
Abstract
Although apoptosis is well recognized as a cell death program with clear anticancer roles, accumulating evidence linking apoptosis with tissue repair and regeneration indicates that its relationship with malignant disease is more complex than previously thought. Here we review how the responses of neighboring cells in the microenvironment of apoptotic tumor cells may contribute to the cell birth/cell death disequilibrium that provides the basis for cancerous tissue emergence and growth. We describe the bioactive properties of apoptotic cells and consider, in particular, how apoptosis of tumor cells can engender a range of responses including pro-oncogenic signals having proliferative, angiogenic, reparatory, and immunosuppressive features. Drawing on the parallels between wound healing, tissue regeneration and cancer, we propose the concept of the “onco-regenerative niche,” a cell death-driven generic network of tissue repair and regenerative mechanisms that are hijacked in cancer. Finally, we consider how the responses to cell death in tumors can be targeted to provide more effective and long-lasting therapies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Cell death
- Apoptosis
- Tumor microenvironment
- Macrophage
- Extracellular vesicle
- Burkitt lymphoma
- Starry-sky
- Angiogenesis
- Onco-regenerative niche
- Anticancer therapy
3.1 Introduction: Imbalances of Cell Birth and Cell Death in Cancer
Cell gain is finely balanced ultimately by cell loss in normal tissue homeostasis. In cancer, by contrast, this balance is lost and cell gain (cell birth) outweighs cell loss (Fig. 3.1). Of possible cell loss mechanisms, which include cell migration, differentiation, and cell death, the latter plays crucial roles in regulating the sizes of normal and malignant cell populations. Although physiological cell death mechanisms have been vigorously pursued over recent years, our knowledge of the molecular mechanisms that regulate regression of cell populations through cell death lags behind that of population expansion through proliferation. Expansion of a rogue tissue through dysregulated imbalance of normal homeostatic mechanisms is the fundamental principle at the root of malignant disease which stems from loss of the key controlling elements of both cell proliferation and cell survival/death.

Cell birth/cell death balances and imbalances and their relationships to the tumor microenvironment. Conceptual examples illustrating the impact of dying cells on tissue microenvironments. (a) Effects of proliferation or apoptosis of single cells. In the case of cell 1, the integration of external and internal mitogenic (green arrow) and apoptotic (red arrow) signals favors proliferation (green dotted arrow) and leads to cell gain. For cell 2, apoptosis signaling predominates, the cell dies (red dotted arrow), and the endpoint is cell loss. Cell 3 initially proliferates and one of its progeny subsequently undergoes apoptosis. In this case cell birth is balanced by cell death, illustrating homeostasis. (b) Examples of homotypic effects of dying cells in multicellular populations. In the upper scenario, cell 4 initially proliferates and one of its progeny undergoes apoptosis. Subsequently, this apoptotic cell provides mitogenic signal(s) to cell 5, a member of the same lineage as cell 4. Cell 5 proliferates, the result being net gain of cells. This illustrates compensatory proliferation. Cell 6 interacts with its relative, cell 7 as illustrated, which results in net loss of cells (“apoptosis-induced apoptosis”) whereas cell 8 interacts with cell 9 in homeostatic mode via compensatory proliferation. (c) Conceptual illustrations of apoptotic cells influencing multicellular populations through heterotypic interactions. Cell 10 initially proliferates and one of its progeny subsequently dies. The latter cell then activates a member of another cell lineage within the tissue—for example, an endothelial cell (E). This cell then leads to a relative of cell 10, cell 11, receiving mitogenic signals (e.g., through improved vascularization) resulting in net gain of cells. In the middle example, death of cell 12 leads to activation of a different lineage (for example, an immune cell) which causes further death of cell 12’s relatives such as cell 13, leading to net cell loss. In the lower example, the death of cell 14 stimulates compensatory proliferation in its relative, cell 15 via activation of a cell of another lineage (for example, a macrophage, M). (d) Application of the mechanisms illustrated in (a–c) to the tumor microenvironment, indicating how death of tumor cells can lead to positive as well as negative signals influencing tumor growth, ultimately with net growth being the outcome
Genetic mutations that result in gain of function of oncoproteins like Bcl-2 and loss of function of tumor suppressors like p53 are classical examples of canonical pathways that lead to inappropriate survival of premalignant and malignant cells, promoting the cell birth/death imbalance in cancer. The resulting imbalance is dynamic: in other words, although cell death mechanisms may well be required to be inhibited in individual cells acquiring cancerous characteristics [1, 2], such mechanisms are often prominent in the populations of transformed cells in established tumors. Thus, it is not the case that regulated cell death mechanisms, notably apoptosis, are switched off in malignant disease. It follows that cell death mechanisms of ultimate advantage to the success of the malignant populations will be retained. Indeed, ‘constitutive’ apoptosis is often highly prominent in aggressive cancers. The critical point for successful establishment and progression of malignant disease is that the cell birth/cell death equation favors cell birth, the net effect being population expansion (Fig. 3.1). It is becoming clear that the dying component in this dynamic disequilibrium is not a passive entity but rather can feed positively into the cell population expansion and malignant evolutionary processes. This unexpected oncogenic property of dying cells is the main subject of this chapter.
Historically, in contrast to cell gain, cell loss has not been widely studied in the context of cancer. In cases where it has, there is a clear message: substantial cell loss either (a) logically virtually balances out cell gain in slow growing tumors; or (b) nonintuitively is associated with aggressive, rapidly growing tumors. Cell loss means that even rapidly growing tumors are actually substantially constrained in their volumetric growth rates, relative to the doubling time of the tumor cells themselves [3].
An example of a relatively ‘balanced’ tumor is basal cell carcinoma (BCC) . One of the most common human tumors, BCC is a slow growing, locally invasive tumor which takes many months to double in size. However, average cell doubling times in BCC are only around 9 days. The relatively slow net expansion of the tumor cell population is likely to be due to cell death providing a significant balancing effect. Notably, histologically overt evidence of apoptosis—pyknotic nuclei—is common in these tumors [4]. Similar balancing effects have been reported for micrometastases [5]. A logical consequence of aggressive tumors outgrowing local anabolic resources is that, in rapidly growing tumors, cell death becomes more prominent as the tumors get progressively bigger, tumor population growth slowing in parallel [6]. Such death has commonly been described as necrotic—as this is easier to discern histologically—but apoptosis (again reported by the hallmark pyknotic nuclei of apoptotic cells) is also a conspicuous feature of tumors that have outgrown nutrient and oxygen supplies [7, 8].
Like mitosis, apoptosis is prominent in a wide variety of established, aggressive tumor types, including non-Hodgkin’s lymphoma (NHL) [9], squamous cell carcinoma [10, 11], transitional cell carcinoma [12], hepatocellular carcinoma [13], and undoubtedly many others. The preferential association of constitutive apoptosis with aggressiveness of malignant disease is reflected in recent work indicating that cleaved caspase-3, the activated form of the apoptosis effector caspase-3, is a risk factor in gastric, ovarian, and cervical cancers [14]. The significance of a relatively high constitutive apoptosis index should not be underestimated since it not only represents a very high level of cell death but also links strongly to poor prognosis. Just as mitotic figures represent only a very brief visualization window on proliferating cells, so apoptosis in histological sections is only fleetingly evident because of the rapidity with which apoptotic cells disappear from microscopic view. Since they are efficiently phagocytosed and digested, apoptotic cells are only identifiable in tissues for around 1–3 hours [15]. Therefore, just as small numbers of mitotic figures are indicative of very rapid proliferation, so small numbers of apoptotic bodies are likely to indicate massive cell death in situ. In certain tumors such as “starry-sky” lymphomas (to be discussed in further detail later in this chapter), mitosis, apoptosis, and phagocytic clearance of apoptotic cells by macrophages can all be routinely observed in standard histological fields (Fig. 3.2). These lymphomas take their description from the infiltrating macrophages, which appear as bright “stars” in a darkly staining “sky” of tumor cells in standard histological sections.

Aggressive “starry-sky” non-Hodgkin’s B-cell lymphoma . Standard hematoxylin and eosin (a) and immunohistochemical preparations (b, c) of the classical starry-sky lymphoma, Burkitt’s lymphoma. (a) The starry-sky macrophages (M) are apparent as areas of brightness against a dark background of tumor cells. The macrophages contain apoptotic tumor cells in various states of degradation. Mitotic figures are also present (circled). (b) Macrophages labeled according to their expression of CD68. (c) Apoptotic cells revealed by in situ end labeling of cleaved DNA. Most are associated with the starry-sky macrophages
Refsum and Berdal stated almost half a century ago: “Tumour growth must be looked upon as dependent on the process(es) of cell proliferation and cell loss, where changes in any of (these processes) can result in profound alterations in the clinical rate of tumour growth. The cancer problem is not only why cancer cells proliferate uncontrolled, but also why so many of these cells die…” [16]. In the ensuing decades, much has been learned about modes of cell loss and their consequences. As we discuss in the following, although it has greater renown for its tumor-suppressive properties, cell loss by apoptosis also has sinister oncogenic attributes.
3.2 A Poisoned Chalice: Tumor-Promoting as well as Tumor-Suppressing Roles of Apoptosis
It has been appreciated for well over a century since Paget’s original ‘seed and soil’ suggestion in 1889 [17] that the tumor microenvironment plays critical roles in the aggressiveness of malignant disease. Tumors are rogue tissues, often with features resembling chronic wounds, described by Dvorak as “wounds that do not heal” [18]. It is beyond doubt that stromal cellular elements, the nontransformed cells of tumors, including fibroblasts, endothelial cells, fat cells, and inflammatory cells, participate in a two-way signaling conversation with the transformed cells in order to produce a deregulated, aggressive, invasive tissue. Less well appreciated are the roles tumor cell death can play in conditioning the tumor microenvironment. Apoptosis appears to act as a bipolar modulator of tumor growth and progression: on the one hand, it is well established that apoptosis functions to prevent oncogenesis, while on the other, as we detail as follows, apoptosis has tumor-promoting properties. But how can apoptosis function both as tumor suppressor and tumor promoter? A comparison between the role of apoptosis in single cells versus its roles in cell populations helps to unify this apparent paradox (Fig. 3.1).
In a single cell, the cell-autonomous consequence of apoptosis is, of course, that the cell is deleted and removed from the population. This cell fate decision is based upon external signals, which interact with the internal context of the cell. This means that apparently identical external signals (for example, a TNF-family member or hypoxic stress) can have different consequences depending on the internal composition of the cell as well as its microenvironment [19]. In the early stages of oncogenesis, the decision to undergo apoptosis is likely to be an important mechanism for removal of individual premalignant cells that have acquired potentially dangerous genetic mutations. However, apoptosis—even of a single cell—cannot be regarded as an isolated event because there are consequences of such an event for the tissue in which that cell resides: for example, phagocytosis and other responses in neighboring cells (vide infra). Furthermore, genetic material from apoptotic tumor cells can be acquired by phagocytes [20–23]. Perhaps the most efficient way of getting rid of a potentially dangerous cell is by jettisoning it to the exterior environment as is seen in the sloughing off of apoptotic, terminally differentiated cells in epithelia. Even here, however, cells in lower layers have potential to respond to their dying upper neighbors. This is illustrated by the retrieval in some mammalian species of dying, epithelial cells by phagocytes, for example, in the guinea pig, dying apical enterocytes are engulfed by responsive macrophages from the lamina propria [24]. Therefore, an individual dying cell can impact profoundly upon its near and distant neighbors.
In a population of tumor cells, while the core apoptosis program is cell autonomous (whether it is triggered through intrinsic or extrinsic mechanisms), and the consequence for individual cells is loss of those cells, the net effect for the tumor cell population as a whole is (a) that it contracts; (b) that it continues to expand, though at a lower rate than if all cells survived; or paradoxically (c), that it grows with the aid of mechanisms triggered by the cell death process itself (Fig. 3.1). For example, as discussed elsewhere in this book (Chap. 4) and later in this chapter, cell death can engender compensatory proliferation, accumulation, and protumor activation of stromal elements and angiogenesis, as well as suppression of antitumor immune responses. In this way, the very act of cell deletion has real and sinister potential to activate a multitude of pathways that support cell gain in outpacing cell loss [25–28].
3.3 Cell Death Modalities Influencing the Tumor Microenvironment: Apoptosis and Other Mechanisms
Multiple cell death routes including apoptosis, necrosis, and autophagy-associated cell death are prominent in malignant disease, even in individual tumors. Indeed, the available evidence suggests that multiple forms of cell death are likely to occur in individual tumors, either constitutively via stress responses, or as a consequence of therapy. As well as apoptosis, necrosis, and autophagy-associated cell death, additional modalities such as anoikis, mitotic catastrophe, entosis, necroptosis, and others are all likely to contribute to the tumor microenvironment to a greater or lesser extent in concert with other cell fate processes including differentiation, proliferation, and senescence.
Caspase-dependent apoptosis is the most widely studied form of programmed cell death in cancer and is our main focus here. The seminal definition of apoptosis by Kerr, Wyllie, and Currie in their classic work was based on morphological features of dying cells in multiple contexts, not least malignant diseases [29]. These authors and others noted the peculiar hallmarks of dying tumor cells: the cell shrinkage, chromatin condensation, and the formation of vesicles and apoptotic bodies containing organelles apparently free from degradation in many different tumor types. Later, biochemical features of apoptosis subdivided this form of programmed cell death into caspase-dependent and caspase-independent categories (see [30]). The most renowned microenvironmental effects of apoptosis are anti-inflammatory and tolerogenic phagocytic clearance responses, which may be hijacked in tumors to provide them with immunological escape mechanisms [31]. However, certain cell death stimuli, notably anthracycline anticancer drugs, induce immunogenic apoptosis [32]. Currently, the microenvironmental effects of apoptosis and other forms of cell death in cancer are largely underestimated and underinvestigated.
3.3.1 Triggering of Apoptosis in Growing Tumors
A generic principle at the root of cell death in malignant disease is environmental stress: the nascent tumor must be located in and/or acquire the appropriate generative niche in which to evolve and grow and it is important that, at the population level, the niche fosters the imbalance of cell birth over cell death that is required for successful tumor growth. In fact it seems likely that many tumors or tumor cell clones fail to achieve persistent net growth and never become clinically problematic. Alternatively, death of such clones may ‘feed’ potentially more aggressive neoplastic or preneoplastic cells. Once established, an aggressively growing tumor rapidly outgrows its environment and it is logical that metabolic cellular stress stimuli—limitations in essential nutrient, growth factor, and oxygen supplies—feature prominently in triggering multiple cell death pathways. Cell death induced by therapy adds an additional dimension as does death due to the relatively inefficient seeding of malignant clones to metastatic sites [33] (Fig. 3.3).

Triggers for tumor cell apoptosis of the tumor microenvironment. Illustrative sources of proapoptotic stimuli in tumors. In addition to apoptosis induced by therapy or antitumor immunity, limited amounts of nutrients and oxygen as a consequence of insufficient angiogenesis are likely to be important in generating proapoptotic signals. Clonal ‘hallmark-deficiency’ refers to clones of tumor cells carrying genetic mutations but fail to acquire hallmark characteristics of cancer cells (for example, as defined by Hanahan and Weinberg [2]) necessary for survival in primary or alternatively metastatic tissue locations. The ‘Onco-regenerative niche’ refers to a putative collection of conserved tissue repair and regeneration mechanisms driven by cell damage and hijacked in cancer (see text for details). AiA: propagation of apoptosis triggering through the process of ‘apoptosis-induced apoptosis’ [34]
Thus, multiple factors are likely to contribute to the cell fate decision-making processes that culminate in apoptosis, as well as other forms of cell death, in tumors [19, 35, 36]. Apoptotic signaling pathways may be initiated at multiple cellular locations including the plasma membrane (e.g., death receptor signaling; absence of growth factors), nucleus (irreparable DNA damage response), endoplasmic reticulum (e.g., unfolded protein response), and mitochondria. The latter organelles are central to the intrinsic apoptosis program, notably its initiation and also contribute to execution and amplification of extrinsic apoptosis pathways. Conditions of oxygen stress, glucose, and amino acid deprivation caused by rapid tumor growth are therefore likely to have fundamental effects on tumor cell populations by eliciting proapoptotic signals at mitochondria and the endoplasmic reticulum [35, 36]. In order to maintain the cell birth/cell death imbalance in favor of tumor growth, it follows that the tumor microenvironment needs to adapt in a dynamic way. As we will discuss, responses to apoptosis play key roles in such adaptation.
3.3.2 Necrosis , Autophagy , and Other Mechanisms
In addition to our main focus on apoptosis, brief discussion of other forms of cell death is also warranted here since multiple death processes can contribute to the tumor microenvironment (see [37] for a recent review of accidental versus regulated forms of cell death). Importantly, different forms of cell death may condition the tumor microenvironment in contrasting ways such as anti-inflammatory versus proinflammatory, tolerogenic versus immunogenic, death promoting versus death inhibiting. The ‘balance’ of these effects has not yet been studied in detail but will undoubtedly impinge significantly on tumor evolution and patient outcome and the underlying mechanisms have potential to provide information on novel therapeutic targeting.
Among cell death processes, the classical antithesis of apoptosis is necrosis. Because of the relative ease by which necrotic death can be visualized histologically, this passive, catastrophic form of cell death was appreciated long before apoptosis or other forms of programmed cell death because it affects contiguous tracts of cells, rendering the process microscopically obvious [38]. In rapidly growing tumors, necrotic lesions are prominent due to the growing tumor population outpacing effective nutrient supplies and gaseous exchange. Such a stressful scenario also elicits other forms of cell death (vide infra). In stark contrast to apoptosis, necrosis is typically portrayed as proinflammatory and immunogenic (although this generalized principle has been challenged, see [39]) and it is by no means clear how necrosis contributes to tumor cell biology. Given the generality of necrosis in aggressive tumors and its association with poor prognosis, it seems likely that it fails to impart a negative influence beyond that of the cells immediately affected. The same may prove to be true of regulated necrosis (necroptosis), which has the same morphological features as necrosis [40].
Based on its origin from cell damage, it has been suggested that the main role of necrosis is to induce tissue repair responses [41] and indeed it has been proposed as a critical inducer of tumorigenesis [42]. Therefore in the context of tumor biology, necrosis, like apoptosis, may also prove to feed into the cell birth/cell death equation in complex ways. Furthermore, necrosis can effectively suppress antitumor T cell immunity [43]. In many, perhaps all, cases, necrotic lesions in tumors are accompanied or presaged by apoptotic or other death modalities. Furthermore, the microenvironmental effects of necrosis may, in principle, be dominantly subverted by the effects of alternative modes of cell death or vice versa. Although the relative dominance of cell death responses in various tissue damage or disease scenarios has not been studied in detail, it is of interest to note that, in terms of activating macrophage migratory responses, at least in the case of the Drosophila hemocyte, apoptosis dominates over acute wounding (laser-induced cell ablation) and developmental growth factor signals [44].
Autophagic cell death is also likely to play important roles in tumor microenvironmental conditioning alongside apoptosis and other forms of cell death [45]. Although autophagy is a cell survival mechanism that is triggered under conditions of stress, including the challenging tissue environment in cancer, its deregulation or failure can lead to cell death. In cancer its divergent roles are context dependent [46]. A most interesting case in point comes from recent metabolic studies in prostate cancer in which it was found that arginine starvation (by arginine deiminase) caused autophagic death of prostate cancer cells deficient in arginosuccinate synthetase. The cell death was characterized by mitochondrial depolarization and breakdown of chromatin in large autophagosomes [47]. Autophagy has been reported to limit inflammation associated with necrosis [48] and downregulate proinflammatory cytokine responses at the level of transcription [49]. In this anti-inflammatory context, autophagy broadly resembles apoptosis in its microenvironmental effects. Although its role in suppressing antitumor immunity (as is the case with apoptosis) has yet to be defined in detail, a recent study suggests that autophagic degradation of nuclear components, notably lamin B1 has tumor-suppressive properties through its capacity to induce oncogene-mediated senescence [50].
3.4 Biological Activities of Apoptotic Tumor Cells
The anti-inflammatory and tolerogenic effects of apoptosis are well established and have been reviewed extensively elsewhere (see Chaps. 2 and 7). By contrast, although it has been known for well over half a century that dying cells can promote tumor growth, this property has been underappreciated. Early studies showed that tumor cells that were destined to die (lethally irradiated or histoincompatible cells) exhibited substantial tumor growth-promoting activities when admixed with small numbers of compatible viable tumor cells in murine transplantable carcinoma, sarcoma, and lymphoma models [51–53]. Host systemic effects were ruled out by the key observation that the admixed populations must be present at the same anatomical site in order to achieve enhanced tumor growth [52]. Initial clarification of underlying mechanisms was provided by the results of investigations of ascites tumors transplanted in diffusion chambers, which separated the host microenvironment from the growing tumor cells (together with admixed damaged cells) by a 0.45 μm membrane. Critically, these studies showed that direct, contact-mediated effects of host cells in the immediate tumor microenvironment were not required in order for the growth-promoting effects of lethally irradiated tumor cells to be revealed. Thus, Ehrlich ascites and L1210 lymphoma cell growth were increased by orders of magnitude in the presence of lethally irradiated cells which would be presumed to have been committed to undergo apoptosis. Heat-killed, necrotic, cells induced only a small, statistically insignificant growth-promoting effect in comparison [54]. Although contributions of soluble factors or extracellular vesicles from host inflammatory or other cells in these models cannot be excluded, these studies suggested a ‘feeder’ principle in the tumor microenvironment emanating from dying or dead cells. In this respect, the dying/dead cells were thought of as a source of nutrients in much the same way as irradiated cells could be used as a source of feeder cells to clone tumor lines like HeLa [55].
These early studies serve to provide a firm grounding for the establishment of the principle that cell death can impart potentially significant oncogenic effects in the microenvironment of tumors. In our opinion, it seems likely, however, that there will prove to be exceptions to this principle and/or alternative, context-dependent effects, which are growth inhibitory and therefore tumor suppressive. In addition, it seems logical that dying cells will prove to have different properties from dead cells. One possibility is that, since dying cells are cleared so efficiently from tissues under normal circumstances (free dead cells never being observed in normal physiology), dead cells engender unwanted effects such as proinflammatory properties. It is noteworthy in the context of cancer that, at least in vitro in the case of hybridoma cells, cell death constrains growth and antibody productivity in the surviving cells [56]. These observations suggest that the positive and negative effects of cell death driven by multiple factors, including type of death, type of dying cell, phase of death, and type of responding cell are likely to be integrated in the tumor microenvironment to determine overall biological outcome.
3.4.1 Mechanisms
Accumulating evidence indicates that the biological activities of apoptotic cells, including effects of relevance to the tumor microenvironment, are mediated through three mechanisms: (1) intercellular contact, (2) production of extracellular vesicles, and (3) release of soluble factors.
Although the specific molecular details are far from clear (see [15, 39] for reviews), the recognition and binding mechanisms that are a prerequisite for the clearance of apoptotic cells through phagocytosis, together with the production of secreted factors directly by apoptotic cells, are associated with additional, nonphagocytic responses of interacting cells (including both phagocytes and nonphagocytes). These range from anti-inflammatory mediators and migratory responses to cell fate decisions such as cell survival, growth, differentiation, and death. Current knowledge is summarized in Table 3.1 and Fig. 3.4.

Responses to apoptosis in the tumor microenvironment . Through direct contact or via release of extracellular vesicles and soluble factors, apoptotic cells communicate with a range of neighboring cells to elicit a multitude of biological responses that have implications for the growth and spread of malignant disease. Responding cells include phagocytes, other cells of the immune system, stromal cells and tumor cells themselves, including cancer stem cells. AiA apoptosis-induced apoptosis
The exposure of phosphatidylserine (PS) is the most renowned plasma membrane moiety in the cell–cell interactions that contribute to these processes. Other alterations in the plasma membranes of apoptotic cells, involving additional lipids as well as carbohydrates, proteins, and nucleic acids, undoubtedly play key roles too. For example, apoptotic cells display increased levels of heat shock proteins, HSP25, HSP60, HSP70, and HSP90 at their surfaces [68] and translocation of HSP60 to the surface of apoptotic cells can promote phagocytosis [71]. Additional plasma membrane changes involve loss of ‘don’t-eat-me’ signals such as the plasma membrane glycoproteins CD31 [63] and CD47 [64], along with exposure of the ‘eat-me’ signal PS. These changes lead to the engulfment of apoptotic cells and are linked to the production of anti-inflammatory mediators by phagocytes. As yet, it is unclear to what extent these changes are linked to other, nonphagocytic, responses to apoptotic cells, either in normal or malignant contexts. It is notable that loss of functional CD47 leads to phagocytosis of cells regardless of their commitment to apoptosis and anti-CD47 antibodies are providing promising biological therapeutics for non-Hodgkin’s lymphoma and acute lymphoblastic leukemia [101, 102].
Although the extracellular vesicles (EVs) produced during apoptosis are often simply referred to as ‘apoptotic bodies,’ the latter term is not well defined and it seems likely that cell stress and apoptosis lead to active release of various categories of EVs ranging in size from small, exosome-like vesicles (30–100 nm in diameter) through microvesicles (~100–1000 nm) to large subcellular vesicles (>1000 nm, perhaps most generally described by the term ‘apoptotic bodies’). Given the well-described capacity for extracellular vesicles to carry or display at their membrane surfaces multiple categories of biological molecules, including transmembrane protein receptors, translational and enzyme systems, DNA, and micro-RNAs [103, 104], it seems probable that EVs produced actively during cell stress and cell death will prove to have multiple biological functions, especially since EVs released in association with apoptosis harbor hundreds of proteins [97, 100]. It is notable and relevant to the concept of apoptosis leading to the production of functional vesicles that EVs can mediate the transfer of biologically active molecules between cells. Furthermore, as a consequence of exposure of PS, which appears to be a general characteristic of EVs , these particles may elicit responses in nonapoptotic cells similar to the effects of apoptotic cells themselves. In this sense, apoptotic cell-derived EVs may act as complex intercellular signaling structures that inform the microenvironment of the apoptotic cell, fostering specific responses. For example, at early stages of activation, neutrophils release EVs which, like apoptotic cells, can inhibit inflammatory responses of macrophages and dendritic cells via the Mer receptor tyrosine kinase [105]. Furthermore, it is known that apoptotic cells can transfer biologically active genetic material, notably oncogenes, into phagocytes [21] and it seems likely that stressed or apoptotic cell-derived EVs have similar properties, especially since some are known to carry cargoes of DNA and/or micro-RNAs. The consequences of tumor cell-derived EVs for cancer pathogenesis and their potential as targets in following disease progression and therapeutic responses are well rehearsed [106]. However, the biological roles of apoptotic cell-derived EVs are ill defined, not least in cancer. To date, our knowledge of the function of apoptotic tumor-cell derived EVs is limited to their ability to activate chemotactic responses in mononuclear phagocytes via the chemokine CX3CL1 (fractalkine) [65] and also by an ICAM-3-dependent mechanism [87]. They may also be involved in stimulating proliferation of rat insulinoma cells [107] and in expressing and processing certain matrix metalloproteinases [27]. It is important to note, however, that in none of these studies have apoptotic EVs been specifically discriminated from EVs produced as a consequence of other processes such as preapoptotic stress. We suggest that future work will uncover numerous additional cargoes and functions of apoptotic tumor cell-derived EVs of fundamental importance to human cancer pathogenesis, diagnosis, prognosis, and therapy. Future work will also uncover the mechanisms through which apoptotic EVs exert their effects, such as by receptor-mediated endocytic or phagocytic events, via membrane fusion or by secretion or diffusion of soluble factors from the vesicles. In the latter context, it has been shown that EVs produced prior to loss of membrane permeability of the originator apoptotic cell body have highly permeable membranes, permitting release of large macromolecules [97].
The release of biologically active soluble factors from apoptotic cells is well established (Table 3.1), especially in the context of inflammation control with factors such as IL-10, transforming growth factor-β1 (TGF-β1), α-defensins and lactoferrin being produced by various types of apoptotic cell [72–74, 76]. Chemotactic ‘find-me’ signals including lysophosphatidylcholine (LPC), CX3CL1, nucleotides, and sphingosine-1-phosphate (S1P) are released by apoptotic cells in order to attract mononuclear phagocytes to sites of apoptosis [39, 108, 109]. Most, if not all, of these factors have pleiotropic properties, endowing apoptotic cells with the propensity to foster multiple responses of relevance to tumor establishment and growth, not least survival and proliferation of viable neighboring tumor cells as well as pro-oncogenic activation of stromal cellular elements of the tumor microenvironment. There seems little doubt that the capacity for release of biologically active moieties by apoptotic cells is currently underestimated. We return to this subject later when we consider compensatory proliferation and mitogen production such as prostaglandin E2 (PGE2) [110] in response to apoptosis.
Finally, in this section, it is worth mentioning an intriguing function of apoptotic cells which appears to be the sequestration of chemokines. Specifically, apoptotic leukocytes can express increased functional CCR5 leading to sequestration of CCL3, CCL4, and CCL5 and this can be modulated positively by proresolution mediators such as lipoxin A4, resolvin E1, and protectin D1, and negatively by proinflammatory mediators such as TNF-α [111]. On this basis it might be speculated that apoptotic cells in the tumor microenvironment could regulate the activity of chemokines through modulation of chemokine receptor expression, which in turn may be controlled on the apoptotic cells by the cytokine milieu of the tumor.
3.4.2 Activities of Stressed Cells Versus Dying Cells
The wide-ranging changes in phenotypes and activities of apoptotic tumor cells together with the responses they consequently elicit demonstrate that apoptosis is far from the silent process it was initially thought to be. It is important, however, to consider the extent to which the microenvironmental conditioning by dying cells in tumors originates from biological mediators produced (a) as a consequence of preapoptotic stress responses or alternatively (b) specifically as a result of activation of the apoptotic program and the cell moving past the ‘point of no return’ in this pathway, widely regarded as mitochondrial outer membrane permeabilization (MOMP). Prototypic examples of stress-response mediators, heat-shock proteins, notably HSP27 and HSP70, are known to inhibit apoptosis and may modulate oncogenesis. For example, HSP70 can inhibit apoptosis signaling through interference with apoptosis protease-activating factor-1 (Apaf-1) and apoptosis-inducing factor (AIF) [112]. Furthermore, modulation by stress signals can induce a state of antitumor immunogenicity in previously tolerogenic apoptotic tumor cells [70]. Importantly, it should be appreciated that cellular stress responses are restorative, being aimed at cell survival rather than cell death [113]. Studies aimed at dissecting the molecular mechanisms responsible for the biological effects of apoptotic cells must carefully uncouple the underlying principles from those resulting from preapoptotic stress.
The associations between cell stress and cell death open up possibilities that common principles underlie the capabilities of stressed and dying cells to influence the pro-oncogenic tumor microenvironment. A tantalizing example here is cellular senescence, an overtly tumor-suppressive mechanism, which appears to be a state of chronic stress, designed to lock cells out of cycle and thereby prevent propagation of premalignant, damaged cells [114]. Potential parallels may exist between apoptotic tumor cells and senescent tumor cells participating in paracrine pro-oncogenic signaling events.
3.5 Responses to Cell Death in the Tumor Microenvironment
The most renowned responses to apoptotic cells emanate from the phagocytes that engulf them, the molecular mechanisms underlying phagocytosis, and anti-inflammatory signaling being the most widely studied, especially in macrophages (reviewed in Chaps. 2 and 9). It has been known for many years that apoptotic cells not only elicit engulfment responses and the production of anti-inflammatory mediators by phagocytes but also angiogenic and growth factors such as vascular endothelial growth factor (VEGF) and hepatocyte growth factor (HGF), respectively. We have previously proposed the ‘3Rs’ of apoptotic cell clearance as recognition, response, and removal [15, 39] and, just as unconventional phagocytes such as epithelial cells, endothelial cells, and fibroblasts respond to their apoptotic cellular neighbors by engulfing them, it follows that a wide variety of different cell types are able to respond in other ways to apoptotic cells in their vicinity through direct intercellular contact and via soluble factors or EVs produced by the dying cells. Apart from phagocytic and anti-inflammatory responses, relatively little is known either about the variety of cells that can respond to their apoptotic neighbors or about the range of such responses (cf. [115–118]). Available evidence, however, indicates that multiple cell types can respond to apoptotic cells and that the nature of such responses can vary widely. While such responses are undoubtedly fundamental to normal physiological systems and normal tissue homeostasis, here, we consider these responses in the context of the microenvironment of malignant tissue.
The macrophage represents a focal point of cell death and cancer. Macrophages are almost invariably found in either normal or diseased tissues at sites of apoptosis and can accumulate in tumors in large numbers: for example, up to 50 % of breast cancer tissue can be comprised of macrophages [119]. The functional plasticity of macrophages makes them highly responsive to their microenvironments. Tumor-associated macrophages (TAMs) have the potential to exert innate antitumor immunity as appears to be the case in colorectal and gastric cancer. This stems from their adopting a proinflammatory phenotype in the tumor microenvironment that inhibits tumor cell proliferation and promotes T cell-mediated antitumor immunity [120]. In most cancers including breast cancer, lymphoma, cervical cancer, squamous carcinomas, and melanoma, however, TAMs are associated with poor prognosis [121]. Information supporting the view that tumor cell apoptosis is capable of driving protumor properties of TAMs has begun to emerge. Thus, in postpartum breast cancer, the phagocytic and anti-inflammatory receptor tyrosine kinase Mer that interacts with apoptotic cells via its ligands Gas-6 and Protein S (which bind to PS exposed on apoptotic cells) promotes cancer progression as a consequence of tumor cell apoptosis [122]. Furthermore, in prostate cancer, the engulfment of apoptotic tumor cells by macrophages induces the upregulation of milk fat globule epidermal growth factor (MFG-E8)—a PS-binding ligand that bridges apoptotic cells with αvβ3 and αvβ5 integrins on phagocytes—and enhancement of signal transducer and activator of transcription 3 (STAT3)-dependent transcription with resultant pro-oncogenic/anti-inflammatory polarization (“M2-like”) of the macrophages [123]. (With respect to macrophage polarization, it is worth noting that ‘canonical’ phenotypes centered around M1 and M2 are unlikely to represent the true picture of the range of activation states of macrophages either in cancer or in tissue repair [27, 124, 125]; here, for convenience, we will use the terms M1-like and M2-like to indicate positioning of macrophages toward a particular activation state.) The results of these studies of prostate and breast cancer demonstrate that multiple receptors of macrophages are involved in apoptotic cell clearance and in anti-inflammatory responsiveness to apoptosis. It will be important in future studies to determine the relative importance of different phagocytic and anti-inflammatory receptors of macrophages in a wide range of cancers in which apoptosis is significant. It is tempting to speculate, for example, that Liver X receptor (LXR) is important in this context especially since LXR signaling has been shown to play key roles in apoptotic cell clearance by mouse macrophages and its associated anti-inflammatory responses and immunological tolerance [126].
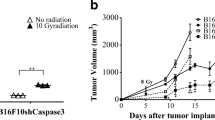
We have recently carried out detailed studies of the role of apoptosis in the pathogenesis of aggressive starry-sky B-cell non-Hodgkin’s lymphoma (NHL), prototypically Burkitt’s lymphoma (BL) [27]. In these tumors frequent apoptosis can readily be observed histologically and the majority of the apoptotic tumor cells are found in association with TAMs, creating the distinctive starry-sky morphology (Fig. 3.2). Inhibition of apoptosis in a murine xenograft model of BL led to impaired tumor growth and substantially reduced tumor cell proliferation. Notably, when tumor cell apoptosis was suppressed, the hypoxic microenvironment of the tumor was enhanced due to impaired blood vessel formation, suggesting a role for tumor cell apoptosis in driving not only proliferation but also angiogenesis. In both patient samples and mouse models of Burkitt’s lymphoma, a strong correlation between tumor cell apoptosis and macrophage numbers was observed indicating that death of tumor cells induces TAM accumulation. A proportion of these TAMs also express proliferative markers suggesting that apoptotic cell conditioning may stimulate the proliferation of starry-sky TAMs, helping to populate the growing tumor with macrophages. In situ gene expression profiling of starry-sky TAMs revealed a protumorigenic activation state characterized by transcriptional activation of anti-inflammatory, proangiogenic, and tissue remodeling pathways. For example, starry-sky TAMs were found to preferentially express high levels of TGF-β1, growth factors including IGF-1 and PDGF-CC, and several genes associated with angiogenesis and metastasis including MMP2, MMP3, MMP12, TIMP2, ANPEP, LGALS3, HMOX1, and GPNMB. In addition, CD91, Mer, Axl, and Gas6 were found among apoptotic cell interaction molecules that were upregulated in starry-sky TAMs providing further evidence for the involvement of the Tyro-Axl-Mer receptor tyrosine kinase axis in TAM activation and extending the variety of phagocytic receptors of apoptotic cells of potential importance for tumor pathogenesis. Apoptotic tumor cells thus direct the activities of TAMs toward the acquisition of multiple and diverse protumorigenic properties that support the growth of tumor cells and subsequent oncogenic progression.
The accumulating evidence therefore indicates that tumor cell apoptosis can elicit specific tumor-promoting responses in breast cancer, prostate cancer, and NHL. A similar argument may also be made for melanoma [27] and recent results also indicate that apoptosis can drive hepatocarcinogenesis [127]. The current state of knowledge suggests that apoptosis of tumor cells may represent a generic, pro-oncogenic stimulus which acts through multiple molecular cell biological mechanisms affecting antitumor immunity, tumor population growth, and angiogenesis. While responses have thus far been focused mainly on the role of macrophages, additional nonmacrophage mechanisms are likely to prove to be important for tumor progression, especially since apoptotic cells are known to have direct effects on angiogenesis [94] and also have the potential to produce immunomodulatory factors, chemokines, cytokines, and mitogens directly (Table 3.1).
The mitogenic capacity of apoptosis is well illustrated by the phenomenon of compensatory proliferation of neighboring cells, first described in Drosophila. Developmental and wounding studies in the fly indicated that the pathways underlying the proliferative responses to apoptosis involve p53, WNT, TGF-β/BMP, JNK, and Hedgehog (Hh) signaling (see [128] for recent review). The pathways are context related and may be dependent or independent of executioner caspase activation. Compensatory proliferation in response to injury has also been described during regenerative responses in other species, including Hydra, Planaria, newts, frogs, zebrafish, and mice. In the latter, PGE2, a key inflammatory and regulatory niche player, is produced to drive compensatory proliferation as a result of caspase-3-dependent activation of calcium-independent phospholipase A2 (iPLA2) [110]. It is conceivable that PGE2 could mediate its regenerative power in this context through activation of β-catenin following binding to the G protein-coupled receptor EP2. Additional plausible molecular players in compensatory proliferation that may be produced downstream of caspase activation and apoptosis include Sdf1, FGF20, and IL11 [128].
Apoptosis-driven compensatory proliferation of hematopoietic stem/progenitor cells (HSCs) has been implicated in thymic lymphoma development in mice subjected to low-dose irradiation. This pathway requires p53, which activates leukocyte apoptosis via the BH3-only proapoptosis protein, PUMA [129, 130]. Thus, PUMA−/− HSCs are protected from cell death but show reduced compensatory proliferation and stress-associated DNA damage with consequent inhibition of thymic lymphoma formation in response to gamma-irradiation. In this way it seems that p53, acting via its apoptosis-promoting properties, appears to promote tumorigenesis induced by DNA damage, an activity of p53 which contradicts its widely accepted antitumor function in securing the demise and deletion of cells carrying potentially oncogenic mutations. A possible alternative explanation in this scenario is that, although PUMA is clearly a mediator of p53-induced cell death in response to radiation, it may also have additional specific roles in thymic lymphomagenesis, especially since PUMA−/− mice still produced many different kinds of tumors (high- and low-grade lymphomas, sarcomas, and carcinomas) in response to irradiation [130].
Recent evidence indicates that, in stark contrast to compensatory proliferation, apoptotic cells can also induce further apoptosis in neighboring cells, thereby propagating the apoptosis signal from a single cell to a population. Thus, genetic studies in Drosophila wing disc development show that apoptosis in one compartment in the tissue can induce apoptosis in a neighboring compartment. The authors appropriately termed this effect “apoptosis-induced-apoptosis (AiA)” and identified the Drosophila TNF homolog Eiger as a key factor, triggered by JNK signaling, in mediating this response [34]. They also demonstrated a similar, TNF-α-mediated orchestration of hair follicle cell death in mice. These observations have obvious implications for the roles of apoptosis both in constraining cancer emergence and in delivering effective therapy.
Although it is well accepted that apoptosis represents a significant control mechanism in the pathogenesis of malignant disease through inhibitory mechanisms that limit cancer outgrowth, it is clear that responses to the process are likely to play important roles in modulating tumor growth and progression in positive as well as negative ways. In considering these effects of apoptotic cells, it is essential also to take into account the nonapoptotic roles of the caspases that execute the apoptosis program [131, 132] as well as features of apoptotic cells such as PS exposure that may have tumor modulatory effects independently of apoptosis. Examples here include the caspase-3-mediated activation of iPLA2-induced activation of migration of ovarian cancer cells [133] and of PS exposure on intratumoral endothelial cells [134] that occur in the absence of apoptosis. As previously proposed, we support the argument that responses to such effects may occur in parallel in the tumor microenvironment whether they are “apoptosis-like” effects or whether they are bona fide effects of the apoptosis program produced by dying cells.
3.6 Apoptosis and Angiogenesis : The Effects of Hypoxia
The microenvironments of malignant tumors are heterogeneous with respect to oxygen tension and hypoxia is a well-known stress factor in tumors. Typically, lethally low partial pressures of oxygen (including anoxia) in rapidly growing cancers lead to necrotic zones of dead tumor cells with cells at the borders of these ischemic regions undergoing hypoxia-induced apoptosis. In tumors in which apoptosis is constitutively prominent such as starry-sky NHL, it seems that the hypoxia caused by the rapidly proliferating tumor cells creates a dynamic cycle of malignant tissue expansion in which the hypoxia induces apoptosis that stimulates angiogenesis, and that this in turn permits further population growth with further hypoxia-induced apoptosis as the tumor cells outgrow their oxygen supply [27]. In this cyclical cause-and-effect feed-forward system, apoptosis of tumor cells may exert a key oncogenic response in endothelial cells which results in neovascularization.
In order for such a system to work, there also needs to be adaptation to hypoxia. Both transformed and nontransformed cells adapt to the various levels of hypoxia found in progressing tumors via the induction of hypoxia-inducible factors (HIF) and NF-κB [135]. These transcription factors are both activated in response to hypoxia following the inactivation of prolyl hydroxylases. HIF proteins function as a heterodimer of alpha and beta subunits but under normoxic conditions the alpha subunits are constitutively marked for degradation as a result of hydroxylation by prolyl hydroxylases; inhibition of these enzymes under hypoxic conditions enables the alpha subunits to translocate to the nucleus, dimerize with beta subunits, and regulate transcription [136]. Similarly, NF-κB is activated under hypoxic conditions via the inhibition of prolyl hydroxylases, leading to a suppression of hydroxylation of IκB kinase-β, which can then phosphorylate the NF-κB inhibitor, IκBα, leading to its degradation and a cessation of NF-κB inhibition [137]. As a consequence of HIF and NF-κB activation, hypoxia regulates a broad range of oncogenic cellular functions at the transcriptional level, including metabolism, proliferation, apoptosis, inflammation, angiogenesis, and tissue remodeling.
Available evidence indicates that, depending on cellular context (as well as oxygen tension level) hypoxia may either trigger or suppress apoptosis, for example, through differential effects of HIF on pro- or antiapoptotic Bcl-2-family protein expression. Furthermore, glycolytic enzymes can be induced by HIF 1α and, at least in certain tumor cell types, this is associated with suppression of apoptosis. It has been suggested that this may be important in the adaptation of tumor cells to aerobic glycolysis, otherwise known as the Warburg effect [138].
Hypoxic regions of tumors are frequently populated with TAMs that generally have protumorigenic properties. For example, in breast cancer, TAMs are most abundantly found in poorly vascularized regions of the tumor [119]. Indeed, hypoxia can regulate the expression of chemokines and their receptors including HIF1-regulated CXCL12/CXCR4 and CXCL8/CXCR8 to help navigate macrophages to regions of low oxygen tension [139, 140]. The exposure of TAMs to hypoxia also modulates their function. For example, in a BALB/c mammary adenocarcinoma model, tumors contained MHC IIhi and MHC IIlow TAMs; the MHC IIhi TAMs expressed higher levels of proinflammatory genes including NOS2, PTGS2, IL1b, IL6, and IL12b whereas the MHC IIlow TAMs expressed more anti-inflammatory genes such as ARG1, CD163, STAB1, and MRC1 [141]. Hypoxic regions of these tumors were predominantly populated with MHC IIlow TAMs, i.e., anti-inflammatory macrophages, which were poor at processing antigen and stimulating T cell-mediated responses. Hypoxic TAMs have also been reported to produce IL-10, which again contributes to the anti-inflammatory microenvironment, promoting evasion of antitumor T cell-based immunity [142]. In addition to promoting TAM enrichment and helping to reduce inflammation in the tumor, hypoxia-conditioned TAMs further promote tumor growth by stimulating neoangiogenesis. This is mediated by the HIF-driven upregulation and release of VEGF and also via the release of various cytokines including CXCL12, CCL2, CXCL8, CXCL1, CXCL13, and CCL5 [119, 142, 143]. Moreover, hypoxia-conditioned macrophages support metastasis by induction of the HIF1α-regulated gene, MIF, leading to enhanced release of MMP-9 [142].
The role of cell death in the hypoxia-associated accumulation and conditioning of TAMs is not yet understood, but given the colocalization of cell death and hypoxia, it seems probable that cell death will prove to be important in helping to drive cellular adaptation in the tumor microenvironment. Future studies investigating the links between (1) hypoxia, (2) regulation of tumor cell apoptosis, (3) TAM accumulation and activation, and (4) endothelial cell activation will substantially improve our knowledge of the fundamental molecular cell biology of cell death-driven oncogenic progression. As we discuss in the next section, we can draw close parallels between these associations and basic tissue repair mechanisms that may be driven by cell death in tumors.
3.7 Apoptosis and Tissue Repair Mechanisms in Cancer: The “Onco-Regenerative Niche”
Given the close parallels between wound healing mechanisms—i.e., normal responses to tissue damage—and inflammatory host responses in cancer, it seems reasonable to expect that cell damage, including stress responses leading to cell death, plays significant roles in both normal tissue repair and in cancer pathogenesis. Indeed as we have discussed earlier, while cancers have been described as “wounds that do not heal,” it may be more accurate to describe them as “wounds that do not stop repairing” (Savill, personal communication). Sites of injury—and consequently sites of repair responses—have been found to be favorable for tumor formation and metastasis including the gastrointestinal tract [144], lung [145], and liver [146]. Since tissue regenerative processes are closely linked to tissue repair, with organs such as the gut, skin, and liver having especially remarkable powers of turnover and regeneration, it seems reasonable to propose the concept of the “onco-regenerative niche” (ORN), a complex, interconnected confraternity of cells and extracellular factors that contribute to malignant disease via conserved, fundamental tissue repair mechanisms. The linkage between cell death and tissue regeneration is well illustrated in mouse models of liver damage and regeneration [147, 148] providing further rationale for consideration of cell death as a key driver mechanism in the ORN.
An important cellular player in the ORN is likely to be the macrophage, since TAMs have widely accepted pro-oncogenic functions—including playing a central role in a cancer stem cell niche [149]—and it has long been recognized that the mononuclear phagocytic arm of the inflammatory response plays critically important roles in wound healing [150, 151]. In the initial stages of the wound healing response, macrophages are believed to help remove damaged tissue and cells and release proinflammatory cytokines, chemokines, and factors such as VEGF that drive subsequent angiogenesis and granulation tissue formation. During the resolution phase, apoptosis is the principal mechanism through which healing wounds are depleted of inflammatory cells, macrophages mediating their safe disposal [150]. Just as phagocytosis of apoptotic tumor cells can alter the phenotype and function of TAMs, macrophages engulfing apoptotic cells in wounds are driven toward an anti-inflammatory phenotype with expression of CD206 and arginase I [150]. The clearance of apoptotic cells by macrophages thus changes the wound microenvironment from proinflammatory to anti-inflammatory thereby limiting excessive tissue damage.
The final stage of wound repair is tissue remodeling. This involves the induction of angiogenesis to restore a normal blood supply to the region and fibroblast-driven extracellular matrix deposition. In tumors, the extracellular matrix is constantly remodeling and new blood vessels must form to sustain additional growth [152, 153]. Myofibroblasts are highly contractible fibroblasts that aid tissue remodeling during wound healing but die by apoptosis when the extracellular matrix is able to support the wound. In tumors they are often found to persist, perhaps due to the ongoing matrix remodeling of tumors and the presence of tumor and stromal-derived growth factors such as TGF-β1, FGF, and PDGF. Myofibroblasts in tumors can be significant sources of growth-promoting and metastatic factors [153] but nothing is yet known of the implications of their apoptosis or their responses to apoptosis in tumors.
Although details of the activation status and effector functions of macrophages in wound repair are awaited, the available evidence suggests strong similarities between activation states that predominate early and later in wound repair and the macrophage polarization that characterizes early and established tumors. Thus, both in cancer and in wound healing, early macrophages may predominantly display proinflammatory phenotypes (“M1-like”) whereas at later times during wound healing and in established tumors, the dominant macrophage activation state is anti-inflammatory/reparatory (“M2-like”) [154, 155] (see also Chap. 9). Notably, studies of diabetic mice in which wound healing is impaired, as is commonly the case in human diabetic patients, indicated that effective clearance of apoptotic cells, including apoptotic neutrophils and endothelial cells, by wound macrophages was closely coupled to effective wound healing. In particular, in line with the aforementioned “M1-like” to “M2-like” transition, proinflammatory wound macrophages appear to be driven to an anti-inflammatory activation state by apoptotic cell clearance mechanisms [156]. Similarly, age-related impairment of wound healing has been suggested to be associated with defective clearance of apoptotic cells by macrophages [157]. These observations highlight the importance of macrophage-dependent clearance of apoptotic cells in the formation of an anti-inflammatory microenvironment, which under normal circumstances is necessary for healing and repair of wounds but in the more sinister setting of cancer, apoptosis may promote tumor cell survival, proliferation, tissue remodeling, neovascularization, and metastasis.
We suggest that cell death plays a driving role in the establishment of the ORN and that responding normal host cells, especially in the environs of cell demise (or as a result of recruitment incited by cell death), perform critical oncogenic functions as a consequence. In this way multiple cellular players in the ORN, such as—in addition to macrophages—endothelial cells and surviving or emerging clones of transformed cells, could interact in tissue repair modes in order to foster tumor establishment, stemness of cancer clones, metastatic spread, and post-therapy relapse. Of direct relevance to this argument is the wound-healing response to cell death via the Mer receptor tyrosine kinase that drives metastasis in postpartum breast cancer [122]. It will be important to establish the critical tissue repair molecules that are central to the ORN. Given their role in wound healing and liver regeneration, notably in the latter their production by apoptotic hepatocytes [147], we speculate that Hedgehog ligands produced by dying tumor cells could provide important molecular components of the ORN. As we discuss in the next section, PGE2 signaling may also be crucial, especially in the ORN that may characterize post-therapeutic disease relapse.
3.8 Opportunities for Improving Cancer Treatment
The pro-oncogenic, as well as anti-oncogenic, properties of tumor cell apoptosis suggest opportunities for novel anticancer therapies. Mainly, such opportunities remain undeveloped, both from the standpoint of, for example, either promoting AiA or inhibiting compensatory proliferation. As detailed elsewhere in this book, both chemotherapeutic and radiotherapeutic treatments induce various modes of cell death including apoptosis. The key requirement for successful therapy is sustained imbalance of the tumor cell birth/cell death equation in favor of cell death. Immunogenic cell death is one way in which this can be achieved [158]. Since TAMs play prominent roles in responding to apoptotic tumor cells in pro-oncogenic modes of accumulation and activation it follows that therapeutic strategies aimed at (1) reducing TAM accumulation, and (2) repolarizing TAMs to an antitumor phenotype to mediate the elimination of tumor cells could also be valuable. Both strategies have potential to limit protumor and anti-inflammatory signaling by TAMs, and to lead to enhanced antitumor immunity, resulting in decreased tumor growth and reduced metastasis.
Therapeutic strategies aimed at limiting the accumulation of TAMs in the tumor microenvironment have focused either on reducing recruitment of monocytes or suppressing macrophage survival in the tumor microenvironment. CCL2 and CSF-1 are abundantly expressed in many tumors and are believed to be among the most important factors contributing to macrophage infiltration [159]. Additionally, many tumors upregulate CSF-1 as a result of cytotoxic or ionizing radiation therapies, which can lead to further accumulation of TAMs [160, 161]. Preclinical murine models targeting CCL2, CSF-1, or their receptors (CCR2 and CSF-1R, respectively) either alone, or in combination with chemotherapy are showing potential at reducing TAM accumulation, and enhanced antitumor immunity leading to decreased tumor growth and reduced metastases [160–164]. A Phase 1B clinical trial using an anti-CCR2 antibody is currently underway combined with standard chemotherapy in pancreatic cancer patients (http://clinicaltrials.gov/show/NCT01413022). Recent work also highlights the importance of the CCL2/CCR2 axis in orchestrating macrophage-driven pulmonary metastasis in murine breast cancer. Notably, a key downstream signaling arm, CCL3/CCR1 was identified, which the authors suggest could prove to be a useful, low-toxicity therapeutic target for treating metastatic disease [165].
Several therapies have been shown to suppress macrophage survival in tumors. Trabectedin, a chemotherapeutic agent, induces apoptosis exclusively in mononuclear phagocytes via the extrinsic apoptotic pathway mediated by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptors, and can deplete macrophages in tumors, thereby contributing to inhibition of tumor growth and reduction in angiogenesis [166]. Liposomal bisphosphonates, including clodronate and zoledronic acid, are also capable of macrophage depletion and have shown to reduce tumor growth and tumor vascularization [167, 168], as well as suppression of bone and muscle metastasis [169] in murine cancer models. Additionally, immunotoxin-conjugated mAbs targeting TAM surface proteins, including for example scavenger receptor-A, CD52, and folate receptor β, have also shown potential at reducing TAM infiltrate and restricting tumor growth in murine models of ovarian cancer and glioblastoma [170–172].
Alternative therapeutic approaches have targeted the plasticity of macrophages and are directed at preventing TAM phenotypic changes that promote tumor growth and/or re-educate TAMs in favor of antitumor and proinflammatory signaling. These strategies are in line with findings that patients with high ratios of M1-like/M2-like TAMs more frequently experience a complete tumor resolution and higher overall survival than patients with low M1-like/M2-like ratios [173, 174]. Common strategies have sought to target TAMs directly to achieve repolarization. NF-κB activation can redirect TAMs to a tumoricidal M1-like phenotype [175] and several agents capable of activating NF-κB have been reported, including Toll-like receptor (TLR) agonists, anti-CD40 mAbs, and anti-IL-10R mAbs. CpG oligodeoxynucleotide (CpG-ODN), a TLR9 ligand can upregulate NF-κB activation in TAMs, leading to the production of IL-12 and TNF [176] and increased expression of MHC Class II, CD86, CD80, CD40, and IFN-γ, while decreasing expression of IL-4Rα, IL-4, and IL-10 [177]. Combining CpG-ODN treatment with anti-IL-10R mAb and monocyte chemoattractant CCL16 was shown to rapidly change the phenotype of infiltrating macrophages and induced the rejection of various preexisting tumors in murine models [176]. CpG-ODN-therapies currently in clinical trials show modest activity but may be improved by identifying molecular characteristics of subgroups of patients that could potentially benefit from this treatment [178–180]. CD40 activation can also reverse immune suppression of macrophages and drive antitumor T cell responses. Addition of anti-CD40 mAb to patients with chemotherapy-naïve, surgically incurable pancreatic ductal adenocarcinoma has been shown to induce an influx of CD40-activated, tumoricidal macrophages and promote the depletion of the tumor stroma [181, 182]. Blockade of the CSF1/CSF1R axis has also been shown to change the TAM phenotype by reducing expression of TAM genes such as MRC1, HMOX1, and ARG1, while upregulating IL1b and led to an increase in survival when given early in disease or increased tumor regression when given at advanced stages of a xenograft model of proneural glioblastoma multiforme [183].
Other strategies to reprogram macrophages that have shown success in preclinical cancer models are intratumoral delivery of IL-21 [184], and low-dose irradiation of macrophages [185], both of which were found to switch macrophage activation to an antitumor, proinflammatory phenotype, which was accompanied by increased infiltration of T cells. Furthermore, two microRNAs, miR-155 [186] and miR-125b [187], have been identified that are associated with proinflammatory activation of macrophages and should be further investigated as therapies for redirecting TAMs.
An approach to alter TAM phenotype of particular relevance to this chapter is the targeting of apoptotic cell-mediated activation of macrophages. Such therapies are generally targeted against PS expressed on the membrane of apoptotic cells. Masking of PS by a mutant form of MFG-E8 has been shown to inhibit phagocytosis of apoptotic cells by macrophages and could also inhibit the enhanced production of IL-10 [188]. Furthermore, addition of annexin V, which binds to PS on apoptotic cells, has been shown to target irradiated lymphoma cells to CD8+ dendritic cells for in vivo clearance, leading to release of proinflammatory cytokines and regression of tumors [189]. Murine and rat models of prostate cancer have also shown success when chemotherapy or irradiation was combined with an anti-PS antibody, 2aG4, a variant of the human antibody bavituximab, compared to either therapy alone [190, 191]. Gene expression analysis showed that TAMs cultured in the presence of 2aG4 increased iNOS, inflammatory cytokines IL-12 and TNFα, and T cell costimulatory molecules (CD80, CD86, and MHC class II), and decreased expression of arginase I, immunosuppressive cytokines IL-10 and TGF-β, and VEGF-A [191]. Bavituximab, a human anti-PS antibody, is currently being tested in a clinical trial [192]. Additionally, chemotherapies that induce immunogenic cell death can limit apoptotic cell activation of macrophages, and instead signal via dendritic cells leading to stimulation of T cells, which improves therapeutic responses [158].
The notion that switching macrophages from M2-like procancer to M1-like anticancer activation states is undoubtedly simplistic and seems likely to be tumor specific or even tumor region specific, depending on the functional subtleties of the activation status of the particular TAMs and their biological activities in modulating tumor growth and progression. A relevant salutary observation is that macrophages activated in vitro with the classical M1, proinflammatory stimuli, IFNγ and TNFα have been reported to promote tissue (skeletal muscle) regeneration [124]. Furthermore, the inflammatory drive of the tumor pathogenesis will be important in approaches that target the polarization state of the macrophage. For example, although the receptor tyrosine kinases (RTKs) Mer and Axl are widely held as having strong oncogenic properties [193], which has led to the development of inhibitors of these RTKs for treatment of solid tumors [194], the oncogenic role of the Tyro, Axl, Mer-RTK axis may be tumor and/or cell context specific. Thus, in inflammation-driven colitis-linked cancer, loss of Mer and Axl actually promotes the underlying oncogenic processes through failure to dampen inflammation [195]. Furthermore, suppression of apoptotic cell clearance via the Tyro, Axl, Mer-RTK axis could also act to promote oncogenesis in an inflammatory cancer context. By contrast, the Tyro, Axl, Mer-RTK axis may activate protumor mechanisms through suppression of antitumor immunity or through driving oncogenic properties in tumor-associated macrophages.
An additional TAM therapeutic approach is to enlist them to target tumor cells, most likely through using their ability to perform antibody-dependent cellular phagocytosis (ADCP) . Recent work by Montalvao et al. has suggested that the effectiveness of rituximab, an anti-CD20 therapy that is proven successful for treating B cell malignancies, is dependent on ADCP by Kupffer cells, the macrophages of the liver [196]. Similarly, antitumor mAb therapy in various murine carcinoma models resulted in rapid phagocytosis of tumor cells by Kupffer cells and inhibition of liver metastasis [197]. As we have already noted earlier, the antagonism of “don’t-eat-me” signals such as CD47 is a potentially highly effective response in cancer therapy that harnesses the power of the phagocytes to engulf and degrade otherwise viable tumor cells.
Cell death induced by anticancer therapeutics may activate host responses that promote relapse. Recent work illustrates this effect especially in murine models of breast cancer and melanoma in which postradiotherapy or postchemotherapy, tumor repopulation was shown to be driven by caspase-3-dependent PGE2 production generated by therapy-induced tumor cell apoptosis [25, 198]. Human studies of several cancer types indeed support the argument that caspase-3 could be an important therapeutic target [14]. In our view, the presumption that inducing cell death in tumors (by whatever means) should be the fundamental aim of cancer therapy is fatally flawed. Given the burgeoning evidence that host responses to tumor cell death—including therapy-induced death—may be firmly rooted in tissue repair and regeneration, it seems timely to reassess the therapeutic “death hit” in terms of the host response, especially since post-therapeutic relapse is such a common event. In addition to sustaining the anticancer effect through stimulating antitumor immunity as is the case with immunogenic cell death, combination therapies to induce tumor cell death and simultaneously to block death-induced development of the ‘relapse niche’ should be considered. Identification of the key molecular targets in this niche will be crucial to the development of effective and lasting novel anticancer therapies.
3.9 Conclusions and Future Perspectives
Far from being passive biological entities that simply signify reduction in malignant cell population sizes, apoptotic cells actively modulate the tumor microenvironment, eliciting multiple host responses, many having pro-oncogenic properties. Therefore, although evasion from apoptosis has accepted roles as an acquired characteristic of a cancer cell, its oncogenic features should be considered as part of the tumor microenvironmental contribution to the development of malignant disease. The surprising effects of apoptosis in promoting as well as inhibiting oncogenesis are reflected by recent evidence that p53, the prototypical tumor suppressor, also has tumor-promoting properties [199].
The notion that apoptosis may provide a generic driver mechanism that elicits tissue repair-like processes unifies the features of wound healing, tissue regeneration, and developmental remodeling that characterize malignant tumors. In this respect, the concept of the ORN constitutes a focus for the identification of future therapeutic targets. The critical question here is what molecular mechanisms determine the nature of the responses that emanate from apoptosis? What features of an apoptotic cell lead to compensatory proliferation, angiogenesis, antitumor immunity, or further cell death? As we have discussed, we are beginning to understand this important question and elucidate the answers. Undoubtedly, through thorough understanding of the molecular cell biology of the systemic as well as microenvironmental responses to apoptosis—and indeed other forms of cell death—in tumors, the future holds great promise for improved diagnosis, patient stratification, and long-lasting anticancer therapies.
References
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Wyllie AH. The biology of cell death in tumours. Anticancer Res. 1985;5:131–6.
Weinstein GD, Frost P. Cell proliferation in human basal cell carcinoma. Cancer Res. 1970;30:724–8.
Holmgren L, O’Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med. 1995;1:149–53.
Clifton KH, Yatvin MB. Cell population growth and cell loss in the MTG-B mouse mammary carcinoma. Cancer Res. 1970;30:658–64.
Revesz L, Klein G. Quantitative studies on the multiplication of neoplastic cells in vivo. II. Growth curves of three ascites lymphomas. J Natl Cancer Inst. 1954;15:253–73.
Lala PK. Evaluation of the mode of cell death in Ehrlich ascites tumor. Cancer. 1972;29:261–6.
Leoncini L, Del Vecchio MT, Megha T, Barbini P, Galieni P, Pileri S, Sabattini E, Gherlinzoni F, Tosi P, Kraft R, et al. Correlations between apoptotic and proliferative indices in malignant non-Hodgkin’s lymphomas. Am J Pathol. 1993;142:755–63.
Ohbu M, Saegusa M, Okayasu I. Apoptosis and cellular proliferation in oesophageal squamous cell carcinomas: differences between keratinizing and nonkeratinizing types. Virchows Archiv. 1995;427:271–6.
Naresh KN, Lakshminarayanan K, Pai SA, Borges AM. Apoptosis index is a predictor of metastatic phenotype in patients with early stage squamous carcinoma of the tongue: a hypothesis to support this paradoxical association. Cancer. 2001;91:578–84.
Jalalinadoushan M, Peivareh H, Azizzadeh Delshad A. Correlation between apoptosis and histological grade of transitional cell carcinoma of urinary bladder. Urol J. 2004;1:177–9.
Sun BH, Zhang J, Wang BJ, Zhao XP, Wang YK, Yu ZQ, Yang DL, Hao LJ. Analysis of in vivo patterns of caspase 3 gene expression in primary hepatocellular carcinoma and its relationship to p21(WAF1) expression and hepatic apoptosis. World J Gastroenterol. 2000;6:356–60.
Hu Q, Peng J, Liu W, He X, Cui L, Chen X, Yang M, Liu H, Liu S, Wang H. Elevated cleaved caspase-3 is associated with shortened overall survival in several cancer types. Int J Clin Exp Pathol. 2014;7:5057–70.
Gregory CD, Pound JD. Cell death in the neighbourhood: direct microenvironmental effects of apoptosis in normal and neoplastic tissues. J Pathol. 2011;223:177–94.
Refsum SB, Berdal P. Cell loss in malignant tumours in man. Eur J Cancer. 1967;3:235–6.
Langley RR, Fidler IJ. The seed and soil hypothesis revisited—the role of tumor-stroma interactions in metastasis to different organs. Int J Cancer. 2011;128:2527–35.
Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–9.
Flusberg DA, Sorger PK. Surviving apoptosis: life-death signaling in single cells. Trends Cell Biol. 2015;25(8):446–58.
Bergsmedh A, Ehnfors J, Kawane K, Motoyama N, Nagata S, Holmgren L. DNase II and the Chk2 DNA damage pathway form a genetic barrier blocking replication of horizontally transferred DNA. Mol Cancer Res. 2006;4:187–95.
Bergsmedh A, Szeles A, Henriksson M, Bratt A, Folkman MJ, Spetz AL, Holmgren L. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc Natl Acad Sci U S A. 2001;98:6407–11.
Ehnfors J, Kost-Alimova M, Persson NL, Bergsmedh A, Castro J, Levchenko-Tegnebratt T, Yang L, Panaretakis T, Holmgren L. Horizontal transfer of tumor DNA to endothelial cells in vivo. Cell Death Differ. 2009;16:749–57.
Holmgren L, Szeles A, Rajnavolgyi E, Folkman J, Klein G, Ernberg I, Falk KI. Horizontal transfer of DNA by the uptake of apoptotic bodies. Blood. 1999;93:3956–63.
Han H, Iwanaga T, Uchiyama Y, Fujita T. Aggregation of macrophages in the tips of intestinal villi in guinea pigs: their possible role in the phagocytosis of effete epithelial cells. Cell Tissue Res. 1993;271:407–16.
Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, O’Sullivan B, He Z, Peng Y, Tan AC, Zhou L, Shen J, Han G, Wang XJ, Thorburn J, Thorburn A, Jimeno A, Raben D, Bedford JS, Li CY. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med. 2011;17:860–6.
Lauber K, Munoz LE, Berens C, Jendrossek V, Belka C, Herrmann M. Apoptosis induction and tumor cell repopulation: the yin and yang of radiotherapy. Radiat Oncol. 2011;6:176.
Ford CA, Petrova S, Pound JD, Voss JJ, Melville L, Paterson M, Farnworth SL, Gallimore AM, Cuff S, Wheadon H, Dobbin E, Ogden CA, Dumitriu IE, Dunbar DR, Murray PG, Ruckerl D, Allen JE, Hume DA, van Rooijen N, Goodlad JR, Freeman TC, Gregory CD. Oncogenic properties of apoptotic tumor cells in aggressive B cell lymphoma. Curr Biol. 2015;25:577–88.
Lauber K, Herrmann M. Tumor biology: with a little help from my dying friends. Curr Biol. 2015;25:R198–201.
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57.
Green DR. Means to an end. Apoptosis and other cell death mechanisms. New York: Cold Spring Harbor Laboratory Press; 2011.
Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–75.
Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell. 2010;140:798–804.
Obenauf AC, Massague J. Surviving at a distance: organ specific metastasis. Trends Cancer. 2015;1:76–91.
Perez-Garijo A, Fuchs Y, Steller H. Apoptotic cells can induce non-autonomous apoptosis through the TNF pathway. eLife. 2013;2, e01004.
Galluzzi L, Bravo-San Pedro JM, Kroemer G. Organelle-specific initiation of cell death. Nat Cell Biol. 2014;16:728–36.
Green DR, Galluzzi L, Kroemer G. Cell biology. Metabolic control of cell death. Science. 2014;345:1250256.
Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico-Petruzzelli M, Baehrecke EH, Bazan NG, Bertrand MJ, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Bredesen DE, Brenner C, Campanella M, Candi E, Cecconi F, Chan FK, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin KM, Di Daniele N, Dixit VM, Dynlacht BD, El-Deiry WS, Fimia GM, Flavell RA, Fulda S, Garrido C, Gougeon ML, Green DR, Gronemeyer H, Hajnoczky G, Hardwick JM, Hengartner MO, Ichijo H, Joseph B, Jost PJ, Kaufmann T, Kepp O, Klionsky DJ, Knight RA, Kumar S, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, Lopez-Otin C, Lugli E, Madeo F, Malorni W, Marine JC, Martin SJ, Martinou JC, Medema JP, Meier P, Melino S, Mizushima N, Moll U, Munoz-Pinedo C, Nunez G, Oberst A, Panaretakis T, Penninger JM, Peter ME, Piacentini M, Pinton P, Prehn JH, Puthalakath H, Rabinovich GA, Ravichandran KS, Rizzuto R, Rodrigues CM, Rubinsztein DC, Rudel T, Shi Y, Simon HU, Stockwell BR, Szabadkai G, Tait SW, Tang HL, Tavernarakis N, Tsujimoto Y, Vanden Berghe T, Vandenabeele P, Villunger A, Wagner EF, Walczak H, White E, Wood WG, Yuan J, Zakeri Z, Zhivotovsky B, Melino G, Kroemer G. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015;22:58–73.
Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–305.
Gregory CD, Pound JD. Microenvironmental influences of apoptosis in vivo and in vitro. Apoptosis. 2010;15:1029–49.
Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–21.
Medzhitov R, Janeway Jr CA. Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298–300.
Vakkila J, Lotze MT. Inflammation and necrosis promote tumour growth. Nat Rev Immunol. 2004;4:641–8.
Gamrekelashvili J, Kruger C, von Wasielewski R, Hoffmann M, Huster KM, Busch DH, Manns MP, Korangy F, Greten TF. Necrotic tumor cell death in vivo impairs tumor-specific immune responses. J Immunol. 2007;178:1573–80.
Moreira S, Stramer B, Evans I, Wood W, Martin P. Prioritization of competing damage and developmental signals by migrating macrophages in the Drosophila embryo. Curr Biol. 2010;20:464–70.
Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15(2):81–94.
White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–10.
Changou CA, Chen YR, Xing L, Yen Y, Chuang FY, Cheng RH, Bold RJ, Ann DK, Kung HJ. Arginine starvation-associated atypical cellular death involves mitochondrial dysfunction, nuclear DNA leakage, and chromatin autophagy. Proc Natl Acad Sci U S A. 2014;111:14147–52.
Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, Nelson DA, Jin S, White E. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64.
Wen Z, Fan L, Li Y, Zou Z, Scott MJ, Xiao G, Li S, Billiar TR, Wilson MA, Shi X, Fan J. Neutrophils counteract autophagy-mediated anti-inflammatory mechanisms in alveolar macrophage: role in posthemorrhagic shock acute lung inflammation. J Immunol. 2014;193:4623–33.
Dou Z, Xu C, Donahue G, Shimi T, Pan JA, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, Catanzaro JM, Daniel Ricketts M, Lamark T, Adam SA, Marmorstein R, Zong WX, Johansen T, Goldman RD, Adams PD, Berger SL. Autophagy mediates degradation of nuclear lamina. Nature. 2015;527:105–9.
Revesz L. Effect of tumour cells killed by x-rays upon the growth of admixed viable cells. Nature. 1956;178:1391–2.
Revesz L. Effect of lethally damaged tumor cells upon the development of admixed viable cells. J Natl Cancer Inst. 1958;20:1157–86.
Ringertz N, Klein E, Revesz L. Growth of small compatible tumor implants in presence of admixed radiation-killed or incompatible tumor cells. Cancer. 1959;12:697–707.
Seelig KJ, Revesz L. Effect of lethally damaged tumour cells upon the growth of admixed viable cells in diffusion chambers. Br J Cancer. 1960;14:126–38.
Puck TT, Marcus PI, Cieciura SJ. Clonal growth of mammalian cells in vitro; growth characteristics of colonies from single HeLa cells with and without a feeder layer. J Exp Med. 1956;103:273–83.
Gregory CD, Pound JD, Devitt A, Wilson-Jones M, Ray P, Murray RJ. Inhibitory effects of persistent apoptotic cells on monoclonal antibody production in vitro: simple removal of non-viable cells improves antibody productivity by hybridoma cells in culture. MAbs. 2009;1:370–6.
Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–16.
Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J Clin Invest. 1998;101:890–8.
Fond AM, Lee CS, Schulman IG, Kiss RS, Ravichandran KS. Apoptotic cells trigger a membrane-initiated pathway to increase ABCA1. J Clin Invest. 2015;125:2748–58.
Emoto K, Toyama-Sorimachi N, Karasuyama H, Inoue K, Umeda M. Exposure of phosphatidylethanolamine on the surface of apoptotic cells. Exp Cell Res. 1997;232:430–4.
Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, Barbour SE, Milstien S, Spiegel S. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 2008;22:2629–38.
Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, Xu Y, Autenrieth IB, Schulze-Osthoff K, Belka C, Stuhler G, Wesselborg S. Apoptotic cells induce migration of phagocytes via caspase-3- mediated release of a lipid attraction signal. Cell. 2003;113:717–30.
Brown S, Heinisch I, Ross E, Shaw K, Buckley CD, Savill J. Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature. 2002;418:200–3.
Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–34.
Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, Melville L, Melrose LA, Ogden CA, Nibbs R, Graham G, Combadiere C, Gregory CD. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood. 2008;112:5026–36.
Leonardi-Essmann F, Emig M, Kitamura Y, Spanagel R, Gebicke-Haerter PJ. Fractalkine-upregulated milk-fat globule EGF factor-8 protein in cultured rat microglia. J Neuroimmunol. 2005;160:92–101.
Miksa M, Amin D, Wu R, Ravikumar TS, Wang P. Fractalkine-induced MFG-E8 leads to enhanced apoptotic cell clearance by macrophages. Mol Med. 2007;13:553–60.
Sapozhnikov AM, Ponomarev ED, Tarasenko TN, Telford WG. Spontaneous apoptosis and expression of cell surface heat-shock proteins in cultured EL-4 lymphoma cells. Cell Prolif. 1999;32:363–78.
Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol. 2000;12:1539–46.
Feng H, Zeng Y, Whitesell L, Katsanis E. Stressed apoptotic tumor cells express heat shock proteins and elicit tumor-specific immunity. Blood. 2001;97:3505–12.
Goh YC, Yap CT, Huang BH, Cronshaw AD, Leung BP, Lai PB, Hart SP, Dransfield I, Ross JA. Heat-shock protein 60 translocates to the surface of apoptotic cells and differentiated megakaryocytes and stimulates phagocytosis. Cell Mol Life Sci. 2011;68:1581–92.
Gao Y, Herndon JM, Zhang H, Griffith TS, Ferguson TA. Antiinflammatory effects of CD95 ligand (FasL)-induced apoptosis. J Exp Med. 1998;188:887–96.
Chen W, Frank ME, Jin W, Wahl SM. TGF-beta released by apoptotic T cells contributes to an immunosuppressive milieu. Immunity. 2001;14:715–25.
Bournazou I, Pound JD, Duffin R, Bournazos S, Melville LA, Brown SB, Rossi AG, Gregory CD. Apoptotic human cells inhibit migration of granulocytes via release of lactoferrin. J Clin Invest. 2009;119:20–32.
Bournazou I, Mackenzie KJ, Duffin R, Rossi AG, Gregory CD. Inhibition of eosinophil migration by lactoferrin. Immunol Cell Biol. 2010;88:220–3.
Miles K, Clarke DJ, Lu W, Sibinska Z, Beaumont PE, Davidson DJ, Barr TA, Campopiano DJ, Gray M. Dying and necrotic neutrophils are anti-inflammatory secondary to the release of alpha-defensins. J Immunol. 2009;183:2122–32.
Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK, Leitinger N, Ravichandran KS. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–6.
Horino K, Nishiura H, Ohsako T, Shibuya Y, Hiraoka T, Kitamura N, Yamamoto T. A monocyte chemotactic factor, S19 ribosomal protein dimer, in phagocytic clearance of apoptotic cells. Lab Invest. 1998;78:603–17.
Knies UE, Behrensdorf HA, Mitchell CA, Deutsch U, Risau W, Drexler HC, Clauss M. Regulation of endothelial monocyte-activating polypeptide II release by apoptosis. Proc Natl Acad Sci U S A. 1998;95:12322–7.
Thery C, Boussac M, Veron P, Ricciardi-Castagnoli P, Raposo G, Garin J, Amigorena S. Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J Immunol. 2001;166:7309–18.
Zirngibl M, Furnrohr BG, Janko C, Munoz LE, Voll RE, Gregory CD, Schett G, Herrmann M. Loading of nuclear autoantigens prototypically recognized by systemic lupus erythematosus sera into late apoptotic vesicles requires intact microtubules and myosin light chain kinase activity. Clin Exp Immunol. 2015;179:39–49.
Blume KE, Soeroes S, Waibel M, Keppeler H, Wesselborg S, Herrmann M, Schulze-Osthoff K, Lauber K. Cell surface externalization of annexin A1 as a failsafe mechanism preventing inflammatory responses during secondary necrosis. J Immunol. 2009;183:8138–47.
Krispin A, Bledi Y, Atallah M, Trahtemberg U, Verbovetski I, Nahari E, Zelig O, Linial M, Mevorach D. Apoptotic cell thrombospondin-1 and heparin-binding domain lead to dendritic-cell phagocytic and tolerizing states. Blood. 2006;108:3580–9.
Soulez M, Sirois I, Brassard N, Raymond MA, Nicodeme F, Noiseux N, Durocher Y, Pshezhetsky AV, Hebert MJ. Epidermal growth factor and perlecan fragments produced by apoptotic endothelial cells co-ordinately activate ERK1/2-dependent antiapoptotic pathways in mesenchymal stem cells. Stem Cells. 2010;28:810–20.
Soulez M, Pilon EA, Dieude M, Cardinal H, Brassard N, Qi S, Wu SJ, Durocher Y, Madore F, Perreault C, Hebert MJ. The perlecan fragment LG3 is a novel regulator of obliterative remodeling associated with allograft vascular rejection. Circ Res. 2012;110:94–104.
Moffatt OD, Devitt A, Bell ED, Simmons DL, Gregory CD. Macrophage recognition of ICAM-3 on apoptotic leukocytes. J Immunol. 1999;162:6800–10.
Torr EE, Gardner DH, Thomas L, Goodall DM, Bielemeier A, Willetts R, Griffiths HR, Marshall LJ, Devitt A. Apoptotic cell-derived ICAM-3 promotes both macrophage chemoattraction to and tethering of apoptotic cells. Cell Death Differ. 2012;19:671–9.
Kobara M, Sunagawa N, Abe M, Tanaka N, Toba H, Hayashi H, Keira N, Tatsumi T, Matsubara H, Nakata T. Apoptotic myocytes generate monocyte chemoattractant protein-1 and mediate macrophage recruitment. J Appl Physiol. 2008;104:601–9.
Renz A, Berdel WE, Kreuter M, Belka C, Schulze-Osthoff K, Los M. Rapid extracellular release of cytochrome c is specific for apoptosis and marks cell death in vivo. Blood. 2001;98:1542–8.
Codina R, Vanasse A, Kelekar A, Vezys V, Jemmerson R. Cytochrome c-induced lymphocyte death from the outside in: inhibition by serum leucine-rich alpha-2-glycoprotein-1. Apoptosis. 2010;15:139–52.
Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–30.
Radic M, Marion T, Monestier M. Nucleosomes are exposed at the cell surface in apoptosis. J Immunol. 2004;172:6692–700.
Bilyy RO, Shkandina T, Tomin A, Munoz LE, Franz S, Antonyuk V, Kit YY, Zirngibl M, Furnrohr BG, Janko C, Lauber K, Schiller M, Schett G, Stoika RS, Herrmann M. Macrophages discriminate glycosylation patterns of apoptotic cell-derived microparticles. J Biol Chem. 2012;287:496–503.
Weihua Z, Tsan R, Schroit AJ, Fidler IJ. Apoptotic cells initiate endothelial cell sprouting via electrostatic signaling. Cancer Res. 2005;65:11529–35.
Sirois I, Raymond MA, Brassard N, Cailhier JF, Fedjaev M, Hamelin K, Londono I, Bendayan M, Pshezhetsky AV, Hebert MJ. Caspase-3-dependent export of TCTP: a novel pathway for antiapoptotic intercellular communication. Cell Death Differ. 2011;18:549–62.
Tennant I, Pound JD, Marr LA, Willems JJ, Petrova S, Ford CA, Paterson M, Devitt A, Gregory CD. Innate recognition of apoptotic cells: novel apoptotic cell-associated molecular patterns revealed by crossreactivity of anti-LPS antibodies. Cell Death Differ. 2013;20:698–708.
Wickman GR, Julian L, Mardilovich K, Schumacher S, Munro J, Rath N, Zander SA, Mleczak A, Sumpton D, Morrice N, Bienvenut WV, Olson MF. Blebs produced by actin-myosin contraction during apoptosis release damage-associated molecular pattern proteins before secondary necrosis occurs. Cell Death Differ. 2013;20:1293–305.
Crescitelli R, Lasser C, Szabo TG, Kittel A, Eldh M, Dianzani I, Buzas EI, Lotvall J. Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J extracellular vesicles. 2013;2.
Atkin-Smith GK, Tixeira R, Paone S, Mathivanan S, Collins C, Liem M, Goodall KJ, Ravichandran KS, Hulett MD, Poon IK. A novel mechanism of generating extracellular vesicles during apoptosis via a beads-on-a-string membrane structure. Nat Commun. 2015;6:7439.
Dieude M, Bell C, Turgeon J, Beillevaire D, Pomerleau L, Yang B, Hamelin K, Qi S, Pallet N, Beland C, Dhahri W, Cailhier JF, Rousseau M, Duchez AC, Levesque T, Lau A, Rondeau C, Gingras D, Muruve D, Rivard A, Cardinal H, Perreault C, Desjardins M, Boilard E, Thibault P, Hebert MJ. The 20S proteasome core, active within apoptotic exosome-like vesicles, induces autoantibody production and accelerates rejection. Sci Trans Med. 2015;7:318ra200.
Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, Jan M, Cha AC, Chan CK, Tan BT, Park CY, Zhao F, Kohrt HE, Malumbres R, Briones J, Gascoyne RD, Lossos IS, Levy R, Weissman IL, Majeti R. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010;142:699–713.
Chao MP, Alizadeh AA, Tang C, Jan M, Weissman-Tsukamoto R, Zhao F, Park CY, Weissman IL, Majeti R. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 2011;71:1374–84.
Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–93.
Buzas EI, Gyorgy B, Nagy G, Falus A, Gay S. Emerging role of extracellular vesicles in inflammatory diseases. Nat Rev Rheumatol. 2014;10:356–64.
Eken C, Martin PJ, Sadallah S, Treves S, Schaller M, Schifferli JA. Ectosomes released by polymorphonuclear neutrophils induce a MerTK-dependent anti-inflammatory pathway in macrophages. J Biol Chem. 2010;285:39914–21.
D’Souza-Schorey C, Clancy JW. Tumor-derived microvesicles: shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev. 2012;26:1287–99.
Bonner C, Bacon S, Concannon CG, Rizvi SR, Baquie M, Farrelly AM, Kilbride SM, Dussmann H, Ward MW, Boulanger CM, Wollheim CB, Graf R, Byrne MM, Prehn JH. INS-1 cells undergoing caspase-dependent apoptosis enhance the regenerative capacity of neighboring cells. Diabetes. 2010;59:2799–808.
Peter C, Wesselborg S, Herrmann M, Lauber K. Dangerous attraction: phagocyte recruitment and danger signals of apoptotic and necrotic cells. Apoptosis. 2010;15(9):1007–28.
Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol. 2015;16:907–17.
Li F, Huang Q, Chen J, Peng Y, Roop DR, Bedford JS, Li CY. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci Signal. 2010;3:ra13–13.
Ariel A, Fredman G, Sun YP, Kantarci A, Van Dyke TE, Luster AD, Serhan CN. Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat Immunol. 2006;7:1209–16.
Garrido C, Gurbuxani S, Ravagnan L, Kroemer G. Heat shock proteins: endogenous modulators of apoptotic cell death. Biochem Biophys Res Commun. 2001;286:433–42.
Baixauli F, Lopez-Otin C, Mittelbrunn M. Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Front Immunol. 2014;5:403.
Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118.
Birge RB, Ucker DS. Innate apoptotic immunity: the calming touch of death. Cell Death Differ. 2008;15:1096–102.
Cocco RE, Ucker DS. Distinct modes of macrophage recognition for apoptotic and necrotic cells are not specified exclusively by phosphatidylserine exposure. Mol Biol Cell. 2001;12:919–30.
Cvetanovic M, Mitchell JE, Patel V, Avner BS, Su Y, van der Saag PT, Witte PL, Fiore S, Levine JS, Ucker DS. Specific recognition of apoptotic cells reveals a ubiquitous and unconventional innate immunity. J Biol Chem. 2006;281:20055–67.
Cvetanovic M, Ucker DS. Innate immune discrimination of apoptotic cells: repression of proinflammatory macrophage transcription is coupled directly to specific recognition. J Immunol. 2004;172:880–9.
Lewis CE, Leek R, Harris A, McGee JO. Cytokine regulation of angiogenesis in breast cancer: the role of tumor-associated macrophages. J Leukoc Biol. 1995;57:747–51.
Ong SM, Tan YC, Beretta O, Jiang D, Yeap WH, Tai JJ, Wong WC, Yang H, Schwarz H, Lim KH, Koh PK, Ling KL, Wong SC. Macrophages in human colorectal cancer are pro-inflammatory and prime T cells towards an anti-tumour type-1 inflammatory response. Eur J Immunol. 2012;42:89–100.
Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, Zhao YW, Wei YQ. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One. 2012;7, e50946.
Stanford JC, Young C, Hicks D, Owens P, Williams A, Vaught DB, Morrison MM, Lim J, Williams M, Brantley-Sieders DM, Balko JM, Tonetti D, Earp HS, Cook RS. Efferocytosis produces a prometastatic landscape during postpartum mammary gland involution. J Clin Invest. 2014;124(11):4737–52.
Soki FN, Koh AJ, Jones JD, Kim YW, Dai J, Keller ET, Pienta KJ, Atabai K, Roca H, McCauley LK. Polarization of prostate cancer-associated macrophages is induced by milk fat globule-EGF factor 8 (MFG-E8)-mediated efferocytosis. J Biol Chem. 2014;289:24560–72.
Novak ML, Weinheimer-Haus EM, Koh TJ. Macrophage activation and skeletal muscle healing following traumatic injury. J Pathol. 2014;232:344–55.
Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, Wynn TA. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20.
A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Diaz M, Gallardo G, de Galarreta CR, Salazar J, Lopez F, Edwards P, Parks J, Andujar M, Tontonoz P, Castrillo A. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–58.
Kondylis V, Polykratis A, Ehlken H, Ochoa-Callejero L, Straub BK, Krishna-Subramanian S, Van TM, Curth HM, Heise N, Weih F, Klein U, Schirmacher P, Kelliher M, Pasparakis M. NEMO prevents steatohepatitis and hepatocellular carcinoma by inhibiting RIPK1 kinase activity-mediated hepatocyte apoptosis. Cancer Cell. 2015;28:582–98.
Fuchs Y, Steller H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol. 2015;16:329–44.
Michalak EM, Vandenberg CJ, Delbridge AR, Wu L, Scott CL, Adams JM, Strasser A. Apoptosis-promoted tumorigenesis: gamma-irradiation-induced thymic lymphomagenesis requires Puma-driven leukocyte death. Genes Dev. 2010;24:1608–13.
Labi V, Erlacher M, Krumschnabel G, Manzl C, Tzankov A, Pinon J, Egle A, Villunger A. Apoptosis of leukocytes triggered by acute DNA damage promotes lymphoma formation. Genes Dev. 2010;24:1602–7.
Miura M. Apoptotic and nonapoptotic caspase functions in animal development. Cold Spring Harb Perspect Biol. 2012;4.
Connolly PF, Jager R, Fearnhead HO. New roles for old enzymes: killer caspases as the engine of cell behavior changes. Front Physiol. 2014;5:149.
Zhao X, Wang D, Zhao Z, Xiao Y, Sengupta S, Xiao Y, Zhang R, Lauber K, Wesselborg S, Feng L, Rose TM, Shen Y, Zhang J, Prestwich G, Xu Y. Caspase-3-dependent activation of calcium-independent phospholipase A2 enhances cell migration in non-apoptotic ovarian cancer cells. J Biol Chem. 2006;281:29357–68.
Ran S, Thorpe PE. Phosphatidylserine is a marker of tumor vasculature and a potential target for cancer imaging and therapy. Int J Radiat Oncol Biol Phys. 2002;54:1479–84.
Riboldi E, Porta C, Morlacchi S, Viola A, Mantovani A, Sica A. Hypoxia-mediated regulation of macrophage functions in pathophysiology. Int Immunol. 2013;25:67–75.
Kaelin Jr WG, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402.
Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, Taylor CT. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci U S A. 2006;103:18154–9.
Sendoel A, Hengartner MO. Apoptotic cell death under hypoxia. Physiology (Bethesda). 2014;29:168–76.
Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, Bernasconi S, Saccani S, Nebuloni M, Vago L, Mantovani A, Melillo G, Sica A. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med. 2003;198:1391–402.
Biswas SK, Allavena P, Mantovani A. Tumor-associated macrophages: functional diversity, clinical significance, and open questions. Semin Immunopathol. 2013;35:585–600.
Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, Mack M, Pipeleers D, In’t Veld P, De Baetselier P, Van Ginderachter JA. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010;70:5728–39.
Obeid E, Nanda R, Fu YX, Olopade OI. The role of tumor-associated macrophages in breast cancer progression (review). Int J Oncol. 2013;43:5–12.
Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86:1065–73.
Hartnett L, Egan LJ. Inflammation, DNA methylation and colitis-associated cancer. Carcinogenesis. 2012;33:723–31.
Walter ND, Rice PL, Redente EF, Kauvar EF, Lemond L, Aly T, Wanebo K, Chan ED. Wound healing after trauma may predispose to lung cancer metastasis: review of potential mechanisms. Am J Respir Cell Mol Biol. 2011;44:591–6.
Wang XW, Hussain SP, Huo TI, Wu CG, Forgues M, Hofseth LJ, Brechot C, Harris CC. Molecular pathogenesis of human hepatocellular carcinoma. Toxicology. 2002;181–182:43–7.
Jung Y, Witek RP, Syn WK, Choi SS, Omenetti A, Premont R, Guy CD, Diehl AM. Signals from dying hepatocytes trigger growth of liver progenitors. Gut. 2010;59:655–65.
Lu WY, Bird TG, Boulter L, Tsuchiya A, Cole AM, Hay T, Guest RV, Wojtacha D, Man TY, Mackinnon A, Ridgway RA, Kendall T, Williams MJ, Jamieson T, Raven A, Hay DC, Iredale JP, Clarke AR, Sansom OJ, Forbes SJ. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat Cell Biol. 2015;17(8):971–83.
Lu H, Clauser KR, Tam WL, Frose J, Ye X, Eaton EN, Reinhardt F, Donnenberg VS, Bhargava R, Carr SA, Weinberg RA. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol. 2014;16:1105–17.
Koh TJ, DiPietro LA. Inflammation and wound healing: the role of the macrophage. Expert Rev Mol Med. 2011;13, e23.
Novak ML, Koh TJ. Phenotypic transitions of macrophages orchestrate tissue repair. Am J Pathol. 2013;183:1352–63.
Schafer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9:628–38.
Byun JS, Gardner K. Wounds that will not heal: pervasive cellular reprogramming in cancer. Am J Pathol. 2013;182:1055–64.
Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22:231–7.
Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–55.
Khanna S, Biswas S, Shang Y, Collard E, Azad A, Kauh C, Bhasker V, Gordillo GM, Sen CK, Roy S. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One. 2010;5, e9539.
Swift ME, Burns AL, Gray KL, DiPietro LA. Age-related alterations in the inflammatory response to dermal injury. J Invest Dermatol. 2001;117:1027–35.
Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72.
Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104:2224–34.
DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, Rugo HS, Hwang ES, Jirstrom K, West BL, Coussens LM. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54–67.
Xu J, Escamilla J, Mok S, David J, Priceman S, West B, Bollag G, McBride W, Wu L. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73:2782–94.
Loberg RD, Ying C, Craig M, Day LL, Sargent E, Neeley C, Wojno K, Snyder LA, Yan L, Pienta KJ. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007;67:9417–24.
Rozel S, Galban CJ, Nicolay K, Lee KC, Sud S, Neeley C, Snyder LA, Chenevert TL, Rehemtulla A, Ross BD, Pienta KJ. Synergy between anti-CCL2 and docetaxel as determined by DW-MRI in a metastatic bone cancer model. J Cell Biochem. 2009;107:58–64.
Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, Mitchem JB, Plambeck-Suess SM, Worley LA, Goetz BD, Wang-Gillam A, Eberlein TJ, Denardo DG, Goedegebuure SP, Linehan DC. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res. 2013;19:3404–15.
Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G, Kato Y, Li J, Pollard JW. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med. 2015;212:1043–59.
Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, Erba E, Uboldi S, Zucchetti M, Pasqualini F, Nebuloni M, van Rooijen N, Mortarini R, Beltrame L, Marchini S, Fuso Nerini I, Sanfilippo R, Casali PG, Pilotti S, Galmarini CM, Anichini A, Mantovani A, D’Incalci M, Allavena P. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23:249–62.
Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjallman AH, Ballmer-Hofer K, Schwendener RA. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. Br J Cancer. 2006;95:272–81.
Coscia M, Quaglino E, Iezzi M, Curcio C, Pantaleoni F, Riganti C, Holen I, Monkkonen H, Boccadoro M, Forni G, Musiani P, Bosia A, Cavallo F, Massaia M. Zoledronic acid repolarizes tumour-associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. J Cell Mol Med. 2010;14:2803–15.
Hiraoka K, Zenmyo M, Watari K, Iguchi H, Fotovati A, Kimura YN, Hosoi F, Shoda T, Nagata K, Osada H, Ono M, Kuwano M. Inhibition of bone and muscle metastases of lung cancer cells by a decrease in the number of monocytes/macrophages. Cancer Sci. 2008;99:1595–602.
Pulaski HL, Spahlinger G, Silva IA, McLean K, Kueck AS, Reynolds RK, Coukos G, Conejo-Garcia JR, Buckanovich RJ. Identifying alemtuzumab as an anti-myeloid cell antiangiogenic therapy for the treatment of ovarian cancer. J Transl Med. 2009;7:49.
Bak SP, Walters JJ, Takeya M, Conejo-Garcia JR, Berwin BL. Scavenger receptor-A-targeted leukocyte depletion inhibits peritoneal ovarian tumor progression. Cancer Res. 2007;67:4783–9.
Nagai T, Tanaka M, Tsuneyoshi Y, Xu B, Michie SA, Hasui K, Hirano H, Arita K, Matsuyama T. Targeting tumor-associated macrophages in an experimental glioma model with a recombinant immunotoxin to folate receptor beta. Cancer Immunol Immunother. 2009;58:1577–86.
Petrillo M, Zannoni GF, Martinelli E, Pedone Anchora L, Ferrandina G, Tropeano G, Fagotti A, Scambia G. Polarisation of tumor-associated macrophages toward M2 phenotype correlates with poor response to chemoradiation and reduced survival in patients with locally advanced cervical cancer. PLoS One. 2015;10, e0136654.
Pantano F, Berti P, Guida FM, Perrone G, Vincenzi B, Amato MM, Righi D, Dell’aquila E, Graziano F, Catalano V, Caricato M, Rizzo S, Muda AO, Russo A, Tonini G, Santini D. The role of macrophages polarization in predicting prognosis of radically resected gastric cancer patients. J Cell Mol Med. 2013;17:1415–21.
Biswas SK, Lewis CE. NF-kappaB as a central regulator of macrophage function in tumors. J Leukoc Biol. 2010;88:877–84.
Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005;65:3437–46.
Buhtoiarov IN, Sondel PM, Wigginton JM, Buhtoiarova TN, Yanke EM, Mahvi DA, Rakhmilevich AL. Anti-tumour synergy of cytotoxic chemotherapy and anti-CD40 plus CpG-ODN immunotherapy through repolarization of tumour-associated macrophages. Immunology. 2011;132:226–39.
Weber JS, Zarour H, Redman B, Trefzer U, O’Day S, van den Eertwegh AJ, Marshall E, Wagner S. Randomized phase 2/3 trial of CpG oligodeoxynucleotide PF-3512676 alone or with dacarbazine for patients with unresectable stage III and IV melanoma. Cancer. 2009;115:3944–54.
Carpentier A, Metellus P, Ursu R, Zohar S, Lafitte F, Barrie M, Meng Y, Richard M, Parizot C, Laigle-Donadey F, Gorochov G, Psimaras D, Sanson M, Tibi A, Chinot O, Carpentier AF. Intracerebral administration of CpG oligonucleotide for patients with recurrent glioblastoma: a phase II study. Neuro Oncol. 2010;12:401–8.
Zent CS, Smith BJ, Ballas ZK, Wooldridge JE, Link BK, Call TG, Shanafelt TD, Bowen DA, Kay NE, Witzig TE, Weiner GJ. Phase I clinical trial of CpG oligonucleotide 7909 (PF-03512676) in patients with previously treated chronic lymphocytic leukemia. Leuk Lymphoma. 2012;53:211–7.
Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ, Vonderheide RH. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–6.
Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, Troxel AB, Sun W, Teitelbaum UR, Vonderheide RH, O’Dwyer PJ. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19:6286–95.
Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, Setty M, Leslie CS, Oei Y, Pedraza A, Zhang J, Brennan CW, Sutton JC, Holland EC, Daniel D, Joyce JA. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–72.
Xu M, Liu M, Du X, Li S, Li H, Li X, Li Y, Wang Y, Qin Z, Fu YX, Wang S. Intratumoral delivery of IL-21 overcomes anti-Her2/Neu resistance through shifting tumor-associated macrophages from M2 to M1 phenotype. J Immunol. 2015;194:4997–5006.
Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, Pfirschke C, Voss RH, Timke C, Umansky L, Klapproth K, Schakel K, Garbi N, Jager D, Weitz J, Schmitz-Winnenthal H, Hammerling GJ, Beckhove P. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24:589–602.
O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104:1604–9.
Chaudhuri AA, So AY, Sinha N, Gibson WS, Taganov KD, O’Connell RM, Baltimore D. MicroRNA-125b potentiates macrophage activation. J Immunol. 2011;187:5062–8.
Asano K, Miwa M, Miwa K, Hanayama R, Nagase H, Nagata S, Tanaka M. Masking of phosphatidylserine inhibits apoptotic cell engulfment and induces autoantibody production in mice. J Exp Med. 2004;200:459–67.
Bondanza A, Zimmermann VS, Rovere-Querini P, Turnay J, Dumitriu IE, Stach CM, Voll RE, Gaipl US, Bertling W, Poschl E, Kalden JR, Manfredi AA, Herrmann M. Inhibition of phosphatidylserine recognition heightens the immunogenicity of irradiated lymphoma cells in vivo. J Exp Med. 2004;200:1157–65.
He J, Yin Y, Luster TA, Watkins L, Thorpe PE. Antiphosphatidylserine antibody combined with irradiation damages tumor blood vessels and induces tumor immunity in a rat model of glioblastoma. Clin Cancer Res. 2009;15:6871–80.
Yin Y, Huang X, Lynn KD, Thorpe PE. Phosphatidylserine-targeting antibody induces M1 macrophage polarization and promotes myeloid-derived suppressor cell differentiation. Cancer Immunol Res. 2013;1:256–68.
Chalasani P, Marron M, Roe D, Clarke K, Iannone M, Livingston RB, Shan JS, Stopeck AT. A phase I clinical trial of bavituximab and paclitaxel in patients with HER2 negative metastatic breast cancer. Cancer Med. 2015;4:1051–9.
Verma A, Warner SL, Vankayalapati H, Bearss DJ, Sharma S. Targeting Axl and Mer kinases in cancer. Mol Cancer Ther. 2011;10:1763–73.
Sheridan C. First Axl inhibitor enters clinical trials. Nat Biotechnol. 2013;31:775–6.
Bosurgi L, Bernink JH, Delgado Cuevas V, Gagliani N, Joannas L, Schmid ET, Booth CJ, Ghosh S, Rothlin CV. Paradoxical role of the proto-oncogene Axl and Mer receptor tyrosine kinases in colon cancer. Proc Natl Acad Sci U S A. 2013;110(32):13091–6.
Montalvao F, Garcia Z, Celli S, Breart B, Deguine J, Van Rooijen N, Bousso P. The mechanism of anti-CD20-mediated B cell depletion revealed by intravital imaging. J Clin Invest. 2013;123:5098–103.
Gul N, Babes L, Siegmund K, Korthouwer R, Bogels M, Braster R, Vidarsson G, Ten Hagen TL, Kubes P, van Egmond M. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J Clin Invest. 2014;124:812–23.
Donato AL, Huang Q, Liu X, Li F, Zimmerman MA, Li CY. Caspase 3 promotes surviving melanoma tumor cell growth after cytotoxic therapy. J Invest Dermatol. 2014;134:1686–92.
Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16:393–405.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Gregory, C.D., Ford, C.A., Voss, J.J.L.P. (2016). Microenvironmental Effects of Cell Death in Malignant Disease. In: Gregory, C. (eds) Apoptosis in Cancer Pathogenesis and Anti-cancer Therapy. Advances in Experimental Medicine and Biology, vol 930. Springer, Cham. https://doi.org/10.1007/978-3-319-39406-0_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-39406-0_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-39404-6
Online ISBN: 978-3-319-39406-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)