
Abstract
The study of polymeric materials at several scales of structure will almost certainly require the use of a number of complementary techniques. This chapter provides an overview of the experimental techniques available for visualizing and evaluating polymer morphology. The chapter sets out to provide sufficient information to enable the reader to appreciate the research work described in the following chapters. Imaging techniques can provide invaluable information of many different scales. For the nanoscale, transmission electron microscopy is the technique of choice but at the expense of a much reduced sampling volume and more involved sample preparation. Recent developments in scanning electron microscopes, particularly in terms of field emission sources mean that nanometre resolution is readily available with the attendant advantages of the ease of screening large areas. However as a surface topology technique we need to develop a contrast, which can be readily achieved using, for example, the etching techniques described in this chapter or through examination of a fractured surface. Scattering techniques probe a volume and in some cases information can be obtained in a time-resolving manner, so that the development of structure can be followed in real time. Neutron scattering provides a different contrast to X-ray scattering which can be useful with halogenated polymers or with metal fillers. Recent developments in pulsed neutron sources have resulted in instruments which can provide data in a single calibrated file over length scales from 0.1 to 10 nm. We also show how indirect methods such as thermal analysis can be used to obtain morphological data.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Isotactic Polypropylene
- Poly Ether Ether Ketone
- Secondary Crystallization
- Ruthenium Tetroxide
- Atactic Polypropylene
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
2.1 Introduction
The study of polymeric materials at several scales of structure will almost certainly require the use of a number of complementary techniques. This chapter provides an overview of the experimental techniques available for visualizing and evaluating polymer morphology. The chapter sets out to provide sufficient information to enable the reader to appreciate the research work described in the following chapters. Imaging techniques can provide invaluable information of many different scales. For the nanoscale, transmission electron microscopy is the technique of choice but at the expense of a much reduced sampling volume and more involved sample preparation. Recent developments in scanning electron microscopes, particularly in terms of field emission sources mean that nanometre resolution is readily available with the attendant advantages of the ease of screening large areas. However, as a surface topology technique we need to develop a contrast, which can be readily achieved using, for example, the etching techniques described in this chapter or through examination of a fractured surface. Scattering techniques probe a volume and in some cases information can be obtained in a time-resolving manner, so that the development of structure can be followed in real time. Neutron scattering provides a different contrast to X-ray scattering which can be useful with halogenated polymers or with metal fillers. Recent developments in pulsed neutron sources have resulted in instruments which can provide data in a single calibrated file over length scales from 0.1 to 10 nm. We also show how indirect methods such as thermal analysis can be used to obtain morphological data.
2.2 Indirect Methods
2.2.1 Differential Scanning Calorimetry
Differential scanning calorimetry (DSC) has two particular historical applications to polymers. One is the study of the glass transition (McKenna 1989) and the other is the study of crystallization and melting. Compared to simple molecular materials, polymer crystallization always involves considerable supercooling and a range of melting points depending on thermal history and relation to the thickness of the polymer lamellae, ideally as described by the Gibbs–Thomson equation. DSC is most often the technique of choice for observing both crystallization and melting and, ideally, there is a relationship between the two described by the Hoffman–Weeks equation (Hoffman and Weeks 1962).
According to Hoffman nucleation theory (see Sect. 1.6.1), if chains in a polymer crystal stayed as they were laid down, the crystal would melt almost immediately above its crystallization temperature (solid black line at 45° in Fig. 2.1); however, following subsequent processes the lamellar thickness increases by a thickening factor β (2 in the case of polyethylene) giving a slope with its reciprocal ɣ (here 1/2) in a Hoffman–Weeks plot. The theory of this is quite complicated, and the simple linear plot has been called into question (Marand et al. 1998). Nevertheless, it suffices as a very practical guide to the use of DSC in relating thermal history of a polymer specimen to lamellar morphology.

Hoffman–Weeks plot for polyethylene crystallized in a fast DSC. The symbol open circle represents melting by fast scan DSC after isothermal crystallization, filled circle represents melting after a constant cooling rate and filled diamond represents the melting of a conventional DSC after isothermal crystallization. The solid black line plots T M = T C (Part of Figure 6 reprinted from Polymer, 55(14), 3186–3194 Toda et al. Copyright 2014 with permission from Elsevier)
Testing this relationship was often limited by the fact that for lower crystallization temperatures or at very fast cooling rates (such as found in many industrial processes), thin crystals were formed which on melting could rapidly recrystallize and remelt at higher temperatures (a process generally referred to as reorganization). With recent advances in technology with fast scanning instruments, such crystals can simply melt once, and lower crystallization temperatures are obtainable in the instrument itself. The Hoffman–Weeks relationship has recently been thus tested and found valid for polyethylene (Toda et al. 2014); in Fig. 2.1 the blue diamonds represent conditions attainable in a traditional DSC and the red circles were obtained in a fast scanning calorimeter.
Many of the morphological phenomena in polymers with their concomitant calorimetric behaviour are well displayed in one material, namely isotactic polystyrene.
Figure 2.2 shows the melting endotherm of an isotactic polystyrene specimen which has been fully crystallized at 175 °C (Plans et al. 1984). The first peak I shows the melting of secondary crystals formed after the main spherulitic growth has taken place. At peak II the main crystal population starts to melt, but the melt is able to recrystallize rapidly on the still partly solid material, giving the exotherm (shaded). The crystals subsequently formed are more stable, and finally they melt giving peak III, at a temperature where any potential recrystallization is too slow to take place over the timescale of the experiment (Plans et al. 1984). Lemstra et al. (1972) previously studied this behaviour and used the DSC not only as an analytical technique, but to prepare small specimens with precise thermal history.

DSC thermogram of pure isotactic polystyrene crystallized at 175 °C for 1 h. An exotherm feature appears between peak II and peak III (Scanning rate: 1.25 °C/min). Adapted with permission from figure 3 in Plans J, MacKnight WJ and Karasz FE, Equilibrium Melting Point Depression for Blends of Isotactic Polystyrene with Poly(2,6-dimethylphenylene oxide), Macromolecules 17:810–814. Copyright (1984) American Chemical Society
Some early authors, particularly those working on poly ether ethyl ketone (PEEK ), interpreted the secondary crystallization peak, which occurs just above the crystallization temperature, as being a primary crystallization peak which was subject to melting and reorganization during the scan. The phenomenon was interpreted and a second explanation presented clearly for other polymers by Yagpharov (1986). By reheating directly from the crystallization temperature, it could be shown that the lower melting peak developed subsequently, deserving the term ‘secondary crystallization’, and that the higher melting peak was a primary peak, possibly but not necessarily subject to reorganization. Figure 2.3 below is from work conducted at the University of Reading, UK using this technique, taken from the thesis of Al Raheil (1987).

PEEK crystallized from times stated (a) cooled after crystallization (b) reheated directly from the crystallization temperature (Reproduced with permission from Al-Raheil, Thesis, 1987)
Any interpretation is, however, dependent upon the effect of a crystal’s environment on the kinetics of reorganization; in the case of PEEK , irradiation can be used to suppress reorganization kinetics (Vaughan and Sutton 1995) whereupon it appears more likely that the first-formed lamellar population is best thought of in terms of a continuous spectrum that spans the range contained between the apparently distinct primary and secondary DSC peaks. Something similar was proposed earlier (Lee and Porter 1987; Lee et al. 1989). But Lee and Porter’s range of experimental observation was far too limited to categorically reject the occurrence of distinct secondary crystallization.
Secondary crystallization is widely accepted today. For example Lee et al. (2004) write:
… we propose that secondary crystallization involves the formation of short-range molecular order in the amorphous layers of the lamellar stacks as well as in the amorphous regions between the lamellar stacks. This short-range ordered structure, which is likely a type of single thin lamella, thin lamellae, or fringed micelle-like order, has a lower electron density than the lamellar crystal formed by primary crystallization.
but in ionomers at least Loo et al. (2005) have ruled out the fringed micelle suggestion.
2.2.1.1 Reorganization and ‘Morphological Melting’
Reorganization is a very common phenomenon and can often cloud the interpretation of DSC data. Partial melting and recrystallization of a polymer specimen, generally known as annealing, gives rise to thicker crystals with a higher melting point. For specimens crystallized rapidly, this process can take place within the time frame of a typical DSC scan, often 10 min at 10 K min−1 in industrial use, typically up to eight times faster in a research laboratory.
A particular case in point is polypropylene. When isotactic polypropylene is crystallized at 145 °C and above, all the material is sufficiently stable not to undergo reorganization at usual scan rates. Typically an endotherm such as in Fig. 2.4 is seen, curve 1 (Weng et al. 2003).

DSC melting endotherms of high crystallinity polypropylene (1) crystallized from the melt for 3 h at 145 °C, (2) partially melting scan (3) fully remelted after the partial melting: From figure 3, Weng J, Olley RH, Bassett DC and Jääskeläinen P, Changes in the Melting Behavior with the Radial Distance in Isotactic Polypropylene Spherulites. J. Polym. Sci. Polym. Phys. 41: 2342–2354. Copyright © 2003 by John Wiley Sons, Inc. Reprinted by permission of John Wiley & Sons, Inc
This can then be partially melted (following curve 2) and the specimen quickly removed and quenched, so that the material has recrystallized in a much lower melting state (curve 3). The morphological effect can be seen in Fig. 2.5. The lower melting peak is seen to relate to daughter lamellae formed between the dominant ones, and especially to highly cross-hatched regions which melt out entirely.

Morphological effects of the recrystallization of isotactic polypropylene at 145 °C (a) radial growth enclosing a leaf-shaped area of cross-hatched material, (b) a similar area after partial melting (From figure 4 in. Weng J, Olley RH, Bassett DC and Jääskeläinen P, Changes in the Melting Behavior with the Radial Distance in Isotactic Polypropylene Spherulites. J. Polym. Sci. Polym. Phys. 41: 2342–2354. Copyright © 2003 by John Wiley Sons, Inc. Reprinted by permission of John Wiley & Sons, Inc.)
If a typical isotactic polypropylene is crystallized to completion at a lower temperature, say 130 °C, and heated at 10 °C/min, only one peak appears, but if heated at 40 or 80 °C/min, a double peak is observed. Even so, for polypropylene crystallized at 130 °C, a traditional DSC cannot heat fast enough, but in unpublished work we have seen that if the specimen is melted in a heating bath at temperature between the two peaks and quenched, then similar morphological effects are observed. Yamada et al. (2003b) also give an example of partial melting to reveal morphological heterogeneity.
However, the advent of superfast DSC shows that at lower crystallization temperatures, the melting difference between the two populations becomes less significant, leading to a one-peak situation (Toda et al. 2014). This technique is ideal for investigating the equilibrium melting point of isotactic polypropylene, but careful work even with traditional DSC (Yamada et al. 2003a) can be used to deduce reorganizational effect and thereby determine this temperature.
2.2.1.2 Thermal Fractionation
Polypropylene is a particularly marked example of how even a relatively homogeneous homopolymer can crystallize isothermally giving a variety of crystal populations with different melting points. A phenomenon generally observed is that the first-formed lamellae, called dominant because they form the basis of the spherulitic architecture, tend to be higher in melting temperature than the subsidiary ones which form later. The spread of melting points in the DSC (Gedde and Jansson 1983) can be accompanied by solvent extraction and gel permeation chromatography (Gedde et al. 1983) to show that in a homopolymer (in this case polyethylene) the crystallization process is accompanied by molecular fractionation, with the lower molecular weights crystallizing more slowly and being concentrated in subsidiary lamellae (Gedde and Jansson 1984).
Fractionation during crystallization occurs in copolymers also, thinking particularly of those where one comonomer constitutes 90 % or more of the monomer mixture. In these there is generally molecular heterogeneity with some molecules having greater comonomer content than others. One example is the commercially available propylene-ethylene ‘random’ copolymers (Weng et al. 2004) but the most significant by industrial volume are the medium and linear low-density polyethylenes. Different groups have developed their own particular thermal fractionation techniques using the DSC to investigate the heterogeneity of these materials (Arnal 2001; Müller and Arnal 2005; Shanks and Amarasinghe 2000; Chen et al. 2001).
It also gives indications as to how much blends of two different grades of polyethylene co-crystallize in blends. Figure 2.6 below is a comparison of a blend of two branched polyethylenes: at left is the melting endotherm after simple cooling, at right after a thermal fractionation technique.

Use of the thermal fractionation technique to study branching polydispersity. (Left) Specific heat curves of LLDPE–VLDPE2 blends after cooling at 10 °C/min; (right) the same after thermal fractionation (Reprinted from Polymer 41: 4579–4587, Shanks RA and Amarasinghe G. Crystallisation of blends of LLDPE with branched VLDPE Copyright (2000), with permission from Elsevier)
The Vaughan and Sutton (1995) paper is an example of how DSC can also be used to probe molecular degradation following irradiation. This can occur without gross apparent changes to morphology as revealed by TEM/etching . The paper immediately following Vaughan and Stevens (1995) does the same for PET, which is similar to the above-mentioned isotactic polystyrene in crystallization behaviour, although the processes occur considerably faster. However, the most salient feature of that work was that PET is easily degraded by relatively small doses of irradiation, especially compared to the highly radiation-resistant PEEK.
2.2.2 Dynamics
Dynamic Mechanical Thermal Analysis (DMTA ) records the temperature-dependence of the viscoelastic properties and determines the modulus of elasticity and the damping values by applying an oscillating force to the sample. Changes in the morphology such as crystallization and phase separation change the viscoelastic properties. For example, Zheng et al. have used DMTA techniques in conjunction with infrared spectroscopy (see Sect. 2.2.3) to study the interactions in semi-interpenetrating polymer networks (Zheng et al. 2013). Dielectric spectroscopy measures the dielectric properties of a sample as a function of frequency. It is based on the interaction of an external oscillating field with the electric dipole moment of the sample. It is a technique for polar molecular systems and has been widely used to study molecular relaxations. Nanocomposites have a significant interfacial area and this has a substantial impact on the dielectric properties and hence dielectric spectroscopy is a very useful tool. A recent example of this concerns the study of a nanosilica/polyethylene system, where due to the lack of strong coupling between the applied field and the materials themselves, water was used as a dielectric probe. Since the solubility of water in polyethylene is extremely low, water molecules tend to associate with residual hydroxyl groups on the nanosilica surface, such that changes in surface structure and interfacial interactions manifest themselves as modified relaxation peaks within the so-called water shell (Lau et al. 2013).
2.2.3 Spectroscopy
Many different spectroscopy techniques can be used to provide structural information concerning polymers. In terms of vibrational spectroscopy, Strobl and Hagedorn (1978) showed how the Raman spectrum of polyethylene could be considered in terms of characteristic bands associated with the crystalline phase, a melt-like amorphous phase and a disordered phase of anisotropic nature. Comparison of structural data obtained in this way with equivalent measures obtained by densitometry and X-ray scattering revealed agreement. Similar approaches have been used to evaluate the crystallinity of isotactic polypropylene (Nielsen et al. 2002).
Nuclear magnetic resonance spectroscopy is a well-established chemical characterization technique widely exploited with polymer solutions to quantify the chemical microstructure of polymer chains (Bovey 2007). Developments in NMR technology and computing led to the development of solid state NMR. This enables site-specific information to be obtained from the solid state. This uses interactions like the dipolar coupling and chemical shift anisotropy to extract information from the solid state. More recently, multidimensional spectroscopy (2D and 3D techniques) correlate a structural parameter such as the chemical shift with parameters that carry information about dynamics and the level of order. Solid state NMR is often seen as strongly connected with crystallography as it provides similar precision information albeit of site-specific interactions and their distances (Schmidt-Rohr and Spiess 1994). As introduced in connection with dielectric spectroscopy, the nature of the interface between polymers and nanoparticles is an area that is of considerable interest and many different spectroscopies have been used to explore this. For example, Miwa et al. (2008) used electron spin resonance (ESR) spectroscopy to examine nanocomposites of poly(methyl acrylate) (PMA) and synthetic fluoromica, in which the PMA had been modified to include a nitroxide spin label. That is, a stable free radical introduced into a material that does not have an intrinsic paramagnetic response. This work demonstrated that, in exfoliated systems, the close proximity of the polymer chains and exfoliated fluoromica reduces the PMA chain mobility within a rigid interface region of thickness 5–15 nm.
2.3 Imaging Methods
Imaging techniques provide invaluable information on the morphology of polymers and nanocomposites. Of course an imaging technique is able to provide information on the spatial distribution of the structure and morphology. Each of the techniques considered here has particular advantages as well as specific sample preparation requirements. It is probable that the techniques employed in a particular project are selected as a consequence of the resolution matching the requirements, otherwise all techniques should be considered useful and effective especially when used in combination.
2.3.1 Light Microscopy
The optical microscope is typically used to reveal structures between 1 μm and 1 mm in size. Although the most intricate details of polymer morphology are revealed by the transmission electron microscope, and to some extent the scanning electron microscope and atomic force microscope, these tiny structures are usually aggregated into larger structures; for example, the spherulites in Fig. 2.7a (left) are composed of lamellae which require electron microscopy for their resolution, but the variation in size and spatial distribution of the spherulites is much more easily studied by optical microscopy.

(a) Spherulites in a thin film of polypropylene (Elaine Ann Perkins, University of Reading, UK, unpublished work, 1985), (b) spherulites in polyhydroxybutyrate-co-valerate (Reprinted with permission from Macromolecules, 2010, 43: 4441–4444 Wang Z, Li Y, Yang J, Gou Q, Wu Y, Wu X, Liu Pand Gu Q (2010) Twisting of Lamellar Crystals in Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) Ring-Banded Spherulites Copyright (2010) American Chemical Society, (c) Schematic of spherulite twisted orientations. Redrawn from Hsieh et al. (2014)
Optical microscopy involves a variety of different techniques, and a survey of these will illustrate the breadth of applications to polymer morphology.
The methods of specimen preparation for optical microscopy are various. In fundamental polymer studies, thin films of polymers can be crystallized between a slide and cover slip. In an apparatus such as a hot stage attached to a microscope, the crystallization process can be followed as it occurs. However, two features must be taken into account, firstly that in specimens where large spherulites form, their centres are confined to a narrow plane and are not distributed in depth as they would in a bulk specimen. Also, nucleation of crystal growth may be enhanced on the surface of the slide and cover slip, and transcrystalline layers, rather than the spherulitic structure of the bulk material, may form.
Most often, sectioning is required to examine structure of the material as a whole. For example, there will be great variation in morphology of an injection moulded item between the rapidly chilled surfaces where the polymer came in contact with the cold mould, and the interior where heat was lost much more slowly. However, sections do not have perfectly flat parallel surfaces; the most common form of damage is periodic compression giving rise to ‘chatter’, though with an inferior blade knife marks parallel to the cutting direction also occur. Interference from these can be greatly reduced by immersing the specimen in a liquid with a refractive index close to that of the polymer. For aliphatic polymers, glycerol (n = 1.47) is often a good general purpose liquid, since it is not taken up except by the most polar of polymers such as hydroxyethyl acrylate. For aromatic polymers such as polystyrene, Traylor (1961) dissolved potassium mercuric iodide in the glycerol to increase its refractive index, and there are now many commercial index matching fluids. Sections can also be compressed in such a way that originally circular features such as the bands in spherulites are distorted into ellipses; such distortion is accompanied by an overall background birefringence.
2.3.1.1 Polarized Optical Microscopy (POM )
One of the earliest and most commonly used microscopical methods of examining polymers is between crossed polarizers. Some of the earliest work was determining the birefringence of fibres, then came the study of spherulites in semi-crystalline polymers. Often the spherulites show a simple ‘Maltese Cross’ pattern where the dark areas show zero-amplitude birefringence, which simply arises from the orientation of the crossed polars. In the polypropylene spherulites (Fig. 2.7a), radial growth has occurred along the a*-axis which is the fastest crystal growth direction, while the b- and c-axis are effectively randomly oriented.
Often, however, the spherulites develop with a twisting orientation (though the individual lamellae are not necessarily twisted) producing a banded pattern when observed under POM . This is illustrated in Fig. 2.7c. For example, in polyethylene the b-axis forms the radial growth direction, but under certain growth conditions the a- and c-axis rotate about the radius with increasing distance from the centre. In polyethylene, the crystals are effectively uniaxial, with the higher refractive index along the chains, namely the c-axis. Looking down at A, the two different refractive indices along b and c cause the crystal to show birefringence. Looking down the c-axis at B, the refractive indices along a and b are effectively equal. This gives a simple alternation of light and dark bands, with the dark representing zero-birefringence extinction. Many other polymers, including many polyesters, are biaxial, and often show double banding (Wang et al. 2010) as observed in Fig. 2.7b.
Figure 2.8 (Kumaraswamy 1999) shows how POM can display orientation and structure gradients in injection moulded materials. These images show polypropylene crystallized in a cell designed to probe the different variables (time, temperature, shear rate) found in this process, and the two parts of the figure illustrate the formation of the characteristic skin-core morphology. The oriented near-skin region appears bright when viewed down the neutral direction (c) and dark when viewed down the flow direction (d) indicating that the skin has crystallites with the cylindrical symmetry characteristic of polypropylene row structures.

Optical micrographs of sections through injection moulded polypropylene from figure 4 in Kumaraswamy, 1999 (c) viewed down the neutral direction (d) viewed down the flow direction Reprinted with permission from (G. Kumaraswamy, A.M. Issaian, and J.A. Kornfield Shear-Enhanced Crystallization in Isotactic Polypropylene. 1. Correspondence between in Situ Rheo-Optics and ex Situ Structure Determination, Macromolecules, 32, 7537–7547). Copyright (1999) American Chemical Society
2.3.1.2 Use of Tint Plates
A bright appearance between vertically and horizontally cross polars indicates that, in the diagonal directions, one refractive index is greater than the other, but does not indicate which is which. The ambiguity can be resolved by use of tint plates, of which the first-order compensation plate (quartz sensitive tint) is the most popular. This shifts one component of the beam by one green wavelength (550–580 nm), so that without a specimen, green light destructively interferes, and mainly red and blue are observed, giving rise to the characteristic magenta colour. One direction of the plate will be marked as slow or positive, and if the higher refractive index is parallel to this, a higher order blue interference colour will be produced, while if it is perpendicular, a lower order yellow colour will be seen.
Spherulites of aliphatic hydrocarbon polymers such as polyethylene generally have their molecular chains with the highest refractive index oriented perpendicular to the radial growth direction. This is the case with the polypropylene spherulites shown in Fig. 2.9a, which were crystallized at 140 °C and consequently contain very few cross-hatched lamellae, so in this respect they are like polyethylene spherulites. The lower refractive index in this view is radial in direction, giving a yellow colour where the radius is parallel to the slow direction of the tint plate; these are therefore termed negative spherulites.

Showing use of Quartz sensitive tint . (Left) polypropylene crystallized at 140 °C (−ve spherulite); (right) various orientations in film transcrystallized at 120 °C (+ve spherulites with −ve beta inclusion) (Reproduced with permission from White HM, University of Reading thesis, 1995)
In Fig. 2.9b, the specimen was crystallized at 120 °C, so that there is a preponderance of cross-hatching lamellae located roughly perpendicular to the dominant radially growing ones. This reverses the birefringence, giving positive spherulites in the majority α-phase. However, the isolated inclusion of the β-phase in the transcrystalline layer (t) does not contain any cross-hatching, and it displays strong birefringence characteristic of a negative spherulite.
One hydrocarbon polymer that gives positive spherulites is isotactic polystyrene; here the effect of aromatic rings with strong polarizability perpendicular to the molecular chains dominates over the polarizability of the chain.
Another form of tint plate commonly used is the mica quarter-wave plate with a retardation of 140–155 nm. This gives a grey background becoming brighter or darker with the birefringence of the specimen, and is suitable for black-and-white imaging.
2.3.1.3 Circularly Polarized Light
Dark areas observed in the use of crossed polarizers by themselves can be due to zero-amplitude or zero-birefringence extinction, and sometimes it is advantageous to remove the zero-amplitude extinction. This can be achieved by including a pair of crossed quarter-wave plates in the optical path (Olley and Bassett 1989). When a spherulitic sample is observed in the way, the dark Maltese cross is no longer part of the picture, and one can see the spherulites as bright objects with their mutual boundaries much more clearly displayed. This is particularly helpful when looking at the early stages of spherulitic growth, where the growing object takes a form often called a ‘hedrite’ or ‘axialite’, or in the special case of isotactic polypropylene, a ‘quadrite’. Figure 2.10 shows a film of polypropylene with quadrites in various orientations, with the top picture displayed in circularly polarized light. The object at top left is seen in one of the ‘cardinal’ views, with the b-axis of the earliest formed crystals pointing towards the observer. Without the Maltese cross, the fourfold rosette form of the object is clearly displayed. Moreover, the dark ‘leaf-like’ structure at the centre is a characteristic feature of the development. However, the general brightness indicates that over most of the object, the optic axis lies in the plane of the film, perpendicular to the b-axis. The two objects beneath appear very dark and sheaf-like in form, but were they developing freely in a bulk specimen they would be the same in form as the one above, but with their b-axis located in the plane of the film, and their optic axis (effectively the c-axis) pointing towards the observer.

Use of circularly polarized light (top) and phase contrast (bottom) to show development of polypropylene spherulites at 150 °C (Reprinted from Polymer 30: 399–409, Olley RH and Bassett DC. On the Development of Polypropylene Spherulites, Copyright (1989), with permission from Elsevier)
Where spherulites of more than one crystal phase are growing together, use of circularly polarized light makes delineating and distinguishing the different types of object much easier (He and Olley 2000).
2.3.1.4 Phase Contrast Microscopy
This refers to the technique invented by Zernike, which makes visible the phase shifts produced in a transparent specimen by local variations in refractive index. For the technique as applied to polymers, see Hemsley (1989), Chapter 2; for a description of how the technique works, see Murphy and Davidson (2012), Chapter 7.
An ideal phase contrast specimen is either a well-prepared section or a thin film melted between slide and cover slip. Much of the problem of knife marks or other surface structure in sections and thin films can be alleviated by ‘oiling out’ with a suitable liquid (Traylor 1961).
The method is very suitable for examining particle dispersions and inclusions, especially the rubbery inclusions in toughened plastics. The method is sensitive, and does not involve polarized light, so can reveal features in specimens with overall or localized orientation which might be lost where there is strong birefringence contrast. An example is the clear revelation of the outlines and some internal structure in the early development of polypropylene spherulites, as in the lower part of Fig. 2.10. However, a halo is produced around the objects picked out, and where there is a large concentration of particles, or in thicker specimens, the overlapping haloes can confuse the picture.
An early application was the examination of polyethylene crystals formed from dilute solution, which are ‘phase objects par excellence’ (Keller 1968). As is generally the case, the technique proved an ideal precursor to electron microscopy (Keller and Bassett 1960). Quite recently, the technique has been used to observe the development of the earliest stages of polypropylene crystals directly in the melt (Yamada et al. 2011).
2.3.1.5 Interference Microscopy (Nomarski)
There is a variety of techniques which come under the heading of interference microscopy, and one early example is the determination of the thickness of single crystals with a two beam microscope (Wunderlich and Sullivan 1962).
However, the most widely used type of interference microscopy is that giving Differential Interference Contrast (DIC ), widely known as Nomarski contrast.
The Nomarski technique is directional, because the very small separation of the two beams, about 1 μm, samples small differences in height along the splitting direction only. So, for example, if one is examining a piece of polypropylene tape whose surface is covered with ridges parallel to the tape length, then if the specimen is rotated so that the ridges are perpendicular to the beam separation, they will appear heavily contrasted, but if they lie parallel, then they will almost disappear.
In the system used by the author, the two beams were separated along a diagonal in the field of view. Figure 2.11 shows how they could be used to determine whether a blemish in an otherwise flat surface was a pit or a dome.

Appearance of domes and pits in an otherwise flat surface under Nomarski: for description see text
Normally the microscope can be adjusted so that, on a flat surface, the two beams were out of phase by 1 wavelength of green light, so that the surface appeared first order purple (the same colour as given by a quartz sensitive tint). Higher or lower order colours appeared according to whether the slope on the element of surface being sampled increased or decreased the phase difference. At right, the microscope is adjusted to give a smaller path difference, grey on the Newton colour scale, and this configuration is suitable for black-and-white photography.
This technique works very well for solution-grown crystals, but much better contrast is achieved if a reflective gold (or carbon) is applied. If the crystals are deposited on mica and carbon coated, they can be floated off for electron microscopy.
Where the surface itself is of interest, this is the technique par excellence. For example, blown polypropylene films used in food packaging sometimes show submillimetre ‘haze rings’, which degrade their appearance. These were studied by, among other techniques, Nomarski reflection, and related to the presence of defects in the polypropylene tube prior to blowing (Olley and Bassett 1994).
One very productive use of Nomarski contrast is examination of etched surfaces prior to electron microscopy. Here one does not want to coat the specimen, so in looking at an etched disc prior to replication, it is better to put it on some black card (surface to be replicated uppermost) to eliminate much of the scattered and reflected light from below.
Figure 2.12 shows (a, left) a micrograph (crossed polars) of section from a specimen of poly ether ether ketone (PEEK) taken as it was being prepared for etching, and (b, right) the etched specimen examined under Nomarski contrast. First to notice is the strong correspondence between the morphology of the section and the etched morphology, though it is not exact because of a small depth difference in the specimen. The asterisks at the top mark a pair of ‘comet-like’ spherulites which are easily discerned in both views.

Section of PEEK film and etched stub from which section was taken (Reprinted with permissions from Al-Raheil IAM, University of Reading thesis, 1987)
This pair was taken during the development of the etching procedure, and besides the spherulitic morphology displays some small artefacts (arrowed) which were eliminated with the reagent as eventually published.
This technique is also very useful for examining replicas of etched specimens after they have been extracted and placed on number grids, enabling one to locate specific features under the transmission electron microscope.
2.3.1.6 Ancillary Techniques
One advantage of optical microscopy is that it is easily used in conjunction with ancillary techniques, where heating, stretching and other processes are applied to the specimen. Various manufacturers supply hot stages in which the specimen can be studied while being heated or cooled, allowing direct observation of crystallization and melting processes, and phase separation in blends (Shabana et al. 2000).
As well as simple mechanical testing, it is also possible to study the evolution of morphology under shear conditions, again both in terms of crystallization (Shen et al. 2013; Chan and Gao 2005) and phase separation and ordering in blends (Lin et al. 2012; Zou et al. 2012; Trindade et al. 2004; Kielhorn et al. 2000).
In the authors’ experience, the optical microscope is an invaluable first port of call, even when the target is the finest level of morphology only resolvable by the transmission electron microscope. It is best to start with a very low-power objective, and to move to higher ones. This practice can show up variations in structure throughout a specimen which might totally be missed when going straight to the higher power technique. In addition, sometimes it becomes apparent that what one is looking at is not what one is looking for; it may reveal films or deposits of foreign material, or even that the specimens may have been mixed up. To quote from Hemsley (1989): ‘In no sense is the light microscope the poor relation of the electron microscope, neither is the latter to be considered by the light specialist only when all else fails …. One hesitates to insist that any examination with an electron microscope should be preceded by light microscopy, but polymer science and technology would benefit if this were a more frequent procedure’.
2.3.2 Scanning Electron Microscopy
Scanning electron microscopy (SEM) is a widely available technique in which a fine electron beam is rastered across the sample surface. The electrons interact with atoms in the surface component of the sample, producing various signals (Fig. 2.13) that can be detected and that contain information about the sample’s surface topography and composition. The electron beam is generally scanned in a raster scan pattern, and the beam’s position is combined with the detected signal to produce an image. The so-called secondary electrons are the most commonly used signal. These are produced by excitation of atoms close to the sample surface or in the gold coating often applied to insulating samples to dissipate the build-up of charge. These electrons are of low energy and hence originate in the surface layers. In contrast, the back scattered electrons, electrons from the incident beam that have been scattered by the atoms in the sample, are of high energy and may come from much deeper in the sample as shown in Fig. 2.13. In this case, the efficiency of scattering depends on the atomic number, the larger the value of Z the greater the scattering. As a consequence, an image formed from back scattered electrons may reveal elemental composition variations which can be helpful in revealing the structure of composites. However, due to the larger interaction volume, the resolution of the image will be lower than that formed using secondary electrons. The electrons in the probe beam can excite characteristic X-rays from the atoms in the sample. Which X-rays are excited will depend upon the accelerating voltage. With a suitable energy dispersive X-ray detector images can be formed from the distribution of particular elements in the sample. Again because the interaction volume is large, the resolution of the image will be lower. Recently techniques have been developed which provide for elemental imaging at the nanoscale.

Interaction of the electron beam in a SEM with the surface layers of the sample. The grey area shows the interaction volume and the location of the origin of the signals used in a SEM to construct an image
2.3.2.1 Contrast
As the SEM is a topological technique, we need to reveal the morphology of the sample. If there are compositional variations, then the images formed by characteristic X-rays or by back scattered electrons may suffice. However, if this is insufficient, another very common approach is to prepare a fractured surface. Here the samples are broken under brittle conditions, often by first freezing in liquid nitrogen, then fracturing to reveal the variation in morphology. For polymer blends and mixtures, the staining technique often used in TEM (discussed under TEM) may be helpful in providing a contrast to discern between materials. Another approach is to etch the surface, preferentially removing material and revealing the underlying morphology. The etching technique is described in the following section.
2.3.2.2 Sandwiching and Embedding Techniques
While often simply exposing a microtomed surface is sufficient for the purpose, sometimes specimens require support, especially thin films which cannot stand by themselves and where the top and bottom layers would be removed by the etchant. Sandwiching techniques have been developed, whereby the top and bottom surfaces are supported and protected by a material which can be etched along with the polymer specimen. An example is a work on woven polypropylene tapes, both before and after consolidation (Jordan et al. 2003).
Fibre and weaves can be embedded in a mounting medium which can be etched along with the polymer composing the fibre (Abo El Maaty et al. 1999). Details of such a technique are shown in the Fig. 2.14 below.

A comparison of two sandwiching techniques , applicable to solid specimens (left) and those with open texture (right). Adapted from Jordan et al. (2003)
In the micrographs below, an original weave is shown in a sandwich. At left, A is the high-impact polystyrene and B is the epoxy resin (Fig. 2.15).

SEM of etched polypropylene weave specimens, showing (left) sandwiching technique (right) different structures of polypropylene tapes in transverse and longitudinal directions (Image taken by Robert Olley, 2003, University of Reading, UK. The specimen was provided by the University of Leeds)
At right, in the regions marked C, one tape is emerging, so an etch cross-section of the tape is seen. At D, another tape is running parallel to the surface.
2.3.3 Etching
Both SEM and TEM (to follow) generally require some preparation to make the morphology visible. The etching technique is equally applicable to both, and so is included here between the two sections.
Etching of polymers is very similar to the process in metallography, where a surface is treated with a chemical agent to reveal the underlying phase or crystalline structure, which are then studied under the electron or optical microscopes. A variety of etching procedures are reviewed in (Bassett et al. 2003).
Most etching techniques involve a liquid etchant. Most commonly used are permanganic reagents, generally consisting of a dilute solution of potassium permanganate in a mixture of sulfuric and orthophosphoric acids, either dry or with a certain amount of water. Different compositions are suitable for different classes of polyethylene (Shahin et al. 1999), other polyolefins (Patel and Bassett 1994; Weng et al. 2003, 2004), isotactic polystyrene (Bassett and Vaughan 1985), PEEK (Olley et al. 1986), etc. This is the most common of the oxidizing etchants, though chromic reagents are also used. Polyesters are generally treated with an alkaline reagent, typically a solution of potassium hydroxide in an alcohol.
Etching of a semi-crystalline polymer can be visualized by a simple model of plate-like crystals embedded in a rubbery matrix. Three etching rates are observed: the fastest etching is the removal of the amorphous polymer constituting the matrix; the broad basal surfaces of the plate-like crystals are almost totally resistant to etching, but the thin side-surfaces are eroded at an intermediate rate (Fig. 2.16). This is described more fully in Olley (1986).

Schematic of etching of a polymer crystal: (a) free molecular end (cilium); (b) tight chain fold; (c) loose chain fold; (d) short stem freed from side surface by chemical attack (Reproduced with Permission from Olley 1986)
The morphology of banded spherulitic polyethylene as in Fig. 2.17 (left) illustrates this well. The orientation of crystals rotate periodically along the spherulite radius; at A the resistant basal surfaces of the crystals are seen standing proud of the general etched plane; at B the lamellae are displayed edge-on, so are somewhat less resistant to the etch. The amorphous regions are more highly etched, and lie below the protruding crystalline parts (Shahin and Olley 2002).

(Left) etched banded spherulitic polyethylene (From figure 6c in, Shahin MM and Olley RH. Novel Etching Phenomena in Poly(3-hydroxy butyrate) and Poly(oxymethylene) Spherulites. J Polym Sci Part B: Polym Phys 40: 124–133. Copyright © 2002 by John Wiley Sons, Inc. Reprinted by permission of John Wiley & Sons, Inc.); (right) Original caption: TEM micrographs showing the morphology of sample BPA 124/20 (20 % LPE). It is clear that distinct boundary regions exist between the lamellar aggregates that grow at 124 °C. J. Mater. Sci.,1997, 32, (17), 4523–31, Structure-property relationships in polyethylene blends: the effect of morphology on electrical breakdown strength. Hosier, I. L., Vaughan, A. S. and Swingler, S. G, Figure 8 Reprinted with kind permission from Springer Science and Business Media
The specimen at right in Fig. 2.17 is an example of crystallizing a polyethylene blend as a proposed cable material under a controlled thermal regime in order to optimize the morphology in regard to electrical breakdown behaviour (Hosier et al. 1997). We have included a TEM image here as the higher resolution is useful in identifying the features. Large spherulitic objects made largely of high-density PE are revealed in a matrix where low-density PE predominates.
Etching is used to display not only semi-crystalline morphology, but also multiphase structure in polymer blends which may be totally non-crystalline. An early example is work on high-impact polystyrene and similar materials by Bucknall et al. (1972). From the abstract: ‘These techniques reveal details of orientation in injection mouldings, of internal structure in composite rubber particles, and of crazing and shear band formation. The etch method avoids the specimen distortion inherent in sectioning’. A more recent example is one following the spinodal decomposition of blends of tetramethyl polycarbonate and polystyrene (Shabana et al. 1993). During blend decomposition, one or more of the components may crystallize, and the crystal morphology as well as the phase structure can be observed, for example, in a phase-separating copolymer of polyethylene naphthoate with polyhydroxybenzoate (Shabana et al. 1996) or a blend of polycaprolactone with oligo-styrene (Shabana et al. 2000). The technique may also be used to reveal texture in liquid crystal polymers (Ford et al. 1990).
2.3.4 Transmission Electron Microscopy
In transmission electron microscopy (TEM) , a beam of electrons is passed through a thin sample, such that an image is formed as a result of absorption or diffraction contrast. In the case of polymers, a combination of disorder and radiation sensitivity means that, of these, absorption contrast is most important, in which case, high resolution images can be generated where image contrast is based on the spatial variation in electron density. In the case of materials such as nanocomposites, the distribution of the nanoparticles can therefore easily be imaged, as a result of the difference in the atomic number between the nanoparticles and the matrix polymer, as shown in Fig. 2.18.

Colloidal silver embedded in an epoxy matrix (Image provided courtesy of Dr Suvi Virtanen/Alun Vaughan, University of Southampton, UK)
However, obtaining useful structural information concerning the lamellar texture of semi-crystalline polymers or the phase distribution in polymer blends or block copolymers is often complicated by the fact that the variation in electron density between the structural components of interest is insufficient to provide meaningful image contrast. In such circumstances, additional sample preparation procedures, beyond ensuring that the sample is sufficiently thin, are required.
2.3.4.1 Inducing Contrast
If etching of polymers, as described above, is equivalent to similar process in metallography, then staining has analogues in biology, where the very same TEM contrast problems exist. Essentially, staining involves exposing a polymeric sample to a chemical reagent, which diffuses into the structure and becomes preferentially located at certain sites. The classic example of this concerns studies of the structure of rubber toughened polymer blends in which osmium tetroxide (OsO4) reacts with and preferentially stains unsaturated rubbery phases. The more aggressive analogue, which consequently has greater utility, is ruthenium tetroxide (RuO4); Trent et al. (1983) provide an excellent overview of the staining methodologies and polymer types where RuO4 has been applied with success. While many different stains have been developed to enhance image contrast in the TEM, a major issue with such reagents concerns their aggressive nature, which raises safety concerns during use and in disposal. In the case of staining reagents such as OsO4 and RuO4 that can be used to stain polymers in the vapour phase, the staining device described by Owen and Vesely (1985) has been found to be extremely convenient. A related issue concerns ‘overstaining’, whereby exposure to the stain introduces artefacts into the specimen (Chou et al. 2002). Figure 2.19 shows images of two different materials stained with RuO4; (a) from Trent et al. (1983) is a spherulitic morphology in a sample of high density polyethylene while (b) is a section from a block copolymer of polyethylene and atactic polypropylene (Hong et al. 2001); the amorphous atactic polypropylene is much more heavily stained overall than the semi-crystalline polyethylene in which the stain has also revealed the fine semi-crystalline lamellar morphology.

TEM micrographs of (a) a spherulite centre in a thin film of high density polyethylene stained with RuO4, (b) section of polyethylene-atactic polypropylene block copolymer stained with RuO4. Reproduced with permission (a) from Figure 15 in Trent, Scheinbeim and Couchman, Macromolecules, 1983, 16: 589–598 (Copyright 1983) American Chemical Society. (b) from Figure 3 in Hong, Copyright (2001) with permission from Elsevier
The etching techniques described above in connection with SEM can also be used for TEM, in conjunction with so-called replication. In this, an accurate facsimile of the sample’s surface topography is produced, frequently in the form of a thin carbon film, which is then coated at an oblique with an electron dense metal (shadowed). As a consequence, the distribution of metal on the surface is non-uniform and directly related to the surface topography of the specimen and, hence, the morphological features present within it. Figure 2.20a shows an equivalent structure, as revealed by etching followed by replication, to the lamellar texture shown in Fig. 2.20b which was prepared by staining with chlorosulfonic acid and sectioning. Fig. 2.20a represents a case of a direct replica, where the carbon and shadowing metal are applied directly to the etched polyethylene. Removal of the replica is typically performed using a polyacrylic glue (Morgan et al. 1999) or polyacrylic acid. Two stage (indirect) replicas are more commonly used; a thin sheet of cellulose acetate is applied to the etched surface, peeled off and coated with shadowing metal and carbon. The replica is then placed on top of a TEM grid and freed by dissolving the cellulose acetate away with acetone. Weng et al. (2003, 2004) showed examples of replicas prepared by this technique.

TEM micrographs of 60 % LPE/LDPE blends cooled at 1 °C/min. (a) Sample prepared through etching and replication Scale bar is 1 μm (b) Stained sections, cut from material fixed with chlorosulfonic acid. Scale bar is 200 nm. Reprinted from Polymer, 40:337–348, Morgan RL, Hill MJ and Barham PJ, Morphology, melting behaviour and co-crystallization in polyethylene blends: the effect of cooling rate on two homogeneously mixed blends, Copyright (1999), with permission from Elsevier
2.3.5 Atomic Force Microscopy
Scanning probe microscopy involves a family of techniques in which atomic force microscopy (AFM) is the most popular which is relevant to this work. Scanning tunnelling microscopy can yield atomistic level images under good conditions. In AFM, the so-called contact mode provides an image of the surface topology. The resolution can approach the nanometre scale under ideal conditions but tip adhesion may limit the usefulness with soft materials such as polymers. The non-contact method involves an oscillating cantilever and under appropriate conditions, images of the spatial distribution of different phases or material can be formed where the contrast essentially arises from the differences in the stiffness of the phases.
Although for the highest resolution care has to be taken to isolate vibrations and screen electric fields, AFM is essentially performed in the laboratory environment. As such, it is possible to perform experiments such as heating and cooling in the AFM and the sample does not require treatment to reveal the morphology as in the case of SEM. Time-resolving studies are now possible with the latest developments and this has been used to good effect by Hobbs et al. to observe the growth of polyethylene crystals (Hobbs 2007). Further developments to the technology have allowed for the use of higher frame rate AFM, termed a video AFM, which has been utilized to good effect to show the growth of polyethylene oxide crystals (Hobbs et al. 2005).
2.4 Scattering Methods
Scattering methods are a collection of powerful techniques for evaluation the molecular organization of materials including polymers. One of the earliest applications of X-ray diffraction at the start of the twentieth century was with polymers. There is considerable diversity in the instrumentation available for use with polymers using both laboratory sources and synchrotron national facilities. The type of instrumentation used relates to the type and scale of the information required. Figure 2.21 shows a schematic of the scales of structures which can be studied using readily available equipment.

Various length scales examinable by scattering methods (Reproduced with the permission of the author of the chapter)
In a typical scattering experiment, the incident beam on a sample is elastically scattered by the sample through an angle, θ, described by the equation |Q| = Q = 4πsin(θ)/λ, where 2θ is the scattering angle, λ is the wavelength of the incident beam and Q is the scattering vector. The angle through which the beam is scattered will determine the structural information that can be discerned as shown Fig. 2.21. For example, small angle scattering can be utilized to determine features on the length scales of several to a hundred nanometres, such as the long period and lamellae thickness, whilst wide angle examines the features from several nanometres down to angstrom levels where it can be used in determination of the crystalline unit cell, crystal size and the polymer chain conformation.
2.4.1 X-Ray Scattering
X-ray scattering arises from electrons where the observed intensity of scattering is dependant on the number of electrons present on an atomic species contained within the sample (I ~ Z 2), hence heavier elements scatter more intensely than lighter ones.
2.4.1.1 Small Angle X-Ray Scattering
Numerous small angle X-ray scattering (SAXS ) experiments have been conducted on extracting information regarding the long period which, with knowledge of the degree of crystallinity, can be used to calculate the crystalline and amorphous thicknesses from SAXS data. This is done by considering the material as a two-phase system of periodic alternatively stacked lamellar crystals and amorphous regions. The methodology utilizes a Fourier transform of the data to extract an interface distribution function (Ruland 1977) or the correlation function (Strobl and Schneider 1980) containing the information on the materials long period. Strobl used the correlation function to extract information from polyethylene SAXS data with further consideration of how inhomogeneities and lamellar curvature would impact the results (Strobl and Schneider 1980). Some knowledge of the crystallinity is required in order to determine the values for the crystal and amorphous thicknesses, which would make the combination of wide angle X-ray scattering and SAXS a useful tool in a thorough analysis of a material. An example of a 2D SAXS pattern, and the corresponding data reduction to a one-dimensional plot of intensity as a function of Q, is provided in Fig. 2.22. The scattering pattern was obtained from an annealed fibre of a crystallizable copolymer of l-lactide, ε-caprolactone and glycolide. The 2D scattering pattern exhibits highly anisotropic features (Channuan et al. 2005).

SAXS curve of a crystallizable copolymers of l-lactide, ε-caprolactone and glycolide. Wavelength of X-rays are 1.4 Å. Reprinted from figure 9a in Polymer 46 (17), 6411–6428 W. Channuan, J. Siripitayananon, R. Molloy, M. Sriyai, F.J. Davis and G.R. Mitchell, The structure of crystallisable copolymers of l-lactide, ε-caprolactone and glycolide, Copyright (2005) with permission from Elsevier
Another instance where SAXS has been a useful tool for polymer characterization relates to the examination of block copolymers. Block copolymers can form structural domains that are on length scales detectable by the small angle scattering technique. A series of scattering peaks occur with each peak position having a separation indicative of the structure formed. Examples of this would be if a peak occurred at Q = 1 Å−1, reflections would occur at 2, 3, 4, 5,… for lamellar domains, whilst body centred cubic will have peak positions √2, √3, √4, √5,… (Hamley and Castelletto 2004).
The use of SAXS as a characterization technique for polymeric materials has been extended to include studies of liquid crystal polymers, polymer blends, nanocomposite materials, polymer gels and solutions (Chu and Hsiao 2001).
2.4.1.2 Wide Angle X-Ray Scattering
Wide angle X-ray scattering (WAXS ) provides information on the unit cells where the data obtained can be utilized in determination of the lattice parameters, hkl reflection planes and hence the lattice structure i.e. face centred cubic, hexagonal, etc. In addition to the unit cell information, the level of crystallinity can be determined by considering the polymer as a two-phase material, amorphous and crystalline. The ratio of the second moment of the data corresponding to the sharp crystalline peaks to the total second moment of the scattering curve provides the degree of crystallinity of the sample from the scattering data. However, suitable corrections for incoherent backgrounds, air scattering, thermal motion and lattice imperfections need to be accounted for (Ruland 1961, 1964). Ruland first applied this method to determine the degree of crystallinity in nylon 6 and 7 (Ruland 1964). An example of a wide angle scattering curve is observed below in Fig. 2.23 for a sample of polyethylene oxide.

WAXS Curve of Polyethylene oxide (Mv—100,000 Da). Data was obtained on a powder X-ray diffractometer with the sample in a powdered form at room temperature. X-ray wavelength used was 1.54 Å
2D WAXS patterns can be used to determine the direction of preferred orientation and the orientation distribution function or a set of orientation parameters, P 2n , which describe the orientation distribution function D(α).
where P 2 = (3cos2 α − 1)/2
In many works, attention is focused on 〈P 2〉, although X-ray scattering is a technique able to provide quantitative estimates of all of the orientation parameters. In contrast, birefringence is only related to 〈P 2〉 as it is a second rank tensor property. Similar restrictions relate to all other second rank properties. For a perfectly aligned sample, the value of 〈P 2〉 is 1, whereas a randomly orientated distribution with no preferential alignment 〈P 2〉 has a value of 0. Methods are available for examination of preferential alignment in semi-crystalline and amorphous materials as described by Mitchell (2016). The methodology developed by Mitchell and co-workers in which the scattering is described in terms of a series of spherical harmonics provides a powerful tool box for the analysis of 2D scattering patterns. These techniques were successfully applied to isotactic polypropylene containing a thermotropic liquid crystal (Mitchell et al. 2005). This same methodology was used by Edwards et al. to examine the orientation of polymer lamellae in PCL electrospun fibres collected onto a rotating collector at differing take-up speeds. An increasing level of orientation was observed in the scattering patterns (Fig. 2.24), shown by the azimuthal arcing of the intensity distribution with increasing collection speed until a critical speed was reached that induced fibre breakage (Edwards et al. 2010). The obtained scattering patterns were described as a convolution of the fibre orientation and crystal orientation, for which fibre misalignment can be corrected for to establish the orientation of the crystals in a perfectly aligned sample. Similar methodologies can be applied to small-angle scattering data.

PCL electrospun fibre scattering for samples collected at differing take-up speeds. Wide angle X-ray scattering patterns (top) for electrospun fibres of PCL prepared using the indicating tangential velocities and the small angle X-ray scattering patterns (bottom) for the same samples. The outer edge of the SAXS patterns corresponds to |Q| = 0.08 Å−1. Reprinted from European Polymer Journal, 46 (6). pp. 1175–1183, Edwards, M. D., Mitchell, G. R., Mohan, S. D. and Olley, R. H. Copyright (2010) Development of orientation during electrospinning of fibres of poly(ε-caprolactone) with permission from Elsevier
The use of WAXS is not limited to the study of semi-crystalline materials but can also be applied to purely amorphous materials. WAXS patterns obtained can contain information on the inter- and intra-chain segments and the trans-gauche conformation of the chains. For example, Lovell et al. performed WAXS on atactic polystyrene showing the scattering contributions from the inter- and intra-chain segments, and estimated the trans-gauche conformational arrangement of the chain by comparison to the scattering one would expect for various trans-gauche configurations. An example of this is given below in Fig. 2.25 for atactic polystyrene (Lovell et al. 1979).

(a) experimental X-ray data (b) model of scattering for a tttt chain, (c) model of scattering for ttgg (where t trans, g gauche) (Reproduced from Lovell et al. 1979 with permission of The Royal Society of Chemistry)
2.4.1.3 SAXS/WAXS Instruments
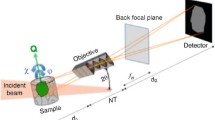
WAXS techniques have become commonly used in characterization of polymer samples with lab scale equipment available for use. Although small angle X-ray lab sources are available, there is significantly higher flux available at synchrotron sources allowing for rapid and in situ studies of samples (Fig. 2.26).

NCD Beamline at ALBA. (a) Image of the WAXS detector (Blue box in the top centre of image) (Picture taken by Sergio Ruiz) and (b) Image of part of the SAXS beamline (Picture taken by Juan Carlos Martίnez). Images provided courtesy of Alba Synchrotron
2.4.2 Neutron Scattering
Neutron scattering occurs from the nucleus of the molecule as opposed to the X-rays which scatter from electrons. As the neutrons scatter from the atomic nucleus, which occupies a significantly smaller volume than that of the electron clouds surrounding atoms, neutron scattering is considered a point scattering technique, hence the observed pattern does not fall off with increasing scattering angle as observed with X-ray scattering. Whilst the observed X-ray intensity is proportional to the atomic number of the scattering atom, neutron scattering lengths exhibit no particular order, with scattering lengths appearing random with atomic species. However, there is a difference in scattering lengths between isotopic species, hence a contrast can be provided by utilizing an isotopic labelling technique.
2.4.2.1 Small Angle Neutron Scattering
Small angle neutron scattering (SANS ) can be utilized to study structural development in a polymer; similar to the SAXS technique, however, the unique isotopic labelling techniques available in a SANS experiment allow for study of the polymer chain dimensions in the bulk and solution state. For example, a hydrogenous polymer matrix that contains perdeuterated polymer chains can provide a contrast which allows the study of the macroscopic chain conformation in the bulk state. The perdeuterated polymer chain is considered to be chemically identical to that of the hydrogenous polymer, hence a study of the polymer in its natural state can be conducted. If a similar experiment were to be conducted with SAXS, it would require the substitution of a heavier element on the polymer chains to provide a scattering contrast, meaning a different chemical species is being studied compared to the normal material. This unique isotopic scattering length difference provided for neutron scattering experiments makes it a powerful tool for the investigation of polymer systems. Various examples are present in the literature where the study of the polymer chain dimensions has been conducted on a variety of polymers. Comparisons of the literature results for these SANS measurements show the theta, glassy and molten polymer state have similar conformation reflecting an ideal random walk as shown in a review collated by Wignall and Melnichenko (2005). For example, both atactic polymers, polystyrene and poly methyl methacrylate were shown to have radii of gyration that varied with the molecular weight of the sample by the relation \( {R}_{\mathrm{g}}=0.275{M}_{\mathrm{W}}^{0.5} \), similar to that measured in the theta state (Wignall and Melnichenko 2005). In addition to bulk materials, thin films and fibres can be studied by SANS to examine the chain conformation. Studies of isotopically labelled electrospun fibres were conducted by Mohan et al. with results showing a small level of preferential alignment of the polymer chain along the fibre axis induced by the electrospinning process, with further extension possible due to mechanical deformation from a rotating collector (Fig. 2.27) (Mohan et al. 2011).

Scattering curves for an electrospun fibre sample of (50/50) hydrogenous/perdueterated polystyrene collected onto a rotating collector. The data has been reduced to a 1D plot of the differential scattering cross section as a function of Q, where the curves represent the scattering both parallel (solid line) and perpendicular (dotted line) to the fibre axis (vertical on page) obtained from the 2D scattering pattern (inset). Inset runs from −0.05 Å−1 < Q < 0.05 Å−1 (Adapted from Mohan et al. 2011 with permission from The Royal Society of Chemistry)
2.4.2.2 Broad Q Neutron Diffraction
Wide angle neutron scattering, commonly referred to as broad Q neutron diffraction, has been performed on various crystalline and amorphous polymers. Exploiting the contrast variation between isotopes as was done in SANS, the broad Q neutron scattering can be used to examine various scattering features. For example, neutron diffraction was performed on amorphous atactic polystyrene, which had selectively deuterated components. This allowed for the examination of the contribution to the scattering from various components on the polymer chain (Fig. 2.28). The structure factor for these scattering patterns is shown below in Fig. 2.28a where it can be seen that the various deuteration levels influence the scattering obtained. Here the scattering at approximately Q ~ 1–1.5 Å−1 arises from scattering between molecules from neighbouring chains, whilst Q > 1.5 Å−1 relates to the correlations from molecules on the same chain. The Fourier transform into a real space correlation function is provided in Fig. 2.28b. The lower values relate to bond lengths present, C–D and C–C, whilst the higher length terms reflect the inter- and intra-chain segment contributions. Figure 2.28b shows a repeating oscillation at approximately 10 Å, 15 Å, 20 Å, 25 Å which relates to the scattering from the phenyl rings along the polymer chain.

(a) Structure factors for selectively deuterated polystyrene . (b) Fourier transform of (a) fully deuterated monomer and (b) partial correlation function relating to backbone atoms (Reproduced from Mitchell et al. 1994, with permission of The Royal Society of Chemistry)
The technique can also be applied to the examination of semi-crystalline polymer systems, for example, Fig. 2.29 shows the WAXS and broad Q neutron diffraction from the same sample of d-PVC. In the X-ray scattering data (Fig. 2.29a) the material appears very disordered, whilst the neutron scattering from the same perdeuterated sample of PVC shows a typical melt structure factor similar to polyethylene although the first peak is somewhat sharper reflecting a higher level of order which perhaps is a consequence of the more polar structure (Mitchell 2011).

(a) Wide-angle X-ray scattering curve for a sample of perdeuterated poly (vinylchloride) (b) Neutron scattering function for the sample subjected to X-ray scattering in the left hand image (Reproduced from Mitchell 2011)
As the intensities of scattered X-rays depend on the atomic number, I ~ Z 2, the scattering for a sample of PVC will be dominated by the scattering from the Cl Atoms (Z = 17), compared with the scattering from C (Z = 6) or H (Z = 1). In contrast, neutron scattering arises from changes in the neutron scattering length b which is determined by the atomic nucleus. For d-PVC b cl = 9.577, b C = 6.646, b D = 6.671, hence the scattering is not dominated over by the presence of the chlorine allowing for a more detailed study of the structure.
2.4.2.3 Neutron Scattering Instruments
Neutrons are produced either in a nuclear reactor (e.g. the Institut Laue Langevin) or at a spallation source (i.e. ISIS Neutron Facility). In the latter most scattering experiments are performed using angular dispersion as in a typical X-ray scattering experiment, whereas at a spallation source, experiments are performed using time of flight techniques which opens up new possibilities as described below. Typically neutron scattering fluxes are lower and the interaction with the sample weaker and hence the neutron scattering signals are much diminished. Much effort has been made to reduce the data collection cycle time, for example, using an area detector (LOQ ISIS first target station Heenan et al. (1997) or many detectors (e.g. NIMROD (Fig. 2.30) second target station ISIS Bowron et al. (2010)).

The NIMROD instrument at ISIS (Image provided courtesy of ISIS Pulsed Neutron Source, STFC, Didcot, UK)
Figure 2.31 shows such data obtainable for a time-resolved study of PCL on the instrument NIMROD at ISIS, STFC, Didcot, UK with a cycle time of 400 s. The scattering data shown ranges from Q = 0.02 to 50 Å−1 and is on a single vertical scale in absolute units.

Time-resolved data obtained on NIMROD for PCL in both the semi-crystalline and melt phases. Inset shows time-resolved data captured during crystallization
2.5 Summary
We have covered a wide range of techniques in this chapter. We can see a continual improvement in the facilities for the study of structure at multiple scales. Simultaneous SAXS and WAXS experiments are now routinely performed at synchrotron-based beam lines, while the NIMROD instruments take this a further step by routinely providing multiple scale data on a single intensity scale. Despite these developments, however exciting, there remains a need to utilize a range of complementary techniques involving both imaging and scattering both to cover the structural scales and to probe the scientific issues.
References
Abo El Maaty MI, Olley RH, Bassett DC (1999) On the internal morphologies of high-modulus polyethylene and polypropylene fibres. J Mater Sci 34:1975–1989
Al Raheil IAM (1987) Development of crystalline morphology in polyethylene and PEEK. PhD Thesis, University of Reading
Arnal ML, Sanchez JJ, Müller AJ (2001) Miscibility of linear and branched polyethylene blends by thermal fractionation: use of the successive selfnucleation and annealing (SSA) technique. Polymer 42:6877–6890
Bassett DC, Vaughan AS (1985) On the lamellar morphology of melt crystallized isotactic polystyrene. Polymer 26(717):725
Bassett DC, Olley RH, Vaughan AS (2003) Specimen preparation for TEM of polymers. In: Pethrick RA, Viney C (eds) Techniques in polymer organisation and morphology characterisation. Experimental methods in polymer characterisation. Wiley, Chichester
Bovey FA (2007) NMR of polymers. eMagRes. Wiley Online
Bowron DT, Soper AK, Jones K, Ansell S, Birch S, Norris J, Perrott L, Riedel D, Rhodes NJ, Wakefield SR, Botti A, Ricci MA, Grazzi F, Zoppi M (2010) NIMROD: the Near and InterMediate Range Order Diffractometer of the ISIS second target station. Rev Sci Instrum 81:033905
Bucknall CB, Drinkwater IC, Keast WE (1972) An etch method for microscopy of rubber-toughened plastics. Polymer 13:115–118
Chan C-K, Gao P (2005) Shear-induced interactions in blends of HMMPE containing a small amount of thermotropic copolyester HBA/HQ/SA. Polymer 46:10890–10896
Channuan W, Siripitayananon J, Molloy R, Sriyai M, Davis FJ, Mitchell GR (2005) The structure of crystallisable copolymers of l-lactide, ε-caprolactone and glycolide. Polymer 46:6411–6428
Chen F, Shanks RA, Amarasinghe G (2001) Crystallisation of single-site polyethylene blends investigated by thermal fractionation techniques. Polymer 42:4579–4587
Chou TM, Prayoonthong P, Aitouchen A, Libera M (2002) Nanoscale artifacts in RuO4-stained poly(styrene). Polymer 43:2085–2088
Chu B, Hsiao BS (2001) Small-angle X-ray scattering of polymers. Chem Rev 101:1727–1762
Edwards MD, Mitchell GR, Mohan SD, Olley RH (2010) Development of orientation during electrospinning of fibres of poly(ε-caprolactone). Eur Polym J 46:1175–1183
Ford JR, Bassett DC, Mitchell DR, Ryan TG (1990) Morphology of a main chain liquid crystal polymer containing semi-flexible coupling chain. Mol Cryst Liq Cryst 180:233–243
Gedde UW, Jansson JF (1983) Molecular fractionation in melt-crystallized polyethylene 1. Differential scanning calorimetry. Polymer 24:1521–1531
Gedde UW, Jansson JF (1984) Molecular fractionation in melt-crystallized polyethylene 3. Microscopy of solvent-treated samples. Polymer 25:1263–1267
Gedde UW, Eklund S, Jansson JF (1983) Molecular fractionation in melt-crystallized polyethylene 2. Effect of solvent-extraction on the structure as studied by differential scanning calorimetry and gel-permeation chromatography. Polymer 24:1532–1540
Hamley IW, Castelletto V (2004) Small-angle scattering of block copolymers in the melt, solution and crystal states. Prog Polym Sci 29:909–948
He Z, Olley RH (2000) On spherulitic forms in an aromatic polyesteramide. Polymer 41:1157–1165
Heenan RK, Penfold J, King SM (1997) SANS at pulsed neutron sources: present and future prospects. J Appl Cryst 30:1140–1147
Hemsley DA (ed) (1989) Applied polymer light microscopy. Springer, Berlin
Hobbs JK (2007) Insights into polymer crystallization from in-situ atomic force microscopy. In: Progress in understanding of polymer crystallization, vol 714. Lecture notes in physics. Springer, Berlin, pp 373–389
Hobbs JK, Vasileva C, Humphris ADL (2005) Real time observation of crystallization in polyethylene oxide with video rate atomic force microscopy. Polymer 46:10226–10236
Hoffman JD, Weeks JJ (1962) Melting process and the equilibrium melting temperature of polychlorotrifluoroethylene. J Res Natl Bur Stand (US) A66: 13
Hong S, Bushelman AA, MacKnight WJ, Gido SP, Lohse DJ, Fetters LJ (2001) Morphology of semicrystalline block copolymers: polyethylene-b-atactic-polypropylene. Polymer 42:5909–5914
Hosier IL, Vaughan AS, Swingler SG (1997) Structure-property relationships in polyethylene blends: the effect of morphology on electrical breakdown strength. J Mater Sci 32(17):4523–4531
Hsieh YT, Ishige R, Higaki Y, Woo EM, Takahara A (2014) Microscopy and microbeam X-ray analyses in poly(3-hydroxybutyrate-co-3-hydroxyvalerate) with amorphous poly(vinyl acetate). Polymer 55:6906–6914
Jordan ND, Bassett DC, Olley RH, Hine PJ, Ward IM (2003) The hot compaction behaviour of woven oriented polypropylene fibres and tapes. II. Morphology of cloths before and after compaction. Polymer 44:1133–1143
Keller A (1968) Polymer crystals. Rep Prog Phys 31:623–704
Keller A, Bassett DC (1960) Complementary light and electron microscope investigations on the habit and structure of crystals, with particular reference to long chain compounds. Proc R Microsc Soc 79:243–261
Kielhorn L, Colby RH, Han CC (2000) Relaxation behavior of polymer blends after the cessation of shear. Macromolecules 33:2486–2496
Kumaraswamy G, Issaian AM, Kornfield JA (1999) Shear-enhanced crystallization in isotactic polypropylene. 1. Correspondence between in situ rheo-optics and ex situ structure determination. Macromolecules 32:7537–7547
Lau KY, Vaughan AS, Chen G, Hosier IL, Holt AF (2013) Absorption current behaviour of polyethylene/silica nanocomposites. Dielectrics 2013(10–12):24–25
Lee Y, Porter RS (1987) Double-melting behavior of poly(ether ether ketone). Macromolecules 20:1336–1341
Lee Y, Porter RS, Lint JS (1989) On the double-melting behavior of poly(ether ether ketone). Macromolecules 22:1756–1760
Lee B, Shin TJ, Lee SW, Yoon J, Kim K, Ree M (2004) Secondary crystallization behavior of poly(ethylene isophthalate-co-terephthalate): time-resolved small-angle X-ray scattering and calorimetry studies. Macromolecules 37:4174–4184
Lemstra PJ, Kooistra T, Challa G (1972) Melting behavior of isotactic polystyrene. J Polym Sci A-2 Polym Phys 10:823–833
Lin D, Cheng H, Zou F, Ning W, Han CC (2012) Morphology evolution of a bisphenol A polycarbonate/poly (styrene-co-acrylonitrile) blend under shear and after shear cessation. Polymer 53:1298–1305
Loo Y-L, Wakabayashi K, Huang YE, Register RA, Hsiao BS (2005) Thin crystal melting produces the low-temperature endotherm in ethylene/methacrylic acid ionomers. Polymer 46:5118–5124
Lovell R, Mitchell GR, Windle A (1979) Wide-angle X-ray scattering study of structural parameters in non-crystalline polymers. Faraday Discuss Chem Soc 68:46–57
Marand H, Xu JN, Srinivas S (1998) Determination of the equilibrium melting temperature of polymer crystals: Linear and nonlinear Hoffman-Weeks extrapolations. Macromolecules 31:8219–8229
McKenna GB (1989) Glass formation and glassy behavior. In: Booth C, Price C (eds) Comprehensive polymer science, vol 2. Polymer properties. Pergamon, Oxford, pp 311–363
Mitchell GR (2011) Chapter 47: neutron diffraction from polymers and other soft matter. In: Imae T, Kanaya T, Furusaka M, Torikai N (eds) Neutrons in soft matter. Wiley, Hoboken
Mitchell GR (2016) Scattering methods for polymer orientation characterisation. Springer, Berlin
Mitchell GR, Rosi-Schwartz B, Ward DJ, Warner M (1994) Local order in polymer glasses and melts. Phil Trans R Soc London A 348:97–115
Mitchell GR, Saengsuwan S, Bualek-Limcharoen S (2005) Evaluation of preferred orientation in multi-component polymer systems using x-ray scattering procedures. Progr Coll Polym Sci 130:149–158
Miwa Y, Drews AR, Schlick S (2008) Unique structure and dynamics of poly(ethylene oxide) in layered silicate nanocomposites: accelerated segmental mobility revealed by simulating ESR spectra of spin-labels, XRD, FTIR, and DSC. Macromolecules 41:4701–4708
Mohan SD, Mitchell GR, Davis FJ (2011) Chain extension in electrospun polystyrene fibres: a SANS study. Soft Matter 7:4397–4404
Morgan RL, Hill MJ, Barham PJ (1999) Morphology, melting behaviour and co-crystallization in polyethylene blends: the effect of cooling rate on two homogeneously mixed blends. Polymer 40:337–348
Müller AJ, Arnal ML (2005) Thermal fractionation of polymers. Prog Polym Sci 30:559–603
Murphy DB, Davidson MW (2012) Fundamentals of light microscopy and electronic imaging, 2nd edn. Wiley-Blackwell, New York
Nielsen AS, Batchelder DN, Pyrz R (2002) Estimation of crystallinity of isotactic polypropylene using Raman spectroscopy. Polymer 43:2671–2676
Olley RH (1986) Selective etching of polymeric materials. Sci Prog 70:17–43
Olley RH, Bassett DC (1989) On the development of polypropylene spherulites. Polymer 30:399–409
Olley RH, Bassett DC (1994) On Surface-Morphology and Drawing of Polypropylene Films. J Macromol Sci Phys B33:209–227
Olley RH, Bassett DC, Blundell DJ (1986) Permanganic etching of PEEK. Polymer 27:344–348
Owen G, Vesely D (1985) An easy and economical method for staining electron microscopy specimens with osmium tetroxide. Proc R Microsc Soc 20:297
Patel D, Bassett DC (1994) On spherulitic crystallization and the morphology of melt-crystallized isotactic poly(4-methylpentene-1). Proc R Soc A 445:577–595
Plans J, MacKnight WJ, Karasz FE (1984) Equilibrium Melting Point Depression for Blends of Isotactic Polystyrene with poly(2,6-dimethylphenylene oxide). Macromolecules 17:810–814
Ruland W (1961) X-ray determination of crystallinity and diffuse disorder scattering. Acta Crystallogr 14:1180–1185
Ruland W (1964) Crystallinity and disorder parameters in nylon 6 and nylon 7. Polymer 5:89–102
Ruland W (1977) Determination of the interface distribution function of lamellar two-phase systems. Coll Polym Sci 255:417
Schmidt-Rohr K, Spiess HW (1994) Multidimensional solid-state NMR and polymers. Elsevier, Amsterdam
Shabana HM, Guo W, Olley RH, Bassett DC (1993) Electron-microscopic observation of spinodal decomposition in blends of tetramethyl polycarbonate and polystyrene. Polymer 34:1313–1315
Shabana HM, Olley RH, Bassett DC, Zachmann H-G (1996) On crystallization and phase separation phenomena in PEN/PHBA copolyesters. J Macromol Sci Phys B35:691–708
Shabana HM, Olley RH, Bassett DC, Jungnickel BJ (2000) Phase separation induced by crystallization in blends of polycaprolactone and polystyrene: an investigation by etching and electron microscopy. Polymer 41:5513–5523
Shahin MM, Olley RH (2002) Novel etching phenomena in poly(3-hydroxy butyrate) and poly(oxymethylene) spherulites. J Polym Sci B Polym Phys 40:124–133
Shahin MM, Olley RH, Blissett MJ (1999) Refinement of etching techniques to reveal lamellar profiles in polyethylene banded spherulites. J Polym Sci Polym Polym Phys 37:2279–2286
Shanks RA, Amarasinghe G (2000) Crystallisation of blends of LLDPE with branched VLDPE. Polymer 41:4579–4587
Shen B, Liang Y, Kornfield JA, Han CC (2013) Mechanism for shish formation under shear flow: an interpretation from an in situ morphological study. Macromolecules 46:1528–1542
Strobl GR, Hagedorn V (1978) Raman spectroscopic method for determining the crystallinity of polyethylene. J Polym Sci Polym Phys 16:1181–1193
Strobl GR, Schneider M (1980) Direct evaluation of the electron density correlation function of partially crystalline polymers. J Polym Sci Polym Phys 18:1343–1359
Toda A, Taguchi K, Nozaki K, Konishi M (2014) Melting behaviors of polyethylene crystals: an application of fast-scan DSC. Polymer 55:3186–3194
Traylor A (1961) A sample preparation technique for the application of phase contrast microscopy to polystyrene-type polymers. Anal Chem 33:1629–1630
Trent JS, Scheinbeim JI, Couchman PR (1983) Ruthenium tetraoxide staining of polymers for electron microscopy. Macromolecules 16(4):589–598
Trindade AC, Godinhoa MH, Figueirinhas JL (2004) Shear induced finite orientational order in urethane/urea elastomers. Polymer 45:5551–5555
Vaughan AS, Stevens GC (1995) On radiation effects in poly(ethylene terephthalate): a comparison with PEEK. Polymer 36:1541–1547
Vaughan AS, Sutton SJ (1995) On radiation effects in oriented PEEK. Polymer 36:1549–1554
Wang Z, Li Y, Yang J, Gou Q, Wu Y, Wu X, Liu P, Gu Q (2010) Twisting of lamellar crystals in poly(3-hydroxybutyrate-co-3-hydroxyvalerate) ring-banded spherulites. Macromolecules 43:4441–4444
Weng J, Olley RH, Bassett DC, Jääskeläinen P (2003) Changes in the melting behavior with the radial distance in isotactic polypropylene spherulites. J Polym Sci Polym Phys 41:2342–2354
Weng J, Olley RH, Bassett DC, Jääskeläinen P (2004) Crystallization of propylene–ethylene random copolymers. J Polym Sci Polym Phys 42:3318–3332
Wignall GD, Melnichenko YB (2005) Recent applications of small-angle neutron scattering in strongly interacting soft condensed matter. Rep Prog Phys 68:1761–1810
Wunderlich B, Sullivan P (1962) Interference microscopy of crystalline high polymers. Determinations of the thickness of single crystals. J Polym Sci 56:19–25
Yagpharov M (1986) Thermal analysis of secondary crystallization in polymers. J Therm Anal Calorim 31:1073–1082
Yamada K, Hikosaka M, Akihiko Toda A, Yamazaki S, Tagashira K (2003a) Equilibrium melting temperature of isotactic polypropylene with high tacticity: 1. Determination by differential scanning calorimetry. Macromolecules 36:4790–4801
Yamada K, Hikosaka M, Toda A, Yamazaki S, Tagashira K (2003b) Equilibrium melting temperature of isotactic polypropylene with high tacticity. 2. Determination by optical microscopy. Macromolecules 36:4802–4812
Yamada K, Kajioka H, Nozaki K, Toda A (2011) Morphology and growth of single crystals of isotactic polypropylene from the melt. J Macromol Sci Phys 50:236–247
Zheng K, Zhang J, Cheng J (2013) Morphology, structure, miscibility, and properties of wholly soy-based semi-interpenetrating polymer networks from soy–oil–polyol-based polyurethane and modified soy protein isolate. Ind Eng Chem Res 52:14335–14341
Zou F, Dong X, Lin D, Liu W, Wang D, Han CC (2012) Morphological and rheological responses to the transient and steady shear flow for a phase-separated polybutadiene/polyisoprene blend. Polymer 53:4818–4826
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Mohan, S., Olley, R.H., Vaughan, A.S., Mitchell, G.R. (2016). Evaluating Scales of Structures. In: Mitchell, G., Tojeira, A. (eds) Controlling the Morphology of Polymers. Springer, Cham. https://doi.org/10.1007/978-3-319-39322-3_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-39322-3_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-39320-9
Online ISBN: 978-3-319-39322-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)