
Abstract
Inflammation and cancer have a long and contentious history. Currently, there are two lenses through which the role of inflammation in cancer can be viewed. Substantial evidence suggests that inflammation can not only propagate, but even initiate cancer pathogenesis. However, emerging studies indicate that inflammation may alternatively enhance host containment and destruction of tumorigenic cells. Herein, we explore how our understanding of inflammation in cancer has evolved, from the first identification of excessive inflammation in tumors two millennia ago to the complex association between inflammation and cancer pathogenesis with the recent emergence of immune-harnessing cancer therapies. We discuss the dynamic roles of various immune cells, cytokines, and specific lipid autacoid signaling in cancer, focusing on fatty acid-derived lipid mediators such as prostaglandin E2. We contrast the pro-tumorigenic and anti-tumorigenic functions of immune cells and lipid mediators, while highlighting how their functions can be dramatically altered by the tumor microenvironment.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Inflammation
- Cancer
- Tumor
- Lipids
- Cytokines
- Immune cells
- Macrophages
- Natural killer cells
- T cells (T lymphocytes)
- B cells (B lymphocytes)
- Dendritic cells
- Inflammatory score
- Prostaglandin E2
- Annexin A1
- C-reactive protein (CRP)
History
The association between inflammation and cancer dates back as early as two millennia ago, when Claudius Galen expanded on Hippocrates’ theory of cancer as an excess of black bile (melancholia) by suggesting that cancer was a swelling resulting from excessive inflammation [1, 2]. In 1861, Dr. Rudolph Virchow, a German pathologist, delivered a twenty-part lecture series at the Pathological Institute of Berlin in which he was the first to describe immune cell infiltrates in tumors. Specifically, he noted white blood cells in tumor tissue and suggested that immune cells release factors that stimulate the proliferation of tumor cells [3]. Thirty years later, Dr. William B. Coley alternatively linked immune stimulation to cancer regression. At the New York’s Academy of Medicine, he reported three cases of tumor regression resulting from inoculating sarcoma patients with a strain of Streptococcus bacterium [4]. Later coined “Coley’s toxin,” inoculation with this bacterium is now known to have elicited tumor regression through tumor necrosis factor-α (TNFα)-mediated activation of cytotoxic immune cells.
Within the last 50 years, seminal progress has been made towards understanding inflammation and cancer. In 1986, Harvard Medical School pathologist Dr. Harold Dvorak elegantly characterized tumors as “wounds that do not heal,” noting that while tumors elicited an immune response, they exhibited persistent inflammation rather than the gradual cessation of acute inflammation associated with wound healing [5]. Drs. Lisa Coussens, Douglas Hanahan, and Zena Werb later demonstrated that premalignant tissue became malignant with the assistance, or “co-conspiracy,” of inflammatory cells, such as mast cells, macrophages, and T lymphocytes [6, 7]. It is now recognized that an inflammatory tumor microenvironment plays a key role in every stage of tumor development. Several immune cell types, including macrophages, neutrophils, mast cells, and regulatory T cells, secrete tumor growth-promoting chemokines, pro-angiogenic factors, and metastasis-promoting extracellular proteases [8, 9]. By 2011, Drs. Douglas Hanahan and Robert Weinberg included inflammation as a defining hallmark of cancer [10].
Despite inflammation commonly being viewed as “the other half of the tumor” [11], a growing body of evidence suggests immune cells and their mediators may play more ambiguous roles, in the prevention of prevent cancer progression. In 1977, Dr. Alberto Mantovani demonstrated that antibody-dependent cellular cytotoxicity (ADCC) inhibited tumor cell growth via both cytostatic (inhibiting cell growth and division) and cytolytic (lysis-inducing) mechanisms [12]. Mantovani’s work became the foundation for future studies characterizing the antitumor potential of immune survelliance. As Coley’s century-old observations suggested, [c]ancer cells express antigens that can be identified as “nonself” by surveillance immune cells, such as natural killer cells and cytotoxic T cells, and can elicit an antitumor immune response. Mantovani’s later studies characterizing the ability of “tumor-associated macrophages” to both stimulate and inhibit tumor growth further highlighted the contrasting pro- and anti-tumorigenic functions of the immune system [13]. In 1987, Dr. Frances Balkwill and her colleagues demonstrated that intraperitoneal treatment with the pro-inflammatory cytokines TNFα and interferon γ (INFγ) prolonged survival of ovarian tumor-bearing mice [14]. Building on Coley’s and Mantovani’s pioneering studies, her findings suggested provoking an inflammatory immune response may alternatively elicit endogenous anti-tumorigenic activity. Balkwill later characterized novel mechanisms through which TNFα can be simultaneously pro- and anti-tumorigenic depending on the specific tumor microenvironment [15].
Self-limited and localized, or acute, inflammation serves as the body’s endogenous defense against the outside world. However, unresolved acute inflammation can become chronic. Chronic inflammation and inflammatory diseases are in fact risk factors for over ten different cancer types: chronic Helicobacter pylori predisposes to stomach cancer, inflammatory bowel diseases predispose to colon cancer, etc. [16]. However, chronic inflammation may not always progress to a related organ system cancer. For example, psoriasis (chronic inflammation of the skin) is not always linked to skin cancer incidence [11]. Moreover, 19 cancers have been associated with prior bacterial, viral, or parasitic infection [16]. Chronic inflammation can even be necessary for cancer pathogenesis, as mice engineered to develop hepatitis (a chronic inflammatory disease of the liver) failed to develop hepatocellular carcinoma when TNFα was neutralized [17]. Importantly, NFκβ , a pro-inflammatory transcription factor critical in innate and adaptive immune activation, was also suppressed in these mice [17]. The Karin laboratory demonstrated that activation of NFκβ perpetuates inflammation and contributes to colon tumor growth, while inhibition of NFκβ downregulates inflammation, resulting in tumor regression [18]. The inflammatory component of malignancies is further evidenced by the clinical effectiveness of anti-inflammatories, including nonsteroidal anti-inflammatory drugs (NSAIDs) such as aspirin, as anticancer therapies [19]. As we strive to untangle the complex, multifaceted relationship between inflammation and cancer, our understanding of the disease has shifted radically. Cancer is increasingly illuminated as a war of self, the outcome hinging on whether our immune systems keep our mutated cells in check or assist in their progression and eventual malignant escape.
Tumor Microenvironment
The tumor microenvironment has become a focus in cancer research as its constituents are the major contributors of tumor-promoting inflammation . Here, we focus specifically on the diverse roles of immune cells, cytokines, lipid mediators, and inflammatory biomarkers that actively mediate inflammation in the tumor microenvironment. While inflammation has traditionally been associated with cancer progression, current studies continue to identify both pro- and anti-tumorigenic mechanisms enacted by our immune system. We explore the pro-tumorigenic mechanisms of infiltrating immune cells, pro-inflammatory cytokines lipid mediators, and pro-inflammatory biomarkers, while examining how these immune cells and their mediators may paradoxically play an anti-tumorigenic role in the context of cancer.
Immune Cell Biomarkers
Both the innate and adaptive immune systems contribute to cancer-associated inflammation. Immune cells such as macrophages, dendritic cells, natural killer cells, T lymphocytes, and B lymphocytes play critical pro-tumorigenic and anti-tumorigenic roles in inflammation-associated cancer pathogenesis.
Macrophages
Macrophages are traditionally viewed as scavengers of the immune system. As monocytes, their immature predecessors, they circulate in the blood until they receive a signal to migrate into tissues, where they mature into macrophages. Macrophages are dual-functional: while they participate in the innate immune response by surveying tissues and destroying antigens via phagolysosomes, they also function as antigen-presenting cells within the adaptive immune system to mount specific T and B cell responses against foreign particles, microbes, or even cancer cells.
While previously thought to be predominantly anti-tumorigenic, our understanding of macrophages in cancer has evolved since the 1980s to encompass their more recently characterized pro-tumorigenic actions. In the 1980s and 1990s, resident liver macrophages known as Kupffer cells were reported to possess anti-tumorigenic activity [20]. Specifically, Kupffer cells were shown to phagocytose tumor cells, and their systemic depletion led to increased metastases [21]. Tumor-bearing rats lacking macrophages, a result of exogenous depletion, were shown to exhibit increased tumor differentiation and decreased survival due to aggressive tumor growth [22]. In contrast, the depletion of monocytes and macrophages in animal tumor models demonstrated reduced tumor incidence, as well as inhibition of tumor growth and angiogenesis [23]. The anti-tumorigenic nature of macrophages is further exemplified by the evolution of tumor cells to evade macrophage detection. Leukemia cell expression of CD47, an anti-apoptotic and autophagic marker, was demonstrated to inhibit macrophage phagocytosis, enabling tumor immune evasion and cancer progression [24].
Macrophages have been characterized in various tumorigenic and metastatic disease settings. For instance, glioblastoma has been associated with increased numbers of circulating monocytes [25]. Further, phagocytosis of circulating breast cancer exosomes by distant macrophages has been shown to increase macrophage secretion of pro-inflammatory cytokines via NFκβ activation [26]. Importantly, this implicates macrophages as potential mediators of metastasis, as breast cancer commonly metastasizes to the lung and brain. Indeed, studies have characterized the pro-metastatic role of macrophages in genetically engineered murine breast cancer models [27].
It is now appreciated that functionally distinct subsets of macrophages exist, and their polarization is influenced by their surroundings. Two of these polarizations are referred to as “M1” and “M2” phenotypes. However, the traditional M1/M2 categorization of macrophages appears to be an oversimplification [28, 29], as recent studies have expanded the role of M2-phenotypic macrophages to include mediating the resolution of inflammation. During the resolution of inflammation, an active process, lipid autacoids stimulate M2 macrophages to efferocytose apoptotic cellular debris [30,31,32,33]. Glioblastoma-associated myeloid cells are characterized as having an “M0,” non-polarized phenotype. However, these non-polarized macrophages have been found to express some characteristic M2 markers, namely TGF-β and IL-10 [25]. M2-polarized macrophages in a non-cancer setting are characterized as anti-inflammatory, participating in wound healing and tissue repair; however, in a cancer setting, they are characteristically viewed as “pro-tumorigenic.” Specifically, M2 macrophages have been shown to promote disease progression in numerous cancers, including lung, breast, and ovarian [34,35,36]. In genetically engineered murine models of lung adenocarcinoma, depletion of M2-polarized alveolar macrophages inhibited tumor growth [34]. Additionally, the polarization of macrophages to their M2 phenotype has been shown to accelerate breast cancer growth [35]. Similarly, M2-phenotype macrophages have been shown to be present in ascites fluid taken from ovarian cancer patients [36]. Interestingly, when these macrophages were cultured with lipopolysaccharide (LPS), an inflammatory stimulus, they adopted an M1 phenotype, exhibiting toll-like receptor activation and upregulating the cytotoxic activity of natural killer (NK) cells [36]. Thus, the reprogramming of M2 macrophages to an M1 phenotype can promote their antitumor activity.
In contrast to M2-polarized macrophages, M1 macrophages have been characterized as anti-tumorigenic. In non-cancer settings, these macrophages mount an inflammatory response via their phagocytic and antigen-presenting activity [30, 31]. Current cancer immune-based therapies include polarizing macrophages to their M1 phenotype. Blocking TGFβ signaling in combination with stimulation of toll-like receptor 7 (characteristic of innate immune activation) results in the polarization of macrophages toward an M1 phenotype with specific antitumor activity [37].
In addition to M1 macrophages, studies have identified CD169+ macrophages, non-phagocytic mediators of immune tolerance, as important anti-tumorigenic cells. While CD169+ macrophages are unable to phagocytose cells and debris, they are of particular importance in immune tolerance [28]. Interestingly in an inflammatory setting, these macrophages have been shown to activate cytotoxic CD8+ T cells with a greater range of targets than dendritic cells [38]. As cancers have a wide variety of constantly mutating antigenic targets, this is a potential mechanism correlating CD169+ macrophages with documented antitumor activity in a variety of cancers, including endometrial carcinoma and melanoma [39,40,41]. CD169+ macrophages inhibit melanoma growth in orthotopic murine models via their ability to bind tumor-derived extracellular vesicles in draining lymph nodes and subsequently present them to B cells [41]. These various host macrophage-mediated mechanisms induce an adaptive immune response against the tumor, thus highlighting enhancement of the immune system as a potential cancer therapy.
Dendritic Cells
Similar to macrophages, dendritic cells exhibit both pro- and anti-tumorigenic activity in cancer. In inflammatory reactions, dendritic cells are the primary antigen-presenting cell. After binding an antigen at the site of inflammation, dendritic cells migrate to lymph nodes and activate antigen-specific immune responses from B and T lymphocytes. Dendritic cells are present in a variety of tumor types, including breast cancer [42, 43]. In a genetically engineered murine model of breast cancer (MMTV-PyMT), dendritic cells were demonstrated to be the most prevalent immune cell in the tumor tissue. Subsequent depletion of dendritic cells ultimately inhibited tumor growth and lung metastasis [44]. While dendritic cells are pro-tumorigenic, they can also possess antitumor activity. The difference in roles may be related to tumor progression. During the initial states of tumor progression, dendritic cells demonstrate antitumor activity. However, with tumor progression, dendritic cells lose their antitumor activity, unable to perform antigen presentation to T cells, and can even become immunosuppressive against T cells [45]. This highlights the adaptability of dendritic cell function within the tumor microenvironment and provides a possible approach for future immunotherapy. In fact, dendritic cells are currently being utilized in cancer vaccination clinical trials [46,47,48]. Tumor-associated dendritic cells exposed to tumor antigens ex vivo are then reintroduced to glioblastoma patients. These results have demonstrated harnessing dendritic cells antigen-presenting activity increases overall and progression-free survival in glioblastoma patients [49].
Natural Killer Cells
Natural killer (NK) cells also play a key role in inflammation and cancer. NK cells mediate antibody-dependent cellular cytotoxicity within the innate immune system. Recent studies have also implicated NK cells in adaptive and memory immunity [50]. NK cells not only promote nonalcoholic steatohepatitis (inflammation and fat accumulation of the liver), but also play a pivotal role in its progression to hepatocellular carcinoma (HCC) [51]. Specifically, NK cells interact with CD8+ T cells to release pro-inflammatory cytokines, contributing to tumor progression [51]. Further, specific subsets of NK cells have been implicated in halting natural host immune responses in breast cancer patients via expression of TGFβ and IL-10 [52].
Conversely, studies have identified various antitumor activities of NK cells. Prostate cancer patients with increased peripheral natural killer cells achieved improved overall survival. [53] Specifically, NK cells in these patients express NKp46, DNAm-1, and NKG2D, which contributed to the lysis of prostate tumor cells [53]. Additionally activation of NK cells in the presence of IL-12 and IL-15 allows for the mounting of cytotoxic response against breast cancer stem cells [54]. Importantly, IL-12 and IL-15 activate NK cells and enhance their cytotoxic activity.
T Cells
The adaptive immune system, comprised of T and B cells, also plays a critical role in inflammation and cancer. T cells, including CD4+ T helper cells and CD8+ T cytotoxic cells, are antigen-specific cells that have a range of known phenotypes [55]. CD4+ T cells assist in B cell antibody class switching, and activation of CD8+ T cells and can have both pro- and anti-tumorigenic activities. CD4+ T cell infiltration is increased in skin dysplasia, as well as in squamous cell carcinoma. Their role in tumor pathogenesis is evident as demonstrated in a CD4+ T cell knockout murine model, in which tumor growth is suppressed [56]. However, it appears the tumorigenic role of CD4+ T cells varies across malignant tissues. Specifically, the loss of CD4+ T cells in the liver has been shown to accelerate tumor growth both in murine models and human patient samples of hepatocarcinogenesis. This mechanism may be due to an increase in hepatocytes’ lipid concentration, resulting in CD4+ T cell death and subsequent disease progression from nonalcoholic fatty liver disease to cancer [57]. T cells with cytotoxic phenotypes (CD8+) are anti-tumorigenic, specifically in genetically engineered murine breast cancer models. Their cytotoxicity against tumor cells is dependent on IL-15, and knocking out IL-15 facilitates a marked increase in tumor growth [58]. Similarly, in genetically engineered murine non-small cell lung cancer models, knocking out CD8+ T cells resulted in stimulation of tumor growth and reduced survival [59]. In recent clinical studies, CD8+ T cells appear to play a key role in antitumor immunity in both pancreatic and colorectal cancers. An increase in both CD8+ T cells and CD4+ T cells resulted in increased overall and disease-free survival in patients with pancreatic ductal carcinoma [60]. Reduced metastasis has been correlated with increased numbers of cytotoxic cells in colorectal cancer patient samples [61]. Similarly, depletion of CD8+ T cells in murine colorectal cancer models resulted in accelerated tumor growth. Thus, T cells represent the flexibility of the adaptive immune system by hindering and promoting tumor growth.
B Cells
B lymphocytic cells, another essential arm of the adaptive immune system, are best known for their role in humoral immunity, providing immune responses from a distance. B cells secrete antigen-specific antibodies, which have a broad range of activities including marking cells for destruction (opsonization), providing a physical barrier on an antigen, or facilitating the creation of immune memory. Consistent with immune cells we have explored previously, B cells also appear to have variable roles in tumor pathogenesis. Most notably, B cells have been implicated in pancreatic cancer progression. The depletion of B cells in mice inhibited orthotopic pancreatic adenocarcinoma tumor progression. The underlying mechanism is believed to be associated with B cells’ role in macrophage polarization, specifically polarizing macrophages to an M2 phenotype [62]. Similarly, pancreatic tumors injected into mice lacking functional B cells exhibited decreased tumor growth as compared to wild-type mice. Alternatively, transplanting B cell-deficient mice with wild-type B cells promote tumor growth, further highlighting the pro-tumorigenic role of B cells [63]. The presence of mature B cells has also been correlated with higher epithelial ovarian tumor grade. Additionally, increased levels of plasma cells, which are terminally differentiated B cells, have been correlated with decreased overall and ovarian cancer-specific survival [64]. While largely pro-tumorigenic, B cells have also been shown to have anti-tumorigenic functions. Gene expression studies have revealed that the presence of B cells in the microenvironment of basal-like breast tumors correlates with increased progression-free survival [65]. While the specific mechanisms through which B cells exert anti-tumorigenic action is unclear, it is evident that B cells can be both anti- and pro-tumorigenic depending on the specific tumor environment.
Cytokine and Chemokine Biomarkers
We will next explore the role of cytokines and chemokines in cancer-associated inflammation. Cytokines and chemokines are small protein signaling molecules that enable crosstalk between immune cells and direct immune cell trafficking, respectively. While cytokines and chemokines in non-cancer settings may be considered pro- or anti-inflammatory, similar to immune cells, their role in tumor progression varies. While there is an enormous number of cytokines and chemokines that have been implicated in cancer, we focus on TNFα, TGFβ, IL-1, IL-6, IL-10, and CCL2.
Tumor Necrosis Factor α
Tumor necrosis factor α (TNFα) is perhaps the most iconic cytokine in cancer-association inflammation. However, as noted by Balkwill, “tumor necrosis factor” is likely a misnomer [15]. While the name “tumor necrosis factor” would lead one to believe it has antitumor activity, recent research has supported a dual role in tumorigenesis for TNFα. TNFα is traditionally implicated in septic shock and is characterized as a pro-inflammatory cytokine. TNFα has also been demonstrated to have both pro- and anti-tumorigenic roles. Its contribution to cancer pathogenesis is believed to be via activation of immune cells, including B cells, which recent studies have shown are likely responsible for skin carcinogenesis. Selectively knocking out TNFα in B cells resulted in a reduction of papillomas in tumor-bearing mice [66]. Similar observations have been made in orthotopic glioblastoma tumor models, in which knocking out TNFα significantly increases survival rates [67]. TNFα has also been shown to be increased in non-small cell lung and pancreatic cancers, and its expression in ovarian carcinoma patient tissue correlates with high-grade serous carcinomas and endometrioid carcinomas [68, 69, 70]. While it would appear TNFα could be an appealing target for cancer therapy, anti-TNFα therapies have failed in the clinic to date [71]. Insight into its therapeutic failure may be due to TNFα’s role as an anti-tumorigenic cytokine. If TNFα is expressed directly by tumor cells, it could then exert an autocrine antitumor activity. Mice injected with tumor cells genetically modified to secrete high levels of TNFα have little to no tumor growth as compared to control. This model has been recapitulated in breast, melanoma, and lung carcinoma models [72]. This in turn could possibly be utilized for future gene therapy, to stimulate ones’ own tumor to exert autologous antitumor activity.
Transforming Growth Factor-β
Transforming growth factor-β (TGF-β) is widely studied for its activation of regulatory T cells and Th17 cells. TGF-β activation of regulatory T cells has been shown to dampen inflammation and activate self-tolerant immune mechanisms. Elevated levels of TGF-β secreted from natural killer cells, along with IL-10, have been reported in breast cancer patients [52]. TGF-β has also been implicated in the progression of cervical squamous cell carcinoma. TGF-β activation has been demonstrated to be the result of thrombospondin-1, an acute phase inflammatory protein, secretion due to the interaction between cancer cells and cancer-associated fibroblasts [73]. TGF-β receptor 1 and 2 knockout mice exhibit markedly decreased pancreatic tumor growth. However, selective knockout of the TGFbeta receptor in epithelial cells significantly promotes pancreatic tumor growth [74]. This highlights the role of the tumor microenvironment in mediating pro- and anti-tumorigenic inflammation signals. Anti-TGF-β therapy in a recent clinical trial proved to have preliminary tumor reduction on advanced melanoma and renal cell carcinoma, with no apparent toxicity [75]. This establishes TGF-β as a putative viable cytokine target for future cancer therapy.
It has been demonstrated in murine pancreatic cancer models that TGF-β may also have antitumor activity. In orthotopic and genetically engineered murine models, pharmacological inhibition of TGF-β signaling in the pancreas using a TGF-β receptor antagonist contributed to pancreatic ductal adenocarcinoma progression [76]. Like other inflammatory mediators in the tumor microenvironment, TGF-β exhibits both pro- and anti-tumorigenic activity and its actions may require further studies.
Interleukin-1
Other cytokines and chemokines have ambiguous roles in inflammation associated with tumor pathogenesis. Within the IL-1 family, IL-1α, IL-1β, and IL-1 receptor antagonist (IL-1Ra) tend to be a focus of many studies. IL-1 is known to enhance CD4+ T cell proliferation and differentiation of B cells in standard inflammatory settings. It is most often associated with induction of fever in the early phase of the acute inflammatory reaction. However, it has been shown that inhibition of IL-1α and IL-1β in murine myeloma models decreases survival rates. In this model, IL-1α and IL-1β increase Th1 cell secretion of pro-inflammatory cytokines, which in turn activate cytotoxic macrophage responses toward tumor cells [77]. IL-1α is the dominant family member in acute inflammation. However, as acute inflammation progresses to become chronic, IL-1β becomes the predominant mediator [78]. As recent studies have demonstrated, IL-1β appears to play an integral role in inflammation and cancer. Decreased IL-1β secretion by tumor cells or stroma correlates with a decrease in progression-free survival in prostate cancer patients, thus highlighting the potential antitumor mechanisms associated with IL-1β in the tumor microenvironment [79]. Whereby, other studies have suggested IL-1β exhibits pro-tumor effects. Specifically, infiltrating neutrophils in a colitis model will secrete IL-1β that in turn contributes to colitis-associated tumorigenesis via upregulating the secretion of IL-6 [80]. IL-1Ra is a competitive antagonist to IL-1α and IL-1β and has been shown to inhibit their pro-inflammatory mechanisms [81]. While IL-1Ra may appear to be an alluring target for cancer therapy, recent data suggests its role in cancer progression is more complex than previously viewed. An increase in IL-1Ra has been correlated with decreased event-free and overall survival in T cell lymphoma patients [82]. Similarly, IL-Ra levels are increased in women with newly diagnosed breast cancers, as compared to breast cancer negative controls [83]. Taken together, the IL-1 cytokine family activity appears to be rather situational in its pro- and anti-tumorigenic activity.
Interleukin-6
Similar to IL-1, IL-6 is another cytokine implicated in acute inflammation and more specifically fever. IL-6 is synthesized and secreted predominantly by macrophages and enhances macrophages’ ability to present to T cells via upregulation of B7 expression following recognition of pathogen-associated molecular patterns. Unlike the IL-1 family mediators, IL-6 has been shown to be predominantly pro-tumorigenic. In 2014, Karin and Taniguchi described IL-6 as one of the “critical lynchpins” associating inflammation and cancer [84]. They implicated IL-6 downstream signaling as contributing to tumor cell survival and proliferation, as well as to inflammation in the tumor microenvironment. Similarly, they highlighted that IL-6 is not only associated with acute inflammation, but also participates in T cell activation throughout chronic inflammation. IL-6 has also been associated with at least 12 cancer types in humans, including but not limited to stomach, pancreatic, liver, intestinal, uterine, breast, lung, esophageal, prostate, bladder, and kidney cancers [84]. In a murine model of pancreatitis (a chronic inflammatory condition), the deletion of IL-6 has led to the recovery of normal pancreatic tissue as compared to wild-type mice, which ultimately develop pancreatic tumors [85]. IL-6 secretion from fibroblasts has even been implicated as a mechanism for angiogenesis, again highlighting the pro-tumorigenic role of IL-6 [86]. IL-6 has been implicated in the progression of both triple-negative breast cancer and pancreatic cancer [70, 85, 87]. Blocking the IL-6 receptor on breast cancer cells has been shown to render the tumor cells unable to adhere to endothelium, which is a key step in metastasis [87]. Thus, IL-6 not only possesses the potential to promote primary tumor growth but also tumor angiogenesis and metastasis.
Interleukin-10
In noncancer settings, IL-10 is traditionally characterized as an anti-inflammatory cytokine, for its inhibition of NFκβ, a transcription factor implicated in both cancer and inflammation. Recent studies, however, have shown that IL-10 may contribute to tumor growth and even cancer therapy resistance. IL-10 is increased in breast tumor tissue, with a corresponding increase in macrophage infiltration [88]. Further, increased secretion of IL-10 by macrophages in the tumor stroma has been associated with drug resistance in breast cancer [88]. This again presents an interesting paradigm, in which a characteristically anti-inflammatory cytokine contributes to tumor growth.
Chemokine Ligand 2
Chemokine ligand 2 (CCL2) , also known as monocyte chemoattractant protein 1 (MCP1) , is involved in macrophage chemotaxis, signaling macrophages to traffic to a specific tissue site. CCL2 plays a critical role in inflammation and cancer, particularly for its role in breast cancer metastasis. CCL2 secreted by tumor cells and macrophages increases metastatic seeding of breast cancer cells via stimulating the secretion of CCL3 [27]. Interestingly, CCL3, also known as macrophage inflammatory protein 1-α (MIP1-α), is known to play a role in acute inflammation, again demonstrating the intertwined role of inflammation and cancer. Further, a recent clinical trial implicated antagonizing CCL2 activity in pancreatic tumor inhibition. Pharmacologically inhibiting CCR2, the receptor for CCL2, in combination with chemotherapy significantly inhibited tumor growth [89]. In this study, inhibition of CCR2 also decreased tumor-associated macrophages and regulatory T cells while increasing CD8+ and CD4+ T cells.
Lipid and Protein Biomarkers
Another lens to view inflammation and cancer is through inflammatory lipids and proteins. Lipids biosynthesized from arachidonic acid are termed eicosanoids, potent locally acting mediators of inflammation (Fig. 7.1). We discuss specifically prostaglandin E2 (PGE2), epoxyeicosatrienoic acids (EETs), omega-3 fatty acids, and leukotrienes, as well as emerging protein biomarkers annexin A1 and C-reactive protein (CRP).

Arachidonic acid metabolism via cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P450 (CYP450) enzymes. Lipids highlighted include PGE2, LTB4, LTC4, LTD4, LTE4, 11,12-EET, and 14,15-EET for their diverse roles in inflammation-associated tumor pathogenesis
Prostaglandin E2
Prostaglandin E2 (PGE2) is a characteristically pro-inflammatory bioactive lipid synthesized from arachidonic acid initially by cyclooxygenase 1 (COX-1) and cyclooxygenase 2 (COX-2) and then by PGE synthase. Both PGE2 and COX-2 expression have been implicated in inflammation and cancer. PGE2 has been most notably characterized in colon cancer, where neutralizing PGE synthase and thus inhibiting PGE2 synthesis decreases colon tumor formation in genetically engineered murine models of colon cancer [90]. Further, levels of PGE2 positively correlate with cancer stem cell markers in colorectal cancer patient tumor samples. PGE2 has also been shown to be pro-metastatic, as administration of PGE2 results in increased tumor and liver metastasis in a genetically engineered model of colorectal cancer [91]. Mechanistically, PGE2 has been demonstrated to promote tumor growth through stimulation of angiogenesis and immune suppression in several cancers, including colon cancer [92]. PGE2 generated by tumor cells additionally stimulates myeloid-derived suppressor cells to inhibit natural killer cells, contributing to immune suppression [93]. In addition to the well characterized activity of PGE2 in colon cancer, the deletion of COX or PGE synthase in melanoma cells resulted in tumor rejection in immunocompetent mice [94]. However, in Rag1 knockout (KO) mice, melanoma tumor cells lacking PGE synthase form growing tumors [94]. As Rag1 KO mice are unable to generate mature T and B lymphocytes, this result highlights the relationship between PGE2, immune cells, and tumor progression. We analyze the characterization of PGE2 within this paradigm in regard to its relationship with omega-3 fatty acids and aspirin in the sections entitled “Omega-3 Fatty Acids and Derivatives” and “Inflammation and Cancer: Clinical Applications,” respectively.
Epoxyeicosatrienoic Acids
Epoxyeicosatrienoic acids (EETs) are locally acting lipid signaling molecules with a short half-life and exhibit both autocrine and paracrine activities. EETs are the products of arachidonic acid metabolism by cytochrome P450 enzymes. EETs are then further metabolized by soluble epoxide hydrolase (sEH) into dihydroxyeicosatrienoic acid (DHET). EETs have been vastly studied in various inflammatory diseases. While EETs are known to mediate proliferation, migration, and inflammation in human tissues, their molecular mechanisms in doing so remain poorly characterized [95]. EETs are known to play a role in coronary arteriole dilation [96], stimulate tissue and organ regeneration [97], promote wound healing [98], delay seizure onset [99], and participate in many other disease processes with an inflammatory component [100]. Interestingly, their role in specific inflammatory diseases appears to be somewhat complex. EETs have been shown to be cardioprotective, inhibit pathogenesis of diabetes, and exert renal and neuronal protection. EETs also promote tumor cell proliferation and regulate host antitumor immunity [101, 102]. Thus, their role in cancer continues to be an active area of study. It has been demonstrated that both exogenous systemic and endogenous endothelium-derived EETs promote not only tumor growth and angiogenesis in murine models but also metastasis and tumor dormancy escape [101]. The EETs pro-tumor activity in these models was in part due to increasing VEGF secretion from endothelium in the tumor microenvironment [101]. In fact, the pro-tumorigenic role of EETs has also been demonstrated in human breast tumor tissue. Increased levels of 14,15-EET correlate with greater malignancy potential in breast cancer patients [103]. While the role of EETs in cancer requires further elucidation, modulation of EET levels via sEH has been purposed as a possible new direction in cancer therapy [95, 104]. Specifically, the promotion of EET metabolism by the endogenous over-expression of sEH in transgenic mice has been shown to reduce tumor burden [101]. In summary, the role of EETs in cancer remains an interesting vantage point whereby to view inflammation and cancer, while also providing insight into possible new therapeutic targets.
Omega-3 Fatty Acids and Derivatives
Omega-3 fatty acids are essential dietary polyunsaturated fatty acids that are metabolized in the human body to other essential lipid metabolites, including prostaglandins, thromboxane, and leukotrienes. Omega-3 fatty acids and their derivatives have recently gained widespread public attention for their potential wide-ranging health benefits. Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) are omega-3 fatty acids that are frequently marketed as dietary supplements to promote heart health. The potential role of these fatty acids in cancer pathogenesis is also of interest, as diets rich in omega-3 fatty acids correlate with a reduction in cancer-related deaths [105]. The anti-inflammatory mechanism to activity of omega-3 fatty acids has been primarily attributed to its metabolites, EPA and DHA. These molecules saturate enzymes that classically metabolize arachidonic acid into pro-inflammatory molecules and instead generate lipid autacoids with more anti-inflammatory characteristics [106]. EPA and DHA can additionally activate peroxisome proliferator-activated receptors (PPARs), transcription factors with known anti-inflammatory effects [106], and, interestingly, antitumor activity [107, 108]. Dietary supplementation of omega-3 fatty acids has been associated with decreased risk of colorectal cancer mortality [109]. Of note, chronic inflammation, such as ulcerative colitis, is a risk factor for colorectal cancer pathogenesis [110].
Omega-3 fatty acid supplementation has also been demonstrated to decrease prostate cancer progression in murine models via a macrophage-mediated mechanism [111]. Dietary DHA in these models decreased tumor-associated macrophage viability, as well as pro-inflammatory cytokines and NFκβ-mediated gene expression. Induction of tumor cell apoptosis may mediate, at least in part, the antitumor activity of omega-3 fatty acids [112]. Studies have further indicated that omega-3 fatty acids are capable of inducing apoptosis in a range of solid cancers in vitro, ranging from gastrointestinal origin to neural tissue, and even hematological cancers [112]. While the underlying mechanism remains to be fully elucidated, it appears that omega-3 fatty acids play a role in inducing both the intrinsic and extrinsic apoptosis pathways [112]. In addition to inducing apoptotic programs, omega-3 fatty acids play an anti-tumorigenic role through increasing tumor cell susceptibility to cytotoxic therapies and are being investigated as adjuvant cancer therapies [113]. Omega-3 fatty acid supplementation was also found to enrich tumor cell membranes in unsaturated fatty acids, in turn making the cells more susceptible to destruction by free radicals [113].
An important process in tumor malignancy is epithelial-mesenchymal transition (EMT). This transformation is critical for tumor cell invasion into neighboring tissues and sets the foundation for tumor metastasis. DHA has proven to be efficacious in inhibiting epithelial-mesenchymal transition in colorectal cancer models [114]. The mechanism of DHA’s direct action in halting this process remains relatively unknown. As previously described, PGE2 and COX-2 are characteristically pro-tumorigenic. Recent studies in endometrial cancer suggest that omega-3 fatty acid supplementation exerts its antitumor activity through downregulating COX-2 expression and thus subsequently decreasing endogenous PGE2 levels [115]. This mechanism has also been demonstrated in a colon cancer model, in which EPA and DHA supplementation increased lipoxin A4 (LXA4), an anti-inflammatory endogenous lipid mediator [116]. While the intricate workings of omega-3 fatty acids still required further research, their potential in cancer therapeutics appears promising.
Leukotrienes
Similar to EETs, leukotrienes are another class of eicosanoid metabolites of arachidonic acid generated by members of the lipoxygenase enzyme family. Leukotrienes are traditionally viewed as pro-inflammatory molecules and have been highly studied in inflammatory lung diseases. Similar to the other lipid mediators, their role in inflammation is being actively elucidated. Leukotrienes are mainly synthesized by leukocytes, contributing to both innate and adaptive immunity responses [117]. In acute inflammatory settings, leukotriene B4 (LTB4) increases leukocyte trafficking, as well as pro-inflammatory cytokines such as IL-6 and TNFα. In fact, it has been demonstrated that cellular secretion of LTB4 is a crucial first step in potentiating inflammation-induced tumorigenesis in a lung cancer model [118]. The mechanism of LTB4 tumorigenic contribution was demonstrated to be mediated by signaling through binding of its receptor BLT1, characteristically expressed on peripheral blood leukocytes [118]. LC-MS-MS-based profiling demonstrated an increase in leukotrienes LTC4 and LTE4 correlates with tumor progression in aggressive murine lung cancer models [119]. Interestingly, in these models, resident alveolar macrophages demonstrated high expression of 5-lipoxygenase (5-LOX) and subsequent increases in LTB4, LTC4, and LTD4 secretion, as compared to infiltrating macrophages which did not produce leukotrienes [119]. This study provides insight into leukotrienes’ locally acting inflammatory mechanisms in a cancer setting. Interestingly, the deletion of 5-LOX in a murine lung cancer model stimulates primary tumor growth and liver metastasis through the regulation of T cells [120].
Recent evidence implicating 5-LOX as an inhibitor of tumor growth suggests that leukotrienes produced by this enzyme potentially enact anti-tumorigenic programs in addition to their well-characterized pro-tumorigenic activities. Inhibition of 5-LOX in murine models has been demonstrated to reduce polyp burden in intestinal mucosa, a known mechanistic step in the APC-driven adenoma-carcinoma sequence of colon cancer [121]. In this model, the inhibition of 5-LOX was further accompanied by a decrease in inflammatory infiltrate, including cytokines and immune cells [121]. Further, the inhibition of 5-LOX has been shown to selectively induce apoptosis in prostate cancer cells via decreased expression of c-Myc mRNA [122]. C-Myc is a commonly mutated gene in various cancers, allowing for unregulated cell proliferation. In pancreatic cancer models, 5-LOX knockout mice were shown to have decreased pancreatic lesions, precursors to pancreatic ductal adenocarcinoma [123]. Thus, while further studies on the role of leukotrienes in cancer are needed, it is apparent that their modulation could provide important insight into cancer pathogenesis and potentially open new therapeutic avenues.
Annexin A1
A protein of growing interest in inflammation and cancer is annexin A1 , an anti-inflammatory protein that inhibits acute inflammation by blocking phospholipase A2. This neutralization results in decreased eicosanoid production and inhibits leukocyte adhesion and infiltration. In breast cancer patients, high levels of annexin A1 have been associated with decreased overall survival and poor breast cancer-specific survival [124, 125]. Similarly, lung cancer patients have been shown to have a significant increase in annexin A1 levels in plasma [126]. Further, annexin A1 mRNA and protein levels are increased in lung tumor tissue as compared to adjacent, noncancerous tissue [126]. Thus, annexin A1, an anti-inflammatory protein, may prove to be an important biomarker for tumor progression.
C-Reactive Protein
C-reactive protein (CRP) is released by macrophages and enhances complement recognition and subsequent phagocytosis by macrophages. CRP is clinically used as a biomarker for various inflammatory diseases, including inflammatory bowel disease, pelvic inflammatory disease, arthritis, autoimmunity, and even heart disease. While C-reactive protein’s role in cancer has remained elusive, it has been shown to be a biomarker for various cancers. Elevated CRP levels are validated biomarkers in both Crohn’s disease and ulcerative colitis, both chronic inflammatory diseases of the colon. Elevated levels of CRP have also been associated with an increased risk of colorectal cancer development [127]. Moreover, increased levels of CRP correlate with decreased overall survival and more aggressive tumor recurrence and metastasis in oral squamous cell carcinomas [128]. CRP levels have also been associated with an increased risk of epithelial ovarian carcinoma and breast cancer [129,130,131]. Increased levels of CRP are correlated with decreased cancer-specific survival in pancreatic cancer patients [132]. While the relationship between CRP and cancer risk and progression remains unclear, research suggests that CRP is largely pro-tumorigenic and could serve as a clinical biomarker for several types of cancer.
Inflammation and Cancer: Clinical Applications
Chronic inflammation is a known risk factor for various cancers. For example, pancreatitis is a known risk factor for pancreatic cancer, ulcerative colitis and Crohn’s disease are known risk factors for colon cancer, and Helicobacter pylori infections are known to increase the risk of stomach cancer. Other associations include cystitis and bladder cancer, Barrett’s esophagus and esophageal carcinoma, and bronchitis and lung cancer [16, 133]. However, the extent to which underlying inflammation contributes to tumor growth remains elusive. Inflammatory reactions appear to have a threshold for which they can either play a pro-tumorigenic role or anti-tumorigenic role. Thus, characterizing the point at which inflammation transitions from facilitating tumor inhibition to promoting disease progression has been the aim of many recent clinical studies. These studies aim to evaluate the “overall inflammatory score” in relation to specific cancers. Further, these clinical studies seek to highlight a potential threshold between acute and chronic inflammation that, when surpassed, could contribute to cancer pathogenesis.
In a recent study, in which the inflammatory score reflected the neutrophil to lymphocyte ratio, an increased neutrophil to lymphocyte ratio is an independent predictive marker or clinical outcomes in head and neck squamous cell carcinoma patients. This increased ratio was also associated with a decrease in overall survival and recurrence-free survival [134]. Another study found men who demonstrated either acute or chronic inflammation in prostate tissue with a negative prostate biopsy have a decreased risk in prostate cancer pathogenesis 2 years post-biopsy. However, 4 years post-biopsy, decreased risk in prostate cancer was found to be positively associated with acute inflammation [135]. In a similar study on prostate cancer, chronic inflammation, defined by the presence of lymphocytes, plasma cells, and macrophages, was associated with lower prostate tumor volume [136]. In a large-scale study evaluating the overall risk of cancer, a combination of CRP levels and leukocyte count was used as an inflammatory score. An increase in the inflammatory score correlated with an increase in prostate, lung, and colorectal cancer risk [137]. A correlation between decreased inflammation and increased cancer survival has also been shown in cervical cancer, in which a decreased platelet to lymphocyte ratio correlates with an increase in overall and disease-free survival in patients [138]. Recent studies further highlight the importance of lymphocyte to monocyte ratio in several cancers. Interestingly, decrease in lymphocyte to monocyte ratio was found to be consistent with decrease in overall survival in colorectal cancer, lung cancer, pancreatic cancer, and Hodgkin’s lymphoma [139]. An additional large-scale, retrospective study demonstrated that a greater ratio of lymphocytes to monocytes resulted in increased overall and disease-free survival in non-small cell lung cancer patients [140].
Thus, while the mechanisms underlying inflammation’s contribution to cancer progression remain an area of ongoing research, it is apparent that inflammation has an intimate connection with tumor pathogenesis. Therefore, it is no surprise that in efforts to prevent cancer progression, widely used anti-inflammatory medications have come into focus as prophylaxis. Aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) have been the subject of small- and large-scale cancer patient studies. Beginning in the late 1980s, there has been a growing body of evidence implicating aspirin’s ability to decrease colorectal cancer risk [141, 142]. Although yet to be elucidated, aspirin’s antitumor mechanism appears to be its multifaceted inhibition of inflammation through COX acetylation. Aspirin acetylation of cyclooxygenase inhibits prostaglandin synthesis, thus decreasing the pro-inflammatory and pro-tumorigenic activities of PGE2 as well as decreasing platelet activation. It has been hypothesized that aspirin also inhibits immune cell activation [142]. Aspirin regiments have been associated with decreased risks of epithelial ovarian cancer and gastric cancer [143,144,145]. Aspirin has also been associated with decreased risk of breast cancer-related death following breast cancer diagnosis [146]. However, retrospective studies assessing aspirin’s anticancer benefits remain controversial. One review concluded that there was no statistically significant correlation between low-dose aspirin use and reduction in overall cancer risk and that aspirin’s chemopreventive activity was limited to colorectal cancer risk [147]. Similarly, other nonsteroidal anti-inflammatory drugs have proved to decrease risk of colorectal and prostate cancer [148,149,150]. The mechanism of NSAIDs within the tumor microenvironment has also been characterized as their ability to inhibit tumorigenic potential and tumor immune tolerance, as well as to enhance immunosurveillance [110]. Interestingly, this activity was found to be mediated in part through the reduction of PGE2 [110]. However, anti-inflammatories such as aspirin and COX-2 inhibitors have faced therapeutic challenges in the clinic, as they cause severe side effects such as kidney toxicity, stomach bleeding, and heart complications. Thus, more efforts to characterize the anti-tumorigenic mechanisms of aspirin and other NSAIDS are needed to develop novel viable clinical therapeutics that harness their broad anticancer activity.
Summary
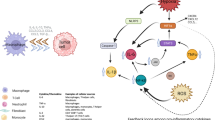
Extensive evidence has established a link between inflammation and cancer, from the first observation of inflammation in tumors hundreds of years ago to current studies that have characterized the malleable and complex roles inflammatory cells and their mediators play in cancer progression. Experimental and clinical studies provide mechanisms that can be harnessed for future cancer therapy, with the aim to halt pro-tumorigenic inflammatory processes or harness the anti-tumorigenic pathways embedded in the human immune system. Many of the lipid mediators discussed above have potential as cancer biomarkers and exhibit a dual role in tumorigenesis, highlighting their diverse biological activity in various tumor microenvironments (Fig. 7.2). The frame through which to view inflammation and cancer is not one of strictly “pro-tumor” or “antitumor”; rather there exists a multitude of environmental influences that ultimately direct the role inflammation plays within the context of cancer. Future studies will be required to elucidate both the pro- and anti-tumorigenic roles inflammation plays, as well as to provide novel therapies that harness the immune system to inhibit or prevent cancer.

Biomarkers in the tumor microenvironment can simultaneously promote (red) and inhibit (blue) tumor pathogenesis, highlighting the fluidity of the role of inflammation in cancer
References
Trinchieri G. Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annu Rev Immunol. 2012;30:677–706. doi:10.1146/annurev-immunol-020711-075008.
Reedy J. Galen on cancer and related diseases. Clio Med. 1975;10:227–38.
Virchow R. Cellular pathology as based upon physiological and pathological histology: twenty lectures delivered in the pathological Institute of Berlin during the months of February, march, and April, 1858. New York: Robert M. De Witt; 1860.
Coley WB II. Contribution to the knowledge of sarcoma. Ann Surg. 1891;14:199–220.
Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–9. doi:10.1056/NEJM198612253152606.
Coussens LM, et al. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999;13:1382–97.
Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell. 2000;103:481–90.
Di Carlo E, et al. The intriguing role of polymorphonuclear neutrophils in antitumor reactions. Blood. 2001;97:339–45.
Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. 2015;15:73–86. doi:10.1038/nri3789.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi:10.1016/j.cell.2011.02.013.
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44. doi:10.1038/nature07205.
Mantovani A, Caprioli V, Gritti P, Spreafico F. Human mature macrophages mediate antibody-dependent cellular cytotoxicity on tumour cells. Transplantation. 1977;24:291–3.
Mantovani A, Bottazzi B, Colotta F, Sozzani S, Ruco L. The origin and function of tumor-associated macrophages. Immunol Today. 1992;13:265–70. doi:10.1016/0167-5699(92)90008-U.
Balkwill FR, Ward BG, Moodie E, Fiers W. Therapeutic potential of tumor necrosis factor-alpha and gamma-interferon in experimental human ovarian cancer. Cancer Res. 1987;47:4755–8.
Balkwill F. Tumor necrosis factor or tumor promoting factor? Cytokine Growth Factor Rev. 2002;13:135–41.
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi:10.1038/nature01322.
Pikarsky E, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6. doi:10.1038/nature02924.
Luo JL, Maeda S, Hsu LC, Yagita H, Karin M. Inhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell. 2004;6:297–305. doi:10.1016/j.ccr.2004.08.012.
Elwood PC, et al. Aspirin in the treatment of cancer: reductions in metastatic spread and in mortality: a systematic review and meta-analyses of published studies. PLoS One. 2016;11:e0152402. doi:10.1371/journal.pone.0152402.
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi:10.1016/j.cell.2010.03.014.
Heuff G, et al. Enhanced tumour growth in the rat liver after selective elimination of Kupffer cells. Cancer Immunol Immunother. 1993;37:125–30.
Oosterling SJ, et al. Macrophages direct tumour histology and clinical outcome in a colon cancer model. J Pathol. 2005;207:147–55. doi:10.1002/path.1830.
Weber C, et al. Macrophage infiltration and alternative activation during wound healing promote MEK1-induced skin carcinogenesis. Cancer Res. 2016;76:805–17. doi:10.1158/0008-5472.CAN-14-3676.
Jaiswal S, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271–85. doi:10.1016/j.cell.2009.05.046.
Gabrusiewicz K, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. 2016;1:e85841. doi:10.1172/jci.insight.85841.
Chow A, et al. Macrophage immunomodulation by breast cancer-derived exosomes requires toll-like receptor 2-mediated activation of NF-kappaB. Sci Rep. 2014;4:5750. doi:10.1038/srep05750.
Kitamura T, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med. 2015;212:1043–59. doi:10.1084/jem.20141836.
Chavez-Galan L, Olleros ML, Vesin D, Garcia I. Much more than M1 and M2 macrophages, there are also CD169(+) and TCR(+) macrophages. Front Immunol. 2015;6:263. doi:10.3389/fimmu.2015.00263.
Van Overmeire E, Laoui D, Keirsse J, Van Ginderachter JA, Sarukhan A. Mechanisms driving macrophage diversity and specialization in distinct tumor microenvironments and parallelisms with other tissues. Front Immunol. 2014;5:127. doi:10.3389/fimmu.2014.00127.
Dey A, Allen J, Hankey-Giblin PA. Ontogeny and polarization of macrophages in inflammation: blood monocytes versus tissue macrophages. Front Immunol. 2014;5:683. doi:10.3389/fimmu.2014.00683.
Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi:10.1016/j.immuni.2014.06.010.
Fullerton JN, Gilroy DW. Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov. 2016;15:551–67. doi:10.1038/nrd.2016.39.
Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101. doi:10.1038/nature13479.
Zaynagetdinov R, et al. Chronic NF-kappaB activation links COPD and lung cancer through generation of an immunosuppressive microenvironment in the lungs. Oncotarget. 2016;7:5470–82. doi:10.18632/oncotarget.6562.
Zonari E, et al. A role for miR-155 in enabling tumor-infiltrating innate immune cells to mount effective antitumor responses in mice. Blood. 2013;122:243–52. doi:10.1182/blood-2012-08-449306.
Bellora F, et al. TLR activation of tumor-associated macrophages from ovarian cancer patients triggers cytolytic activity of NK cells. Eur J Immunol. 2014;44:1814–22. doi:10.1002/eji.201344130.
Peng J, et al. Inhibition of TGF-beta signaling in combination with TLR7 ligation re-programs a tumoricidal phenotype in tumor-associated macrophages. Cancer Lett. 2013;331:239–49. doi:10.1016/j.canlet.2013.01.001.
Bernhard CA, Ried C, Kochanek S, Brocker T. CD169+ macrophages are sufficient for priming of CTLs with specificities left out by cross-priming dendritic cells. Proc Natl Acad Sci U S A. 2015;112:5461–6. doi:10.1073/pnas.1423356112.
Ohnishi K, et al. Prognostic significance of CD169-positive lymph node sinus macrophages in patients with endometrial carcinoma. Cancer Sci. 2016;107:846–52. doi:10.1111/cas.12929.
Saito Y, et al. Prognostic significance of CD169+ lymph node sinus macrophages in patients with malignant melanoma. Cancer Immunol Res. 2015;3:1356–63. doi:10.1158/2326-6066.CIR-14-0180.
Pucci F, et al. SCS macrophages suppress melanoma by restricting tumor-derived vesicle-B cell interactions. Science. 2016;352:242–6. doi:10.1126/science.aaf1328.
Palucka K, Coussens LM, O’Shaughnessy J. Dendritic cells, inflammation, and breast cancer. Cancer J. 2013;19:511–6. doi:10.1097/PPO.0000000000000007.
Tran Janco JM, Lamichhane P, Karyampudi L, Knutson KL. Tumor-infiltrating dendritic cells in cancer pathogenesis. J Immunol. 2015;194:2985–91. doi:10.4049/jimmunol.1403134.
Lohela M, et al. Intravital imaging reveals distinct responses of depleting dynamic tumor-associated macrophage and dendritic cell subpopulations. Proc Natl Acad Sci U S A. 2014;111:E5086–95. doi:10.1073/pnas.1419899111.
Scarlett UK, et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J Exp Med. 2012;209:495–506. doi:10.1084/jem.20111413.
Prue RL, et al. A phase I clinical trial of CD1c (BDCA-1)+ dendritic cells pulsed with HLA-A*0201 peptides for immunotherapy of metastatic hormone refractory prostate cancer. J Immunother. 2015;38:71–6. doi:10.1097/CJI.0000000000000063.
Kranz LM, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534:396–401. doi:10.1038/nature18300.
Cornelissen R, et al. Extended tumor control after dendritic cell vaccination with low-dose cyclophosphamide as adjuvant treatment in patients with malignant pleural mesothelioma. Am J Respir Crit Care Med. 2016;193:1023–31. doi:10.1164/rccm.201508-1573OC.
Phuphanich S, et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother. 2013;62:125–35. doi:10.1007/s00262-012-1319-0.
Geiger TL, Sun JC. Development and maturation of natural killer cells. Curr Opin Immunol. 2016;39:82–9. doi:10.1016/j.coi.2016.01.007.
Wolf MJ, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. 2014;26:549–64. doi:10.1016/j.ccell.2014.09.003.
Ostapchuk YO, et al. Peripheral blood NK cells expressing HLA-G, IL-10 and TGF-beta in healthy donors and breast cancer patients. Cell Immunol. 2015;298:37–46. doi:10.1016/j.cellimm.2015.09.002.
Pasero C, et al. Highly effective NK cells are associated with good prognosis in patients with metastatic prostate cancer. Oncotarget. 2015;6:14360–73. doi:10.18632/oncotarget.3965.
Yin T, et al. Human cancer cells with stem cell-like phenotype exhibit enhanced sensitivity to the cytotoxicity of IL-2 and IL-15 activated natural killer cells. Cell Immunol. 2016;300:41–5. doi:10.1016/j.cellimm.2015.11.009.
Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol. 2013;13:309–20. doi:10.1038/nri3442.
Daniel D, et al. Immune enhancement of skin carcinogenesis by CD4+ T cells. J Exp Med. 2003;197:1017–28. doi:10.1084/jem.20021047.
Ma C, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531:253–7. doi:10.1038/nature16969.
Dadi S, et al. Cancer immunosurveillance by tissue-resident innate lymphoid cells and innate-like T cells. Cell. 2016;164:365–77. doi:10.1016/j.cell.2016.01.002.
Ganesan AP, et al. Tumor-infiltrating regulatory T cells inhibit endogenous cytotoxic T cell responses to lung adenocarcinoma. J Immunol. 2013;191:2009–17. doi:10.4049/jimmunol.1301317.
Ino Y, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer. 2013;108:914–23. doi:10.1038/bjc.2013.32.
Mlecnik B, et al. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci Transl Med. 2016;8:327ra326. doi:10.1126/scitranslmed.aad6352.
Gunderson AJ, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discov. 2016;6:270–85. doi:10.1158/2159-8290.CD-15-0827.
Pylayeva-Gupta Y, et al. IL35-producing B cells promote the development of pancreatic neoplasia. Cancer Discov. 2016;6:247–55. doi:10.1158/2159-8290.CD-15-0843.
Lundgren S, Berntsson J, Nodin B, Micke P, Jirstrom K. Prognostic impact of tumour-associated B cells and plasma cells in epithelial ovarian cancer. J Ovarian Res. 2016;9:21. doi:10.1186/s13048-016-0232-0.
Iglesia MD, et al. Prognostic B-cell signatures using mRNA-seq in patients with subtype-specific breast and ovarian cancer. Clin Cancer Res. 2014;20:3818–29. doi:10.1158/1078-0432.CCR-13-3368.
Schioppa T, et al. B regulatory cells and the tumor-promoting actions of TNF-alpha during squamous carcinogenesis. Proc Natl Acad Sci U S A. 2011;108:10662–7. doi:10.1073/pnas.1100994108.
Kusne Y, et al. Targeting aPKC disables oncogenic signaling by both the EGFR and the proinflammatory cytokine TNFalpha in glioblastoma. Sci Signal. 2014;7:ra75. doi:10.1126/scisignal.2005196.
Gupta M, Babic A, Beck AH, Terry K. TNF-alpha expression, risk factors, and inflammatory exposures in ovarian cancer: evidence for an inflammatory pathway of ovarian carcinogenesis? Hum Pathol. 2016;54:82–91. doi:10.1016/j.humpath.2016.03.006.
Liao C, et al. Association between Th17-related cytokines and risk of non-small cell lung cancer among patients with or without chronic obstructive pulmonary disease. Cancer. 2015;121(Suppl 17):3122–9. doi:10.1002/cncr.29369.
Blogowski W, et al. Selected cytokines in patients with pancreatic cancer: a preliminary report. PLoS One. 2014;9:e97613. doi:10.1371/journal.pone.0097613.
Roberts NJ, Zhou S, Diaz LA Jr, Holdhoff M. Systemic use of tumor necrosis factor alpha as an anticancer agent. Oncotarget. 2011;2:739–51. doi:10.18632/oncotarget.344.
Dondossola E, et al. Self-targeting of TNF-releasing cancer cells in preclinical models of primary and metastatic tumors. Proc Natl Acad Sci U S A. 2016;113:2223–8. doi:10.1073/pnas.1525697113.
Nagura M, et al. Invasion of uterine cervical squamous cell carcinoma cells is facilitated by locoregional interaction with cancer-associated fibroblasts via activating transforming growth factor-beta. Gynecol Oncol. 2015;136:104–11. doi:10.1016/j.ygyno.2014.11.075.
Principe DR, et al. TGFbeta signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res. 2016;76:2525–39. doi:10.1158/0008-5472.CAN-15-1293.
Morris JC, et al. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS One. 2014;9:e90353. doi:10.1371/journal.pone.0090353.
Zhao Z, Xi H, Xu D, Li C. Transforming growth factor beta receptor signaling restrains growth of pancreatic carcinoma cells. Tumour Biol. 2015;36:7711–6. doi:10.1007/s13277-015-3466-3.
Haabeth OA, Lorvik KB, Yagita H, Bogen B, Corthay A. Interleukin-1 is required for cancer eradication mediated by tumor-specific Th1 cells. Oncoimmunology. 2016;5:e1039763. doi:10.1080/2162402X.2015.1039763.
Dinarello CA, van der Meer JW. Treating inflammation by blocking interleukin-1 in humans. Semin Immunol. 2013;25:469–84. doi:10.1016/j.smim.2013.10.008.
Rodriguez-Berriguete G, et al. Clinical significance of both tumor and stromal expression of components of the IL-1 and TNF-alpha signaling pathways in prostate cancer. Cytokine. 2013;64:555–63. doi:10.1016/j.cyto.2013.09.003.
Wang Y, et al. Neutrophil infiltration favors colitis-associated tumorigenesis by activating the interleukin-1 (IL-1)/IL-6 axis. Mucosal Immunol. 2014;7:1106–15. doi:10.1038/mi.2013.126.
Palomo J, Dietrich D, Martin P, Palmer G, Gabay C. The interleukin (IL)-1 cytokine family--balance between agonists and antagonists in inflammatory diseases. Cytokine. 2015;76:25–37. doi:10.1016/j.cyto.2015.06.017.
Gupta M, et al. Comprehensive serum cytokine analysis identifies IL-1RA and soluble IL-2Ralpha as predictors of event-free survival in T-cell lymphoma. Ann Oncol. 2016;27:165–72. doi:10.1093/annonc/mdv486.
Patel SK, et al. Inflammatory biomarkers, comorbidity, and neurocognition in women with newly diagnosed breast cancer. J Natl Cancer Inst. 2015;107. doi: 10.1093/jnci/djv131 .
Taniguchi K, Karin M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin Immunol. 2014;26:54–74. doi:10.1016/j.smim.2014.01.001.
Zhang Y, et al. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res. 2013;73:6359–74. doi:10.1158/0008-5472.CAN-13-1558-T.
Nagasaki T, et al. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br J Cancer. 2014;110:469–78. doi:10.1038/bjc.2013.748.
Geng Y, et al. Phenotypic switch in blood: effects of pro-inflammatory cytokines on breast cancer cell aggregation and adhesion. PLoS One. 2013;8:e54959. doi:10.1371/journal.pone.0054959.
Yang C, et al. Increased drug resistance in breast cancer by tumor-associated macrophages through IL-10/STAT3/bcl-2 signaling pathway. Med Oncol. 2015;32:352. doi:10.1007/s12032-014-0352-6.
Nywening TM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17:651–62. doi:10.1016/S1470-2045(16)00078-4.
Montrose DC, et al. The role of PGE2 in intestinal inflammation and tumorigenesis. Prostaglandins Other Lipid Mediat. 2015;116–117:26–36. doi:10.1016/j.prostaglandins.2014.10.002.
Wang D, Fu L, Sun H, Guo L, DuBois RN. Prostaglandin E2 promotes colorectal cancer stem cell expansion and metastasis in mice. Gastroenterology. 2015;149:1884–95. doi:10.1053/j.gastro.2015.07.064.
Xu L, et al. COX-2 inhibition potentiates antiangiogenic cancer therapy and prevents metastasis in preclinical models. Sci Transl Med. 2014;6:242ra284. doi:10.1126/scitranslmed.3008455.
Mao Y, et al. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res. 2014;20:4096–106. doi:10.1158/1078-0432.CCR-14-0635.
Zelenay S, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. 2015;162:1257–70. doi:10.1016/j.cell.2015.08.015.
Panigrahy D, Greene ER, Pozzi A, Wang DW, Zeldin DC. EET signaling in cancer. Cancer Metastasis Rev. 2011;30:525–40. doi:10.1007/s10555-011-9315-y.
Larsen BT, et al. Epoxyeicosatrienoic and dihydroxyeicosatrienoic acids dilate human coronary arterioles via BK(ca) channels: implications for soluble epoxide hydrolase inhibition. Am J Physiol Heart Circ Physiol. 2006;290:H491–9. doi:10.1152/ajpheart.00927.2005.
Panigrahy D, et al. Epoxyeicosanoids promote organ and tissue regeneration. Proc Natl Acad Sci U S A. 2013;110(33):13528–33. doi:10.1073/pnas.1311565110.
Sander AL, et al. Cytochrome P450-derived epoxyeicosatrienoic acids accelerate wound epithelialization and neovascularization in the hairless mouse ear wound model. Langenbeck’s Arch Surg. 2011;396:1245–53. doi:10.1007/s00423-011-0838-z.
Inceoglu B, et al. Epoxy fatty acids and inhibition of the soluble epoxide hydrolase selectively modulate GABA mediated neurotransmission to delay onset of seizures. PLoS One. 2013;8:e80922. doi:10.1371/journal.pone.0080922.
Zhang G, Kodani S, Hammock BD. Stabilized epoxygenated fatty acids regulate inflammation, pain, angiogenesis and cancer. Prog Lipid Res. 2014;53:108–23. doi:10.1016/j.plipres.2013.11.003.
Panigrahy D, et al. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J Clin Invest. 2012;122:178–91. doi:10.1172/JCI58128.
Wang D, Dubois RN. Epoxyeicosatrienoic acids: a double-edged sword in cardiovascular diseases and cancer. J Clin Invest. 2012;122:19–22. doi:10.1172/JCI61453.
Wei X, et al. Elevated 14,15- epoxyeicosatrienoic acid by increasing of cytochrome P450 2C8, 2C9 and 2J2 and decreasing of soluble epoxide hydrolase associated with aggressiveness of human breast cancer. BMC Cancer. 2014;14:841. doi:10.1186/1471-2407-14-841.
Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol. 2013;53:37–58. doi:10.1146/annurev-pharmtox-011112-140244.
Bell GA, et al. Intake of long-chain omega-3 fatty acids from diet and supplements in relation to mortality. Am J Epidemiol. 2014;179:710–20. doi:10.1093/aje/kwt326.
Yates CM, Calder PC, Ed Rainger G. Pharmacology and therapeutics of omega-3 polyunsaturated fatty acids in chronic inflammatory disease. Pharmacol Ther. 2014;141:272–82. doi:10.1016/j.pharmthera.2013.10.010.
Panigrahy D, et al. PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc Natl Acad Sci U S A. 2008;105:985–90. doi:10.1073/pnas.0711281105.
Kaipainen A, et al. PPARalpha deficiency in inflammatory cells suppresses tumor growth. PLoS One. 2007;2:e260. doi:10.1371/journal.pone.0000260.
Song M, et al. Marine omega-3 polyunsaturated fatty acid intake and survival after colorectal cancer diagnosis. Gut. 2016. doi:10.1136/gutjnl-2016-311990.
Wang D, DuBois RN. The role of anti-inflammatory drugs in colorectal cancer. Annu Rev Med. 2013;64:131–44. doi:10.1146/annurev-med-112211-154330.
Liang P, et al. Effect of dietary omega-3 fatty acids on tumor-associated macrophages and prostate cancer progression. Prostate. 2016;76:1293–302. doi:10.1002/pros.23218.
D’Eliseo D, Velotti F. Omega-3 fatty acids and cancer cell cytotoxicity: implications for multi-targeted cancer therapy. J Clin Med. 2016;5:15. doi:10.3390/jcm5020015.
Nabavi SF, et al. Omega-3 polyunsaturated fatty acids and cancer: lessons learned from clinical trials. Cancer Metastasis Rev. 2015;34:359–80. doi:10.1007/s10555-015-9572-2.
D’Eliseo D, et al. Epithelial-to-mesenchymal transition and invasion are upmodulated by tumor-expressed granzyme B and inhibited by docosahexaenoic acid in human colorectal cancer cells. J Exp Clin Cancer Res. 2016;35:24. doi:10.1186/s13046-016-0302-6.
Pan J, et al. Elevation of omega-3 polyunsaturated fatty acids attenuates PTEN-deficiency induced endometrial cancer development through regulation of COX-2 and PGE2 production. Sci Rep. 2015;5:14958. doi:10.1038/srep14958.
Zhang C, Yu H, Ni X, Shen S, Das UN. Growth inhibitory effect of polyunsaturated fatty acids (PUFAs) on colon cancer cells via their growth inhibitory metabolites and fatty acid composition changes. PLoS One. 2015;10:e0123256. doi:10.1371/journal.pone.0123256.
Di Gennaro A, Haeggstrom JZ. The leukotrienes: immune-modulating lipid mediators of disease. Adv Immunol. 2012;116:51–92. doi:10.1016/B978-0-12-394300-2.00002-8.
Satpathy SR, et al. Crystalline silica-induced leukotriene B4-dependent inflammation promotes lung tumour growth. Nat Commun. 2015;6:7064. doi:10.1038/ncomms8064.
Poczobutt JM, et al. Eicosanoid profiling in an orthotopic model of lung cancer progression by mass spectrometry demonstrates selective production of leukotrienes by inflammatory cells of the microenvironment. PLoS One. 2013;8:e79633. doi:10.1371/journal.pone.0079633.
Poczobutt JM, et al. Deletion of 5-lipoxygenase in the tumor microenvironment promotes lung cancer progression and metastasis through regulating T cell recruitment. J Immunol. 2016;196:891–901. doi:10.4049/jimmunol.1501648.
Gounaris E, et al. Zileuton, 5-lipoxygenase inhibitor, acts as a chemopreventive agent in intestinal polyposis, by modulating polyp and systemic inflammation. PLoS One. 2015;10:e0121402. doi:10.1371/journal.pone.0121402.
Sarveswaran S, Chakraborty D, Chitale D, Sears R, Ghosh J. Inhibition of 5-lipoxygenase selectively triggers disruption of c-Myc signaling in prostate cancer cells. J Biol Chem. 2015;290:4994–5006. doi:10.1074/jbc.M114.599035.
Knab LM, et al. Ablation of 5-lipoxygenase mitigates pancreatic lesion development. J Surg Res. 2015;194:481–7. doi:10.1016/j.jss.2014.10.021.
Bhardwaj A, et al. Annexin A1 preferentially predicts poor prognosis of basal-like breast cancer patients by activating mTOR-S6 signaling. PLoS One. 2015;10:e0127678. doi:10.1371/journal.pone.0127678.
Sobral-Leite M, et al. Annexin A1 expression in a pooled breast cancer series: association with tumor subtypes and prognosis. BMC Med. 2015;13:156. doi:10.1186/s12916-015-0392-6.
Rong B, et al. Elevated serum annexin A1 as potential diagnostic marker for lung cancer: a retrospective case-control study. Am J Transl Res. 2014;6:558–69.
Ananthakrishnan AN, et al. Serum inflammatory markers and risk of colorectal cancer in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2014;12:1342–8. doi:10.1016/j.cgh.2013.12.030.
Chen IH, et al. Using SCC antigen and CRP levels as prognostic biomarkers in recurrent oral cavity squamous cell carcinoma. PLoS One. 2014;9:e103265. doi:10.1371/journal.pone.0103265.
Ose J, et al. Inflammatory markers and risk of epithelial ovarian cancer by tumor subtypes: the EPIC cohort. Cancer Epidemiol Biomark Prev. 2015;24:951–61. doi:10.1158/1055-9965.EPI-14-1279-T.
Trabert B, et al. Pre-diagnostic serum levels of inflammation markers and risk of ovarian cancer in the prostate, lung, colorectal and ovarian cancer (PLCO) screening trial. Gynecol Oncol. 2014;135:297–304. doi:10.1016/j.ygyno.2014.08.025.
Guo L, et al. C-reactive protein and risk of breast cancer: a systematic review and meta-analysis. Sci Rep. 2015;5:10508. doi:10.1038/srep10508.
Szkandera J, et al. Validation of C-reactive protein levels as a prognostic indicator for survival in a large cohort of pancreatic cancer patients. Br J Cancer. 2014;110:183–8. doi:10.1038/bjc.2013.701.
Greene ER, Huang S, Serhan CN, Panigrahy D. Regulation of inflammation in cancer by eicosanoids. Prostaglandins Other Lipid Mediat. 2011;96:27–36. doi:10.1016/j.prostaglandins.2011.08.004.
Charles KA, et al. Systemic inflammation is an independent predictive marker of clinical outcomes in mucosal squamous cell carcinoma of the head and neck in oropharyngeal and non-oropharyngeal patients. BMC Cancer. 2016;16:124. doi:10.1186/s12885-016-2089-4.
Moreira DM, et al. Baseline prostate inflammation is associated with a reduced risk of prostate cancer in men undergoing repeat prostate biopsy: results from the REDUCE study. Cancer. 2014;120:190–6. doi:10.1002/cncr.28349.
Moreira DM, Nickel JC, Andriole GL, Castro-Santamaria R, Freedland SJ. Chronic baseline prostate inflammation is associated with lower tumor volume in men with prostate cancer on repeat biopsy: results from the REDUCE study. Prostate. 2015;75:1492–8. doi:10.1002/pros.23041.
Morrison L, et al. Inflammatory biomarker score and cancer: a population-based prospective cohort study. BMC Cancer. 2016;16:80. doi:10.1186/s12885-016-2115-6.
Zheng RR, et al. Cervical cancer systemic inflammation score: a novel predictor of prognosis. Oncotarget. 2016;7:15230–42. doi:10.18632/oncotarget.7378.
Gu L, et al. Prognostic role of lymphocyte to monocyte ratio for patients with cancer: evidence from a systematic review and meta-analysis. Oncotarget. 2016;7:31926–42. doi:10.18632/oncotarget.7876.
Hu P, et al. Prognostic significance of systemic inflammation-based lymphocyte- monocyte ratio in patients with lung cancer: based on a large cohort study. PLoS One. 2014;9:e108062. doi:10.1371/journal.pone.0108062.
Kune GA, Kune S, Watson LF. Colorectal cancer risk, chronic illnesses, operations, and medications: case control results from the Melbourne colorectal cancer study. Cancer Res. 1988;48:4399–404.
Drew DA, Cao Y, Chan AT. Aspirin and colorectal cancer: the promise of precision chemoprevention. Nat Rev Cancer. 2016;16:173–86. doi:10.1038/nrc.2016.4.
Baandrup L, Kjaer SK, Olsen JH, Dehlendorff C, Friis S. Low-dose aspirin use and the risk of ovarian cancer in Denmark. Ann Oncol. 2015;26:787–92. doi:10.1093/annonc/mdu578.
Trabert B, et al. Aspirin, nonaspirin nonsteroidal anti-inflammatory drug, and acetaminophen use and risk of invasive epithelial ovarian cancer: a pooled analysis in the ovarian cancer association consortium. J Natl Cancer Inst. 2014;106:djt431. doi:10.1093/jnci/djt431.
Ye X, et al. Frequency-risk and duration-risk relationships between aspirin use and gastric cancer: a systematic review and meta-analysis. PLoS One. 2013;8:e71522. doi:10.1371/journal.pone.0071522.
Fraser DM, Sullivan FM, Thompson AM, McCowan C. Aspirin use and survival after the diagnosis of breast cancer: a population-based cohort study. Br J Cancer. 2014;111:623–7. doi:10.1038/bjc.2014.264.
Chubak J, et al. Aspirin for the prevention of cancer incidence and mortality: systematic evidence reviews for the U.S. preventive services task force. Ann Intern Med. 2016;164:814–25. doi:10.7326/M15-2117.
Friis S, Riis AH, Erichsen R, Baron JA, Sorensen HT. Low-dose aspirin or nonsteroidal anti-inflammatory drug use and colorectal cancer risk: a population-based, case-control study. Ann Intern Med. 2015;163:347–55. doi:10.7326/M15-0039.
Nan H, et al. Association of aspirin and NSAID use with risk of colorectal cancer according to genetic variants. JAMA. 2015;313:1133–42. doi:10.1001/jama.2015.1815.
Vidal AC, et al. Aspirin, NSAIDs, and risk of prostate cancer: results from the REDUCE study. Clin Cancer Res. 2015;21:756–62. doi:10.1158/1078-0432.CCR-14-2235.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Sulciner, M.L., Gilligan, M.M., Zetter, B.R., Panigrahy, D. (2017). Inflammation and Cancer: The Role of Lipid Signaling in the Continuum Between Two Ends of the Tumor Spectrum. In: Akslen, L., Watnick, R. (eds) Biomarkers of the Tumor Microenvironment. Springer, Cham. https://doi.org/10.1007/978-3-319-39147-2_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-39147-2_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-39145-8
Online ISBN: 978-3-319-39147-2
eBook Packages: MedicineMedicine (R0)