
Abstract
Yersinia outer protein P (YopP) is injected by Y. enterocolitica into host cells thereby inducing apoptotic and necrosis-like cell death in dendritic cells (DC). Here we show the pathways involved in DC death caused by the catalytic activity of YopP. Infection with Yersinia enterocolitica, translocating catalytically active YopP into DC, triggered procaspase-8 cleavage and c-FLIPL degradation. YopP-dependent caspase-8 activation was, however, not mediated by tumor necrosis factor (TNF) receptor family members since the expression of both CD95/Fas/APO-1 and TRAIL-R2 on DC was low, and DC were resistant to apoptosis induced by agonistic anti-CD95 antibodies or TNF-related apoptosis-inducing ligand (TRAIL). Moreover, DC from TNF-Rp55−/− mice were not protected against YopP-induced cell death demonstrating that TNF-R1 is also not involved in this process. Activation of caspase-8 was further investigated by coimmunoprecitation of FADD from Yersinia-infected DC. We found that both cleaved caspase-8 and receptor interacting protein 1 (RIP1) were associated with the Fas-associated death domain (FADD) indicating the formation of an atypical death-inducing signaling complex (DISC). Furthermore, degradation of RIP mediated by the Hsp90 inhibitor geldanamycin significantly impaired YopP-induced cell death. Altogether our findings indicate that Yersinia-induced DC death is independent of death domain containing receptors, but mediated by RIP and caspase-8 at the level of DISC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Gram-negative rod Yersinia enterocolitica causes diarrhoea and occasionally systemic infections in human and rodents [1]. Yersinia outer proteins (Yops) injected by a type three secretion system into host cells are essential for Yersinia-mediated pathogenicity [2]. YopJ of Y. pseudotuberculosis, also called Yersinia outer protein P (YopP) in Y. enterocolitica, was postulated to have protease activity toward ubiquitin-like proteins [3] with a presumptive cysteine protease catalytic triad His109-Glu128-Cys172 [3–5]. Recently, YopP/J was shown to be an acetyltransferase and deubiquitinase thereby inhibiting mitogen-activated protein kinase and nuclear factor (NF)-κB signaling pathways [6, 7]. YopP/J enhances the apoptotic potential of lipopolysaccharide (LPS) in antigen presenting cells by targeting inhibitory κB kinase (IKK)-β and TGF-β-activated kinase-1 (TAK-1) resulting in impaired activation of the anti-apoptotic NF-κB and subsequent cell death [8–13]. In dendritic cells (DC) YopP causes apoptotic cell death through caspase cleavage but also triggers at the same time a caspase-independent necrosis-like cell death [10, 14, 15].
Several studies have proposed an upstream role of caspase-8 in YopP-induced cell death. For example, macrophages transfected with dominant negative Fas-associated death domain (FADD) or the caspase-8 inhibitor crmA were protected against YopP-induced cell death [12, 16]. In addition, YopP might activate the mitochondrial apoptosis pathway by the proteolytic cleavage of the caspase-8 substrate Bid [4, 15]. However, DC overexpressing anti-apoptotic Bcl-2 were not protected against Yersinia-induced cell death suggesting that YopP does not induce DC cell death primarily via the mitochondrial death pathway [15].
The upstream signaling events in YopP-mediated activation of caspase-8 leading to cell death of antigen presenting cells are largely unknown. Caspase-8 is the initiator caspase of death domain containing receptors the activation of which causes cell death via the activation of the caspase cascade by proteolytic cleavage and activation of the downstream effector caspases 3, 6 and 7 [17]. This pathway is typically activated upon binding of the death receptor ligands tumor necrosis factor (TNF)-alpha, CD95L/FasL/APO-1L and TRAIL to the death receptors TNF-R1, CD95/Fas/APO-1 and TRAIL-R, respectively. Binding of the ligand to the corresponding death receptor leads to the rapid recruitment of the adaptor proteins FADD or TNF-R-associated death domain (TRADD) which is mediated by the intracellular death domain (DD) of the death receptors [18, 19]. FADD contains a death effector domain (DED) by which one of two DEDs within procaspase-8 are bound inducing the autocatalytic activation of procaspase-8 [20].
The caspase-8 activating complex which consists of the death receptors CD95 or TRAIL-R1, -R2, the adaptor protein FADD, and caspase-8 is called death-inducing signaling complex (DISC) [21–23]. Apoptosis initiated by death receptors is regulated at the level of DISC by the cellular Fas-associated death domain-like interleukin-1-β-converting enzyme (FLICE)-inhibitory protein long form (c-FLIPL), a potent inhibitor of caspase-8 activation which interacts with FADD by its two DEDs [24–27]. c-FLIPL is a catalytically inactive homologue of caspase-8 which blocks caspase-8 activity within the DISC by forming a proteolytically inactive heterodimer with caspase-8. FLIPL is processed by caspase-8 to a p43 cleavage product within the DISC which specifically interacts with TRAF2 and subsequently induces activation of the NF-κB signaling pathway, thereby acting as an anti-apoptotic molecule [28–31]. Beside FLIPL the receptor interacting protein (RIP) can be recruited to the DISC by its DED domain triggering an alternative necrosis-like form of cell death on the one hand [32, 33] and the activation of NF-κB on the other hand [34–36].
Here we studied the impact of death receptor signaling via TNF-R1, CD95 and TRAIL-R2 on cell death of DC induced by the catalytic activity of YopP. We show that death receptor activation is unlikely to be involved but that both caspase-8 activation and RIP play an important role in YopP-induced DC death.
Materials and methods
Bacterial strains and plasmids
The mutant strains of Y. enterocolitica and plasmids used in this study are listed in Table 1. To prepare bacteria for the infection, overnight cultures grown at 27°C were diluted 1:20 in fresh Luria-Bertani broth supplemented with the appropriate antibiotics and grown for 1.5–2 h at 37°C. After washing the bacteria with PBS the bacterial concentration was measured densitometrically at 600 nm.
Cell culture and infection of cells
Bone marrow-derived DC were obtained from 6–8 week old BALB/c, C57BL/6 (Harlan, Borchen, Germany) or TNF-Rp55−/− mice (The Jackson Laboratory, Bar Habor, USA) [39] mice as previously described [40]. Briefly, immature DC were generated from bone marrow-derived cells by cultivating them in supplemented RPMI medium (Biochrom, Berlin, Germany) containing 200 U of GM-CSF/ml for 8–10 days.
After seeding in medium lacking antibiotics, cells were infected at a multiplicity of infection (MOI) of 10:1 unless otherwise noted and centrifuged for 5 min at 400 g. One hour post-infection extracellular bacteria were killed by the addition of gentamicin (100 μg/ml) (Sigma, Taufkirchen, Germany). To inhibit caspases and RIP, respectively, cells were incubated with the pancaspase inhibitor Z-Val-Ala-Asp-fluoromethyl ketone (zVAD-fmk, 100 μM) (Bachem, Heidelberg, Germany) and geldanamycin (1 μg/ml) (Sigma), respectively.
Thymocytes were prepared from freshly dissected thymus of BALB/c mice by sieving thymus through a 70 μm-rated cell strainer (BD Biosciences, Heidelberg, Germany) and cultivating cells in RPMI 1640 medium supplemented with 10% FCS, 1% HEPES, 2 mM glutamin, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Stimulation of cells via death receptors
To stimulate DC and thymocytes via the CD95 receptor cells were incubated for 10 min with stimulating anti-CD95 antibodies clone Jo2 (BD Pharmingen, Heidelberg, Germany) (1–5 μg) in 50 μl PBS on ice. Cells were then transferred into medium, shifted to 37°C and further incubated as indicated in the text. Cross-linking of anti-CD95 antibodies was performed by incubation with fluorescein (FITC)-conjugated anti-Armenian hamster antibodies (BD Pharmingen, Heidelberg, Germany) (1 μg/ml) for 20 min. To induce cell death via TRAIL-R2, mouse recombinant TRAIL (Alexis, Grünberg, Germany) was added to DC and Jurkat cells, respectively, in a final concentration of 500 ng/ml.
TNF-α ELISA
5 × 105 DC were cultivated in 48-well-plates in 250 μl DC medium lacking antibiotics and infected with Y. enterocolitica. LPS (4 μg/ml) used as a positive control was from S. typhimurium (Sigma, Taufkirchen, Germany). At the indicated time points post-infection, cell culture supernatants were taken and TNF-α-ELISA (OptEIA Mouse TNF ELISA Set from BD Biosciences, Heidelberg, Germany) was performed as recommended by the manufacturer`s instructions.
Assessment of cell death by flow cytometry
The loss of cell membrane integrity was measured by incubating the cells with propidium iodide (PI) (Calbiochem, Bad Soden, Germany) (50 ng/ml) for 10 min on ice. The mitochondrial transmembrane potential ΔΨm was determined by incubating cells with tetramethyl-rhodamine ethyl ester (TMRE, Molecular Probes, Leiden, Netherlands) (40 nM) for 15 min at 37°C. Cells were analyzed by flow cytometry on a FACSCalibur (BD Biosciences, Heidelberg, Germany) using WinMDI version 2.8 software (J. Trotter, The Scripps Institute, La Jolla, CA). To assess the DNA content of nuclei, cell pellets were resuspended in lysis buffer containing PI and nuclei were analyzed by flow cytometry [41].
Expression of death receptors
All antibodies were purchased from BD Pharmingen (Heidelberg, Germany) unless noted otherwise. To measure CD95 expression, DC from BALB/c or thymocytes were incubated with anti-CD95 antibody clone Jo2 (1 μg) in 50 μl PBS for 30 min on ice and then labeled for 20 min on ice with a secondary fluorescein (FITC)-conjugated anti-Armenian hamster antibody. Cells incubated only with secondary FITC-conjugated antibody were used as a control for unspecific binding. DR5 antibody (MD5-1) (abcam, Cambridge, UK) and secondary FITC-conjugated anti-Armenian hamster antibody were used to determine the expression of TRAIL-R2 on DC. PE-coupled HL3 (anti-CD11c) and FITC-conjugated L3T4 (anti-CD4) antibodies were used to label DC and thymocytes, respectively. Cells were analyzed by flow cytometry. Mean fluorescence intensities were determined after gating on CD11chigh cells (DC) and CD4high cells (thymocytes), respectively.
Caspase activity
For the measurement of caspase-3 activity 5 × 105 DC were lysed, incubated with the fluorogenic substrate N-acetyl-Asp-Glu-Val-Asp-aminomethylcoumarin (DEVD-AMC, 50 μM) (Bachem, Heidelberg, Germany) and the release of aminomethylcoumarin, which corresponds to the DEVDase activity of caspase-3 in the cell extracts, was determined by fluorometry as described recently [14].
Transfection of cells
1 × 107 DC were transfected with AMAXA biosystems Cell Line Nucleofactor® Kit V (Köln, Germany) as recommended by the manufacturer’s instructions. 5 μg plasmid DNA (control pcDNA3 and P-CA plasmid [11], see Table 1), respectively, were added and electroporated with an AMAXA electroporator (programme T-16) (AMAXA biosystems, Köln, Germany). After addition of 400 μl medium, cells were incubated for 5 h at 37°C. Efficiency of transfection was determined by cotransfection with plasmid DNA encoding the gfp gene. Cell viability was determined by analysing the amount of PI-negative cells by flow cytometry.
Immunoblotting
To carry out immunoblots for caspase-8, c-FLIP and RIP 2–5 × 106 DC were lysed with buffer containing 50 mM Tris pH7.4, 50 mM NaCl, 1 mM EDTA and 0.5% NP, and protein concentration was determined by the BCA protein assay (Pierce, Bonn, Germany). YopP-FLAG was detected intracellularly in cytosolic and mitochondrial fractions obtained by a method of Samali et al. [42] and modified as described recently [15]. The separation of cell extracts into mitochondrial and cytosolic fractions was controlled by using the mouse monoclonal antibody to mtHsp70 (mitochondrial heat shock protein 70 kDa) (Dianova, Dörentrup, Germany) and mouse monoclonal antibody to GAPDH (Glyseraldehyde-3-phosphate dehydrogenase) (Biozol, Eching, Germany).
50–100 μg protein of samples and 10 μl of immunoprecipitate samples, respectively, were separated by SDS-PAGE and electrotransferred to nitrocellulose membranes (Schleicher & Schüll, Dassel, Germany). After blocking with 5% skim milk for 2 h the membranes were incubated with rat monoclonal antibody to caspase-8 (Alexis, Grünberg, Germany), mouse monoclonal antibody to FLAG (Sigma, Taufkirchen, Germany), rat monoclonal antibody to FLIP (Dave-2) (Alexis, Grünberg, Germany), rabbit polyclonal antibody to IKKβ (Santa Cruz Biotechnology, Heidelberg, Germany), mouse monoclonal antibody to FADD (Immunotech, Hamburg, Germany), or mouse monoclonal antibody to RIP (BD Transduction Laboratories, Heidelberg, Germany). Immunoreactive bands were visualized by incubation with peroxidase-conjugated secondary antibodies (anti-rabbit and anti-mouse from Dako, Hamburg, Germany, anti-rat from Dianova, Dörentrup, Germany) using enhanced chemoluminescence reagents (ECL, Amersham Biosciences, Freiburg, Germany).
Coimmunoprecipitation
1 × 108 DC were infected with Y. enterocolitica (pYV+, P-wt or P-CA) at MOI of 50:1 for 0.5 h and 1 h, respectively. Cells were collected, washed with PBS, and lysed with a buffer containing 0.5% Nonidet-P40, 40 μM DTT, 50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, pH 7.5 and complete protease inhibitor coctail (Roche, Mannheim, Germany) for 30 min at 4°C. To reduce the unspecific protein content, lysates were incubated with 2 μg mouse IgG1 negative control (Serotec, Wiesbaden, Germany) for 30 min and subsequently with 60 μl Protein G Sepharose beads (Sigma, Taufkirchen, Germany) for 1 h at 4°C. After this precleaning step, supernatants were incubated with 2 μg anti-FLAG (IgG1) antibody overnight at 4°C and then with Protein G Sepharose beads for additional 2 h at 4°C. Beads were recovered by centrifugation, washed three times with lysis buffer, resuspended in PBS and transferred into columns of an immunoprecipitation kit (Sigma, Taufkirchen, Germany). After centrifugation of the columns (12.000 g, 30 s) beads remaining in the columns were resuspended in 40 μl 1× Laemmli buffer and boiled for 5 min at 95°C. Protein samples for analysis by SDS-PAGE and immunoblotting were obtained by recentrifugation of columns.
DISC analysis by immunoprecipitation
5 × 107 DC or thymocytes were infected with Y. enterocolitica (P-wt, P-CA) at MOI of 10:1 for 1 h or incubated with anti-CD95 antibodies, respectively. Cells were collected, washed with PBS, and lysed with 1 × IP buffer (immunoprecipitation kit, Sigma, Taufkirchen, Germany) completed with protease inhibitor cocktail as recommended by the manufacturer’s instructions for 30 min. After transfer of the supernatants into the colums of the kit, 1 μg of mouse-anti-FADD antibody (IgG1) (Immunotech, Hamburg, Germany) was added and incubated overnight at 4°C. Subsequently 40 μl Protein G Sepharose beads from the kit were added and incubated for 2 h at 4°C. Beads were washed five times with 1 × IP buffer and once with 0.1 × IP buffer by centrifugation of columns at 12.000 g for 30 s. Beads remaining in the columns were resuspended in 40 μl 1 × Laemmli buffer and boiled for 5 min at 95°C. Protein samples for analysis by SDS-PAGE and immunoblotting were obtained by recentrifugation of the columns.
Statistical analysis
Values are expressed as means ± SEM. To evaluate the difference in means between two groups, the two-tailed Student’s t-test was used. Significance was defined as a p-value less than 0.05.
Results
Catalytic activity of YopP is essential for the induction of cell death in mouse DC
To address the role of the YopP catalytic activity in the induction of DC death, DC were infected with the Yersinia wild-type strain (pYV+), the YopP-deficient mutant YopP−, the catalytically active (P-wt) or the catalytically inactive Yersinia mutant (P-CA) and cell death was determined by the uptake of PI via disrupted cellular membranes and the formation of hypodiploid nuclei.
Figure 1A demonstrates that upon infection with the Yersinia strains injecting catalytically active YopP, pYV+ (left panel) or P-wt (right panel), the percentage of PI-positive cells rapidly increased and reached similar levels 4 h post-infection (53% and 54% PI-positive cells, respectively). YopP-dependent leakage of cellular membranes was observed as early as 90 min after infection. In contrast, the percentage of PI-positive cells infected with YopP− or P-CA was similar to that of untreated cells (14% and 19%, respectively vs. 13% in uninfected cells) after 4 h indicating that DC death is caused by catalytically active YopP. The results were confirmed by the investigation of the nuclear leakage of DNA from DC 24 h post-infection with Yersinia. Whereas infection with pYV+ or P-wt induced the formation of hypodiploid apoptotic nuclei, infection with YopP− or P-CA did not (Fig. 1B). These data indicate that the catalytic activity of YopP is required for the induction of cell death in murine DC.

Yersinia-induced cell death is mediated by catalytically active YopP. DC were infected with the Yersinia wild-type strain (pYV+), the YopP-deficient (YopP−), the catalytically active (P-wt) and the catalytically inactive (P-CA) mutant strains, respectively. (A) Flow cytometric analysis of DC stained with propidium iodide (PI) 0–240 min after infection. (B) Flow cytometric analysis of nuclei 24 h after infection. Medium (Med) = untreated cells. Data are mean values ± SEM from three individual experiments. *p < 0.05, **p < 0.025, ***p < 0.002 compared to YopP− and P-CA mutant, respectively
Caspase-8 cleavage occurs early upon YopP-induced death of DC [14, 15]. Moreover, LPS via TLR4 triggers YopP-dependent caspase-8 activation in the early phase of infection [14]. A link between the TLR4 pathway and DISC components like FADD, caspase-8 and RIP via the TLR4 adaptor molecule TRIF has been identified in macrophages [12, 16, 43]. However, we have demonstrated that DC from TRIF knock out mice were not protected against LPS/YopP-induced cell death [14]. Hence, we next investigated whether death receptors like TNF-R1, CD95 or TRAIL-R2, which are the classical mediators of caspase-8 activation within the DISC, might contribute to caspase-8 activation and thus to YopP death-inducing activity in DC.
TNF-R1, CD95 or TRAIL-R2 signaling do not contribute to YopP-induced DC death
TNF-α production by macrophages has been observed as early as 15 min after the stimulation with LPS [44]. This pleiotropic cytokine generally triggers inflammatory responses through NF-κB activation. However, in cells that are defective in NF-κB, TNF-α stimulation leads to apoptosis via TNF-R1 [45, 46]. As YopP impairs NF-κB-activation in DC and macrophages about 90 min post Yersinia infection [8–10], we wondered, whether an early, LPS-mediated autocrine TNF-α production combined with a delayed YopP-mediated inhibition of NF-κB-activation of DC, would explain YopP-induced cell death in DC. Analysis of TNF-α-levels by ELISA in supernatants of Yersinia-infected DC (Fig. 2A) indicated that TNF-α was produced as early as 45 min after P-CA infection and its production increased up to 240 min post-infection (>2,000 pg/ml). DC treated with LPS produced similar amounts of TNF-α, whereas uninfected DC did not. By contrast, TNF-α production was significantly reduced upon infection with the YopP-wt mutant suggesting that secretion of TNF-α does not contribute to YopP-induced cell death. To confirm these results, DC deficient in TNF-R1 (TNF-Rp55−/−) were infected with Yersinia P-wt or P-CA and cell death rates were determined by PI-uptake and analysis by flow cytometry. As shown in Fig. 2B, DC from TNF-R1-deficient mice were as sensitive to P-wt-induced cell death as DC from wild-type mice (C57BL/6) (47% vs. 50% PI-positive DC) indicating that TNF-R1 signaling is not involved in the process.

YopP-dependent suppression of TNF-α secretion and expression of death receptors during Yersinia infection. DC were infected with Yersinia (P-wt, P-CA). (A) TNFα levels in the cell culture supernatants of DC from C57BL/6 mice 15–240 min post-infection determined by ELISA. LPS (4 μg/ml) was used as a positive control. Data are mean values from two individual experiments performed in duplicates. (B) Flow cytometric analysis of DC from TNF-Rp55-deficient mice (TNF-Rp55−/−) and wild-type mice (C57BL/6) stained with propidium iodide (PI) 6 h post-infection. Data are mean values ± SEM from three individual experiments. Medium = untreated cells. (C) Expression of CD95 and TRAIL-R2, respectively, on DC analyzed by flow cytometry 6 h after infection with Yersinia (P-wt, P-CA) and staining with anti-CD95 or anti-TRAIL-R2 antibodies and FITC-labelled secondary antibodies (gray). Given percentage values result from gating on CD11chigh/CD95high cells (upper panel) or CD11chigh/TRAIL-R2high cells (lower panel) after subtraction of the correspondent percentage values resulting from cells stained only with secondary antibodies (white). Data are representative of 3 independent experiments
Since bacterial pathogens may induce apoptosis in mammalian cells by upregulation of CD95 expression upon infection [47], we investigated the expression of CD95 after Yersinia infection. As shown in Fig. 2C, the expression of CD95 on surface of both uninfected and Yersinia-infected DC was low indicating that CD95 is not essential for YopP-induced cell death in DC. Likewise, DC were resistant to apoptosis induction by agonistic anti-CD95 antibodies, even when cross-linked by secondary antibodies (data not shown). In contrast, thymocytes freshly isolated from mice expressed significant amounts of CD95 (81.8%) and were highly sensitive to CD95-induced cell death indicated by the percentage of cells with low inner transmembrane mitochondrial potential after staining with TMRE (data not shown). These data suggest that mouse DC are largely resistant to CD95-induced apoptosis.
Beside TNF or CD95L, TRAIL also activates caspase-8 when triggering apoptosis. To elucidate a potential role of the TRAIL system in YopP-induced cell death, expression of TRAIL-R2 on DC was determined. As shown in Fig. 2C, expression of TRAIL-R2 was low on DC which were kept in medium for 4 h (6.4%). DC infection with either Yersinia P-wt or P-CA increased by twofold the expression of TRAIL-R2 (about 15%). However, this increase in TRAIL-R2 expression is most likely due to Yersinia LPS since similar results were obtained when DC were incubated with heat-killed Yersinia (data not shown). Moreover, both uninfected DC and infected DC (P-wt, P-CA) were resistant to TRAIL-induced apoptosis indicated by the percentage of cells with low inner transmembrane mitochondrial potential after staining with TMRE (data not shown) suggesting that TRAIL-R2 signaling is not involved in YopP-induced cell death.
YopP causes cleavage of FLIP and activation of caspases
To analyse the mechanisms by which catalytically active YopP causes caspase activation independently of death receptors, DC were infected with Yersinia P-wt and P-CA, respectively, and immunoblots for caspase-8 and c-FLIP from cell extracts were performed. As shown in Fig. 3A, a 18 kDa caspase-8 fragment was detected 60 min post-infection with P-wt, whereas procaspase-8 was not cleaved upon P-CA infection. These data indicate that the catalytic activity of YopP is essential for caspase-8 activation in DC. Accordingly, c-FLIPL was rapidly cleaved to its p43 fragment by 30 min and completely degraded 120 min post-infection with P-wt (Fig. 3A). By contrast, in cells infected with the proteolytic inactive mutant, P-CA, the uncleaved p55 full length c-FLIPL was detected up to 240 min post-infection, which suggests that YopP-dependent cleavage of c-FLIPL depends on caspase-8 activation in Yersinia-infected DC. The 43 kDa c-FLIPL fragment detectable also in P-CA-infected cells seems to be the result of a LPS-mediated processing of c-FLIPL, as we have observed this fragment in DC treated with LPS (data not shown). Hence, LPS by activation of NF-κB might enhance the expression levels of c- FLIPL and thereby cause spontaneous processing of c-FLIPL [31, 48].

Activation of caspases and cleavage of c-FLIP upon Yersinia infection. DC from BALB/c mice were infected with the Yersinia (P-wt, P-CA). (A) At the indicated time points post-infection, cells were lysed and immunoblots for caspase-8 and c-FLIP were performed. (B) DC lysates were incubated with the fluorogenic caspase-3 substrate Ac-DEVD-AMC and means of fluorescence units (FU)/min (determined in duplicates) were analyzed by fluorometry. Data are mean values ± SEM from three individual experiments. Medium = untreated cells
In agreement with caspase-8 processing and activation, caspase-3 activation occurred early between 30 and 45 min post-infection with P-wt, and further increased 60 min post-infection (Fig. 3B). By contrast, in uninfected DC or in cells infected with the catalytically inactive Yersinia mutant P-CA, caspase-3 activity was much lower, indicating that full caspase activation requires YopP catalytic activity in DC.
YopP triggers the formation of a caspase-8-FADD-RIP complex without interacting with caspase-8
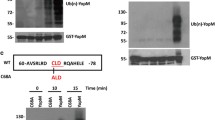
To identify possible targets of YopP mediating caspase activation in DC, we analyzed the subcellular location of YopP. For this purpose, DC were transfected with plasmid DNA encoding the FLAG-tagged, catalytically inactive YopP (P-CA). The YopP-CA mutant was chosen to avoid toxicity caused by active YopP and electroporation. The efficiency of transfection after 5 h was 34% as indicated by the cotransfection with a GFP-plasmid (data not shown). Cytosolic and mitochondrial fractions were then used for immunoblotting. As shown in Fig. 4A, YopP-FLAG was detected in the cytosolic, but not in the mitochondrial fraction.

Subcellular localization of YopP, analysis of interaction between YopP and caspase-8 and DISC formation upon Yersinia infection. (A) DC from BALB/c mice were transfected with plasmid DNA encoding P-CA-FLAG or control vector plasmid. 5 h post-transfection cytosolic (cytosol) and mitochondrial (mitochondria) cell fractions were prepared and FLAG-immunoblots were carried out. (B) DC from BALB/c mice were infected (MOI 50:1) with the Yersinia mutants (pYV+, P-wt, P-CA) for 0.5 and 1 h, respectively. Subsequently, YopP was immunoprecipitated by anti-FLAG-antibodies and Protein G Sepharose beads (line 1–4 and 6) and immunoblots (IB) for FLAG, caspase-8 and IKKβ were performed from immunoprecipitates (IP). Lysates of DC infected for 1 h with Yersinia P-wt and P-CA (MOI 10:1) (line 5 and 7) were used as controls. (C) DC from BALB/c mice were infected with Yersinia (P-wt, P-CA) and FADD was immunoprecipitated (IP) to carry out immunoblots (IB) for FADD, caspase-8, and RIP. Lysates of DC infected for 1 h with Yersinia P-wt and P-CA (MOI 10:1) were used as controls. (D) Left panel: Mouse thymocytes were treated for 1 h with or without anti-CD95 antibodies Jo2 (5 μg) and FADD was immunoprecipitated as described in (C). Right panel: Lysates of thymocytes treated with or without anti-CD95 antibodies Jo2 (5 μg) for 120 min were used as a control. Data are representative of three independent experiments
To investigate whether cytosolic YopP directly interacts with caspase-8, DC were infected with Yersinia P-wt and P-CA (MOI 50, line 1–4 + 6), respectively, and YopP-FLAG was precipitated from DC lysates and subjected to immunoblotting. As shown in Fig. 4B, YopP-FLAG was detected in immunoprecipitates from P-wt and from P-CA-infected DC (line 3–4 + 6) as well as in lysates of infected DC (MOI 10, line 5 + 7), but not in immunoprecipitates from Yersinia pYV+-infected cells carrying YopP without FLAG-tag (line 1 + 2). By contrast, caspase-8 was only detected in DC lysates (line 5 and 7), but not in immunoprecipitates (line 1–4 and 6), suggesting that YopP does not form a complex with caspase-8. IKKβ was present in lysates of infected cells as well as in immunoprecipitates from P-CA and P-wt infected DC (line 3–7) confirming that catalytically active as well as catalytically inactive YopP bind to IKKβ as reported previously [49].
As YopP did not directly bind to caspase-8, we investigated whether YopP by its catalytic activity might cause a formation of the DISC independently of death receptors and thereby promote caspase-8 activation. To investigate DISC formation, cells were infected with Yersinia P-wt and P-CA, respectively, and the adaptor protein FADD was immunoprecipitated. As shown in Fig. 4C, FADD (26 kDa) was detected by immunoblotting in immunoprecipitates as well as in lysates of infected DC. Cleaved caspase-8 (p41/43) colocalised with FADD in P-wt-infected DC indicating the formation of a DISC. Furthermore, these results are concordant with recent reports showing that cells transfected with dominant negative FADD were resistant to YopP-induced apoptosis [12, 16]. However, full processing of caspase-8 to the p18 cleavage product was not detected. As a control, mouse thymocytes, which are known to trigger DISC formation upon agonistic anti-Fas antibody Jo2 stimulation [50], were treated with or without anti-CD95 antibody Jo2. After immunoprecipitation of FADD, cleaved caspase-8 (p41/43) was detected as a component of CD95 DISC in thymocytes stimulated with anti-CD95 (Fig. 4D).
Nevertheless, since YopP can trigger the formation of a caspase-8-FADD complex and involves both caspase-dependent and -independent signaling events [15], we wondered whether the death domain-containg kinase RIP plays a role in this process. Immunoblotting analysis of immunoprecipitated FADD revealed that cleaved RIP (p34 and p39) colocalises with FADD and cleaved caspase-8 in P-wt infected DC (Fig. 4C) indicating that YopP induces caspase-8 and RIP cleavage at the DISC level. The bands arising at about 25 kDa and 50 kDa in all IP-conditions (line 1–9) most likely attribute to heavy and light chains of the anti-FADD-antibodies used for immunoprecipitation.
Suppression of RIP protects against YopP-induced DC death
RIP is stabilized by the heat shock protein Hsp90. Geldanamycin disrupts this binding and consequently leads to RIP degradation. We therefore analyzed the effect of geldanamycin on YopP-induced cell death in DC. Preincubation of DC with geldanamycin reduced RIP protein expression level to 50–70% (Fig. 5A) and significantly protected DC from P-wt-induced cell death, while spontaneous cell death observed in P-CA-infected DC or incubated with medium alone was not affected (Fig. 5B). Preincubation of DC with geldanamycin slightly increases cell death in P-CA-infected DC. This effect might be caused by LPS of bacteria. Inhibition of caspase-dependent signaling by use of the pancaspase inhibitor zVAD-fmk even increased geldanamycin-induced protection towards Yersinia P-wt-induced cell death, highlighting the crucial involvement of caspases in YopP-induced cell death. The slower migrating form of RIP, indicated by an asterisk in Fig. 5A, could correspond to the autophosphorylated RIP proposed to initiate RIP signaling [51].

Geldanamycin impaires expression of RIP and reduces YopP-dependent DC death. DC were preincubated for 2 h and 4 h, respectively, with the RIP inhibitor geldanamycin (1 μg/ml) and additionally treated with or without zVAD-fmk (100 μg/ml) for 1 h. (A) RIP immunoblots from DC treated with or without geldanamycin and zVAD-fmk, respectively. * = phospho-RIP. (B) DC were infected with Yersinia (P-wt, P-CA) for 4 h, stained with propidium iodide (PI) and analyzed by flow cytrometry. Medium = uninfected cells. Data are mean values ± SEM from three individual experiments. * = p < 0.006, ** = p < 0.001 compared to the corresponding condition without zVAD.fmk
Discussion
DC have a unique function as antigen presenting cells to capture antigen, undergo terminal differentiation called maturation and migrate to lymph nodes which leads to the initiation of T cell responses [52]. Yersinia outer protein P mediates apoptotic and necrosis-like cell death in DC, thereby evading DC functions [10, 14, 15]. Several studies have proposed an upstream role of caspase-8 in YopP-induced cell death of DC and macrophages [4, 12, 14–16]. However, the mechanisms of YopP-mediated caspase-8 activation remained largely unknown, in particular, the possible involvement of death receptor signaling pathways.
In this study we have analyzed the impact of the YopP catalytic activity on the induction of cell death in DC. Infection of DC with catalytically active or inactive Yersinia mutant strains showed that the catalytic activity of YopP is essential for the initiation of DC death similarly to that described for macrophages [4].
It has been reported that autocrine production of TNF-α in macrophages after the stimulation with LPS rapidly induced apoptotic cell death (3–6 h) [44]. In accordance to this, we observed that DC secreted large amounts of TNF-α into the supernatants already 45 min after the treatment with LPS. This was also observed when DC were infected with the catalytically inactive Yersinia mutant P-CA. In contrast, TNF-α secretion by DC infected with the cell death-inducing Yersinia mutant P-wt was significantly diminished, in particular at early time points (45–60 min) post-infection indicating that autocrine TNF-α secretion does not account for cell death in DC. There are two possible explanations for the suppression of TNF-α secretion upon infection with Yersinia P-wt. First, Yersinia YopP-mediated inhibition of NF-κB activation could lead to impaired production of TNF-α [8–10]. Second, DC death occurs already 90 min post-infection with Yersinia P-wt and thus prevents LPS-induced TNF-α secretion. In agreement with the above-mentioned results, DC from TNF-Rp55−/− mice failed to resist YopP-induced cell death, although it was reported that these mice were less susceptible to Y. enterocolitica infection as observed by a lower percentage of apoptotic splenocytes compared to wild-type mice [53].
In cells deficient of NF-κB activity, i.e., Yersinia-infected cells [9, 10], TNF-α induces apoptosis via complex II formation [45, 46]. This complex is built after internalization of TNF-R1, dissociation of RIP and TRAF2, and recruitment of FADD and caspase-8 via TRADD protein [45, 46, 54]. However, complex II is unlikely to be causative for YopP-induced DC death since we have demonstrated that DC from TNF-Rp55−/− mice were as sensitive as their wild-type counterpart to YopP-induced cell death. In addition, using recombinant ligands of the TNF family, neither CD95 nor TRAIL-R2 were involved in the cytolytic activity of YopP. Accordingly, although mouse bone-marrow derived DC express the death receptor CD95, they were resistant to Fas-mediated killing by the agonistic mAb, Jo-2 as described elsewhere [55, 56]. The expression of CD95 on DC is dependent on DC maturation as it was shown that 14% of mouse BMDC expressed CD95 at day 8 of cultivation [57] which is in accordance with the CD95 expression (13%) of DC used in this study.
The TNF-related apoptosis-inducing ligand (TRAIL) mainly induces apoptosis in transformed (tumor) cells [58, 59] sparing healthy, non-transformed cells [60–62]. However, some reports indicate that TRAIL may also trigger cell death in primary cells, i.e., mouse thymocytes [63, 64]. Cells normally resistant to TRAIL-induced apoptosis underwent apoptosis after infection with respiratory syncytial or influenza viruses [65, 66]. Mouse DC express low amounts of TRAIL-R2 [67]. But its expression can be upregulated in some mouse cancer cell lines by LPS-stimulation [68], as well as in mouse DC upon infection with Y. enterocolitica (our study). The increase in TRAIL-R2 expression on DC was not sufficient to trigger TRAIL-induced cell death in these cells, and thus it appears that the TRAIL pathway is unlikely involved in YopP-induced cell death.
Nevertheless, our data point to a critical role of caspase-8 in triggering the YopP-induced apoptosis. Previous publications indicate that overexpression of a dominant negative version of FADD or the caspase-8 inhibitor crmA protects macrophages from YopP-induced cell death [12, 16]. Accordingly, we have demonstrated here that the catalytic activity of YopP is essential for the activation of procaspase-8 and -3. Together with the observation that both caspase-3 activation and c-FLIPL (p55) disappearance occur early in the infection, it is likely that caspase-8 activation is an upstream event in the cell death process upon infection with the catalytically active Yersinia YopP. How the FADD-caspase-8 platform occurs in P-wt-infected DC for the activation of caspase-8 remains to be determined.
It has been reported that the Chlamydia protein associating with death receptors (CADD) directly interacts with death receptors [69]. However, YopP does not interact directly with FADD in macrophages [4] nor with caspase-8 in DC (this study). Nevertheless, receptor independent signaling platforms could contribute to the control of YopP-induced cell death which may explain our findings that both caspase-8 and RIP are required for YopP-induced cell death. Indeed, it has recently been shown that caspase-8 is not only required for macrophage differentiation and for the control of NF-κB activation, but also that its activation and regulation involves a multimolecular platform composed of FADD, caspase-8 and RIP that was shown to be independent of the classical death receptors such as Fas, TNF-R or TRAIL receptors [70]. Interestingly, YopP injection triggers the formation of a caspase-8-FADD-RIP complex. Moreover, both caspase-8 and RIP cleavage products were detected within this complex indicating activation of caspase-8 and cleavage of RIP within this complex is dependent on the catalytic activity of YopP. RIP cleavage is an important process in TNF-induced apoptosis since the 39 kDa C-terminal cleavage product of RIP enhances the interaction between TRADD and FADD and attenuates NF-κB-activation and thereby amplifies the activation of caspase-8 [71–73]. Hence, it is conceivable that YopP-dependent DC death is augmented by cleavage of RIP at the level of DISC, since LPS was reported to trigger cleavage of RIP in macrophages when activation of NF-κB is inhibited [43].
DED-containing proteins have been implicated in death receptor-mediated necrotic cell death [74]. In the absence of caspase activity these pathways require the kinase activity of RIP, and therefore use phosphorylation rather than proteolysis as the primary mode of cell death trigger [33]. We found that RIP obviously mediates YopP-dependent DC death, in particular caspase-independent DC death as we observed that geldanamycin-mediated RIP degradation rescued DC from YopP induced cell death both in cells treated without and with the pancaspase inhibitor zVAD-fmk. This result is in agreement with the increasing evidence that caspase inhibition occasionally turns programmed cell death from apoptosis into necrosis [75]. Since RIP deficient mice die within the early postnatal period [76], we were not able to use DC from these mice to determine the exact role of RIP in Yersinia-induced DC death.
Altogether, we provide a strong evidence that DC death induced by catalytically active YopP is not mediated by death receptors but involves caspase-8 for the triggering of apoptosis as well as RIP for caspase-independent DC death at the level of DISC. YopP/J by its catalytic triad His109-Glu128-Cys172 catalyzes serine/threonin acetylation [6, 7] thereby mediating the inactivation of mitogen-activated protein (MAP) kinase kinases (MKK). Given that YopP-mediated inhibition of MAP kinases promoted cell death of DC (own unpublished data), it is conceivable, that acetylation of MKK by YopP mediates DISC formation in DC. This hypothesis is supported by a study of Yeh et al. showing that MKK1 antagonizes FADD-mediated apoptosis by the induced expression of FLIP [77].
Conclusion
Yersinia outer protein P by its catalytic activity induces the formation of an atypical death receptor induced signaling complex (DISC) independently of death receptors in primary DC. This complex allows apoptosis triggering through caspase-8 activation and necrosis via receptor-interacting protein (RIP). Our results strongly support former findings demonstrating that YopP-induced cell death is impaired by dominant negative FADD expression, and provide a molecular explanation on how apoptotic and necrosis-like features arise in YopP-induced DC death.
References
Naktin J, Beavis KG (1999) Yersinia enterocolitica and Yersinia pseudotuberculosis. Clin Lab Med 19:523–536, vi
Cornelis GR, Boland A, Boyd AP et al (1998) The virulence plasmid of Yersinia, an antihost genome. Microbiol Mol Biol Rev 62:1315–1352
Orth K, Xu Z, Mudgett MB et al (2000) Disruption of signaling by Yersinia effector YopJ, a ubiquitin-like protein protease. Science 290:1594–1597
Denecker G, Declercq W, Geuijen CA et al (2001) Yersinia enterocolitica YopP-induced apoptosis of macrophages involves the apoptotic signaling cascade upstream of bid. J Biol Chem 276:19706–19714
Zhang Y, Ting AT, Marcu KB, Bliska JB (2005) Inhibition of MAPK and NF-kappa B pathways is necessary for rapid apoptosis in macrophages infected with Yersinia. J Immunol 174:7939–7949
Mukherjee S, Keitany G, Li Y et al (2006) Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312:1211–1214
Mittal R, Peak-Chew SY, McMahon HT (2006) Acetylation of MEK2 and I kappa B kinase (IKK) activation loop residues by YopJ inhibits signaling. Proc Natl Acad Sci USA 103:18574–18579
Ruckdeschel K, Harb S, Roggenkamp A et al (1998) Yersinia enterocolitica impairs activation of transcription factor NF-kappaB: involvement in the induction of programmed cell death and in the suppression of the macrophage tumor necrosis factor alpha production. J Exp Med 187:1069–1079
Ruckdeschel K, Mannel O, Richter K et al (2001) Yersinia outer protein P of Yersinia enterocolitica simultaneously blocks the nuclear factor-kappa B pathway and exploits lipopolysaccharide signaling to trigger apoptosis in macrophages. J Immunol 166:1823–1831
Erfurth SE, Grobner S, Kramer U et al (2004) Yersinia enterocolitica induces apoptosis and inhibits surface molecule expression and cytokine production in murine dendritic cells. Infect Immun 72:7045–7054
Haase R, Richter K, Pfaffinger G, Courtois G, Ruckdeschel K (2005) Yersinia outer protein P suppresses TGF-beta-activated kinase-1 activity to impair innate immune signaling in Yersinia enterocolitica-infected cells. J Immunol 175:8209–8217
Haase R, Kirschning CJ, Sing A et al (2003) A dominant role of Toll-like receptor 4 in the signaling of apoptosis in bacteria-faced macrophages. J Immunol 171:4294–4303
Thiefes A, Wolf A, Doerrie A et al (2006) The Yersinia enterocolitica effector YopP inhibits host cell signalling by inactivating the protein kinase TAK1 in the IL-1 signalling pathway. EMBO Rep 7:838–844
Grobner S, Schulz S, Soldanova I et al (2007) Absence of Toll-like receptor 4 signaling results in delayed Yersinia enterocolitica YopP-induced cell death of dendritic cells. Infect Immun 75:512–517
Grobner S, Autenrieth SE, Soldanova I et al (2006) Yersinia YopP-induced apoptotic cell death in murine dendritic cells is partially independent from action of caspases and exhibits necrosis-like features. Apoptosis 11:1959–1968
Ruckdeschel K, Mannel O, Schrottner P (2002) Divergence of apoptosis-inducing and preventing signals in bacteria-faced macrophages through myeloid differentiation factor 88 and IL-1 receptor-associated kinase members. J Immunol 168:4601–4611
Leist M, Jaattela M (2001) Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol 2:589–598
Ashkenazi A, Dixit VM (1998) Death receptors: signaling and modulation. Science 281:1305–1308
Strasser A, O’Connor L, Dixit VM (2000) Apoptosis signaling. Annu Rev Biochem 69:217–245
Muzio M, Chinnaiyan AM, Kischkel FC et al (1996) FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 85:817–827
Kischkel FC, Hellbardt S, Behrmann I et al (1995) Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J 14:5579–5588
Peter ME, Krammer PH (2003) The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ 10:26–35
Lee KH, Feig C, Tchikov V et al (2006) The role of receptor internalization in CD95 signaling. EMBO J 25:1009–1023
Irmler M, Thome M, Hahne M et al (1997) Inhibition of death receptor signals by cellular FLIP. Nature 388:190–195
Sharp DA, Lawrence DA, Ashkenazi A (2005) Selective knockdown of the long variant of cellular FLICE inhibitory protein augments death receptor-mediated caspase-8 activation and apoptosis. J Biol Chem 280:19401–19409
Chang DW, Xing Z, Pan Y et al (2002) c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J 21:3704–3714
Krueger A, Baumann S, Krammer PH, Kirchhoff S (2001) FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol Cell Biol 21:8247–8254
Kataoka T, Tschopp J (2004) N-terminal fragment of c-FLIP(L) processed by caspase-8 specifically interacts with TRAF2 and induces activation of the NF-kappaB signaling pathway. Mol Cell Biol 24:2627–2636
Kataoka T, Budd RC, Holler N et al (2000) The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and Erk signaling pathways. Curr Biol 10:640–648
Kreuz S, Siegmund D, Rumpf JJ et al (2004) NFkappaB activation by Fas is mediated through FADD, caspase-8, and RIP and is inhibited by FLIP. J Cell Biol 166:369–380
Dohrman A, Kataoka T, Cuenin S, Russell JQ, Tschopp J, Budd RC (2005) Cellular FLIP (long form) regulates CD8+ T cell activation through caspase-8-dependent NF-kappa B activation. J Immunol 174:5270–5278
Chan FK, Shisler J, Bixby JG et al (2003) A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem 278:51613–51621
Holler N, Zaru R, Micheau O et al (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1:489–495
Lewis J, Devin A, Miller A et al (2000) Disruption of hsp90 function results in degradation of the death domain kinase, receptor-interacting protein (RIP), and blockage of tumor necrosis factor-induced nuclear factor-kappaB activation. J Biol Chem 275:10519–10526
Hur GM, Lewis J, Yang Q et al (2003) The death domain kinase RIP has an essential role in DNA damage-induced NF-kappa B activation. Genes Dev 17:873–882
Yu L, Alva A, Su H et al (2004) Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304:1500–1502
Heesemann J, Laufs R (1983) Construction of a mobilizable Yersinia enterocolitica virulence plasmid. J Bacteriol 155:761–767
Ruckdeschel K, Richter K, Mannel O, Heesemann J (2001) Arginine-143 of Yersinia enterocolitica YopP crucially determines isotype-related NF-kappaB suppression and apoptosis induction in macrophages. Infect Immun 69:7652–7662
Pfeffer K, Matsuyama T, Kundig TM et al (1993) Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 73:457–467
Lutz MB, Kukutsch N, Ogilvie AL et al (1999) An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods 223:77–92
Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C (1991) A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods 139:271–279
Samali A, Cai J, Zhivotovsky B, Jones DP, Orrenius S (1999) Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J 18:2040–2048
Ma Y, Temkin V, Liu H, Pope RM (2005) NF-kappaB protects macrophages from lipopolysaccharide-induced cell death: the role of caspase-8 and receptor-interacting protein. J Biol Chem 280:41827–41834
Xaus J, Comalada M, Valledor AF et al (2000) LPS induces apoptosis in macrophages mostly through the autocrine production of TNF-alpha. Blood 95:3823–3831
Muppidi JR, Tschopp J, Siegel RM (2004) Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity 21:461–465
Micheau O, Tschopp J (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114:181–190
Jendrossek V, Grassme H, Mueller I, Lang F, Gulbins E (2001) Pseudomonas aeruginosa-induced apoptosis involves mitochondria and stress-activated protein kinases. Infect Immun 69:2675–2683
Lens SM, Kataoka T, Fortner KA et al (2002) The caspase-8 inhibitor c-FLIP(L) modulates T-cell receptor-induced proliferation but not activation-induced cell death of lymphocytes. Mol Cell Biol 22:5419–5433
Zhou H, Monack DM, Kayagaki N et al (2005) Yersinia virulence factor YopJ acts as a deubiquitinase to inhibit NF-kappa B activation. J Exp Med 202:1327–1332
Hueber AO, Bernard AM, Herincs Z, Couzinet A, He HT (2002) An essential role for membrane rafts in the initiation of Fas/CD95-triggered cell death in mouse thymocytes. EMBO Rep 3:190–196
Lee TH, Shank J, Cusson N, Kelliher MA (2004) The kinase activity of Rip1 is not required for tumor necrosis factor-alpha-induced IkappaB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J Biol Chem 279:33185–33191
Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392:245–252
Zhao YX, Lajoie G, Zhang H, Chiu B, Payne U, Inman RD (2000) Tumor necrosis factor receptor p55-deficient mice respond to acute Yersinia enterocolitica infection with less apoptosis and more effective host resistance. Infect Immun 68:1243–1251
Schneider-Brachert W, Tchikov V, Neumeyer J et al (2004) Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity 21:415–428
Ashany D, Savir A, Bhardwaj N, Elkon KB (1999) Dendritic cells are resistant to apoptosis through the Fas (CD95/APO-1) pathway. J Immunol 163:5303–5311
Yokota A, Oikawa A, Matsuda C, Shinohara N, Eshima K (2003) Cell-mediated fas-based lysis of dendritic cells which are apparently resistant to anti-Fas antibody. Microbiol Immunol 47:285–293
McLellan AD, Terbeck G, Mengling T et al (2000) Differential susceptibility to CD95 (Apo-1/Fas) and MHC class II-induced apoptosis during murine dendritic cell development. Cell Death Differ 7:933–938
Cretney E, Shanker A, Yagita H, Smyth MJ, Sayers TJ (2006) TNF-related apoptosis-inducing ligand as a therapeutic agent in autoimmunity and cancer. Immunol Cell Biol 84:87–98
Murata T, Tsuboi M, Hikita K, Kaneda N (2006) Protective effects of neurotrophic factors on tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis of murine adrenal chromaffin cell line tsAM5D. J Biol Chem 281:22503–22516
Gura T (1997) How TRAIL kills cancer cells, but not normal cells. Science 277:768
Walczak H, Miller RE, Ariail K et al (1999) Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med 5:157–163
Takeda K, Yamaguchi N, Akiba H et al (2004) Induction of tumor-specific T cell immunity by anti-DR5 antibody therapy. J Exp Med 199:437–448
Ursini-Siegel J, Zhang W, Altmeyer A et al (2002) TRAIL/Apo-2 ligand induces primary plasma cell apoptosis. J Immunol 169:5505–5513
Finnberg N, Gruber JJ, Fei P et al (2005) DR5 knockout mice are compromised in radiation-induced apoptosis. Mol Cell Biol 25:2000–2013
Ishikawa E, Nakazawa M, Yoshinari M, Minami M (2005) Role of tumor necrosis factor-related apoptosis-inducing ligand in immune response to influenza virus infection in mice. J Virol 79:7658–7663
Kotelkin A, Prikhod’ko EA, Cohen JI, Collins PL, Bukreyev A (2003) Respiratory syncytial virus infection sensitizes cells to apoptosis mediated by tumor necrosis factor-related apoptosis-inducing ligand. J Virol 77:9156–9172
Sato K, Nakaoka T, Yamashita N et al (2005) TRAIL-transduced dendritic cells protect mice from acute graft-versus-host disease and leukemia relapse. J Immunol 174:4025–4033
Luo JL, Maeda S, Hsu LC, Yagita H, Karin M (2004) Inhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell 6:297–305
Stenner-Liewen F, Liewen H, Zapata JM, Pawlowski K, Godzik A, Reed JC (2002) CADD, a Chlamydia protein that interacts with death receptors. J Biol Chem 277:9633–9636
Rebe C, Cathelin S, Launay S et al (2007) Caspase-8 prevents sustained activation of NF-{kappa}B in monocytes undergoing macrophagic differentiation. Blood 109:1442–1450
Lin Y, Devin A, Rodriguez Y, Liu ZG (1999) Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev 13:2514–2526
Kim JW, Choi EJ, Joe CO (2000) Activation of death-inducing signaling complex (DISC) by pro-apoptotic C-terminal fragment of RIP. Oncogene 19:4491–4499
Martinon F, Holler N, Richard C, Tschopp J (2000) Activation of a pro-apoptotic amplification loop through inhibition of NF-kappaB-dependent survival signals by caspase-mediated inactivation of RIP. FEBS Lett 468:134–136
Jaattela M, Tschopp J (2003) Caspase-independent cell death in T lymphocytes. Nat Immunol 4:416–423
Vercammen D, Brouckaert G, Denecker G et al (1998) Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med 188:919–930
Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P (1998) The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 8:297–303
Yeh JH, Hsu SC, Han SH, Lai MZ (1998) Mitogen-activated protein kinase kinase antagonized fas-associated death domain protein-mediated apoptosis by induced FLICE-inhibitory protein expression. J Exp Med 188:1795–1802
Acknowledgment
This work was supported by grants of the Eberhard-Karls-University of Tübingen (fortüne 1109, IZKF 1459 (Medizinische Fakultät)) to SG and the DFG (AU 102) to IBA.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gröbner, S., Adkins, I., Schulz, S. et al. Catalytically active Yersinia outer protein P induces cleavage of RIP and caspase-8 at the level of the DISC independently of death receptors in dendritic cells. Apoptosis 12, 1813–1825 (2007). https://doi.org/10.1007/s10495-007-0100-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-007-0100-x