
Abstract
The ATP-binding cassette (ABC) transporters form one of the largest families of proteins in living organisms. They are overrepresented in bacteria where they are involved in the influx or efflux of various molecules. Although bacterial drug efflux transporters were initially discovered as ion-motive-driven pumps, evidence has accumulated since the mid-1990s that members of the ABC superfamily can play a prominent role in drug resistance mechanisms. Yet, the implication of drug efflux ABC transporters in clinical settings is still lagging behind for most bacterial pathogens. Thanks to the accumulation of three-dimensional structures, our knowledge of the functioning mechanisms of drug efflux transporters has progressed tremendously in the recent years, but many questions still remain. In this chapter, we will summarize the current view of the structures and transport mechanisms of drug efflux ABC transporters with an emphasis on multidrug bacterial efflux pumps. Unsolved mysteries about these fascinating transporters will also be mentioned.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- ABC transporters
- Multidrug resistance
- P-Glycoprotein
- ATP switch model
- Constant contact model
- Drug binding
- BmrA
- LmrA
- MacAB
- MsbA
- PatAB
- Sav1866
1 Introduction
The ATP-binding cassette (ABC) transporters form a large superfamily of proteins that use the energy of ATP binding and hydrolysis to translocate a wide variety of solutes across biological membranes [1]. (Although this chapter is focused on bacterial transporters, some references to eukaryotic transporters will also be included when relevant to this topic.) Apart from a few exceptions [2], the importers are only present in prokaryotes, while exporters are found in all organisms. The minimal functional unit contains two transmembrane domains (TMDs) and two nucleotide-binding domains (NBDs) [3, 4]. These domains can be synthesized as four separate polypeptides or as various combinations of three, two, or a single polypeptide. The NBDs energize the transporter by binding and hydrolyzing ATP, while the TMDs are responsible for the specificity and translocation pathway of the substrates. Therefore the NBD primary sequences are fairly well conserved, while the TMD sequences and topology, in particular the number of transmembrane helices, are highly variable depending on the solute transported. In addition, the majority of the importers require an extracellular substrate-binding protein that delivers the substrate to the transporter [5].
2 Multidrug Transporters
2.1 Homodimers
The first bacterial multidrug resistance (MDR) ABC transporter was discovered in Lactococcus lactis and was named LmrA (Lactococcus multidrug resistance ATP) [6]. LmrA functions as a homodimer [7], each monomer being made of one TMD containing six predicted transmembrane helices and one ABC domain. LmrA was initially chosen for investigation based on its homology with the human MDR transporter P-glycoprotein. It was later shown to complement the P-glycoprotein human gene in eukaryotic cells [8]. The overexpression of LmrA in a drug-hypersusceptible strain of Escherichia coli induced a resistance phenotype to several structurally unrelated compounds, i.e., ethidium, daunorubicin, rhodamine 6G, and tetraphenylphosphonium [6]. In addition, the accumulation of daunorubicin in inverted membrane vesicles was dependent on ATP hydrolysis and inhibited by the other substrates and reserpine, a classical inhibitor of drug efflux pumps.
A Bacillus subtilis transporter homologous to LmrA was shown to transport a variety of drugs (e.g., Hoechst 33342, doxorubicin, and 7-aminoactinomycin D) when overexpressed in inverted E. coli membrane vesicles. Originally known as YvcC [9], this homodimeric transporter [10] was renamed BmrA (Bacillus multidrug resistance ATP) [11]. Later, a B. subtilis strain resistant to the antibiotic cervimycin C was isolated and shown to strongly upregulate the expression of bmrA due to a promoter mutation [12].
Two other drug transporters, Sav1866 from Staphylococcus aureus and MsbA from various bacteria, will be described in more details below.
2.2 Heterodimers
While LmrA and BmrA are typical homodimers, other drug transporters are heterodimers. The latter transporters include LmrCD in L. lactis [13], BmrCD in B. subtilis [14], PatAB in Streptococcus pneumoniae [15], and SmdAB in Serratia marcescens [16]. In these heterodimers, one of the ATP-binding sites is degenerated [17, 18] with conserved residues such as the glutamate adjacent to the Walker B motif, the histidine of the H-loop, and/or some residues in the signature motif of the opposite NBD being naturally substituted by non-consensual residues. Consequently, the functioning mechanism of these heterodimers is asymmetric with the degenerated NDB being poorly active in ATP hydrolysis. A similar scenario occurs in eukaryotic transporters that bear two nonequivalent NBDs (e.g., multidrug resistance protein MRP1, cystic fibrosis transmembrane conductance regulator CFTR, or antigen peptide transporter TAP1/TAP2) [19, 20].
LmrCD was shown to be a major MDR transporter in L. lactis [21, 22] and its expression is under the control of the transcriptional repressor LmrR [23, 24]. Binding of drugs to LmrR reduces its affinity for LmrCD promoter thereby inducing the expression of the MDR transporter [25–27].
BmrCD is a B. subtilis transporter whose expression is stimulated by various antimicrobial agents [14], especially protein synthesis inhibitors. The latter drugs were recently shown to induce the expression of BmrCD through a ribosome-mediated transcriptional attenuation mechanism [28]. When overexpressed in E. coli membranes, BmrCD transports several drugs such as Hoechst 33342, doxorubicin, and mitoxantrone [14]. Its efficient expression and purification was exploited for several structural and functional studies [29–31].
The implication of the S. pneumoniae transporter PatAB in MDR was first demonstrated when inactivation of its genes induced an increased susceptibility to several antimicrobial agents: acriflavine, berberine, ethidium bromide, and norfloxacin [32]. After exposing a laboratory strain to ciprofloxacin, a multidrug-resistant strain was isolated in which PatA and PatB genes were upregulated [33]. Such upregulation by fluoroquinolones was also found in clinical isolates [34–36]. Importantly, disruption of PatA and PatB genes overexpressed in many clinical isolates restored drug susceptibility, either completely for ethidium bromide or partially for fluoroquinolones [35]. Several mechanisms were described for PatAB upregulation: disruption of a transcriptional attenuator [37], gene duplication [38], or promoter region and internal mutations [39]. Studies of the transporter overexpressed in E. coli show that only the heterodimer is functional for drug efflux [40].
Other transporters whose function is less characterized were shown to have drug transport capabilities, such as TmrAB in Thermus thermophilus [18] or TM287/TM288 in Thermotoga maritima [41]. A recent study suggests that TmrAB may be a glycolipid flippase analogous to the transporter MsbA [42].
2.3 Other Drug ABC Exporters with a Different Topology
DrrAB from Streptomyces peucetius exports the anticancer antibiotics daunorubicin and doxorubicin that this microbe produces. It was long thought to be a narrow-spectrum drug transporter until a recent biochemical characterization indicated its ability to also transport the Hoechst 33342 and ethidium bromide [43]. In contrast to LmrA and BmrA, where each monomer is a TMD fused to an NBD, DrrA is a single NBD subunit, while DrrB is a TMD subunit predicted to contain eight transmembrane helices [44].
MacAB-TolC from E. coli was characterized as a macrolide-specific tripartite efflux pump [45]. Homologues are present in various Gram-negative bacteria. MacB is an ABC transporter from the cytoplasmic membrane with an inverted topology: an N-terminal NBD is fused to a C-terminal TMD containing four predicted transmembrane helices. TolC is an outer membrane channel protein, while MacA is a membrane fusion protein that interacts with both partner proteins. Nanomolar affinity interactions occur between TolC and MacA and between MacA and MacB [46]. MacB is a dimer [47] whose ATPase activity is strongly stimulated by MacA [48]. Several drug-unrelated physiological functions have been proposed, the latest being protoporphyrin efflux [49].
3 Structure of the Nucleotide-Binding Domains, Consensus Motifs, and the ATP Sandwich Dimer
The NBDs of ABC transporters are well conserved, both in sequence with several motifs and in structure [50]. HisP, which is the ATPase subunit from a bacterial histidine importer, was the first NBD crystallized [51]. The domain had an L shape and is made of three subdomains (Fig. 4.1) [52]. One is a RecA-like subdomain present in many ATPases and that carries the Walker A and Walker B motifs [53, 54] as originally described in numerous ATPases (e.g., the Fo-F1) [55]. The former, also known as the P-loop, has the consensus sequence GX2GXGKT/S (where X is any residue; see Fig. 4.1). Some backbone amino groups within this motif, and especially the ε-amino group of the conserved lysine, stabilize the bound nucleotide by making hydrogen bonds with the β- and γ-phosphate oxygen atoms. The Walker B motif is usually made of four hydrophobic residues that form a β-strand and is terminated by a conserved Asp. This Asp residue coordinates the catalytic Mg2+ cofactor by hydrogen bonding via a water molecule.

Structure of the nucleotide-binding domains. (a) Schematic view of the conserved elements present in the NBD of exporters (see the text for details). The color-coding is the same as in (b). (b) The dimer of NBDs trapped in a transient ATP-bound state. The two identical NBDs of LolD (PDB code MJ0796) are shown here, and one is colored in gray and the other is colored in pale green, wheat, and pale yellow for the β-, RecA-, and α-helical subdomains, respectively. The conserved motifs are shown for one monomer in red (Walker A), orange (Walker B), blue (H-loop), magenta (Q-loop), gray (ABC signature), green (x-loop), and cyan (D-loop). The tyrosine which is part of the A-loop and stacks the adenine ring of ATP is shown in stick representation, like the two ATP molecules trapped at the NBD interface, and colored by elements (C, green; O, red; N, blue; and P, orange). An ATPase inactive mutant was used here (Glu171Gln) allowing the stabilization of this transient ATP-bound conformation. This figure was made with PyMOL using the PDB code 1L2T [65]
In ABC transporters with NBDs bearing consensual signatures motifs, the Walker B motif is immediately followed by an invariant Glu residue whose position in the three-dimensional (3D) structure is reminiscent of the catalytic Glu found in other ATPases [53, 56, 57]. The involvement of this residue as a catalytic base, as initially proposed based on mutagenesis and kinetic studies [58], has been later substantiated by the 3D structure of the maltose transporter [59], and this seemed to nail down the original controversy about this residue [60]. Additional motifs are present in the RecA-like subdomain including: (i) The Q-loop, a stretch of ~ eight amino acids starting with a conserved Gln and joining the RecA-like and α-helical subdomains, makes part of the interface with the TMDs. A conformational switch of the Gln residue during the catalytic cycle, engaging the MgATP and moving away after ATP hydrolysis, may be involved in transmitting conformational changes between NBDs and TMDs. (ii) The H-loop contains a conserved His that acts as a linchpin in ATP hydrolysis by interacting with the γ-phosphate of ATP and the catalytic Glu [61]. (iii) The D-loop carries usually the conserved sequence “SALD,” a distinctive feature of ABC proteins located downstream of the Walker B motif. When the NBDs are sufficiently close to each other, the D-loop establishes a complex hydrogen bond and electrostatic network with the Walker A motif and H-loop of the opposite NBD. Because of its position at the dimer interface, the “D-loop” originally referred to “dimer” [62], while it was later alluded to the invariant aspartic acid of the motif. In the NBD dimers, the D-loops also connect and stabilize the catalytic Glu and attacking water [59, 63]. By contacting the ATP-binding sites both in cis and in trans, the D-loops are likely to play a major role in the communication between the active sites, the control of ATP hydrolysis, and also the directionality and energy of the transport as shown recently [64].
The two other subdomains are specific to the ABC family. One is the α-helical subdomain, which contains the family signature motif. Its sequence usually starts with LSGGQ and belongs to a loop located at the N-terminus of an α-helix. The role of this motif had remained elusive for a long time until the 3D structures of ABC dimers were solved [62, 65]. Hence, in all ABC family members, the NBDs associate transiently in a head-to-tail dimer in which the ATP molecules are sandwiched between the Walker A motif of one domain and the signature motif of the other domain. The LSGGQ sequence makes extensive hydrogen bonds with the ATP and is required for ATP hydrolysis. The role of this motif is likely similar to the arginine finger present in some P-loop GTPases, which stabilize the active site of the opposite domain (see Fig. 4.1). Another motif present in this subdomain is the x-loop, defined as TEVGERG sequence in Sav1866 (see below). It is only present in exporters and precedes the signature motif in the α-helical subdomain [66]. Its name refers to the fact that it interacts with both intracellular loops. Based on its proximity with the signature motif, it has been hypothesized to transmit conformational changes between the ATP-binding site and the TMDs [67–70].
The other subdomain is called the β-subdomain. It encompasses the A-loop which is located upstream of the Walker A motif and bears a conserved aromatic (A) residue that stacks against the adenine ring of the nucleotide, helping to stabilize it [53, 71]. While providing extremely valuable insights into the mechanism of ABC transporters, the structures of isolated NBDs lead to flawed interpretations of catalytic mechanisms since the TMDs impose some structural constraints and alter the geometry of the catalytic sites [59, 72, 73].
4 Structures of Whole Drug Exporters
Crystallized exporters were captured in mainly two opposite conformational states: outward facing and inward facing (Fig. 4.2).

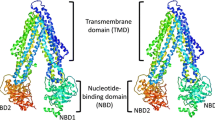
3D structures of selected drug exporters from ABC family. The N-terminal half of P-glycoprotein (P-gp), TM287, and one monomer of Sav1866 are colored in green, while the C-terminal half of P-gp, TM288, and the other monomer of Sav1866 are shown in blue. When present, AMP-PNP is shown in red stick representation
Some structures originally contained major errors and were later corrected [74, 75]. Sav1866 is a multidrug transporter from S. aureus [76], and its crystal structure caused the retraction of erroneous MsbA structures [77] and thus revealed for the first time the correct architecture of an ABC exporter [66]. Sav1866 is a homodimer, and each protomer is made of six transmembrane helices located at the N-terminal side of the transporter and one C-terminal NBD (Fig. 4.2). Sav1866 was crystallized in an outward-facing conformation in which the NBDs are in close contact (closed state). Although this conformation was observed with ADP bound in the catalytic sites, this state most likely represents an ATP-bound state. Accordingly, a second structure with AMP-PNP bound instead was virtually identical [78]. The two monomers exhibit an extensive twist, and the domains of each monomer significantly contact those of the other monomer (Fig. 4.2). In this state, a central cavity was formed at the interface of the two TMDs. This cavity was shielded from the cytoplasm and the inner leaflet of the lipid bilayer, but accessible from the outer leaflet and the extracellular space (outward-facing conformation). The transmembrane helices are connected via short extracellular loops and long intracellular loops that protrude and extend the helical transmembrane bundles (Fig. 4.2). Consequently, the NBDs are located 25 Å away from the membrane. The TMD interface with the NBDs mostly involves the so-called coupling helices of the intracellular domains ICD1 and ICD2. The coupling helix 1 of ICD1 is located between transmembrane helices 2 and 3 and interacts mostly with the NBD of its own monomer. The coupling helix 2 of ICD2 located between transmembrane helices 4 and 5 interacts only with the opposite monomer (Fig. 4.3). This trans interaction of ICD2 is the trademark of the ABC exporters. Yet, the coupling helix 2 is rather similar to the coupling helix of the importers since it docks into a groove at the interface between the RecA-like and α-helical subdomains of the NBDs (Fig. 4.3). In this closed outward-facing conformation, the interaction of the two NBDs is similar to the transient head-to-tail conformation of isolated NBDs.

Close view of the interface NBD/TMD in Sav1866 (PDB code 2ONJ). One monomer is shown in green, while transmembrane helices 4–5 and coupling helix 2 (CH2) from the other monomer are colored in blue. The Q-loop is colored in orange, while the x-loop is shown in yellow. AMP-PNP is visible in red sticks
MsbA is a lipid A flippase [79], but it also has the ability to transport some drugs [80, 81]. Depending on its origin and the crystallization conditions, three different conformations were obtained for this exporter: one which is very similar to the Sav1866 structure (closed AMP-PNP bound or ADP-Vi state, MsbA of Salmonella enterica serovar Typhimurium), one where the two NBDs are close to each other but not yet engaged in a tight interaction (closed apo state, MsbA of Vibrio cholerae), and finally one with the two NBDs widely separated in the so-called open state with an inward-facing conformation (MsbA of E. coli [EcMsbA]) [75]. In all the conformations, the transmembrane helices 4–5 and the associated coupling helix 2 cross over the homodimer interface and contact the opposite subunit. However, intracellular loop 1 loses contact with the opposite subunit in the open apo state. The open conformation has been subjected to controversy, but similar conformations were obtained for the mouse or Caenorhabditis elegans P-glycoproteins [82, 83], yet not as widely open as in the structure of EcMsbA (i.e., the NBDs are separated by ~ 25–30 Å in the P-glycoprotein vs. ~ 50 Å in EcMsbA). Interestingly, a structure of a flippase was recently solved in three different conformations: two open with various separations of the NBDs (44 Å and 30 Å) and one closed in an ADP-bound outward-occluded conformation [84]. Again, the two open inward-facing structures were suspected to be biased by the presence of detergent or crystal lattice contacts. However, the 3D structure of BmrA obtained in a lipidic environment was consistent with the open structures of P-glycoproteins [85], and this suggests that the presence of detergent in the X-ray crystallography experiments was not forcing the structure of the transporters in abnormal conformations.
The structure of an antimicrobial peptide exporter, McjD, offers presumably the first view of an intermediate conformation of the catalytic cycle of exporters, in an outward-occluded state [86]. While the two NBDs of McjD are still engaged in an ATP-bound conformation similar to that found in Sav1866, the TMD moiety shows a different organization of the transmembrane helices. This creates an internal cavity not accessible to either side of the membrane and that could possibly accommodate the transported molecule, i.e., the microcin J25.
The first structure of a heterodimer was obtained for TM287/TM288 [41]. It showed an inward-facing conformation at the membrane level, but the NBDs were only partially disengaged with significant contact being maintained at the degenerate ATP-binding site where an AMP-PNP molecule was still bound. However, even in the apo form of TM287/TM288, its two NBDs keep the same interaction/orientation at the degenerate site [87]. Hence, the authors raised the possibility that AMP-PNP is a poor ATP analogue for heterodimeric ABC transporters. Of note, AMP-PNP and AMP-PCP also failed to generate a closed NBD conformation in the homodimer of ABCB10 [88].
In addition to the crystal structures, the structure of the heterodimer TmrAB in a nucleotide-free state was obtained at a subnanometric scale by cryo-EM. It revealed an inward-facing conformation, yet a contact was maintained between the two NBDs at the level of the two Ct-helices, one in each NBD [89]. Therefore, it is possible that a full physical separation between the two NBDs, in the nucleotide-free state, is a prerogative of homodimers or full-length transporters bearing two consensual ATP-binding sites.
5 An Alternating Access Mechanism
An alternating access mechanism seems the prevailing process in ABC transporters. It involves switching between two opposite conformations in which the substrate-binding site is alternatively accessible to one side of the membrane [75, 90–92]. Substrate binding on the inner or outer membrane leaflet and release on the opposite side are coordinated by the catalytic events, i.e., ATP binding, hydrolysis, and product release. Several studies on drug transporters suggest a lower affinity for drugs in the outward-facing conformation thereby explaining their release outside the cell [7, 93, 94]. The different conformational structures of MsbA lead to a transition model [75] in which pivoting of transmembrane helices 4–5 around the extracellular loops 2 and 3 brings the NBDs near each other; in this configuration, the NBDs are not properly aligned since the two Walker A motifs are facing each other, and a sliding movement of the NBDs along the interface would be required to align each Walker A motif with each LSGGQ motif, thereby pulling transmembrane helices 3–6 away from transmembrane helices 1–2. The newly formed outward opening is created between transmembrane helices 1 and 3, whereas the inward opening was formed between transmembrane helices 4 and 6. Recent molecular dynamic studies suggest twisting of the NBDs during the catalytic cycle of MsbA [95] and P-glycoprotein [96]. Furthermore, a misalignment of the NBDs was also observed in the crystal structures of ABCB10 [88], a putative transporter of heme precursors [97, 98]. In contrast, the NBDs in the heterodimer TM287/TM288 are partially interacting and are correctly aligned for canonical dimer formation [87]. Further experimental validation will be required to determine whether these conformational differences are physiologically relevant and truly reflect mechanistic differences between transporters. A variation of the classical alternating access mechanism has been recently proposed for the lipid-linked oligosaccharide flippase PglK of Campylobacter jejuni [84]. In this model, although the transporter can adopt inward- and outward-facing conformations, the substrate directly binds the outward-facing state and is flipped upon ATP hydrolysis.
6 Drug-Binding Sites
The most remarkable feature of MDR pumps is their ability to transport a wide variety of structurally dissimilar drugs. X-ray structures of murine P-glycoprotein revealed a large internal cavity open to both the cytoplasm and the membrane inner leaflet, with a wide separation between the two NBDs [74, 82]. This configuration generates the presence of two portals at the level of the inner membrane leaflet. The first is located between transmembrane helices 4 and 6 on one side and the second between transmembrane helices 10 and 12 on the other side (Fig. 4.4). Of note, an N-terminal helical hairpin occludes one of these portals in the crystal structure of the C. elegans P-glycoprotein, but the overall shape of the two proteins is otherwise similar [83]. Drugs could reach access to the transport pathway from the aqueous phase [99–101] or through these portals within the membrane. Because many drug substrates partition and concentrate in the membranes [102], it is likely that drugs usually enter the transporter through the membrane inner leaflet. Consistent with this, Jin et al. observed a 100- to 4,000-fold increase in drug apparent affinity when studying the drug-stimulated ATPase activity of P-glycoprotein in membranes as compared to detergent [83].

Structure of P-glycoprotein in complex with two QZ59-SSS molecules (PDB 4M2T). The N-terminal half of P-glycoprotein is shown in green cartoon representation, and the C-terminal half is shown in transparent blue surface (TM transmembrane helix). The two drugs are colored in red and circled by black spheres. The putative drug-binding cavity is indicated with the yellow triangle
The identification of the drug-binding site(s) in P-glycoprotein has been the goal of many studies (see a review in [103]). It was early recognized that a drug-binding site was localized within the TMDs [104]. Binding and kinetic analysis suggested the presence of several drug-binding sites [105–107]. Based on kinetic studies, Shapiro and collaborators proposed the existence of three drug-binding sites in P-glycoprotein, named H (Hoechst), R (rhodamine), and P (prazosin and progesterone) sites [108, 109]. Both the H and R sites are competent for transport, while the P site is an allosteric site. The R site preferentially binds rhodamine 123 and anthracyclines; the H site preferentially binds Hoechst 33342, quercetin, and colchicine; the P site binds preferentially prazosin and progesterone. The existence of two different H and R sites in P-glycoprotein was also evidenced by Förster resonance energy transfer (FRET) studies from Sharom’s laboratory [110, 111]. A positive cooperative effect between the R and H sites was observed: the addition of a small concentration of a drug that binds to one site stimulates the transport of the substrate bound to the other site. Such reciprocal drug transport stimulation was also later observed with LmrA [7] and BmrA [11]. Shapiro and Ling also reported that other drugs such as vinblastine, etoposide, and actinomycin D compete with both H and R sites. Cysteine-scanning mutagenesis and thiol-reactive drugs such as dibromobimane, methanethiosulfonate-rhodamine, and methanethiosulfonate-verapamil were extensively employed to localize drug-binding sites in P-glycoprotein [112]. A common drug-binding pocket was found at the interface between the TMDs that can accommodate at least two drugs [113].
Murine P-glycoprotein was co-crystallized with two stereoisomers of cyclic hexapeptide inhibitors, cyclic-tris-(R)-valineselenazole (QZ59-RRR) and cyclic-tris-(S)-valineselenazole (QZ59-SSS). Either one molecule of QZ59-RRR or two molecules of QZ59-SSS were found in the central cavity of P-glycoprotein (Fig. 4.4) [74, 82].
The drug-binding cavity contains nine aromatic residues that are identical in human and murine P-glycoprotein but not conserved in C. elegans P-glycoprotein. There are no charged residues in the drug-binding pocket of mammalian P-glycoprotein structures, in contrast to C. elegans P-glycoprotein and MsbA, which has 16 charged residues pointing directly toward the substrate translocation pathway [74]. Knowing the position of the two cyclic peptides QZ59-RRR and QZ59-SSS in the central cavity of the mouse P-glycoprotein, Martinez et al. sought to localize the H and R sites by assessing whether these peptides compete with the transport of Hoechst 33342 and daunorubicin and by performing molecular docking simulations [114]. They proposed the location of the H site along the central cavity and the QZ59-SSS molecule closer to the center of the membrane, with the R site at a deeper position in the cavity, overlapping the location of the QZ59-SSS molecule most embedded in the structure (see also [103]). Another group proposed similar locations for H and R sites, and the potent inhibitors tariquidar and elacridar bind to P-glycoprotein sites that coincide or overlap with these sites [115]. Importantly, the suggested R site is consistent with the cross-linking studies with methanethiosulfonate-rhodamine [112]. However, daunorubicin-binding site in MsbA was mapped at a different location, closer to the inner leaflet of the membrane [116]. Since the physiological substrate of MsbA is lipid A, it is conceivable that drugs opportunistically accommodate to the binding pocket of the transporter, possibly in a different location from typical multidrug transporters [117].
7 Basal ATPase Activity in Multidrug Transporters
Multidrug transporters typically display a high basal ATPase activity, which for bacterial transporters is often moderately stimulated by drugs [11, 30, 118, 119]. This behavior contrasts for instance with the well-coupled peptide exporter complex TAP [120] or some ABC importers [121–123]. Nevertheless, several lines of evidence suggest that drugs binding facilitate the dimerization of the NBDs thereby stimulating ATP hydrolysis [124, 125]. These observations are reminiscent of the mechanism of ATPase stimulation in other ABC transporters by allocrites [126], partner proteins [127], or proteins delivering the solute to importers [128–130].
Several plausible explanations could account for this seemingly “futile” ATPase activity in drug transporters. First, it might be due to nonoptimal conditions of purification or reconstitution procedures; given that this behavior is widespread among differently purified multidrug transporters, this seems unlikely. Second, it might result from the transport of lipids that could stimulate the ATP hydrolysis of the transporters [42, 80, 131, 132]. Third, it might be an intrinsic property of drug transporters, as suggested by a thermodynamic analysis of P-glycoprotein activity [133]. Ernst, Schmitt, and collaborators have proposed an elegant hypothesis: the kinetic substrate selection model [134, 135]. The basal ATPase activity may have the advantage of maximizing the number of transporters competent for substrate binding in inward-facing conformations. This model proposes that the time spent in the inward- or outward-facing states affects substrate selection and explains how two substrates with identical affinities, but dissimilar kon and koff, can be transported with different efficiencies. MDR transporters have the unique ability to recognize a huge variety of structurally dissimilar substrates. If the ATPase activities of these transporters were tightly coupled to the drug extrusion process, then only the substrates capable of stimulating the ATPase activity would be transported. Possibly, a tight coupling would only be achieved at the expense of substrate diversity. Being capable to switch rapidly between the two opposite conformations, inward facing (i.e., in a conformation allowing to capture a noxious compound if present) and outward facing in an ATPase active conformation, even in the absence of a drug, might be the price to pay to make sure that any bound drug will be rapidly expelled out of the cell before being released from the transporter in an on and off process. Given the apparently relatively low affinity for many drugs, a fast rate of ATPase activity (coupled or not with the transport process) might overcome the rate constant of the drug release (koff). Thus, wasting some energy in the absence of a drug might ensure the polyspecificity for many unwanted molecules and their efficient efflux once captured by the transporter.
8 Transport Mechanisms and Structural Flexibility of Multidrug Transporters
During the catalytic cycle, the two NBDs of ABC transporters engage and disengage with each other [129, 136]. Because the two ATP-binding sites are localized at the interface of the two monomers, ATP binding promotes the formation of a closed conformation [137, 138]. Although the interface of dimerization was a matter of debate for some time [139], the head-to-tail orientation first envisioned by Jones and George [140] was observed in the crystal structure of the ABC protein Rad50 involved in DNA double-strand break repair [62]. Later, this arrangement was validated with the crystal structure of an NBD dimer stabilized by the mutation of the catalytic glutamate [65] and the photocleavage of both the Walker A and LSGGQ motifs by the transition state analogue orthovanadate in the maltose transporter [141]. The latter observation also indicated that ATP hydrolysis occurs only in the closed conformation. Consistent with this, mutations in the LSGGQ motif strongly alter the ATPase activity of ABC transporters [142, 143]. The release of Pi and/or ADP destabilizes the dimer such that the NBDs move apart from each other. In addition to the interdomain movement, the RecA-like and α-helical subdomains within each NBD rotate toward each other upon ATP binding and move outward in the post-hydrolysis stage [52, 139, 144]. Hence, the energy of ATP binding and hydrolysis is coupled to conformational changes in the TMD thereby mediating alternating access of the substrate-binding site to each side of the membrane.
Several inward-facing conformations of P-glycoprotein exhibiting different degrees of domain separation were crystallized [74, 83, 145] hence suggesting a highly flexible protein. The distance between the α-carbons of the Walker A cysteines in the mouse or C. elegans P-glycoprotein varied between 38 and 53 Å [103]. These observations are consistent with the flexibility reported in the apo states of LmrA [146] and BmrA [147] in detergent. This flexibility evidenced by high rates of H/D exchange in the apo state of BmrA is presumably caused by multiple conformations of the two ICDs, thereby allowing some freedom of rotation of the NBD [147]. In line with this, Cys-Cys cross-links were obtained for BmrA between the NBD and ICD1 that suggests the existence of additional, possibly transient, conformations of BmrA in the resting state [148]. Additionally, it was possible to cross-link two Cys residues, one in each Walker A motif of the P-glycoprotein suggesting that the two NBDs adopt alternate orientations in the resting state [149]. P-Glycoprotein structures have been suspected to exhibit nonphysiological conformations due to the absence of lipid bilayer and nucleotides. However, Wen et al. recently showed that, in intact lipid bilayers and in the presence or absence of nucleotides, P-glycoprotein adopts a wider range of conformations (both longer and shorter) compared to the original mouse P-glycoprotein crystal structure [150]. The authors suggested that this flexibility might originate from a high number of Gly and Pro residues thereby causing kinking and/or unwinding within the TMDs. Such flexibility may be advantageous to accommodate substrates of various sizes and chemical properties.
The mode and extent of NBD separation in MDR transporters is, however, still under debate [151], and two main hypotheses describing the mechanism of action of ABC transporters have been proposed: the ATP switch model [152] and the constant contact model [153].
8.1 The ATP Switch Model
In the switch model [152], which is also referred as the processive clamp model [154], the NBDs are proposed to dimerize upon ATP binding, sequentially hydrolyze ATP, and completely separate upon release of Pi and/or ADP. The ATP-dependent dimerization generates the outward-facing state, during which the drugs are translocated from the inner to the outer membrane leaflet, while ATP hydrolysis and release of hydrolysis products reset the transporter to the inward-facing conformation. This model largely relies on the available structures of ABC transporters in nucleotide-free or nucleotide-bound conformations. Many experimental data support this model. ATP binding promotes association of isolated NBDs [155] and ATP hydrolysis induces their dissociation [156]. In the context of intact transporters, biophysical and cross-linking studies suggest large-scale movements in MsbA [136, 157–159]. Moreover, ATP binding promotes large conformational changes in LmrA [146] and BmrA [160]. The main concern regarding this model is that apo states, as studied in biochemical experiments, may not be physiologically relevant. Given the prevalence of the nucleotide in cells, it has been proposed that transporters will likely have ATP bound in vivo [161]. Yet, cells and microorganisms in particular have to face stressful conditions that will strongly deplete ATP concentrations (see the discussion in [85]). Moreover, even in optimal laboratory conditions and for fast-growing E. coli bacteria, the ATP level can vary greatly among a bacterial population that originates from a single clone [162]. Two other points are worth considering. First, in which conformational state ADP is released from the transporter? If ADP is released in the open state, then the transporter will return to this state before ATP can bind again, regardless of its concentration in the cell. Second, it is not the ATP concentration itself that really matters but rather the kon of ATP binding. If this rate is slow as compared to the rate of transition between the closed state (just after ATP hydrolysis) and the open state, then the transporter will be able to return to the open state before ATP binds again. Considering all these parameters, the apo state should not be so infrequent for multidrug transporters, in particular in bacteria. In order to test the presence of the inward-facing conformation of P-glycoprotein in mammalian cells, Loo and Clarke [163] placed reporter cysteines in extracellular loops close enough to form a disulfide bond in this conformation but widely separated in the outward-facing conformation. Spontaneous cross-linking strongly suggested the existence, at least transiently in cells, of the inward-facing conformation in which the NBDs are open.
8.2 The Constant Contact Model
An alternative model has been proposed by Jones and George [153], in which the NBDs remain in contact throughout the catalytic cycle. This model should not be mistaken with a constant peripheral interaction between NBDs, as, for instance, in many ABC importers. In this constant contact model, ATP hydrolysis occurs alternately at each site, with one site able to open and exchange hydrolysis products, while the other ATP-bound site remains closed. Hydrolysis of ATP promotes an opening at that site by an outward rotation of the RecA-like subdomain relative to the helical subdomain [164]. This model built on earlier P-glycoprotein work from Senior and collaborators [165]. They proposed an alternating hydrolysis of the NBDs based notably on the observation that both sites were equally active and that orthovanadate-induced ADP trapping in one catalytic site was sufficient to inhibit ATP hydrolysis in both sites [166]. The occlusion of one nucleotide during the transition state has indeed been observed in several proteins: P-glycoprotein [165], BmrA [58], LmrA [7], and maltose transporter [167]. In contrast, two nucleotides were shown trapped in the heterodimeric TmrAB transporter [18]. The asymmetry observed in structural [168], biochemical [169–171] and molecular dynamic studies [164, 172] is often interpreted in favor of the constant contact model.
Another argument cited in favor of this model is that P-glycoprotein retains an ATPase activity when the NBDs are covalently linked together [173–175]. A single molecule FRET analysis of reconstituted P-glycoprotein rather supports a model where the NBDs do not completely dissociate from one another during steady state catalysis although, given the broad distance distribution recorded in all ligand conditions, full dissociation of the NBDs cannot be entirely excluded and may occur during some of the cycles [176]. This model involves an alternating catalysis in which ATP hydrolysis and Pi release are coupled to drug transport.
Lastly, the NBDs in the crystal structures of TM287/TM288 remained in contact, but with coupling helices separated by 15 Å, which is sufficient to make the substrate-binding cavity accessible from inside without the need for NBD full disengagement [87].
9 Concluding Remarks
It should be noted that alternate models are rarely discussed but could be as plausible as the models discussed above. For instance, one can imagine a scenario in which the binding of two ATP molecules generate an outward-facing conformation, as in the switch model, but the hydrolysis of one ATP molecule is sufficient to destabilize the dimer thereby implying a catalytic asymmetry. Whether the two catalytic sites in homodimeric ABC exporters hydrolyze ATP simultaneously, sequentially, in an alternating or stochastic manner has not yet been settled. In the isolated NBDs, MJ0796, the hydrolysis of one molecule of ATP is sufficient to allow the physical disengagement of the two NBDs [156]. Although a stoichiometry of two ATP molecules per substrate transported has been found for the OpuA importer [177], a stoichiometry of one ATP molecule was determined for P-glycoprotein [178]. Owing to the structural and functional diversity of ABC transporters, there might not be a single unified mechanism for all members. For instance, one of two catalytic sites is poorly active in heterodimeric ABC exporters, and such transporters may employ a different catalytic cycle than the homodimeric transporters. Recently, Mchaourab and colleagues proposed that the power stroke for drug export by BmrC/BmrD is the ATP hydrolysis step, in contrast to homodimeric exporters like MsbA where the transport process will occur during the NBD dimerization driven by ATP binding [31].
The recent 3D structure of ABCG5/ABG8, the human sterol exporter exemplifies again the diversity of this family. It shows a unique structure with some traits similar to importers like the lack of cross talk afforded by a coupling helix [179].
References
ter Beek J, Guskov A, Slotboom DJ (2014) Structural diversity of ABC transporters. J Gen Physiol 143:419–435. doi:10.1085/jgp.201411164
Lee M, Choi Y, Burla B, Kim YY, Jeon B, Maeshima M, Yoo JY, Martinoia E et al (2008) The ABC transporter AtABCB14 is a malate importer and modulates stomatal response to CO2. Nat Cell Biol 10:1217–1223. doi:10.1038/ncb1782
Jones PM, O’Mara ML, George AM (2009) ABC transporters: a riddle wrapped in a mystery inside an enigma. Trends Biochem Sci 34:520–531. doi:10.1016/j.tibs.2009.06.004
Rees DC, Johnson E, Lewinson O (2009) ABC transporters: the power to change. Nat Rev Mol Cell Biol 10:218–227. doi:10.1038/nrm2646
Rice AJ, Park A, Pinkett HW (2014) Diversity in ABC transporters: type I, II and III importers. Crit Rev Biochem Mol Biol 49:426–437. doi:10.3109/10409238.2014.953626
van Veen HW, Venema K, Bolhuis H, Oussenko I, Kok J, Poolman B, Driessen AJ, Konings WN (1996) Multidrug resistance mediated by a bacterial homolog of the human multidrug transporter MDR1. Proc Natl Acad Sci U S A 93:10668–10672
van Veen HW, Margolles A, Muller M, Higgins CF, Konings WN (2000) The homodimeric ATP-binding cassette transporter LmrA mediates multidrug transport by an alternating two-site (two-cylinder engine) mechanism. EMBO J 19:2503–2514. doi:10.1093/emboj/19.11.2503
van Veen HW, Callaghan R, Soceneantu L, Sardini A, Konings WN, Higgins CF (1998) A bacterial antibiotic-resistance gene that complements the human multidrug-resistance P-glycoprotein gene. Nature 391:291–295. doi:10.1038/34669
Steinfels E, Orelle C, Dalmas O, Penin F, Miroux B, Di Pietro A, Jault JM (2002) Highly efficient over-production in E. coli of YvcC, a multidrug-like ATP-binding cassette transporter from Bacillus subtilis. Biochim Biophys Acta 1565:1–5. doi:10.1016/S0005-2736(02)00515-1
Dalmas O, Do Cao MA, Lugo MR, Sharom FJ, Di Pietro A, Jault JM (2005) Time-resolved fluorescence resonance energy transfer shows that the bacterial multidrug ABC half-transporter BmrA functions as a homodimer. Biochemistry 44:4312–4321. doi:10.1021/bi0482809
Steinfels E, Orelle C, Fantino JR, Dalmas O, Rigaud JL, Denizot F, Di Pietro A, Jault JM (2004) Characterization of YvcC (BmrA), a multidrug ABC transporter constitutively expressed in Bacillus subtilis. Biochemistry 43:7491–7502. doi:10.1021/bi0362018
Krugel H, Licht A, Biedermann G, Petzold A, Lassak J, Hupfer Y, Schlott B, Hertweck C et al (2010) Cervimycin C resistance in Bacillus subtilis is due to a promoter up-mutation and increased mRNA stability of the constitutive ABC-transporter gene bmrA. FEMS Microbiol Lett 313:155–163. doi:10.1111/j.1574-6968.2010.02143.x
Lubelski J, Mazurkiewicz P, van Merkerk R, Konings WN, Driessen AJ (2004) ydaG and ydbA of Lactococcus lactis encode a heterodimeric ATP-binding cassette-type multidrug transporter. J Biol Chem 279:34449–34455. doi:10.1074/jbc.M404072200
Torres C, Galian C, Freiberg C, Fantino JR, Jault JM (2009) The YheI/YheH heterodimer from Bacillus subtilis is a multidrug ABC transporter. Biochim Biophys Acta 1788:615–622. doi:10.1016/j.bbamem.2008
Marrer E, Schad K, Satoh AT, Page MG, Johnson MM, Piddock LJ (2006) Involvement of the putative ATP-dependent efflux proteins PatA and PatB in fluoroquinolone resistance of a multidrug-resistant mutant of Streptococcus pneumoniae. Antimicrob Agents Chemother 50:685–693. doi:10.1128/AAC.50.2.685-693.2006
Matsuo T, Chen J, Minato Y, Ogawa W, Mizushima T, Kuroda T, Tsuchiya T (2008) SmdAB, a heterodimeric ABC-Type multidrug efflux pump, in Serratia marcescens. J Bacteriol 190:648–654. doi:10.1128/JB.01513-07
Lubelski J, van Merkerk R, Konings WN, Driessen AJ (2006) Nucleotide-binding sites of the heterodimeric LmrCD ABC-multidrug transporter of Lactococcus lactis are asymmetric. Biochemistry 45:648–656. doi:10.1021/bi051276s
Zutz A, Hoffmann J, Hellmich UA, Glaubitz C, Ludwig B, Brutschy B, Tampe R (2011) Asymmetric ATP hydrolysis cycle of the heterodimeric multidrug ABC transport complex TmrAB from Thermus thermophilus. J Biol Chem 286:7104–7115. doi:10.1074/jbc.M110.201178
Hinz A, Tampe R (2012) ABC transporters and immunity: mechanism of self-defense. Biochemistry 51:4981–4989. doi:10.1021/bi300128f
Qin L, Zheng J, Grant CE, Jia Z, Cole SP, Deeley RG (2008) Residues responsible for the asymmetric function of the nucleotide binding domains of multidrug resistance protein 1. Biochemistry 47:13952–13965. doi:10.1021/bi801532g
Lubelski J, de Jong A, van Merkerk R, Agustiandari H, Kuipers OP, Kok J, Driessen AJ (2006) LmrCD is a major multidrug resistance transporter in Lactococcus lactis. Mol Microbiol 61:771–781. doi:10.1111/j.1365-2958.2006.05267.x
Zaidi AH, Bakkes PJ, Lubelski J, Agustiandari H, Kuipers OP, Driessen AJ (2008) The ABC-type multidrug resistance transporter LmrCD is responsible for an extrusion-based mechanism of bile acid resistance in Lactococcus lactis. J Bacteriol 190:7357–7366. doi:10.1128/JB.00485-08
Agustiandari H, Lubelski J, van den Berg van Saparoea HB, Kuipers OP, Driessen AJ (2008) LmrR is a transcriptional repressor of expression of the multidrug ABC transporter LmrCD in Lactococcus lactis. J Bacteriol 190:759–763. doi:10.1128/JB.01151-07
Agustiandari H, Peeters E, de Wit JG, Charlier D, Driessen AJ (2011) LmrR-mediated gene regulation of multidrug resistance in Lactococcus lactis. Microbiology 157:1519–1530. doi:10.1099/mic.0.048025-0
Madoori PK, Agustiandari H, Driessen AJ, Thunnissen AM (2009) Structure of the transcriptional regulator LmrR and its mechanism of multidrug recognition. EMBO J 28:156–166. doi:10.1038/emboj.2008.263
Takeuchi K, Tokunaga Y, Imai M, Takahashi H, Shimada I (2014) Dynamic multidrug recognition by multidrug transcriptional repressor LmrR. Sci Rep 4:6922. doi:10.1038/srep06922
van der Berg JP, Madoori PK, Komarudin AG, Thunnissen AM, Driessen AJ (2015) Binding of the lactococcal drug dependent transcriptional regulator LmrR to its ligands and responsive promoter regions. PLoS One 10:e0135467. doi:10.1371/journal.pone.0135467
Reilman E, Mars RA, van Dijl JM, Denham EL (2014) The multidrug ABC transporter BmrC/BmrD of Bacillus subtilis is regulated via a ribosome-mediated transcriptional attenuation mechanism. Nucleic Acids Res 42:11393–11407. doi:10.1093/nar/gku832
Dezi M, Di Cicco A, Bassereau P, Levy D (2013) Detergent-mediated incorporation of transmembrane proteins in giant unilamellar vesicles with controlled physiological contents. Proc Natl Acad Sci U S A 110:7276–7281. doi:10.1073/pnas.1303857110
Galian C, Manon F, Dezi M, Torres C, Ebel C, Levy D, Jault JM (2011) Optimized purification of a heterodimeric ABC transporter in a highly stable form amenable to 2-D crystallization. PLoS One 6:e19677. doi:10.1371/journal.pone.0019677
Mishra S, Verhalen B, Stein RA, Wen PC, Tajkhorshid E, McHaourab HS (2014) Conformational dynamics of the nucleotide binding domains and the power stroke of a heterodimeric ABC transporter. eLife 3:e02740. doi:10.7554/eLife.02740
Robertson GT, Doyle TB, Lynch AS (2005) Use of an efflux-deficient Streptococcus pneumoniae strain panel to identify ABC-class multidrug transporters involved in intrinsic resistance to antimicrobial agents. Antimicrob Agents Chemother 49:4781–4783. doi:10.1128/AAC.49.11.4781-4783.2005
Marrer E, Satoh AT, Johnson MM, Piddock LJ, Page MG (2006) Global transcriptome analysis of the responses of a fluoroquinolone-resistant Streptococcus pneumoniae mutant and its parent to ciprofloxacin. Antimicrob Agents Chemother 50:269–278. doi:10.1128/aac.50.1.269-278.2006
El Garch F, Lismond A, Piddock LJ, Courvalin P, Tulkens PM, Van Bambeke F (2010) Fluoroquinolones induce the expression of patA and patB, which encode ABC efflux pumps in Streptococcus pneumoniae. J Antimicrob Chemother 65:2076–2082. doi:10.1093/jac/dkq287
Garvey MI, Baylay AJ, Wong RL, Piddock LJ (2011) Overexpression of patA and patB, which encode ABC transporters, is associated with fluoroquinolone resistance in clinical isolates of Streptococcus pneumoniae. Antimicrob Agents Chemother 55:190–196. doi:10.1128/AAC.00672-10
Lupien A, Billal DS, Fani F, Soualhine H, Zhanel GG, Leprohon P, Ouellette M (2013) Genomic characterization of ciprofloxacin resistance in a laboratory-derived mutant and a clinical isolate of Streptococcus pneumoniae. Antimicrob Agents Chemother 57:4911–4919. doi:10.1128/aac.00418-13
Baylay AJ, Piddock LJ (2015) Clinically relevant fluoroquinolone resistance due to constitutive overexpression of the PatAB ABC transporter in Streptococcus pneumoniae is conferred by disruption of a transcriptional attenuator. J Antimicrob Chemother 70:670–679. doi:10.1093/jac/dku449
Baylay AJ, Ivens A, Piddock LJ (2015) A novel gene amplification causes upregulation of the PatAB ABC transporter and fluoroquinolone resistance in Streptococcus pneumoniae. Antimicrob Agents Chemother 59:3098–3108. doi:10.1128/AAC.04858-14
Lupien A, Gingras H, Bergeron MG, Leprohon P, Ouellette M (2015) Multiple mutations and increased RNA expression in tetracycline-resistant Streptococcus pneumoniae as determined by genome-wide DNA and mRNA sequencing. J Antimicrob Chemother 70:1946–1959. doi:10.1093/jac/dkv060
Boncoeur E, Durmort C, Bernay B, Ebel C, Di Guilmi AM, Croize J, Vernet T, Jault JM (2012) PatA and PatB form a functional heterodimeric ABC multidrug efflux transporter responsible for the resistance of Streptococcus pneumoniae to fluoroquinolones. Biochemistry 51:7755–7765. doi:10.1021/bi300762p
Hohl M, Briand C, Grutter MG, Seeger MA (2012) Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation. Nat Struct Mol Biol 19:395–402. doi:10.1038/nsmb.2267
Bechara C, Noll A, Morgner N, Degiacomi MT, Tampe R, Robinson CV (2015) A subset of annular lipids is linked to the flippase activity of an ABC transporter. Nat Chem 7:255–262. doi:10.1038/nchem.2172
Li W, Sharma M, Kaur P (2014) The DrrAB efflux system of Streptomyces peucetius is a multidrug transporter of broad substrate specificity. J Biol Chem 289:12633–12646. doi:10.1074/jbc.M113.536136
Gandlur SM, Wei L, Levine J, Russell J, Kaur P (2004) Membrane topology of the DrrB protein of the doxorubicin transporter of Streptomyces peucetius. J Biol Chem 279:27799–27806. doi:10.1074/jbc.M402898200
Kobayashi N, Nishino K, Yamaguchi A (2001) Novel macrolide-specific ABC-type efflux transporter in Escherichia coli. J Bacteriol 183:5639–5644. doi:10.1128/JB.183.19.5639-5644.2001
Lu S, Zgurskaya HI (2012) Role of ATP binding and hydrolysis in assembly of MacAB-TolC macrolide transporter. Mol Microbiol 86:1132–1143. doi:10.1111/mmi.12046
Lin HT, Bavro VN, Barrera NP, Frankish HM, Velamakanni S, van Veen HW, Robinson CV, Borges-Walmsley MI et al (2009) MacB ABC transporter is a dimer whose ATPase activity and macrolide-binding capacity are regulated by the membrane fusion protein MacA. J Biol Chem 284:1145–1154. doi:10.1074/jbc.M806964200
Tikhonova EB, Devroy VK, Lau SY, Zgurskaya HI (2007) Reconstitution of the Escherichia coli macrolide transporter: the periplasmic membrane fusion protein MacA stimulates the ATPase activity of MacB. Mol Microbiol 63:895–910. doi:10.1111/j.1365-2958.2006.05549.x
Turlin E, Heuck G, Simoes Brandao MI, Szili N, Mellin JR, Lange N, Wandersman C (2014) Protoporphyrin (PPIX) efflux by the MacAB-TolC pump in Escherichia coli. Microbiol Open 3:849–859. doi:10.1002/mbo3.203
Oswald C, Holland IB, Schmitt L (2006) The motor domains of ABC-transporters. What can structures tell us? Naunyn Schmiedebergs Arch Pharmacol 372:385–399. doi:10.1007/s00210-005-0031-4
Hung LW, Wang IX, Nikaido K, Liu PQ, Ames GF, Kim SH (1998) Crystal structure of the ATP-binding subunit of an ABC transporter. Nature 396:703–707. doi:10.1038/25393
Karpowich N, Martsinkevich O, Millen L, Yuan YR, Dai PL, MacVey K, Thomas PJ, Hunt JF (2001) Crystal structures of the MJ1267 ATP binding cassette reveal an induced-fit effect at the ATPase active site of an ABC transporter. Structure 9:571–586. doi:10.1016/S0969-2126(01)00617-7
Geourjon C, Orelle C, Steinfels E, Blanchet C, Deleage G, Di Pietro A, Jault JM (2001) A common mechanism for ATP hydrolysis in ABC transporter and helicase superfamilies. Trends Biochem Sci 26:539–544. doi:10.1016/S0968-0004(01)01907-7
Ye J, Osborne AR, Groll M, Rapoport TA (2004) RecA-like motor ATPases-lessons from structures. Biochim Biophys Acta 1659:1–18. doi:10.1016/j.bbabio.2004.06.003
Walker JE, Saraste M, Runswick MJ, Gay NJ (1982) Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J 1:945–951
Muneyuki E, Noji H, Amano T, Masaike T, Yoshida M (2000) F0F1-ATP synthase: general structural features of ‘ATP-engine’ and a problem on free energy transduction. Biochim Biophys Acta 1458:467–481. doi:10.1016/S0005-2728(00)00095-5
Thomsen ND, Berger JM (2008) Structural frameworks for considering microbial protein- and nucleic acid-dependent motor ATPases. Mol Microbiol 69:1071–1090. doi:10.1111/j.1365-2958.2008.06364.x
Orelle C, Dalmas O, Gros P, Di Pietro A, Jault JM (2003) The conserved glutamate residue adjacent to the Walker-B motif is the catalytic base for ATP hydrolysis in the ATP-binding cassette transporter BmrA. J Biol Chem 278:47002–47008. doi:10.1074/jbc.M308268200
Oldham ML, Chen J (2011) Snapshots of the maltose transporter during ATP hydrolysis. Proc Natl Acad Sci U S A 108:15152–15156. doi:10.1073/pnas.1108858108
Senior AE (2011) Reaction chemistry ABC-style. Proc Natl Acad Sci U S A 108:15015–15016. doi:10.1073/pnas.1111863108
Zaitseva J, Jenewein S, Jumpertz T, Holland IB, Schmitt L (2005) H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB. EMBO J 24:1901–1910. doi:10.1038/sj.emboj.7600657
Hopfner KP, Karcher A, Shin DS, Craig L, Arthur LM, Carney JP, Tainer JA (2000) Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell 101:789–800. doi:10.1016/S0092-8674(00)80890-9
Jones PM, George AM (2012) Role of the D-loops in allosteric control of ATP hydrolysis in an ABC transporter. J Phys Chem A 116:3004–3013. doi:10.1021/jp211139s
Grossmann N, Vakkasoglu AS, Hulpke S, Abele R, Gaudet R, Tampe R (2014) Mechanistic determinants of the directionality and energetics of active export by a heterodimeric ABC transporter. Nat Commun 5:5419. doi:10.1038/ncomms6419
Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ, Hunt JF (2002) ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol Cell 10:139–149. doi:10.1016/S1097-2765(02)00576-2
Dawson RJ, Locher KP (2006) Structure of a bacterial multidrug ABC transporter. Nature 443:180–185. doi:10.1038/nature05155
Becker JP, Van Bambeke F, Tulkens PM, Prevost M (2010) Dynamics and structural changes induced by ATP binding in SAV1866, a bacterial ABC exporter. J Phys Chem B 114:15948–15957. doi:10.1021/jp1038392
Damas JM, Oliveira AS, Baptista AM, Soares CM (2011) Structural consequences of ATP hydrolysis on the ABC transporter NBD dimer: molecular dynamics studies of HlyB. Protein Sci 20:1220–1230. doi:10.1002/pro.650
Kluth M, Stindt J, Droge C, Linnemann D, Kubitz R, Schmitt L (2015) A mutation within the extended X loop abolished substrate-induced ATPase activity of the human liver ATP-binding cassette (ABC) transporter MDR3. J Biol Chem 290:4896–4907. doi:10.1074/jbc.M114.588566
Oancea G, O’Mara ML, Bennett WF, Tieleman DP, Abele R, Tampe R (2009) Structural arrangement of the transmission interface in the antigen ABC transport complex TAP. Proc Natl Acad Sci U S A 106:5551–5556. doi:10.1073/pnas.0811260106
Ambudkar SV, Kim IW, Xia D, Sauna ZE (2006) The A-loop, a novel conserved aromatic acid subdomain upstream of the Walker A motif in ABC transporters, is critical for ATP binding. FEBS Lett 580:1049–1055. doi:10.1016/j.febslet.2005.12.051
Bukowska MA, Hohl M, Geertsma ER, Hurlimann LM, Grutter MG, Seeger MA (2015) A transporter motor taken apart: flexibility in the nucleotide binding domains of a heterodimeric ABC exporter. Biochemistry 54:3086–3099. doi:10.1021/acs.biochem.5b00188
De Marcos LC, Dietrich D, Johnson B, Baldwin SA, Holdsworth MJ, Theodoulou FL, Baker A (2009) The NBDs that wouldn’t die: a cautionary tale of the use of isolated nucleotide binding domains of ABC transporters. Commun Integr Biol 2:97–99. doi:10.4161/cib.7621
Li J, Jaimes KF, Aller SG (2014) Refined structures of mouse P-glycoprotein. Protein Sci 23:34–46. doi:10.1002/pro.2387
Ward A, Reyes CL, Yu J, Roth CB, Chang G (2007) Flexibility in the ABC transporter MsbA: alternating access with a twist. Proc Natl Acad Sci U S A 104:19005–19010. doi:10.1073/pnas.0709388104
Velamakanni S, Yao Y, Gutmann DA, van Veen HW (2008) Multidrug transport by the ABC transporter Sav 1866 from Staphylococcus aureus. Biochemistry 47:9300–9308. doi:10.1021/bi8006737
Chang G, Roth CB, Reyes CL, Pornillos O, Chen YJ, Chen AP (2006) Retraction. Science 314:1875. doi:10.1126/science.314.5807.1875b
Dawson RJ, Locher KP (2007) Structure of the multidrug ABC transporter Sav 1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett 581:935–938. doi:10.1016/j.febslet.2007.01.073
Zhou Z, White KA, Polissi A, Georgopoulos C, Raetz CR (1998) Function of Escherichia coli MsbA, an essential ABC family transporter, in lipid A and phospholipid biosynthesis. J Biol Chem 273:12466–12475. doi:10.1074/jbc.273.20.12466
Reuter G, Janvilisri T, Venter H, Shahi S, Balakrishnan L, van Veen HW (2003) The ATP binding cassette multidrug transporter LmrA and lipid transporter MsbA have overlapping substrate specificities. J Biol Chem 278:35193–35198. doi:10.1074/jbc.M306226200
Woebking B, Reuter G, Shilling RA, Velamakanni S, Shahi S, Venter H, Balakrishnan L, van Veen HW (2005) Drug-lipid A interactions on the Escherichia coli ABC transporter MsbA. J Bacteriol 187:6363–6369. doi:10.1128/JB.187.18.6363-6369.2005
Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT et al (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323:1718–1722. doi:10.1126/science.1168750
Jin MS, Oldham ML, Zhang Q, Chen J (2012) Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 490:566–569. doi:10.1038/nature11448
Perez C, Gerber S, Boilevin J, Bucher M, Darbre T, Aebi M, Reymond JL, Locher KP (2015) Structure and mechanism of an active lipid-linked oligosaccharide flippase. Nature 524:433–438. doi:10.1038/nature14953
Fribourg PF, Chami M, Sorzano CO, Gubellini F, Marabini R, Marco S, Jault JM, Levy D (2014) 3D cryo-electron reconstruction of BmrA, a bacterial multidrug ABC transporter in an inward-facing conformation and in a lipidic environment. J Mol Biol 426:2059–2069. doi:10.1016/j.jmb.2014.03.002
Choudhury HG, Tong Z, Mathavan I, Li Y, Iwata S, Zirah S, Rebuffat S, van Veen HW et al (2014) Structure of an antibacterial peptide ATP-binding cassette transporter in a novel outward occluded state. Proc Natl Acad Sci U S A 111:9145–9150. doi:10.1073/pnas.1320506111
Hohl M, Hurlimann LM, Bohm S, Schoppe J, Grutter MG, Bordignon E, Seeger MA (2014) Structural basis for allosteric cross-talk between the asymmetric nucleotide binding sites of a heterodimeric ABC exporter. Proc Natl Acad Sci U S A 111:11025–11030. doi:10.1073/pnas.1400485111
Shintre CA, Pike AC, Li Q, Kim JI, Barr AJ, Goubin S, Shrestha L, Yang J et al (2013) Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. Proc Natl Acad Sci U S A 110:9710–9715. doi:10.1073/pnas.1217042110
Kim J, Wu S, Tomasiak TM, Mergel C, Winter MB, Stiller SB, Robles-Colmanares Y, Stroud RM et al (2015) Subnanometre-resolution electron cryomicroscopy structure of a heterodimeric ABC exporter. Nature 517:396–400. doi:10.1038/nature13872
Khare D, Oldham ML, Orelle C, Davidson AL, Chen J (2009) Alternating access in maltose transporter mediated by rigid-body rotations. Mol Cell 33:528–536. doi:10.1016/j.molcel.2009.01.035
van Wonderen JH, McMahon RM, O’Mara ML, McDevitt CA, Thomson AJ, Kerr ID, MacMillan F, Callaghan R (2014) The central cavity of ABCB1 undergoes alternating access during ATP hydrolysis. FEBS J 281:2190–2201. doi:10.1111/febs.12773
Zou P, McHaourab HS (2009) Alternating access of the putative substrate-binding chamber in the ABC transporter MsbA. J Mol Biol 393:574–585. doi:10.1016/j.jmb.2009.08.051
Martin C, Higgins CF, Callaghan R (2001) The vinblastine binding site adopts high- and low-affinity conformations during a transport cycle of P-glycoprotein. Biochemistry 40:15733–15742. doi:10.1021/bi011211z
Ramachandra M, Ambudkar SV, Chen D, Hrycyna CA, Dey S, Gottesman MM, Pastan I (1998) Human P-glycoprotein exhibits reduced affinity for substrates during a catalytic transition state. Biochemistry 37:5010–5019. doi:10.1021/bi973045u
Moradi M, Tajkhorshid E (2013) Mechanistic picture for conformational transition of a membrane transporter at atomic resolution. Proc Natl Acad Sci U S A 110:18916–18921. doi:10.1073/pnas.1313202110
Wise JG (2012) Catalytic transitions in the human MDR1 P-glycoprotein drug binding sites. Biochemistry 51:5125–5141. doi:10.1021/bi300299z
Bayeva M, Khechaduri A, Wu R, Burke MA, Wasserstrom JA, Singh N, Liesa M, Shirihai OS et al (2013) ATP-binding cassette B10 regulates early steps of heme synthesis. Circ Res 113:279–287. doi:10.1161/CIRCRESAHA.113.301552
Qiu W, Liesa M, Carpenter EP, Shirihai OS (2015) ATP binding and hydrolysis properties of ABCB10 and their regulation by glutathione. PLoS One 10:e0129772. doi:10.1371/journal.pone.0129772
Loo TW, Bartlett MC, Clarke DM (2004) The drug-binding pocket of the human multidrug resistance P-glycoprotein is accessible to the aqueous medium. Biochemistry 43:12081–12089. doi:10.1021/bi049045t
Loo TW, Bartlett MC, Clarke DM (2009) Identification of residues in the drug translocation pathway of the human multidrug resistance P-glycoprotein by arginine mutagenesis. J Biol Chem 284:24074–24087. doi:10.1074/jbc.M109.023267
Poelarends GJ, Konings WN (2002) The transmembrane domains of the ABC multidrug transporter LmrA form a cytoplasmic exposed, aqueous chamber within the membrane. J Biol Chem 277:42891–42898. doi:10.1074/jbc.M206508200
Gatlik-Landwojtowicz E, Aanismaa P, Seelig A (2006) Quantification and characterization of P-glycoprotein-substrate interactions. Biochemistry 45:3020–3032. doi:10.1021/bi051380+
Chufan EE, Sim HM, Ambudkar SV (2015) Molecular basis of the polyspecificity of P-glycoprotein (ABCB1): recent biochemical and structural studies. Adv Cancer Res 125:71–96. doi:10.1016/bs.acr.2014.10.003
Loo TW, Clarke DM (1999) The transmembrane domains of the human multidrug resistance P-glycoprotein are sufficient to mediate drug binding and trafficking to the cell surface. J Biol Chem 274:24759–24765. doi:10.1074/jbc.274.35.24759
Ayesh S, Shao YM, Stein WD (1996) Co-operative, competitive and non-competitive interactions between modulators of P-glycoprotein. Biochim Biophys Acta 1316:8–18. doi:10.1016/0925-4439(96)00008-7
Garrigos M, Mir LM, Orlowski S (1997) Competitive and non-competitive inhibition of the multidrug-resistance-associated P-glycoprotein ATPase–further experimental evidence for a multisite model. Eur J Biochem 244:664–673. doi:10.1111/j.1432-1033.1997.00664.x
Tamai I, Safa AR (1991) Azidopine noncompetitively interacts with vinblastine and cyclosporin A binding to P-glycoprotein in multidrug resistant cells. J Biol Chem 266:16796–16800
Shapiro AB, Ling V (1997) Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur J Biochem 250:130–137. doi:10.1111/j.1432-1033.1997.00130.x
Shapiro AB, Fox K, Lam P, Ling V (1999) Stimulation of P-glycoprotein-mediated drug transport by prazosin and progesterone. Evidence for a third drug-binding site. Eur J Biochem 259:841–850. doi:10.1046/j.1432-1327.1999.00098.x
Lugo MR, Sharom FJ (2005) Interaction of LDS-751 with P-glycoprotein and mapping of the location of the R drug binding site. Biochemistry 44:643–655. doi:10.1021/bi0485326
Qu Q, Sharom FJ (2002) Proximity of bound Hoechst 33342 to the ATPase catalytic sites places the drug binding site of P-glycoprotein within the cytoplasmic membrane leaflet. Biochemistry 41:4744–4752. doi:10.1021/bi0120897
Loo TW, Clarke DM (2002) Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J Biol Chem 277:44332–44338. doi:10.1074/jbc.M208433200
Loo TW, Bartlett MC, Clarke DM (2003) Simultaneous binding of two different drugs in the binding pocket of the human multidrug resistance P-glycoprotein. J Biol Chem 278:39706–39710. doi:10.1074/jbc.M308559200
Martinez L, Arnaud O, Henin E, Tao H, Chaptal V, Doshi R, Andrieu T, Dussurgey S et al (2014) Understanding polyspecificity within the substrate-binding cavity of the human multidrug resistance P-glycoprotein. FEBS J 281:673–682. doi:10.1111/febs.12613
Pajeva IK, Sterz K, Christlieb M, Steggemann K, Marighetti F, Wiese M (2013) Interactions of the multidrug resistance modulators tariquidar and elacridar and their analogues with P-glycoprotein. ChemMedChem 8:1701–1713. doi:10.1002/cmdc.201300233
Smriti, Zou P, McHaourab HS (2009) Mapping daunorubicin-binding sites in the ATP-binding cassette transporter MsbA using site-specific quenching by spin labels. J Biol Chem 284:13904–13913. doi:10.1074/jbc.M900837200
Siarheyeva A, Sharom FJ (2009) The ABC transporter MsbA interacts with lipid A and amphipathic drugs at different sites. Biochem J 419:317–328. doi:10.1042/BJ20081364
Infed N, Hanekop N, Driessen AJ, Smits SH, Schmitt L (2011) Influence of detergents on the activity of the ABC transporter LmrA. Biochim Biophys Acta 1808:2313–2321. doi:10.1016/j.bbamem.2011.05.016
Seeger MA, Mittal A, Velamakanni S, Hohl M, Schauer S, Salaa I, Grutter MG, van Veen HW (2012) Tuning the drug efflux activity of an ABC transporter in vivo by in vitro selected DARPin binders. PLoS One 7:e37845. doi:10.1371/journal.pone.0037845
Herget M, Kreissig N, Kolbe C, Scholz C, Tampe R, Abele R (2009) Purification and reconstitution of the antigen transport complex TAP: a prerequisite for determination of peptide stoichiometry and ATP hydrolysis. J Biol Chem 284:33740–33749. doi:10.1074/jbc.M109.047779
Ames GF, Nikaido K, Wang IX, Liu PQ, Liu CE, Hu C (2001) Purification and characterization of the membrane-bound complex of an ABC transporter, the histidine permease. J Bioenerg Biomembr 33:79–92. doi:10.1023/A:1010797029183
Davidson AL, Shuman HA, Nikaido H (1992) Mechanism of maltose transport in Escherichia coli: transmembrane signaling by periplasmic binding proteins. Proc Natl Acad Sci U S A 89:2360–2364
Vigonsky E, Ovcharenko E, Lewinson O (2013) Two molybdate/tungstate ABC transporters that interact very differently with their substrate binding proteins. Proc Natl Acad Sci U S A 110:5440–5445. doi:10.1073/pnas.1213598110
Doshi R, van Veen HW (2013) Substrate binding stabilizes a pre-translocation intermediate in the ATP-binding cassette transport protein MsbA. J Biol Chem 288:21638–21647. doi:10.1074/jbc.M113.485714
Loo TW, Bartlett MC, Clarke DM (2003) Drug binding in human P-glycoprotein causes conformational changes in both nucleotide-binding domains. J Biol Chem 278:1575–1578. doi:10.1074/jbc.M211307200
Geng J, Sivaramakrishnan S, Raghavan M (2013) Analyses of conformational states of the transporter associated with antigen processing (TAP) protein in a native cellular membrane environment. J Biol Chem 288:37039–37047. doi:10.1074/jbc.M113.504696
Modali SD, Zgurskaya HI (2011) The periplasmic membrane proximal domain of MacA acts as a switch in stimulation of ATP hydrolysis by MacB transporter. Mol Microbiol 81:937–951. doi:10.1111/j.1365-2958.2011.07744.x
Alvarez FJ, Orelle C, Huang Y, Bajaj R, Everly RM, Klug CS, Davidson AL (2015) Full engagement of liganded maltose-binding protein stabilizes a semi-open ATP-binding cassette dimer in the maltose transporter. Mol Microbiol 98:878–894. doi:10.1111/mmi.13165
Orelle C, Ayvaz T, Everly RM, Klug CS, Davidson AL (2008) Both maltose-binding protein and ATP are required for nucleotide-binding domain closure in the intact maltose ABC transporter. Proc Natl Acad Sci U S A 105:12837–12842. doi:10.1073/pnas.0803799105
Sippach M, Weidlich D, Klose D, Abe C, Klare J, Schneider E, Steinhoff HJ (2014) Conformational changes of the histidine ATP-binding cassette transporter studied by double electron-electron resonance spectroscopy. Biochim Biophys Acta 1838:1760–1768. doi:10.1016/j.bbamem.2014.02.010
Margolles A, Putman M, van Veen HW, Konings WN (1999) The purified and functionally reconstituted multidrug transporter LmrA of Lactococcus lactis mediates the transbilayer movement of specific fluorescent phospholipids. Biochemistry 38:16298–16306
van Helvoort A, Smith AJ, Sprong H, Fritzsche I, Schinkel AH, Borst P, van Meer G (1996) MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell 87:507–517. doi:10.1016/S0092-8674(00)81370-7
Al-Shawi MK, Polar MK, Omote H, Figler RA (2003) Transition state analysis of the coupling of drug transport to ATP hydrolysis by P-glycoprotein. J Biol Chem 278:52629–52640. doi:10.1074/jbc.M308175200
Ernst R, Kueppers P, Stindt J, Kuchler K, Schmitt L (2010) Multidrug efflux pumps: substrate selection in ATP-binding cassette multidrug efflux pumps–first come, first served? FEBS J 277:540–549. doi:10.1111/j.1742-4658.2009.07485.x
Gupta RP, Kueppers P, Schmitt L, Ernst R (2011) The multidrug transporter Pdr5: a molecular diode? Biol Chem 392:53–60. doi:10.1515/BC.2011.011
Cooper RS, Altenberg GA (2013) Association/dissociation of the nucleotide-binding domains of the ATP-binding cassette protein MsbA measured during continuous hydrolysis. J Biol Chem 288:20785–20796. doi:10.1074/jbc.M113.477976
Moody JE, Millen L, Binns D, Hunt JF, Thomas PJ (2002) Cooperative, ATP-dependent association of the nucleotide binding cassettes during the catalytic cycle of ATP-binding cassette transporters. J Biol Chem 277:21111–21114. doi:10.1074/jbc.C200228200
Zoghbi ME, Fuson KL, Sutton RB, Altenberg GA (2012) Kinetics of the association/dissociation cycle of an ATP-binding cassette nucleotide-binding domain. J Biol Chem 287:4157–4164. doi:10.1074/jbc.M111.318378
Yuan YR, Blecker S, Martsinkevich O, Millen L, Thomas PJ, Hunt JF (2001) The crystal structure of the MJ0796 ATP-binding cassette. Implications for the structural consequences of ATP hydrolysis in the active site of an ABC transporter. J Biol Chem 276:32313–32321. doi:10.1074/jbc.M100758200
Jones PM, George AM (1999) Subunit interactions in ABC transporters: towards a functional architecture. FEMS Microbiol Lett 179:187–202. doi:10.1111/j.1574-6968.1999.tb08727.x
Fetsch EE, Davidson AL (2002) Vanadate-catalyzed photocleavage of the signature motif of an ATP-binding cassette (ABC) transporter. Proc Natl Acad Sci U S A 99:9685–9690. doi:10.1073/pnas.152204499
Buchaklian AH, Klug CS (2006) Characterization of the LSGGQ and H motifs from the Escherichia coli lipid A transporter MsbA. Biochemistry 45:12539–12546. doi:10.1021/bi060830a
Tombline G, Bartholomew L, Gimi K, Tyndall GA, Senior AE (2004) Synergy between conserved ABC signature Ser residues in P-glycoprotein catalysis. J Biol Chem 279:5363–5373. doi:10.1074/jbc.M311964200
Orelle C, Alvarez FJ, Oldham ML, Orelle A, Wiley TE, Chen J, Davidson AL (2010) Dynamics of α-helical subdomain rotation in the intact maltose ATP-binding cassette transporter. Proc Natl Acad Sci U S A 107:20293–20298. doi:10.1073/pnas.1006544107
Ward AB, Szewczyk P, Grimard V, Lee CW, Martinez L, Doshi R, Caya A, Villaluz M et al (2013) Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc Natl Acad Sci U S A 110:13386–13391. doi:10.1073/pnas.1309275110
Hellmich UA, Lyubenova S, Kaltenborn E, Doshi R, van Veen HW, Prisner TF, Glaubitz C (2012) Probing the ATP hydrolysis cycle of the ABC multidrug transporter LmrA by pulsed EPR spectroscopy. J Am Chem Soc 134:5857–5862. doi:10.1021/ja211007t
Mehmood S, Domene C, Forest E, Jault JM (2012) Dynamics of a bacterial multidrug ABC transporter in the inward- and outward-facing conformations. Proc Natl Acad Sci U S A 109:10832–10836. doi:10.1073/pnas.1204067109
Dalmas O, Orelle C, Foucher AE, Geourjon C, Crouzy S, Di Pietro A, Jault JM (2005) The Q-loop disengages from the first intracellular loop during the catalytic cycle of the multidrug ABC transporter BmrA. J Biol Chem 280:36857–36864. doi:10.1074/jbc.M503266200
Urbatsch IL, Gimi K, Wilke-Mounts S, Lerner-Marmarosh N, Rousseau ME, Gros P, Senior AE (2001) Cysteines 431 and 1074 are responsible for inhibitory disulfide cross-linking between the two nucleotide-binding sites in human P-glycoprotein. J Biol Chem 276:26980–26987. doi:10.1074/jbc.M010829200
Wen PC, Verhalen B, Wilkens S, McHaourab HS, Tajkhorshid E (2013) On the origin of large flexibility of P-glycoprotein in the inward-facing state. J Biol Chem 288:19211–19220. doi:10.1074/jbc.M113.450114
George AM, Jones PM (2012) Perspectives on the structure-function of ABC transporters: the switch and constant contact models. Prog Biophys Mol Biol 109:95–107. doi:10.1016/j.pbiomolbio.2012.06.003
Higgins CF, Linton KJ (2004) The ATP switch model for ABC transporters. Nat Struct Mol Biol 11:918–926. doi:10.1038/nsmb836
Jones PM, George AM (2009) Opening of the ADP-bound active site in the ABC transporter ATPase dimer: evidence for a constant contact, alternating sites model for the catalytic cycle. Proteins 75:387–396. doi:10.1002/prot.22250
van der Does C, Tampe R (2004) How do ABC transporters drive transport? Biol Chem 385:927–933. doi:10.1515/BC.2004.121
Zoghbi ME, Altenberg GA (2014) ATP binding to two sites is necessary for dimerization of nucleotide-binding domains of ABC proteins. Biochem Biophys Res Commun 443:97–102. doi:10.1016/j.bbrc.2013.11.050
Zoghbi ME, Altenberg GA (2013) Hydrolysis at one of the two nucleotide-binding sites drives the dissociation of ATP-binding cassette nucleotide-binding domain dimers. J Biol Chem 288:34259–34265. doi:10.1074/jbc.M113.500371
Borbat PP, Surendhran K, Bortolus M, Zou P, Freed JH, McHaourab HS (2007) Conformational motion of the ABC transporter MsbA induced by ATP hydrolysis. PLoS Biol 5:e271. doi:10.1371/journal.pbio.0050271
Doshi R, Woebking B, van Veen HW (2010) Dissection of the conformational cycle of the multidrug/lipidA ABC exporter MsbA. Proteins 78:2867–2872. doi:10.1002/prot.22813
Zou P, Bortolus M, McHaourab HS (2009) Conformational cycle of the ABC transporter MsbA in liposomes: detailed analysis using double electron-electron resonance spectroscopy. J Mol Biol 393:586–597. doi:10.1016/j.jmb.2009.08.050
Orelle C, Gubellini F, Durand A, Marco S, Levy D, Gros P, Di Pietro A, Jault JM (2008) Conformational change induced by ATP binding in the multidrug ATP-binding cassette transporter BmrA. Biochemistry 47:2404–2412. doi:10.1021/bi702303s
Gottesman MM, Ambudkar SV, Xia D (2009) Structure of a multidrug transporter. Nat Biotechnol 27:546–547. doi:10.1038/nbt0609-546
Yaginuma H, Kawai S, Tabata KV, Tomiyama K, Kakizuka A, Komatsuzaki T, Noji H, Imamura H (2014) Diversity in ATP concentrations in a single bacterial cell population revealed by quantitative single-cell imaging. Sci Rep 4:6522. doi:10.1038/srep06522
Loo TW, Clarke DM (2014) Cysteines introduced into extracellular loops 1 and 4 of human P-glycoprotein that are close only in the open conformation spontaneously form a disulfide bond that inhibits drug efflux and ATPase activity. J Biol Chem 289:24749–24758. doi:10.1074/jbc.M114.583021
Jones PM, George AM (2011) Molecular-dynamics simulations of the ATP/apo state of a multidrug ATP-binding cassette transporter provide a structural and mechanistic basis for the asymmetric occluded state. Biophys J 100:3025–3034. doi:10.1016/j.bpj.2011.05.028
Tombline G, Senior AE (2005) The occluded nucleotide conformation of P-glycoprotein. J Bioenerg Biomembr 37:497–500. doi:10.1007/s10863-005-9498-4
Urbatsch IL, Sankaran B, Bhagat S, Senior AE (1995) Both P-glycoprotein nucleotide-binding sites are catalytically active. J Biol Chem 270:26956–26961
Sharma S, Davidson AL (2000) Vanadate-induced trapping of nucleotides by purified maltose transport complex requires ATP hydrolysis. J Bacteriol 182:6570–6576. doi:10.1128/JB.182.23.6570-6576.2000
Zaitseva J, Oswald C, Jumpertz T, Jenewein S, Wiedenmann A, Holland IB, Schmitt L (2006) A structural analysis of asymmetry required for catalytic activity of an ABC-ATPase domain dimer. EMBO J 25:3432–3443. doi:10.1038/sj.emboj.7601208
Mittal A, Bohm S, Grutter MG, Bordignon E, Seeger MA (2012) Asymmetry in the homodimeric ABC transporter MsbA recognized by a DARPin. J Biol Chem 287:20395–20406. doi:10.1074/jbc.M112.359794
Sauna ZE, Kim IW, Nandigama K, Kopp S, Chiba P, Ambudkar SV (2007) Catalytic cycle of ATP hydrolysis by P-glycoprotein: evidence for formation of the E.S. reaction intermediate with ATP-gamma-S, a nonhydrolyzable analogue of ATP. Biochemistry 46:13787–13799. doi:10.1021/bi701385t
Siarheyeva A, Liu R, Sharom FJ (2010) Characterization of an asymmetric occluded state of P-glycoprotein with two bound nucleotides: implications for catalysis. J Biol Chem 285:7575–7586. doi:10.1074/jbc.M109.047290
Aittoniemi J, de Wet H, Ashcroft FM, Sansom MS (2010) Asymmetric switching in a homodimeric ABC transporter: a simulation study. PLoS Comput Biol 6:e1000762. doi:10.1371/journal.pcbi.1000762
Loo TW, Bartlett MC, Detty MR, Clarke DM (2012) The ATPase activity of the P-glycoprotein drug pump is highly activated when the N-terminal and central regions of the nucleotide-binding domains are linked closely together. J Biol Chem 287:26806–26816. doi:10.1074/jbc.M112.376202
Loo TW, Clarke DM (2014) Identification of the distance between the homologous halves of P-glycoprotein that triggers the high/low ATPase activity switch. J Biol Chem 289:8484–8492. doi:10.1074/jbc.M114.552075
Verhalen B, Wilkens S (2011) P-glycoprotein retains drug-stimulated ATPase activity upon covalent linkage of the two nucleotide binding domains at their C-terminal ends. J Biol Chem 286:10476–10482. doi:10.1074/jbc.M110.193151
Verhalen B, Ernst S, Borsch M, Wilkens S (2012) Dynamic ligand-induced conformational rearrangements in P-glycoprotein as probed by fluorescence resonance energy transfer spectroscopy. J Biol Chem 287:1112–1127. doi:10.1074/jbc.M111.301192
Patzlaff JS, van der Heide T, Poolman B (2003) The ATP/substrate stoichiometry of the ATP-binding cassette (ABC) transporter OpuA. J Biol Chem 278:29546–29551. doi:10.1074/jbc.M304796200
Eytan GD, Regev R, Assaraf YG (1996) Functional reconstitution of P-glycoprotein reveals an apparent near stoichiometric drug transport to ATP hydrolysis. J Biol Chem 271:3172–3178. doi:10.1074/jbc.271.6.3172
Lee JY, LN Kinch, DM Borek, J Wang, J Wang, IL Urbatsch, XS Xie, NV Grishin, et al., (2016) Crystal structure of the human sterol transporter ABCG5/ABCG8. Nature 533:561–564. doi:10.1038/nature17666
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Orelle, C., Jault, JM. (2016). Structures and Transport Mechanisms of the ABC Efflux Pumps. In: Li, XZ., Elkins, C., Zgurskaya, H. (eds) Efflux-Mediated Antimicrobial Resistance in Bacteria. Adis, Cham. https://doi.org/10.1007/978-3-319-39658-3_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-39658-3_4
Published:
Publisher Name: Adis, Cham
Print ISBN: 978-3-319-39656-9
Online ISBN: 978-3-319-39658-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)