11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog 9(6):e1003400. doi:10.1371/journal.ppat.1003400
Caulfield AJ, Walker ME, Gielda LM, Lathem WW (2014) The Pla protease of Yersinia pestis degrades fas ligand to manipulate host cell death and inflammation. Cell Host Microbe 15(4):424–434. doi:10.1016/j.chom.2014.03.005
Chain PS, Carniel E, Larimer FW, Lamerdin J, Stoutland PO, Regala WM, Georgescu AM, Vergez LM, Land ML, Motin VL, Brubaker RR, Fowler J, Hinnebusch J, Marceau M, Medigue C, Simonet M, Chenal-Francisque V, Souza B, Dacheux D, Elliott JM, Derbise A, Hauser LJ, Garcia E (2004) Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis. Proc Natl Acad Sci USA 101(38):13826–13831. doi:10.1073/pnas.0404012101
Chung LK, Philip NH, Schmidt VA, Koller A, Strowig T, Flavell RA, Brodsky IE, Bliska JB (2014) IQGAP1 is important for activation of caspase-1 in macrophages and is targeted by Yersinia pestis type III effector YopM. MBio 5(4):e01402–e01414. doi:10.1128/mBio.01402-14
Cornelis GR (2006) The type III secretion injectisome. Nat Rev Microbiol 4(11):811–825. doi:nrmicro1526 [pii] 10.1038/nrmicro1526
Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4(5):313–321. doi:10.1038/nchembio.83
Denecker G, Declercq W, Geuijen CA, Boland A, Benabdillah R, van Gurp M, Sory MP, Vandenabeele P, Cornelis GR (2001) Yersinia enterocolitica YopP-induced apoptosis of macrophages involves the apoptotic signaling cascade upstream of bid. J Biol Chem 276(23):19706–19714
Dessein R, Gironella M, Vignal C, Peyrin-Biroulet L, Sokol H, Secher T, Lacas-Gervais S, Gratadoux JJ, Lafont F, Dagorn JC, Ryffel B, Akira S, Langella P, Nunez G, Sirard JC, Iovanna J, Simonet M, Chamaillard M (2009) Toll-like receptor 2 is critical for induction of Reg3 beta expression and intestinal clearance of Yersinia pseudotuberculosis. Gut 58(6):771–776. doi:10.1136/gut.2008.168443
Dewoody R, Merritt PM, Houppert AS, Marketon MM (2011) YopK regulates the Yersinia pestis type III secretion system from within host cells. Mol Microbiol 79(6):1445–1461. doi:10.1111/j.1365-2958.2011.07534.x
Dube PH, Revell PA, Chaplin DD, Lorenz RG, Miller VL (2001) A role for IL-1 alpha in inducing pathologic inflammation during bacterial infection. Proc Natl Acad Sci USA 98(19):10880–10885. doi:10.1073/pnas.191214498
Durand EA, Maldonado-Arocho FJ, Castillo C, Walsh RL, Mecsas J (2010) The presence of professional phagocytes dictates the number of host cells targeted for Yop translocation during infection. Cell Microbiol 12(8):1064–1082. doi:10.1111/j.1462-5822.2010.01451.x
Erfurth SE, Grobner S, Kramer U, Gunst DS, Soldanova I, Schaller M, Autenrieth IB, Borgmann S (2004) Yersinia enterocolitica induces apoptosis and inhibits surface molecule expression and cytokine production in murine dendritic cells. Infect Immun 72(12):7045–7054
Evdokimov AG, Anderson DE, Routzahn KM, Waugh DS (2001) Unusual molecular architecture of the Yersinia pestis cytotoxin YopM: a leucine-rich repeat protein with the shortest repeating unit. J Mol Biol 312(4):807–821. doi:10.1006/jmbi.2001.4973
Grabenstein JP, Marceau M, Pujol C, Simonet M, Bliska JB (2004) The response regulator PhoP of Yersinia pseudotuberculosis is important for replication in macrophages and for virulence. Infect Immun 72(9):4973–4984
Green DR, Ferguson T, Zitvogel L, Kroemer G (2009) Immunogenic and tolerogenic cell death. Nat Rev Immunol 9(5):353–363. doi:10.1038/nri2545
Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, Goktuna SI, Neuenhahn M, Fierer J, Paxian S, Van Rooijen N, Xu Y, O’Cain T, Jaffee BB, Busch DH, Duyster J, Schmid RM, Eckmann L, Karin M (2007) NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 130(5):918–931. doi:10.1016/j.cell.2007.07.009
Grobner S, Autenrieth SE, Soldanova I, Gunst DS, Schaller M, Bohn E, Muller S, Leverkus M, Wesselborg S, Autenrieth IB, Borgmann S (2006) Yersinia YopP-induced apoptotic cell death in murine dendritic cells is partially independent from action of caspases and exhibits necrosis-like features. Apoptosis 11(11):1959–1968. doi:10.1007/s10495-006-0189-3
Grobner S, Adkins I, Schulz S, Richter K, Borgmann S, Wesselborg S, Ruckdeschel K, Micheau O, Autenrieth IB (2007) Catalytically active Yersinia outer protein P induces cleavage of RIP and caspase-8 at the level of the DISC independently of death receptors in dendritic cells. Apoptosis 12(10):1813–1825. doi:10.1007/s10495-007-0100-x
Gurung P, Malireddi RK, Anand PK, Demon D, Walle LV, Liu Z, Vogel P, Lamkanfi M, Kanneganti TD (2012) Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-beta (TRIF)-mediated caspase-11 protease production integrates Toll-like receptor 4 (TLR4) protein- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J Biol Chem 287(41):34474–34483. doi:10.1074/jbc.M112.401406
Gurung P, Anand PK, Malireddi RK, Vande Walle L, Van Opdenbosch N, Dillon CP, Weinlich R, Green DR, Lamkanfi M, Kanneganti TD (2014) FADD and Caspase-8 Mediate Priming and Activation of the Canonical and Noncanonical Nlrp3 Inflammasomes. J Immunol 192(4):1835–1846. doi:10.4049/jimmunol.1302839
Haase R, Kirschning CJ, Sing A, Schrottner P, Fukase K, Kusumoto S, Wagner H, Heesemann J, Ruckdeschel K (2003) A dominant role of Toll-like receptor 4 in the signaling of apoptosis in bacteria-faced macrophages. J Immunol 171(8):4294–4303
He S, Liang Y, Shao F, Wang X (2011) Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA 108(50):20054–20059. doi:10.1073/pnas.1116302108
Heath WR, Carbone FR (2001) Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol 19:47–64. doi:10.1146/annurev.immunol.19.1.47
Hentschke M, Berneking L, Belmar Campos C, Buck F, Ruckdeschel K, Aepfelbacher M (2010) Yersinia virulence factor YopM induces sustained RSK activation by interfering with dephosphorylation. PLoS One 5(10). doi:10.1371/journal.pone.0013165
Hinnebusch BJ, Rosso ML, Schwan TG, Carniel E (2002) High-frequency conjugative transfer of antibiotic resistance genes to Yersinia pestis in the flea midgut. Mol Microbiol 46(2):349–354
Holmstrom A, Rosqvist R, Wolf-Watz H, Forsberg A (1995) Virulence plasmid-encoded YopK is essential for Yersinia pseudotuberculosis to cause systemic infection in mice. Infect Immun 63(6):2269–2276
Holmstrom A, Petterson J, Rosqvist R, Hakansson S, Tafazoli F, Fallman M, Magnusson KE, Wolf-Watz H, Forsberg A (1997) YopK of Yersinia pseudotuberculosis controls translocation of Yop effectors across the eukaryotic cell membrane. Mol Microbiol 24(1):73–91
Jung C, Meinzer U, Montcuquet N, Thachil E, Chateau D, Thiebaut R, Roy M, Alnabhani Z, Berrebi D, Dussaillant M, Pedruzzi E, Thenet S, Cerf-Bensussan N, Hugot JP, Barreau F (2012) Yersinia pseudotuberculosis disrupts intestinal barrier integrity through hematopoietic TLR-2 signaling. J Clin Invest 122(6):2239–2251. doi:10.1172/JCI58147
Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479(7371):117–121. doi:10.1038/nature10558
Koberle M, Klein-Gunther A, Schutz M, Fritz M, Berchtold S, Tolosa E, Autenrieth IB, Bohn E (2009) Yersinia enterocolitica targets cells of the innate and adaptive immune system by injection of Yops in a mouse infection model. PLoS Pathog 5(8):e1000551. doi:10.1371/journal.ppat.1000551
Kwit N, Nelson C, Kugeler K, Petersen J, Plante L, Yaglom H, Kramer V, Schwartz B, House J, Colton L, Feldpausch A, Drenzek C, Baumbach J, DiMenna M, Fisher E, Debess E, Buttke D, Weinburke M, Percy C, Schriefer M, Gage K, Mead P (2015) Human Plague—United States, 2015. MMWR Morb Mortal Wkly Rep 64(33):918–919
Kwuan L, Adams W, Auerbuch V (2013) Impact of host membrane pore formation by the Yersinia pseudotuberculosis type III secretion system on the macrophage innate immune response. Infect Immun 81(3):905–914. doi:10.1128/IAI.01014-12
LaRock CN, Cookson BT (2012) The Yersinia virulence effector YopM binds caspase-1 to arrest inflammasome assembly and processing. Cell Host Microbe 12(6):799–805. doi:10.1016/j.chom.2012.10.020
Lathem WW, Price PA, Miller VL, Goldman WE (2007) A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science 315(5811):509–513
Lemaitre N, Sebbane F, Long D, Hinnebusch BJ (2006) Yersinia pestis YopJ suppresses tumor necrosis factor alpha induction and contributes to apoptosis of immune cells in the lymph node but is not required for virulence in a rat model of bubonic plague. Infect Immun 74(9):5126–5131
Lilo S, Zheng Y, Bliska JB (2008) Caspase-1 activation in macrophages infected with Yersinia pestis KIM requires the type III secretion system effector YopJ. Infect Immun 76(9):3911–3923. doi:IAI.01695-07 [pii] 10.1128/IAI.01695-07
Maldonado-Arocho FJ, Green C, Fisher ML, Paczosa MK, Mecsas J (2013) Adhesins and host serum factors drive Yop translocation by yersinia into professional phagocytes during animal infection. PLoS Pathog 9(6):e1003415. doi:10.1371/journal.ppat.1003415
Man SM, Tourlomousis P, Hopkins L, Monie TP, Fitzgerald KA, Bryant CE (2013) Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1beta production. J Immunol 191(10):5239–5246. doi:10.4049/jimmunol.1301581
Marketon MM, DePaolo RW, DeBord KL, Jabri B, Schneewind O (2005) Plague bacteria target immune cells during infection. Science 309(5741):1739–1741. doi:10.1126/science.1114580
McCoy MW, Marre ML, Lesser CF, Mecsas J (2010) The C-terminal tail of Yersinia pseudotuberculosis YopM is critical for interacting with RSK1 and for virulence. Infect Immun 78(6):2584–2598. doi:10.1128/IAI.00141-10
McDonald C, Vacratsis PO, Bliska JB, Dixon JE (2003) The yersinia virulence factor YopM forms a novel protein complex with two cellular kinases. J Biol Chem 278(20):18514–18523. doi:10.1074/jbc.M301226200
McPhee JB, Mena P, Bliska JB (2010) Delineation of regions of the Yersinia YopM protein required for interaction with the RSK1 and PRK2 host kinases and their requirement for interleukin-10 production and virulence. Infect Immun 78(8):3529–3539. doi:10.1128/IAI.00269-10
McPhee JB, Mena P, Zhang Y, Bliska JB (2012) Interleukin-10 induction is an important virulence function of the Yersinia pseudotuberculosis type III effector YopM. Infect Immun 80(7):2519–2527. doi:10.1128/IAI.06364-11
Meinzer U, Barreau F, Esmiol-Welterlin S, Jung C, Villard C, Leger T, Ben-Mkaddem S, Berrebi D, Dussaillant M, Alnabhani Z, Roy M, Bonacorsi S, Wolf-Watz H, Perroy J, Ollendorff V, Hugot JP (2012) Yersinia pseudotuberculosis effector YopJ subverts the Nod2/RICK/TAK1 pathway and activates caspase-1 to induce intestinal barrier dysfunction. Cell Host Microbe 11(4):337–351. doi:10.1016/j.chom.2012.02.009
Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A (2010) Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A 107(7):3076–3080. doi:10.1073/pnas.0913087107
Mills SD, Boland A, Sory MP, van der Smissen P, Kerbourch C, Finlay BB, Cornelis GR (1997) Yersinia enterocolitica induces apoptosis in macrophages by a process requiring functional type III secretion and translocation mechanisms and involving YopP, presumably acting as an effector protein. Proc Natl Acad Sci USA 94(23):12638–12643
Minnich SA, Rohde HN (2007) A rationale for repression and/or loss of motility by pathogenic Yersinia in the mammalian host. Adv Exp Med Biol 603:298–310. doi:10.1007/978-0-387-72124-8_27
Mittal R, Peak-Chew SY, McMahon HT (2006) Acetylation of MEK2 and I kappa B kinase (IKK) activation loop residues by YopJ inhibits signaling. Proc Natl Acad Sci USA 103(49):18574–18579
Monack DM, Mecsas J, Ghori N, Falkow S (1997) Yersinia signals macrophages to undergo apoptosis and YopJ is necessary for this cell death. Proc Natl Acad Sci USA 94(19):10385–10390
Monack DM, Mecsas J, Bouley D, Falkow S (1998) Yersinia-induced apoptosis in vivo aids in the establishment of a systemic infection of mice. J Exp Med 188(11):2127–2137
Mukherjee S, Keitany G, Li Y, Wang Y, Ball HL, Goldsmith EJ, Orth K (2006) Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312(5777):1211–1214
Munoz-Pinedo C (2012) Signaling pathways that regulate life and cell death: evolution of apoptosis in the context of self-defense. Adv Exp Med Biol 738:124–143. doi:10.1007/978-1-4614-1680-7_8
Nemeth J, Straley SC (1997) Effect of Yersinia pestis YopM on experimental plague. Infect Immun 65(3):924–930
Palmer LE, Hobbie S, Galan JE, Bliska JB (1998) YopJ of Yersinia pseudotuberculosis is required for the inhibition of macrophage TNF-alpha production and downregulation of the MAP kinases p38 and JNK. Mol Microbiol 27(5):953–965
Pechous RD, Sivaraman V, Stasulli NM, Goldman WE (2016) Pneumonic plague: the darker side of Yersinia pestis. Trends Microbiol 24(3):190–197. doi:10.1016/j.tim.2015.11.008
Peters KN, Dhariwala MO, Hughes Hanks JM, Brown CR, Anderson DM (2013) Early apoptosis of macrophages modulated by injection of Yersinia pestis YopK promotes progression of primary pneumonic plague. PLoS Pathog 9(4):e1003324. doi:10.1371/journal.ppat.1003324
Philip NH, Brodsky IE (2012) Cell death programs in Yersinia immunity and pathogenesis. Front Cell Infect Microbiol 2:149. doi:10.3389/fcimb.2012.00149
Philip NH, Dillon CP, Snyder AG, Fitzgerald P, Wynosky-Dolfi MA, Zwack EE, Hu B, Fitzgerald L, Mauldin EA, Copenhaver AM, Shin S, Wei L, Parker M, Zhang J, Oberst A, Green DR, Brodsky IE (2014) Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-kappaB and MAPK signaling. Proc Natl Acad Sci USA 111(20):7385–7390. doi:10.1073/pnas.1403252111
Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA (2012) TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. doi:10.1016/j.cell.2012.07.007
Ratner D, Orning MP, Starheim KK, Marty-Roix R, Proulx MK, Goguen JD, Lien E (2016) Manipulation of IL-1beta and IL-18 production by Yersinia pestis effectors YopJ and YopM and redundant impact on virulence. J Biol Chem. doi:10.1074/jbc.M115.697698
Rebeil R, Ernst RK, Jarrett CO, Adams KN, Miller SI, Hinnebusch BJ (2006) Characterization of late acyltransferase genes of Yersinia pestis and their role in temperature-dependent lipid A variation. J Bacteriol 188(4):1381–1388. doi:10.1128/JB.188.4.1381-1388.2006
Rosadini CV, Zanoni I, Odendall C, Green ER, Paczosa MK, Philip NH, Brodsky IE, Mecsas J, Kagan JC (2015) A single bacterial immune evasion strategy dismantles both MyD88 and TRIF signaling pathways downstream of TLR4. Cell Host Microbe 18(6):682–693. doi:10.1016/j.chom.2015.11.006
Rosenberg JC (1968) Doctors afield: Alexandre Yersin. N Engl J Med 278(5):261–263. doi:10.1056/NEJM196802012780507
Ruckdeschel K, Richter K (2002) Lipopolysaccharide desensitization of macrophages provides protection against Yersinia enterocolitica-induced apoptosis. Infect Immun 70(9):5259–5264
Ruckdeschel K, Machold J, Roggenkamp A, Schubert S, Pierre J, Zumbihl R, Liautard JP, Heesemann J, Rouot B (1997) Yersinia enterocolitica promotes deactivation of macrophage mitogen-activated protein kinases extracellular signal-regulated kinase-1/2, p38, and c-Jun NH2-terminal kinase. Correlation with its inhibitory effect on tumor necrosis factor-alpha production. J Biol Chem 272(25):15920–15927
Ruckdeschel K, Harb S, Roggenkamp A, Hornef M, Zumbihl R, Kohler S, Heesemann J, Rouot B (1998) Yersinia enterocolitica impairs activation of transcription factor NF-kappaB: involvement in the induction of programmed cell death and in the suppression of the macrophage tumor necrosis factor alpha production. J Exp Med 187(7):1069–1079
Ruckdeschel K, Richter K, Mannel O, Heesemann J (2001) Arginine-143 of Yersinia enterocolitica YopP crucially determines isotype-related NF-kappaB suppression and apoptosis induction in macrophages. Infect Immun 69(12):7652–7662
Ruckdeschel K, Pfaffinger G, Haase R, Sing A, Weighardt H, Hacker G, Holzmann B, Heesemann J (2004) Signaling of apoptosis through TLRs critically involves toll/IL-1 receptor domain-containing adapter inducing IFN-beta, but not MyD88, in bacteria-infected murine macrophages. J Immunol 173(5):3320–3328
Ruter C, Silva MR, Grabowski B, Lubos ML, Scharnert J, Poceva M, von Tils D, Flieger A, Heesemann J, Bliska JB, Schmidt MA (2014) Rabbit monoclonal antibodies directed at the T3SS effector protein YopM identify human pathogenic Yersinia isolates. Int J Med Microbiol 304(3–4):444–451. doi:10.1016/j.ijmm.2014.02.003
Schesser K, Spiik AK, Dukuzumuremyi JM, Neurath MF, Pettersson S, Wolf-Watz H (1998) The yopJ locus is required for Yersinia-mediated inhibition of NF-kappaB activation and cytokine expression: YopJ contains a eukaryotic SH2-like domain that is essential for its repressive activity. Mol Microbiol 28(6):1067–1079
Schoberle TJ, Chung LK, McPhee JB, Bogin B, Bliska JB (2016) Uncovering an important role for YopJ in the inhibition of Caspase-1 in activated macrophages and promoting Yersinia pseudotuberculosis virulence. Infect Immun. doi:10.1128/IAI.00843-15
Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514(7521):187–192. doi:10.1038/nature13683
Shin S, Brodsky IE (2015) The inflammasome: learning from bacterial evasion strategies. Semin Immunol 27(2):102–110. doi:10.1016/j.smim.2015.03.006
Sivaraman V, Pechous RD, Stasulli NM, Eichelberger KR, Miao EA, Goldman WE (2015) Yersinia pestis activates both IL-1beta and IL-1 receptor antagonist to modulate lung inflammation during pneumonic plague. PLoS Pathog 11(3):e1004688. doi:10.1371/journal.ppat.1004688
Thorslund SE, Edgren T, Pettersson J, Nordfelth R, Sellin ME, Ivanova E, Francis MS, Isaksson EL, Wolf-Watz H, Fallman M (2011) The RACK1 signaling scaffold protein selectively interacts with Yersinia pseudotuberculosis virulence function. PLoS ONE 6(2):e16784. doi:10.1371/journal.pone.0016784
Torchinsky MB, Garaude J, Martin AP, Blander JM (2009) Innate immune recognition of infected apoptotic cells directs T(H)17 cell differentiation. Nature 458(7234):78–82. doi:10.1038/nature07781
Vance RE, Isberg RR, Portnoy DA (2009) Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe 6(1):10–21. doi:10.1016/j.chom.2009.06.007
Vazquez-Torres A, Jones-Carson J, Baumler AJ, Falkow S, Valdivia R, Brown W, Le M, Berggren R, Parks WT, Fang FC (1999) Extraintestinal dissemination of Salmonella by CD18-expressing phagocytes. Nature 401(6755):804–808. doi:10.1038/44593
Viboud GI, Bliska JB (2005) Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol 59:69–89. doi:10.1146/annurev.micro.59.030804.121320
Vladimer GI, Marty-Roix R, Ghosh S, Weng D, Lien E (2013) Inflammasomes and host defenses against bacterial infections. Curr Opin Microbiol 16(1):23–31. doi:10.1016/j.mib.2012.11.008
Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, Mocarski ES, Pouliot K, Chan FK, Kelliher MA, Harris PA, Bertin J, Gough PJ, Shayakhmetov DM, Goguen JD, Fitzgerald KA, Silverman N, Lien E (2014) Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc Natl Acad Sci USA 111(20):7391–7396. doi:10.1073/pnas.1403477111
Wesley IV, Bhaduri S, Bush E (2008) Prevalence of Yersinia enterocolitica in market weight hogs in the United States. J Food Prot 71(6):1162–1168
Wren BW (2003) The yersiniae–a model genus to study the rapid evolution of bacterial pathogens. Nat Rev Microbiol 1(1):55–64
Wynosky-Dolfi MA, Snyder AG, Philip NH, Doonan PJ, Poffenberger MC, Avizonis D, Zwack EE, Riblett AM, Hu B, Strowig T, Flavell RA, Jones RG, Freedman BD, Brodsky IE (2014) Oxidative metabolism enables Salmonella evasion of the NLRP3 inflammasome. J Exp Med 211(4):653–668. doi:10.1084/jem.20130627
Zauberman A, Cohen S, Mamroud E, Flashner Y, Tidhar A, Ber R, Elhanany E, Shafferman A, Velan B (2006) Interaction of Yersinia pestis with macrophages: limitations in YopJ-dependent apoptosis. Infect Immun 74(6):3239–3250
Zauberman A, Tidhar A, Levy Y, Bar-Haim E, Halperin G, Flashner Y, Cohen S, Shafferman A, Mamroud E (2009) Yersinia pestis endowed with increased cytotoxicity is avirulent in a bubonic plague model and induces rapid protection against pneumonic plague. PLoS ONE 4(6):e5938. doi:10.1371/journal.pone.0005938
Zhang Y, Ting AT, Marcu KB, Bliska JB (2005) Inhibition of MAPK and NF-kappa B pathways is necessary for rapid apoptosis in macrophages infected with Yersinia. J Immunol 174(12):7939–7949
Zheng Y, Lilo S, Brodsky IE, Zhang Y, Medzhitov R, Marcu KB, Bliska JB (2011) A Yersinia effector with enhanced inhibitory activity on the NF-kappaB pathway activates the NLRP3/ASC/caspase-1 inflammasome in macrophages. PLoS Pathog 7(4):e1002026. doi:10.1371/journal.ppat.1002026
Zheng Y, Lilo S, Mena P, Bliska JB (2012) YopJ-induced caspase-1 activation in Yersinia-infected macrophages: independent of apoptosis, linked to necrosis, dispensable for innate host defense. PLoS ONE 7(4):e36019. doi:10.1371/journal.pone.0036019
Zhou H, Monack DM, Kayagaki N, Wertz I, Yin J, Wolf B, Dixit VM (2005) Yersinia virulence factor YopJ acts as a deubiquitinase to inhibit NF-kappa B activation. J Exp Med 202(10):1327–1332
Zimbler DL, Schroeder JA, Eddy JL, Lathem WW (2015) Early emergence of Yersinia pestis as a severe respiratory pathogen. Nat Commun 6:7487. doi:10.1038/ncomms8487
Zwack EE, Snyder AG, Wynosky-Dolfi MA, Ruthel G, Philip NH, Marketon MM, Francis MS, Bliska JB, Brodsky IE (2014) Inflammasome activation in response to the Yersinia Type III secretion system requires hyperinjection of translocon proteins YopB and YopD. MBio 6(1). doi:10.1128/mBio.02095-14
Acknowledgments
We would like to thank Sunny Shin and members of the Brodsky and Shin labs for scientific discussion. Work in the Brodsky lab is supported by the NIH-NIAID and a BWF Investigator in the Pathogenesis of Infectious Disease Award.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Philip, N.H., Zwack, E.E., Brodsky, I.E. (2016). Activation and Evasion of Inflammasomes by Yersinia . In: Backert, S. (eds) Inflammasome Signaling and Bacterial Infections. Current Topics in Microbiology and Immunology, vol 397. Springer, Cham. https://doi.org/10.1007/978-3-319-41171-2_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-41171-2_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-41170-5
Online ISBN: 978-3-319-41171-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)