
Abstract
The CRISPR revolution that began in 2013 has been adopted and embraced by many researchers worldwide, including the mouse molecular genetics community. CRISPR represents one of only a few radical and transformative shifts in transgenic technologies over the past 30 years. This chapter discusses the paradigm shift that CRISPR technology has brought about in the field of mouse genome editing.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
During the past three decades, techniques and procedures in genome manipulation developed and evolved primarily using the laboratory mouse as a model system, mainly because of the availability of murine embryonic stem (ES) cells . ES cells from no species other than the mouse were as robust and efficient for usurping homologous recombination (HR) to induce targeted genetic changes in the mammalian genome. Methods for targeted genomic manipulation without the use of mouse ES cells were practically nonexistent. In the last few years, many techniques involving “designer nucleases” such as zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats/Crispr-associated 9 (CRISPR/Cas9) have been developed that enable species-agnostic genome editing without the need for ES cells. These new techniques have breached the species barrier and seamlessly made their way into the genome-editing arenas of many other species.
Among the methods that use designer nucleases (also known as sequence-specific endonucleases), the CRISPR/Cas9 system has become the most popular. Its technical simplicity, rapidity of designing and performing experiments, and high success rate have been well documented in almost every species in which it has been tried to date. In the pre-CRISPR era, ES cells served as critical reagents enabling giant strides in the field, including the development of tens of thousands of reagents to systematically knock out (KO) mouse genes. Now, however, their utility is becoming overshadowed by new technologies as the mouse genome engineering community is shifting heavily toward CRISPR-based genome-editing approaches. In this chapter, we discuss how this novel technology has impacted the field.
Traditional Mouse Genome Engineering Technologies in the Pre-CRISPR Era
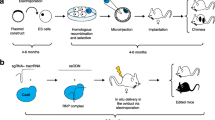
The development of mouse genetic engineering began in the 1980s by attempting to transfer exogenous DNA (genes) into a genome for developing “transgenic (Tg)” animals and to delete/inactivate endogenous genes for developing “knockout (KO)” animals . Transgenesis was achieved by microinjecting purified Tg DNA (transgenes) into fertilized zygotes and subsequently by transferring the zygotes to pseudo-pregnant recipient animals to generate live animals. To accomplish gene knockouts, however, simple injection of DNA into zygotes (as done in the case of transgenics) would not be enough: it was necessary to develop a special tool, the ES cells. Using ES cells, the endogenous genes were first modified through the HR process (which occurs during DNA repair in cells); in the second step the ES cells containing the modified gene were injected into blastocyst embryos about 3 days old to generate chimeric animals [1]. The resulting chimeric animals would contain cells originating from two sources: the cells derived from the embryo host blastocyst and those from exogenously injected ES cells. Breeding of the chimera to a wild-type mouse would result in vertical transmission of the gene-modified allele (the ES cell-derived mutant allele) to heterozygous offspring. Intercrossing between heterozygous offspring would result in production of homozygous, fully ES cell-derived mice. The third type of genetically engineered animal model is a Knockin (knock-in, KI), which refers to (usually) targeted insertion of DNA with a desired type of genetic change. Generation of KI mice also required ES cells, and follows similar steps as just outlined.
The traditional Tg and KO/KI techniques prevailed for nearly three decades and have helped generate thousands of mouse disease models. However, these time-tested methods have a few limitations that are discussed next (sections “Traditional Tg Techniques and Their Limitations” to “Microinjection and Its Limitations”).
Traditional Tg Techniques and Their Limitations
Traditional Tg mice are generated by direct microinjection of Tg DNA (that consists of elements such as promoter, cDNA, transcription terminator, etc.) into the pronuclei of 0.5-day-old fertilized eggs (also known as zygotes), followed by their subsequent transfer to oviducts of pseudo-pregnant mice. The live offspring obtained are called founder (G0) animals if they contain the DNA of interest, which are then bred to establish the Tg line. The detailed descriptions of designing and generating KO/KI mice are reviewed by Haruyama and Kulkarni (Haruyama et al. [2]).
The traditional Tg mice generation methods have a few inherent limitations: (1) random integration of the transgene where local regulatory elements could affect its expression, and/or the transgene itself can disrupt or affect the expression of local genes, and (2) integration at multiple sites or multiple copy integration, which occasionally result in unreliable expression or transgene silencing [3]. Because of such pitfalls, several Tg G0 lines are screened for desired expression before the lines are established for further experiments, a tedious but necessary step using random integration-based Tg mice generation projects [4].
Traditional KO/KI Techniques and Their Limitations
The traditional KO/KI models were generated through the use of ES cells that allow HR to replace or insert a genetically engineered DNA copy of a recombinant DNA construct that is designed and built for each KO/KI project. The process, in brief, includes four major steps: (1) construction of molecular targeting construct; (2) electroporation of the targeting construct into ES cells followed by positive/negative selection of correctly targeted clones; (3) microinjection of ES cell clones into blastocysts and transfer into pseudo-pregnant recipients to generate chimeras; and (4) breeding of chimeras with wild-type mouse to obtain a germline-transmitted mouse line. Detailed descriptions of designing and generating KO/KI mice are reviewed by Hall et al. [5].
Traditional KO/KI mice generation methods also have a few inherent limitations. First, ES cells must retain pluripotency to populate germ cells and vertically transmit the induced mutation to the next generation. Second, germline-competent ES cells are available for only a very few genetic backgrounds, not for many of the commonly used mouse strains. Animal models that cannot be generated in a pure genetic background for many of those strains must undergo many generations of backcross breeding to achieve congenesis [6]. Third, efficient insertion of the targeting construct requires long regions of DNA, or homology arms, flanking each end of the intended target, which can be difficult to achieve for certain genes. Fourth, the targeting constructs need to contain additional elements, such as positive selection (e.g., neomycin or puromycin resistance genes) and negative selection (e.g., thymidine kinase or diphtheria toxin) markers to select single-cell clones that contain the correctly inserted DNA. Fifth, the insertion of long positive selection markers [or certain genetic elements such as flippase (Flp) or Cre recombinase sites that flank the positive selection cassettes] may result in unintentional interference of the regulatory elements near the gene locus. Sixth, the KO/KI strategy would be difficult if a conditional KO needs to be developed for single-exon genes. Seventh, not all chimeras result in germ line-transmitted offspring. Eighth, traditional gene targeting can generally only be used to generate no more than one gene KO/KI in an experiment. Last, design and generation of KO/KI animal models is labor intensive, requires extensive amounts of time, and is quite expensive.
Microinjection and Its Limitations
Both Tg and KO/KI techniques require microinjection directly into mouse embryos. Although tedious, labor intensive, and expensive, the microinjection technique has been used as the gold standard for more than three decades for developing genetically engineered mouse models. The desired DNA cassette is microinjected into zygotes for generation of Tg mice, whereas gene-targeted ES cells are microinjected into blastocysts for generation of KO/KI mice. Zygotes or embryos are produced from females that are superovulated and mated with stud males. To ensure a sufficient number of Tg G0 lines or chimeras, typically 100 or more eggs or 50 or more embryos are injected for Tg or KO/KI projects, respectively. The manipulated embryos need to be surgically transferred into pseudo-pregnant females to generate live offspring.
In general, microinjection has been an integral step in mouse gene targeting projects, but its two major limitations are that it requires sophisticated equipment and well-trained and experienced personnel to perform the procedure. Typically, microinjection equipment costs about $100,000–$200,000, and microinjection (and associated embryo-handling techniques) requires significant practice to perfect. At least a couple years of regular practice are required for a researcher to learn and be proficient in performing microinjection. Also, one needs continued practice to retain technical proficiency.
CRISPR/Cas9 and Mouse Genome Editing
Since 2013, the CRISPR-mediated genome editing has revolutionized almost every field of biology. Briefly, it uses a single guide RNA (sgRNA) with a 20-nucleotide sequence complementary to the target site in the genome, to bring a Cas9 nuclease to the site and make a double-stranded DNA break. This break is then repaired through an error-prone cellular DNA repair process called nonhomologous end-joining (NHEJ), resulting in gene disruption. The cut site can also be repaired by providing a short DNA oligonucleotide with homology regions of about 30–60 nucleotides, or by providing a repair template with long homology arms (typically about 0.5–2 kb or longer). Such donor DNAs are inserted through the less efficient repair processes, such as homology-directed repair (HDR) [7] for single-stranded short templates or HR when using double-stranded templates.
CRISPR technology can also be regarded as a “disruptor” because it has changed the basic format of how Tg and animal genome engineering experiments are performed, which has essentially remained unchanged during the past three decades. Additionally, CRISPR has superseded the other two genome-editing methods, ZFNs and TALENs, which prevailed for about 3 to 5 years before the CRISPR era. Because of its simplicity, relative ease, and rapidity to manipulate the genome, the CRISPR/Cas9 technique has propelled many technology developers to think outside the box and devise novel and innovative features that make it versatile and adaptable to diverse fields of research. As we usher in a new era of genome engineering driven by CRISPR-related techniques, section “CRISPR Technology and the Paradigm Shifts in Mouse Genome Engineering” discusses the paradigm shifts in the field.
Box 1: Limitations of Traditional Mouse Genetic Engineering Technologies and the Paradigm Shifts Created by CRISPR/Cas9 Genome Editing
Traditional genetic engineering approaches | The CRISPR/Cas9 genome-editing approach |
|---|---|
ES cells are absolutely essential for the generation of KO/KI models (costly and time consuming) | Can generate KO/KI models without the need for ES cells (cost- and time saving) |
Techniques can be limited to certain strains where ES cells are available, particularly for KO/KI models | Can develop KO/KI models under any strain background |
Difficult to generate KO/KI models without inserting additional elements in the genome | Can generate most KO/KI models without inserting additional elements in the genome |
Generation of homozygous KO/KI mutants in G0 stage is not possible and G0 animals need to be intercrossed to obtain homozygotes before they are used for phenotyping | Generation of homozygous KO/KI mutants in G0 stage is readily possible, and they can be used for direct phenotyping in some cases (for instance, any visible traits, hematological phenotypes) |
Except in case of advanced techniques [8, 9], multiplexing (multiple Tg lines or multiple genes KO/KI) is difficult | Multiplexing is readily possible |
Except in case of advanced techniques [8, 9], pronuclear injection of Tg DNA will get integrated randomly, often more than one copy and/or occasionally at multiple locations | Targeted insertion of single copy at Cas9 cut site is possible |
Large-scale genome modifications (deletions or replacements) are difficult | Large-scale genome modifications are readily possible |
Microinjection is a critical step; each embryo must be injected manually and transferred back into recipient females | Electroporation can replace the microinjection step and many embryos can be processed simultaneously [10–12]. More advanced approaches (GONAD) can even obviate the need for ex vivo embryo handling [13] |
CRISPR Technology and the Paradigm Shifts in Mouse Genome Engineering
Clearly, the CRISPR system has impacted traditional Tg and KO technologies. Many Tg mouse labs and core facilities across the globe have added CRISPR to their toolbox. The paradigm shifts in mouse genome editing that are listed in Box 1 are discussed next.
-
1.
Ability to bypass the use of ES cells. Two important features of ES cells that make them critical reagents for genome engineering are (1) they maintain pluripotency during culturing and gene targeting and (2) they enable high competency in HR. Historically, mouse ES were the only ES cells with these two features that showed robust performance. Attempts to establish ES cells for other species (except rats) have failed to date. One of the biggest paradigm shifts that CRISPR has caused in the field is its ability to bypass the need for ES cells. This, along with simplicity and lower cost, is the main reason why CRISPR has been so widely applicable in creating gene KO models in any species. Even in mice, with the advent of CRISPR, ES cells that served as valuable tools in generating thousands of mouse models during the past two to three decades are now being superseded by the use of CRISPR [14].
-
2.
Ability to generate KO/KI mice on any genetic background. Because CRISPR-mediated gene editing can be used directly on zygotes to edit genes, practically any strain of mouse can be used for generating KO/KI models. Previously, the field relied on the availability of strain-specific ES cells for developing mouse models. Although better-quality ES cells for the C57BL/6N strain (the most popular in disease research) were developed during the past decade [15], for many years mouse KO technology predominantly relied on ES cells derived from sub-strains on the 129 genetic background. G0 lines generated using 129 ES cells injected into a different genetic background (e.g., C57BL/6) are a mixed strain background that required backcrossing to the desired genetic background for many generations before the model could be used for experiments. The CRISPR system readily offers solutions to such limitations, as it is applicable to any strain, thus obviating the need for backcrossing.
-
3.
Ability to generate point mutations without any other genetic disruptions. For generation of simple KI models such as creating point mutations to mimic human disease or restoring the function of mutant proteins, the CRISPR/Cas9 system offers distinct advantages over traditional methods. Specifically, point mutations can be inserted without the need to include extra DNA near the locus, such as a positive selection cassette when using ES cell-based methods. Occasionally, the presence of such extra elements near the locus may affect gene expression by disrupting adjacent yet unknown regulatory elements, etc.
-
4.
Ability to generate homozygous mutant mice in F 0 generation. With traditional approaches using either ES cells or random transgenesis, it was not possible to obtain homozygous G0 animals. CRISPR-generated models can produce homozygous mutations in the G0 generation and can be used for a quick phenotypic analysis, albeit mosacisim and possible off-target effects must be considered as confounders. Nevertheless, in some cases, phenotypic screening of G0 progeny can provide significant cost and time savings, when compared to the use of other methods that require breeding to achieve homozygosity.
-
5.
Ability to generate multiple mutations in one microinjection experiment . Despite their popular use in the pre-CRISPR era, traditional KO/KI approaches are inadequate in the following aspects: (1) it is practically impossible to simultaneously generate KO mutations for more than one gene at a time; (2) germline transmission of the mutant allele is often not guaranteed; and (3) the models would be only heterozygous initially. In comparison, CRISPR-mediated gene editing offers giant solutions to these limitations. Indeed, the generation of up to five KO mutant models has been reported in one session [16] with CRISPR, where previously such a task would take more than 3 to 4 years because of the time-consuming breeding steps after generating individual KO mice. The cost for such traditional KO projects would be severalfold more than that of CRISPR-based approaches because of the lengthy steps involved. Using CRISPR, it is not uncommon to obtain homozygous alleles for some mutations.
-
6.
Large-scale genome modifications . Although mouse models of large chromosomal deletions and insertions of hundreds of kilobases have been developed using traditional ES cell-based approaches [17, 18], clearly such projects need enormous amounts of resources and time to accomplish, because they were performed through a series of complex and successive modifications. Using certain advanced CRISPR-based strategies, such large-scale insertions and deletions are now possible, making the system highly cost effective [19].
-
7.
Cytoplasmic microinjection . The traditional Tg models are developed by injecting Tg DNA into pronuclei because the injected DNA is intended to be inserted into the genome. Because the CRISPR system constitutes a sgRNA and a Cas9 endonuclease, pronuclear injection might not be required (which can be a difficult skill to master). Further, pronuclei in certain strains of mice are not easy to visualize for microinjection. In many cases, cytoplasmic injection seems to be sufficient [20], especially in cases where simple indel mutants are to be generated without the need for coinjection of complex donor DNA templates. When combined with a donor DNA template for genomic insertion, it is necessary to deliver injection mixture to the pronucleus to ensure insertion efficiency. Simultaneous cytoplasmic and pronuclear injection has become a popular strategy in many labs for CRISPR-based genome-editing applications that suit both NHEJ and HR mechanisms.
-
8.
Novel delivery approaches. CRISPR tools can be delivered to embryos without the need for microinjection or ex vivo handling of embryos. Although microinjection has been used as the gold standard for more than three decades for developing genetically engineered mouse models, there has been constant effort by many researchers to develop microinjection-independent methods because of the inherent limitations of microinjection (covered in the section “Microinjection and Its Limitations”). The advent of CRISPR readily enabled the development of an electroporation technique that can be performed on several embryos at a time, instead of manually injecting them one by one [10–12]. A step further is a new technique developed by us called Genome Editing via Oviductal Nucleic Acids Delivery (GONAD ) [13]. GONAD allows direct electroporation of CRISPR tools into embryos in situ without the need for embryo isolation and handling ex vivo. Thus, GONAD serves to bypass all major bottlenecks of animal transgenesis: isolation, microinjection, and surgical transfer of embryos into pseudo-pregnant mice [13, 21, 22].
-
9.
Germline transmission potential can be high compared to traditional methods-derived chimeras. Because the traditional ES cell-based approach relies on the pluripotency and germline transmission potential of ES cells, some chimeras may not result in passage of the targeted allele to the next generation of offspring. Reasons for failure of germline transmission include low contribution of ES-derived germ cells in chimeric mice, or loss of ES cell pluripotency. Although mosaicism remains a potential disadvantage, the germ cells of CRISPR-generated G0 mice are expected to contain CRISPR-induced mutation(s). Therefore, the chances of germline transmission of a CRISPR-induced mutant allele to the next generation of offspring is high. Furthermore, certain CRISPR-generated G0 mice may contain two or more types of mutations at a given locus, which can be segregated by breeding. Even though the segregation process seems complicated in certain cases, multiple different mutations at the given locus offer more options to study the phenotype using multiple alleles.
The Current Challenges of CRISPR/Cas9-Mediated Mouse Genome Engineering
Poor Efficiency of Insertion of Sequences at Cas9 Cut Sites
Although there are a few reports that demonstrate the insertion of longer DNA cassettes at Cas9-cut sites, increasing the overall efficiency of insertion is an area that needs further development. Despite its widespread use, to achieve targeted insertion at many loci still remains challenging. Although one of the problems may be less efficient guide sequences, the overall low insertion efficiency may also be attributed to the loci (e.g., extent of chromatin density) and donor DNA design (e.g., extent of genomic homology to target region). Additional strategies are necessary to make the CRISPR system suitable for efficient insertion of large DNA cassettes and for generating models more complex than indels on a routine basis.
Challenges in Developing Conditional KO Models
Conditional KO mouse models with two loxP sites flanking the target exon/s is a standard approach followed in traditional ES cell-based applications. Many labs have been trying to develop conditional KO models using CRISPR. Insertion of two loxP sites can be achieved through one of two ways: (1) using a double-stranded (ds) DNA donor containing short homology arms (~1 kb) and two loxP sites flanking the target region [23], or (2) using two separate ssODNs (single-stranded oligodeoxynucleotides) encoding loxP sites in the middle and ultrashort (~60-base-pair) flanking homology arms corresponding to the desired genomic sites and inserting them through two separate CRISPR cuts in the genome [24]. High-efficiency insertion of two loxP sites in cis orientation remains elusive and challenging. The reasons for this are (1) the two independent gRNAs should be efficient in causing double-strand breaks at their target sites; if one fails to cut, the process will not result in the desired alleles; (2) even if both guides work efficiently, NHEJ is still favored, causing the two flanking ends to join together and excluding the intermediary piece of DNA; and (3) challenges in genotyping of correctly inserted loxP sites, specifically using the two ssODNs approach (see the section “Challenges Associated with Genotyping”).
Off-Target Effects
Because the gRNA recognition sequence is only 20 nucleotides long and certain mismatches are tolerated when gRNA binds to genomic DNA, use of CRISPR can result in unintentional off-target cleavages. Some of the initial studies, done in cell culture systems, cautioned that off-target effects could be a major concern with the use of CRISPR technology [25, 26]. Certain strategies have been described to minimize or eliminate off-target cleavages. (1) The Cas9 nickase (nCas9 or Cas9n) approach [27, 28] that uses a mutated Cas9 which can create a nick instead of a double-strand break; by using paired nickases, two nicks are created using two gRNAs close to the target site. (2) Delivery of Cas9 in the form of mRNA or protein instead of plasmid; continued Cas9 expressed from plasmid DNA would result in an abundance of Cas9 protein over a much longer period than needed, resulting in the potential for more off-target cuts than when using Cas9 mRNA or protein. It was presumed that off-target cleavages would be high, based on the observations made in cell culture systems. However, some recent reports demonstrate that off-target effects are minimal or nil in mouse models generated through the CRISPR system [29]; one of the main reasons for this is the use of Cas9 mRNA or protein [30, 31]. Further, the concern about off-target cleavages in mouse models can be addressed by backcrossing G0 mice to segregate mutations through successive breeding steps.
Challenges Associated with Genotyping
Genotyping of CRISPR-generated offspring is another major challenge because it can generate many unexpected outcomes such as imprecise insertion of the donor template, and co-occurrence of more than two types of alleles (also known as mosaicism). It may require careful analysis of many offspring generated from G0 mice to segregate and establish the desired mutations. Also, genotyping may not be easy in certain cases using a simple PCR assay and it may require sequencing of every offspring. To avoid such complications, one can consider choosing gRNAs close to restriction endonuclease (RE ) recognition sites or to include an RE site in their donor template to aid in designing RFLP-PCR (restriction fragment length polymorphism)-based genotyping. Genotyping in case of the loxP ssODN approach is particularly challenging to ensure correct insertion of loxP sites on the same allele. Specifically, genotypic discrimination of correct targeting can be challenging if (a) the two loxP sites are far apart and it is difficult to amplify the entire floxed region by PCR (for confirmation by RFLP), or if (b) if the G0 animals are not homozygous for at least one of the insertion sites. In such scenarios, Southern blotting becomes necessary for accurate confirmation of loxP insertions on the same allele.
Future Impact of CRISPR/Cas9 on Manipulating the Mouse Genome
Clearly, CRISPR/Cas9 has revolutionized many fields of biology, including mouse genome manipulation. Newer CRISPR tools and improved strategies are constantly being added. A few more CRISPR nucleases were recently discovered [32, 33] that offer additional features, refinements, and capabilities to the CRISPR genome-editing toolbox. Such improvements can have a significant impact on both traditional Tg and KO/KI technologies.
Impact on Random Tg Technologies
The majority of Tg mouse models generated to date are of random Tg type; in many cases, such projects fail to obtain reliable TgG0 lines with high efficiency. If CRISPR/Cas9 can be further improved to efficiently insert larger DNA cassettes into the genome at safe harbor sites (e.g., ROSA26), it is very likely that the community will shift to “CRISPR transgenesis” and eventually random integration-based Tg mice production may become obsolete. Although there are not many reports of successful insertion of longer DNA cassettes with CRISPR/Cas9, certain strategies described recently promise targeted transgenesis of larger cassettes [31, 34–36].
Impact on KO/KI Technologies
As already noted, the mouse molecular genetics field has been transitioning rapidly to using CRISPR/Cas9 for making point mutation KIs. Although it is demonstrated that conditional KO models can be generated using the CRISPR system [24], it has not yet become a commonly used method because of the inherent difficulties associated with inserting two loxP sites flanking the target site in cis. It is likely that, in the near future, technical advances will evolve to develop conditional KO models easily and efficiently.
Impact on Microinjection Technique
Last, if in vitro zygote electroporation and GONAD techniques become popular, CRISPR-based methods do not need the specialized microinjection setup or specially skilled personnel, which can allow many researchers to perform genome-editing experiments, in contrast to specialized core facilities that performed traditional, microinjection-based genome editing. GONAD is a promising new method for delivering CRISPR reagents directly to zygotes within the oviducts through electroporation. Compared to microinjection, GONAD requires a higher concentration of reagents to ensure embryonic uptake and activity. Its wide applicability and use in future is likely to result in faster and novel evolution and refining of the CRISPR technique itself, facilitating a transformation in the field of genome editing.
Conclusion
Traditional mouse genome manipulation techniques, established over the past three decades, have been used to develop thousands of mouse models. The recent addition of genome-editing tools such as ZFNs, TALENs, and CRISPR/Cas9 have resulted in a rapid transformation in the landscape of genome manipulation. In particular, CRISPR/Cas9 has been widely adopted during the past 2 to 3 years, and its simplicity and applicability across species has made the process faster, more cost effective, and versatile. It has also helped technology developers to devise newer methodologies that would have been practically impossible in the pre-CRISPR era. Research is underway to find additional CRISPR endonuclease molecules, and newer strategies to improve DNA insertion efficiency and to facilitate and improve the insertion of longer DNA sequences. All such improvements would enable this simple and ingenious method of gene editing to revolutionize biomedical research in the years to come.
References
Behringer R, Gertsenstein M, Nagy KV, Nagy A. Manipulating the mouse embryo: a laboratory manual. Cold Spring Harbor: CSHL Press; 2014.
Haruyama N, Cho A, Kulkarni AB. Overview: engineering transgenic constructs and mice. Curr Protoc Cell Biol. 2009;Chap. 19:Unit 19.10.
Garrick D, Fiering S, Martin DI, Whitelaw E. Repeat-induced gene silencing in mammals. Nat Genet. 1998;18:56–9.
Ohtsuka M, Miura H, Sato M, Kimura M, Inoko H, Gurumurthy CB. PITT: pronuclear injection-based targeted transgenesis, a reliable transgene expression method in mice. Exp Anim. 2012;61:489–502.
Hall B, Limaye A, Kulkarni AB. Overview: generation of gene knockout mice. Curr Protoc Cell Biol. 2009;Chap. 19:Unit 19.12 19.12.1–17.
Gurumurthy CB, Joshi PS, Kurz SG, Ohtsuka M, Quadros RM, Harms DW, Lloyd KCK. Validation of simple sequence length polymorphism regions of commonly used mouse strains for marker assisted speed congenics screening. Int J Genomics. 2015;2015:735845.
Peng Y, Clark KJ, Campbell JM, Panetta MR, Guo Y, Ekker SC. Making designer mutants in model organisms. Development. 2014;141:4042–54.
Ohtsuka M, Ogiwara S, Miura H, Mizutani A, Warita T, Sato M, Imai K, Hozumi K, Sato T, Tanaka M, Kimura M, Inoko H. Pronuclear injection-based mouse targeted transgenesis for reproducible and highly efficient transgene expression. Nucleic Acids Res. 2010;38, e198.
Ohtsuka M, Miura H, Mochida K, Hirose M, Hasegawa A, Ogura A, Mizutani R, Kimura M, Isotani A, Ikawa M, Sato M, Gurumurthy CB. One-step generation of multiple transgenic mouse lines using an improved Pronuclear Injection-based Targeted Transgenesis (i-PITT). BMC Genomics. 2015;16:274.
Kaneko T, Sakuma T, Yamamoto T, Mashimo T. Simple knockout by electroporation of engineered endonucleases into intact rat embryos. Sci Rep. 2014;4:6382.
Kaneko T, Mashimo T. Simple genome editing of rodent intact embryos by electroporation. PLoS One. 2015;10, e0142755.
Hashimoto M, Takemoto T. Electroporation enables the efficient mRNA delivery into the mouse zygotes and facilitates CRISPR/Cas9-based genome editing. Sci Rep. 2015;5:11315.
Takahashi G, Gurumurthy CB, Wada K, Miura H, Sato M, Ohtsuka M. GONAD: genome-editing via Oviductal Nucleic Acids Delivery system: a novel microinjection independent genome engineering method in mice. Sci Rep. 2015;5:11406.
Skarnes WC. Is mouse embryonic stem cell technology obsolete? Genome Biol. 2015;16:109.
Pettitt SJ, Liang Q, Rairdan XY, Moran JL, Prosser HM, Beier DR, Lloyd KC, Bradley A, Skarnes WC. Agouti C57BL/6N embryonic stem cells for mouse genetic resources. Nat Methods. 2009;6:493–5.
Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–8.
Yu Y, Bradley A. Mouse genomic technologies: engineering chromosomal rearrangements in mice. Nat Rev Genet. 2001;2:780–90.
Scheer N, Kapelyukh Y, Chatham L, Rode A, Buechel S, Wolf CR. Generation and characterization of novel cytochrome P450 Cyp2c gene cluster knockout and CYP2C9 humanized mouse lines. Mol Pharmacol. 2012;82:1022–9.
Yoshimi K, Kunihiro Y, Kaneko T, Nagahora H, Voigt B, Mashimo T. ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat Commun. 2016;7:10431.
Horii T, Arai Y, Yamazaki M, Morita S, Kimura M, Itoh M, Abe Y, Hatada I. Validation of microinjection methods for generating knockout mice by CRISPR/Cas-mediated genome engineering. Sci Rep. 2014;4:4513.
Gurumurthy CB, Takahashi G, Wada K, Miura H, Sato M, Ohtsuka M. GONAD: a novel CRISPR/Cas9 genome editing method that does not require ex vivo handling of embryos. Curr Protoc Hum Genet. 2016;88:15.8.1–12.
Sato M, Ohtsuka M, Watanabe S, Gurumurthy CB. Nucleic acids delivery methods for genome editing in zygotes and embryos: the old, the new, and the old-new. Biol Direct. 2016;11(1):16.
Lee AY, Lloyd KCK. Conditional targeting of Ispd using paired Cas9 nickase and a single DNA template in mice. FEBS Open Bio. 2014;4:637–42.
Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–9.
Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–6.
Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31:839–43.
Ran FA, Hsu PD, Lin C-Y, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–9.
Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, Wang L, Hodgkins A, Iyer V, Huang X, Skarnes WC. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods. 2014;11:399–402.
Iyer V, Shen B, Zhang W, Hodgkins A, Keane T, Huang X, Skarnes WC. Off-target mutations are rare in Cas9-modified mice. Nat Methods. 2015;12:479.
Harms DW, Quadros RM, Seruggia D, Ohtsuka M, Takahashi G, Montoliu L, Gurumurthy CB. Mouse genome editing using the CRISPR/Cas system. Curr Protoc Hum Genet. 2014;83:15.7.1–27.
Aida T, Chiyo K, Usami T, Ishikubo H, Imahashi R, Wada Y, Tanaka KF, Sakuma T, Yamamoto T, Tanaka K. Cloning-free CRISPR/Cas system facilitates functional cassette knock-in in mice. Genome Biol. 2015;16:87.
Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F. Cpf1 Is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–71.
Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, Minakhin L, Joung J, Konermann S, Severinov K, Zhang F, Koonin EV. Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol Cell. 2015;60:385–97.
Nakade S, Tsubota T, Sakane Y, Kume S, Sakamoto N, Obara M, Daimon T, Sezutsu H, Yamamoto T, Sakuma T, Suzuki KT. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat Commun. 2014;5:5560.
Quadros RM, Harms DW, Ohtsuka M, Gurumurthy CB. Insertion of sequences at the original provirus integration site of mouse ROSA26 locus using the CRISPR/Cas9 system. FEBS Open Bio. 2015;5:191–7.
Miura H, Gurumurthy CB, Sato T, Sato M, Ohtsuka M. CRISPR/Cas9-based generation of knockdown mice by intronic insertion of artificial microRNA using longer single-stranded DNA. Sci Rep. 2015;5:12799.
Acknowledgments
C.B.G. acknowledges funding support by the NIH grants: Institutional Development Award (IDeA) from the National Institutes for General Medical Sciences grant number P20GM103471 (NIGMS) and NIH Office of Research Infrastructure Programs (ORIP) grant number R24OD018546 (ORIP/DPCPSI). M.O. acknowledges the funding support by the 2014 Tokai University School of Medicine Research Aid and Grant-in-Aid for challenging Exploratory Research (15 K14371) from Japan Society for the Promotion of Science (JSPS).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Gurumurthy, C.B., Quadros, R.M., Sato, M., Mashimo, T., Lloyd, K.C.K., Ohtsuka, M. (2016). CRISPR/Cas9 and the Paradigm Shift in Mouse Genome Manipulation Technologies. In: Turksen, K. (eds) Genome Editing. Springer, Cham. https://doi.org/10.1007/978-3-319-34148-4_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-34148-4_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-34146-0
Online ISBN: 978-3-319-34148-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)