
Abstract
Solid tumors necessitate vascularization for metabolic support and metastasis, relying on the process of angiogenesis to form new blood vessels. However, the constant stimulation of endothelial cells from pro-angiogenic soluble factors and mechanical forces creates a tumor vasculature that is structurally and functionally abnormal. Most anti-angiogenic therapies have focused on targeting VEGF signaling to pursue the tumor vasculature. However, these anti-VEGF therapies have been met with limited success in clinical trials. Hence, recent studies have started to investigate the role of mechanical signaling in tumor angiogenesis as it occurs in a mechanically dynamic environment. This chapter focuses on mechanosensitive ion channels that belong to the transient receptor potential (TRP) superfamily, with special emphasis on the role of TRPV4 in the endothelium, as well as deregulation of TRPV4 signaling within the tumor endothelium, and its potential as a target for normalization of tumor vasculature to improve cancer therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
The oxygen and nutrient demands of solid tumors make them dependent on angiogenesis for new blood vessels to grow and metastasize. However, the continuous needs of the tumor create vessels that are structurally and functionally abnormal [1, 2] and result in a hypoxic environment due to insufficient delivery of nutrients and reduced waste clearance [3]. Tumor angiogenesis is driven by numerous factors in attempts of sprouting new vessels [4] which include acidosis, inflammatory cytokines, as well as the activation of oncogenes that occur at the cellular level, upstream of VEGF and other tumor promoting hormones [5, 6]. Importantly, the tumor extracellular matrix (ECM) becomes stiffer due to the release of plasma components through the leaky tumor vessels as well as dynamic remodeling of these components by tumor and stromal cells. Further, an increase in interstitial fluid pressure in tumors creates turbid blood flow [7] impeding the delivery of cancer drugs. Together, these mechanical forces create an abnormal micromechanical environment to which the tumor vasculature does not respond properly. Therefore, recent studies have started to target the mechanical aspects of the tumor microenvironment for cancer therapies, with specific interest on ion channels. The transient receptor potential (TRP) family of ion channels has already gained much attention in cancer, especially those that exhibit mechanosensitive properties. This chapter will cover the abnormal tumor vasculature, and aims to examine the role of mechanosensitive ion channels involved in the aberrant environment, with a special focus on TRPV4.
Tumor Vasculature
As with any organ, the endothelium can be subjected to dysfunction and failure. Such is the case of the tumor vasculature, the structure of which is controlled by irregular intrinsic and extrinsic factors. These abnormalities generate microvasculature that contain regions of heterogeneity, with areas within the tumor being “hyper” or “hypo” vascular in both function and density [8, 9]. At the cellular level, pro-angiogenic factors generate the weakening of VE-cadherin-mediated endothelial cell (EC) junctions, which in turn distort the vessel wall structure and promote EC migration [10]. Pericytes that cover the endothelium, generally create a stable and mature vascular network, but in tumors have been found reduced in number, and loosely attached to the EC, causing vessels to become immature and leaky. This effect is the result of increased growth factor signaling, such as vascular endothelial growth factor (VEGF) , which has been found to impede the adherence of pericytes to the surrounding endothelium [11–13]. Overall, these new vessels are not only tortuous and dilated, but lack structural support [8], endorsing dysfunctional blood vessels that promote erratic blood flow [8, 14] and hyper-permeability that allow protein and fluid extravasation into the extracellular space, all of which increase matrix deposition [8]. Additionally, the growing tumor mass often times leads to the compression and eventual collapsing of the existing vessels, further diminishing blood supply and increasing hypoxic and acidic conditions within the tumor [8, 15]. Altogether, the structural and functional aberrations of the tumor vasculature are prompted by the persistent needs of the tumor cells to vascularize and the constant stimulation of angiogenesis.
Tumor Angiogenesis
The progression of solid tumors relies heavily on angiogenesis and requires the endothelium to switch from a quiescent, or resting, phenotype to one that is more invasive, termed the “angiogenic switch ”[16]. To begin this process, the basement membrane must be injured or wounded, triggering destruction and hypoxia of the surrounding tissue. Pro-angiogenic factors are released to stimulate EC to migrate, proliferate, and stabilize. However, in tumor angiogenesis, not only are pro-angiogenic factors up-regulated, but angiogenic inhibitors must be down-regulated to maintain adequate stimulation of angiogenesis [17]. Overall, the local equilibrium of the tumor microenvironment is unbalanced, and considering the abnormalities of the tumor vasculature, makes it pertinent to address the complexities associated with tumor EC (TEC) themselves.
Tumor Endothelial Cells (TEC)
Altogether, the structural and functional aberrations of the tumor vasculature promote drastic genetic and morphological changes within the cells [18]. These factors can then generate tumor cells that demonstrate a more hostile phenotype with potential for metastasis [19]. It is now known to a large extent that the tumor endothelium is defective, with TEC showing distinct irregularities with respect to shape and size when compared to normal endothelial cells (NEC). These cells tend to have long cytoplasmic projections that can extend across the lumen, with the tips of some TEC protruding into the lumen, creating intercellular gaps within the vessel wall that most often result in the leakage or pooling of blood [20].
At the molecular level, the tumor endothelium, unlike the normal endothelium, express a host of genes, recently identified as transmembrane proteins called tumor endothelial markers (TEM) [21, 22]. In-depth studies on TEC isolated from different carcinomas, show that these cells do not undergo senescence, are resistant to serum starvation and apoptosis, and are structurally abnormal compared to NEC. TEC isolated from mouse xenograft tumors were found to have variable DNA content, not only between its normal counterpart cell types, but between individual cells as well, indicating the existence of heterogeneity within TEC. These cells have larger nuclei and exhibit characteristic cytogenic and structural abnormalities, such as aneuploidy and chromosomal aberrations including deletions, non-reciprocal translocations, and abnormal centrosomes [23–26]. Furthermore, TEC isolated from mouse prostate tumors were found to express both hematopoietic and mesenchymal stem cell markers, verifying the heterogeneity of these cells. Additionally, it was found that these cells were also able to undergo unusual mesenchymal differentiation into cartilage and bone-like tissues in conditioned medium, confirming the ability of TEC to adapt to their surrounding environment [27]. Based on these studies, it can be interpreted that the cellular and molecular aberrations of TEC can invariably contribute to the abnormal angiogenic process and ultimately tumor growth and metastasis. However, when grown in defined endothelial medium, TEC (derived from mouse prostate tumors) express endothelial markers and show morphological features similar to NEC [27]. Interestingly, we have demonstrated that TEC show aberrant mechanosensing and abnormal angiogenesis in vitro, suggesting deregulation of mechanosensing mechanisms in these cells [28]. A fascinating question, that has yet to be considerably studied, pertains to the origin of these TEC. A variety of studies [29, 30] have highlighted that the source is from stem-cell tumor cells, to “circulating CD34+/VEGFR-2+ endothelial progenitor cells”; however, there has been limited progress in conclusively identifying the source of these cells, making it difficult to develop any specific targets as part of an anti-angiogenic strategy.
Current Approaches to Target Tumor Angiogenesis
Since first described by Dr. Folkman [3], the concept of angiogenesis and anti-angiogenic therapies has revolutionized the way cancer has been studied and clinically treated. Inhibition of angiogenic activators such as VEGF, placental growth factor (PIGF) , fibroblast growth factor (FGF) [31] and associated growth factor signaling mechanisms, have provided new avenues in successfully treating as well as studying tumor angiogenesis. Anti-VEGF therapies, including anti-VEGF neutralizing monoclonal antibodies and receptor tyrosine kinase inhibitors (RTKIs) , dominate current approaches in treating malignant tumors [32, 33], initially achieving great success. One such study found that treating patients of metastatic colorectal cancer with anti-VEGF antibody, Bevacizumab , in combination with systemic chemotherapy, increased patient survival [34], which was later attributed to tumor ‘vascular normalization.’ The principle of vascular normalization seeks to restore the balance of angiogenic factors within the tumor microenvironment to regulate vessel growth and maturity [8, 35]. This allows for improved delivery of chemotherapeutic agents by reestablishing a more normal vascular network. While anti-VEGF-mediated approaches hold promise, it has a narrow window in terms of the transient nature of the resulting ‘normal’ vessel. Furthermore, these growth factor mediated therapies have become redundant, ineffective, and in some instances, detrimental to the treatment of cancer, owing to inherent or acquired drug resistance, the potential for metastatic capability, and the general absence of predictive markers to monitor tumor responses in select patient populations [36]. These findings were further substantiated in a recent finding that demonstrated a rapid decrease in delivery of chemotherapy to the tumor after anti-VEGF therapy in non-small cell lung cancer (NSCLC) patients using PET imaging [37].
Limitations in targeting soluble factors have led to the idea that other factors, such as mechanical forces, may be contributing to the abnormal tumor vasculature. In fact, defects in mechanotransduction, the conversion of mechanical forces into biochemical signals, have been reported as the basis of diverse pathological conditions [38, 39]. A number of studies have also described a balance of underlying mechanical forces as driving factors in sensitizing capillary EC to angiogenic growth factors, to form functional networks of blood vessels. Furthermore, EC are exposed and respond to mechanical forces such as shear stress and cyclic strain imposed by blood flow. In fact, it was postulated that local micromechanical forces modulate the endothelial response to growth factors [40]. One of the fastest responses of EC to mechanical forces is Ca2+ influx through mechanosensitive ion channels. Ion channels have already been the subject of various reviews, as they have recently been found up-regulated/down-regulated or simply dysfunctional in different pathological diseases. However, the role of mechanosensitive ion channels in physiological or pathological angiogenesis is largely unknown. Thus, targeting ion channels in the mechanically dynamic tumor microenvironment may lead to novel therapies among the abnormal tumor vasculature.
TRP Channels
The onset of neovascularization and subsequent tumor progression is associated with the generation of cell populations that differ phenotypically, which often arise from a deregulation of key signaling pathways and mutations or deletion of several proteins. Transient receptor potential (TRP) channels represent a superfamily of proteins that have, over the years, been understood to affect or be affected by a variety of pathological conditions, including cancer. In addition to the transcriptional regulation of TRPs by hormones and growth factors produced by the tumor microenvironment, alternative splicing of genes leads to the generation of protein isoforms with altered functions and variations in subcellular localization. Such effects have been documented in diverse tumor types where a decrease in Ca2+ influx may be attributed to decreased expression of TRP channels [41].
A growing body of evidence has identified members of the TRP family, mainly members of TRPC (canonical), TRPM (melastatin) and TRPV (vanilloid), as key regulators of mechanotransduction [42–45]. TRP channels have a profound effect on EC function, and any dysregulation of these channels can result in EC dysfunction [46]. Because TRP channels are found in the plasma membrane, they are easily and directly accessible to the blood stream, which make these channels potential molecular targets when vascular diseases arise. Below, we specifically describe mechanosensitive TRP channels expressed in the endothelium, most of which are involved in a variety of cancers. Although we have tried to incorporate all relevant studies of these channels in tumor angiogenesis, much of this field remains vastly unexplored, with potential to investigate new targets.
TRPC
TRP canonical channels (TRPC), the founding member of the TRPs, are made up of seven family members (1–7) that are expressed in the endothelium [47], as well as the surrounding smooth muscle cells [48]. TRPC channels support endothelial function such as vascular regulation, permeability, and the endothelial-derived nitric oxide (NO) mediated vasorelaxation of smooth muscle cells. While all of these essential functions may be necessary for the angiogenic process, TRPC1 and TRPC6 have been implicated in mediating mechanotransduction. Studies propose TRPC1 is a stretch-activated channel while others have confirmed TRPC6 is activated by pressure as well as osmotically or mechanically induced plasma membrane stretch [47]. While the role of TRPC1 in angiogenesis has been confirmed, most of the evidence suggests that the mechanosensitive properties may not be obligatory, but that TRPC1 may act alongside other TRPC channels to carry out mechanotransduction. Additionally, TRPC1 and TRPC6 have been found to respond to inflammatory agonists resulting in EC cell shape changes [49, 50]. TRPC6 has also been found in human pulmonary arterial endothelial cells (HPAEC) to control endothelial contraction, cell shape, and permeability [49, 51]. In regards to the cancer environment, many TRPC channels have been found to exist among several different types of cancer tissue, including breast cancer [52], ovarian cancer [53, 54], hepatoma [55], prostate cancer [56], basal cell carcinoma [57], renal cell carcinoma [58], malignant gliomas [59], glioblastoma [60], gastric tumors [61], and lung cancer [62]. However, there have not been any studies to date that suggest TRPC channels are directly involved in the tumor vasculature .
TRPM
TRP melastatin channels (TRPM) are the largest and most diverse among the TRP superfamily. Made up of eight family members (1–8), these channels are widely expressed in endothelial cells and vascular smooth muscle [47], making them important for normal vascular function. When it comes to vascular mechanosensing, both TRPM4 and TRPM7 have been found to contribute to mechanotransduction. In cerebral artery myocytes, TRPM4 is activated by membrane stretch [63] and in cerebral artery smooth muscle cells, take part in mediating membrane depolarization and myogenic vascular tone [64]. Furthermore, some studies have suggested that TRPM4 activation by stretch may be secondary to Ca2+ responses [63–65]. Additionally, TRPM7, which acts as an ion channel and a functional kinase, can be activated by cell stretch and swelling to carry out mechanotransduction [66]. TRPM7 is highly permeable to Mg2+, which contributes to the role TRPM7 plays in angiogenesis and vascular remodeling due to the diverse effect of Mg2+ on EC function [67]. Pathologically, some studies suggest that TRPM4 may be involved in decreased NO production [68], while TRPM7 plays a role in angiogenesis and oxidative stress induced cell death [47, 69]. Recent TRPM7 studies in ovarian cancer have found that TRPM7 is needed for cancer cell growth, migration, and invasion [70], but TRPM4 nor TRPM7 channels have been found to affect the tumor endothelium .
TRPP and TRPA
The TRP polycystin (TRPP) ion channel family is well known in polycystic kidney disease (PKD) [71]. Expressed ubiquitously among vertebrates, these channels are involved in mechanotransduction due to TRPP1 and TRPP2 channel activation by shear stress [47]. These channels are important for flow-induced vascular response and NO production [47, 72].
TRPA1 is the only mammalian member to belong to the TRP ankyrin (TRPA) family. Although predominately expressed in nociceptive neurons of the peripheral ganglia and the mechanosensory epithelium of the inner ear [73], this mechanosensitive ion channel has recently been found expressed in the endothelium as well as the surrounding perivascular cells in cerebral circulation [74]. Overall, TRPA1 mainly plays a role in nociception [75], mechanotransduction [76], and thermal [77], and oxygen sensing [78]. The only studies of TRPA1 in the endothelium were performed by Earley and colleagues [79], which found activated TRPA1 stimulated endothelium-dependent smooth muscle cell hyperpolarization and vasodilation. Pathologically, TRPA1 is involved in acute and chronic pain and possibly chronic inflammatory diseases , and only few studies have been performed in regards to TRPA1 in cancer [80, 81].
TRPV1
The TRP vanilloid (TRPV) family of channels has received the most attention in regards to mechanotransduction in the vasculature, specifically TRPV1 and TRPV4, both of which are expressed in the endothelium [47]. The first family member, TRPV1, is activated by capsaicin [82], anandamide, arachidonic acid (AA) [83], PIP2 hydrolysis [84], acidity, and noxious heat (T > 43 °C) [82], and is important for NO production and endothelium-dependent vascular relaxation [85, 86]. TRPV1 has been implicated to mediate the later stages of angiogenesis from various studies. In human umbilical vascular EC (HUVEC) , TRPV1 activation inhibits VEGF-induced proliferation, DNA synthesis, and capillary-like tube formation. Additionally, activation of TRPV1 via capsaicin also inhibited VEGF-induced vessel formation and vessel sprouting in mouse Matrigel plug and rat aortic ring assays, respectively [87]. When it comes to mechanosensation, TRPV1 was originally discovered to take part in inflammatory thermal hyperalgesia, nociception, and pain sensation [88, 89]. TRPV1 expression has been found altered in cancers of the prostate [90, 91], colon, pancreas [92, 93], and bladder. Expression is increased in all of these except bladder cancer, in which the cancerous urothelium causes a decrease in TRPV1 due to cells becoming more de-differentiated as the carcinoma cells progress to a more aggressive state [92]. Moreover, TRPV1 in cancer has been often associated with pain, especially due to TRPV1 expression in neurons [94]. These findings make TRPV1 a good target for pharmacological inhibition of cancer pain in several types of cancers .
TRPV4 : A Novel Target for Cancer Therapy
In recent years, TRPV4 has emerged as a widely accepted mechanosensor, contributing significantly to the process of mechanotransduction. TRPV4 is a non-specific Ca2+ permeable channel activated by a variety of physical and chemical stimuli such as temperature, hypotonicity, phorbol esters, endocannabinoids, arachidonic acid (AA), and epoxy eicosatrienoic acids (EETs) [95–97]. The TRPV4 protein is composed of a cytosolic N-terminal region, six transmembrane domains including the pore region, and an intracellular C-terminal tail (Fig. 12.1). The N-terminal region contains the ankyrin repeat domains (ARD) , which consists of six ankyrin repeats (ANK) (ANK1–6) [98, 99]. A proline-rich domain (PRD) has been implicated in the mechanosensitivity of the TRPV4 channel, preceding the first ankyrin repeat. Within this PRD, proline residues at positions 142, 143, and 144, interact with pacsin 3, a protein implicated in vesicular membrane transport, endocytosis, and cytoskeleton reorganization [100, 101]. The TRPV4 C-terminal tail contains additional functional domains such as a TRP box, a calmodulin-binding site, and a binding site for cytoskeletal proteins such as MAP7, actin, and tubulin [102, 103].

Schematic of the structure of the mechanosensitive ion channel TRPV4. The TRPV4 channel is a tetramer and each subunit is composed of a cytosolic N-terminal region and six transmembrane domains, including the pore region, and an intracellular C-terminal tail. The N-terminal region contains the ankyrin repeat domain (ARD) , which consists of six ankyrin repeats
TRPV4 is ubiquitously expressed among various tissues including lung, liver, heart, trachea, and the vascular endothelium [104–107]. Structurally similar to the osm-9 gene found in Caenorhabditis elegans , mammalian TRPV4 is thought to share functional properties in mechanosensation. Previous studies found that when human embryonic kidney (HEK) 293 cells transfected with TRPV4 were exposed to shear stress, the increase in intracellular Ca2+ was due to TRPV4 activation, which was inhibited by TRPV antagonist, ruthenium red [108]. Liedtke et al. [109] found that sensory neurons in C. elegans responded to osmotic and mechanical stimuli via TRPV4 channels, providing in vivo support of the mechanosensitive properties of TRPV4. The first evidence indicating TRPV4 as a mechanosensor in the endothelium was reported by Kohler and coworkers [110], where they showed that shear stress-induced Ca2+ influx and vasodilation were mediated by TRPV4, which was confirmed by their later study using TRPV4-null mice [111]. A separate study has also revealed that TRPV4-mediated Ca2+ influx is critical for flow-induced release of mitochondrial reactive oxygen species (ROS) and vasodilation in human coronary arteries [112]. We and others have reported that TRPV4-dependent Ca2+ influx plays critical role in agonist (acetylcholine)-induced vasodilation [74, 113, 114]. Together, these studies demonstrate that TRPV4 plays an important role in the mechanical force and agonist-induced regulation of vascular tone. TRPV4 also plays a role in lung microvascular EC in which elevated lung hydrostatic pressures caused EC Ca2+ influx, an increase in vascular permeability and lung edema; these effects were abolished upon treatment with TRPV4 inhibitors and in TRPV4KO mice [106, 115].
Although the role of TRPV4 as a mechanosensor has been confirmed in the endothelium and other tissue and organ systems, the molecular mechanisms by which TRPV4 transduces mechanical signals is not well known. Both cell and ECM-generated mechanical forces have been critical signals that dictate normal vessel growth and patterning [40, 116, 117]. A critical event that takes place during neovascularization involves the directional migration of EC towards angiogenic stimuli, characterized by the realignment of the actin cytoskeleton and EC reorientation. Endothelial cells have been shown to reorient perpendicular to the direction of cyclic strain; but align parallel to flow in response to shear stress [118]. This reorientation response of EC to cyclic strain was found to be regulated by stretch-activated (SA) channels [119], however the specific identity of the SA channel or molecular mechanism(s) underlying this reorientation response is not known.
We set out to investigate the precise stretch-activated Ca2+ channel involved in regulating EC reorientation and our studies revealed that mechanical strain applied to integrins resulted in a rapid influx of Ca2+ via TRPV4 in EC. We further identified TRPV4 channels as the specific SA channel that mediates integrin-to-integrin signaling required for the cyclic strain-induced reorientation of EC [120] (Fig. 12.2). Specifically, we demonstrated that cyclic strain-induced TRPV4-mediated Ca2+ influx activates PI3K which in turn stimulates the activation of additional β1 integrins that may regulate Rho/Rac signaling required for the reorganization of the actin cytoskeleton and EC reorientation (Fig. 12.3). We further showed that mechanical strain application on β1 integrins activates TRPV4 through its interaction with a transmembrane CD98 protein, located in focal adhesions [121]. Based on these findings, we postulated that the transfer of mechanical force between cell surface molecules (β1 integrins, CD98 and TRPV4) localizes mechanotransduction almost instantaneously within focal adhesions [121] and may regulate many complex cell and tissue behaviors. The importance of TRPV4 as a mechanosensor in EC was further validated when we demonstrated that a siRNA-mediated knockdown of TRPV4 in EC resulted in the failure of these cells to reorient perpendicular to the direction of applied cyclic strain [120] (Fig. 12.2d). To our surprise, when tumor-derived endothelial cells (TEC) were exposed to cyclic strain, these cells failed to reorient [28]. This aberrant mechanosensitive response exhibited by TEC was also found manifested in cell-spreading experiments. These cells exhibited uncontrolled spreading on ECM substrates of increasing rigidity (such as within a tumor), as opposed to NEC that stopped spreading at the highest stiffness. Further, in vitro angiogenesis assays revealed that TEC exhibited abnormal angiogenic behavior which was demonstrated to be mediated by high basal active-Rho and Rho-kinase (ROCK) [28].

TRPV4 channels mediate cyclic strain-induced endothelial cell reorientation. (a) Fluorescence images of endothelial cells (EC) subjected to 0 or 10 % uniaxial cyclic strain (2 h, 1 Hz). Cells were cultured on fibronectin-coated flexible silicone membranes, fixed, and stained with Alexa488-phalloidin to visualize actin stress fibers. Arrow indicates the direction of applied strain. Scale bar: 25 μm. (b) Quantification of cell alignment in control and strain exposed cells as the percentage of cells oriented 90 ± 30° (aligned) relative to the direction of applied strain (p < 0.0006); error bars indicated S.E.M. (c) Immunofluorescence images of EC subjected to 0 or 10 % uniaxial cyclic strain. Cells were stained for vinculin (green) and actin stress fibers (magenta) to show that application of strain causes enhanced recruitment of vinculin to large focal adhesions, that co-localize with the ends of reinforced stress fibers (shown in white). Scale bar: 25 μm. (d) siRNA knockdown of TRPV4 significantly inhibited cyclic strain-induced EC reorientation, compared to siRNA knockdown of TRPV2 or TRPC1. The quantification of cell alignment was measured as the percentage of cells oriented 90 ± 30° (aligned) relative to the direction of applied strain in control (white bars) and strain exposed (black bars) in human EC. (*p < 0.0025) (reprinted from Thodeti et al., Circ. Res. 104:1123–1130, 2009; Fig. 1 and Fig. 6D [120])

TRPV4 mediated mechanotransduction in endothelial cells. A schematic model showing TRPV4-dependent mechanical signaling in normal endothelial cells. Application of mechanical force (cyclic stretch or ECM stiffness) to integrins activates ultra-rapid calcium influx through TRPV4 via interaction with a transmembrane protein CD98. The released Ca2+ activates additional integrins via PI3K. This integrin-to-integrin signaling may further regulate downstream Rho/Rac pathways necessary for reorganization of the actin cytoskeleton and reorientation of EC [120]
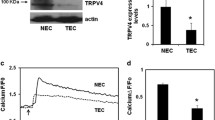
Since TEC failed to reorient to applied cyclic strain, a phenomenon reminiscent to NEC subjected to siRNA-mediated TRPV4 silencing, we postulated that aberrant mechanosensitivity in TEC may be caused by altered TRPV4-dependent signaling. Indeed, our recent work has demonstrated that TRPV4 expression and function are impaired in TEC [122]. Specifically, we found a significant reduction in TRPV4 expression (~40 %), while functional assays revealed a 40–50 % decrease in Ca2+ influx in TEC when stimulated with specific TRPV4 agonists, GSK1016790A (GSK) and 4α-PDD (Fig. 12.4). An important downstream consequence of tumor growth is an increase in matrix rigidity, owing to the leakage of plasma components from highly permeable blood vessels, and increased synthesis/degradation of matrix components [123]. This increase in ECM stiffness has been shown to influence TEC spreading, migration, and tube formation. Because we have previously shown that TEC display abnormal mechanosensitivity to substrate stiffness [28], and TRPV4 was found functionally impaired, we overexpressed TRPV4 to determine whether we could restore TEC mechanosensitivity towards varying ECM rigidity. We found that overexpression of TRPV4 reduced the abnormal spreading exhibited by TEC on the highest stiffness gelatin gels (2 kPa; comparable to the stiffness of tumors). The ability of cells to migrate is reliant on the mechanosensing efficiency of the cell in response to ECM rigidity, which is an important step in the angiogenic process [124]. We therefore investigated the migratory ability of TEC and found that these cells exhibit abnormal migration (40 μm/h); and restoring TRPV4-dependent mechanosensitivity normalized TEC migration consistent with the migration of NEC (10 μm/h). Additionally, overexpression of TRPV4 in TEC was able to decrease the high basal Rho activity previously observed [28], as well as normalize the abnormal angiogenesis exhibited in both 2D and 3D in vitro angiogenesis assays. We further demonstrated that these effects were similarly achieved by modulating TRPV4 activity pharmacologically using a specific TRPV4 activator, GSK1016790A. These findings suggest that TRPV4 signaling regulates tumor angiogenesis via Rho-dependent mechanosensing mechanisms. Finally, to determine the functional role of TRPV4 in tumor angiogenesis in vivo, we induced syngeneic tumors in TRPV4KO mice by subcutaneously injecting LLC (mouse Lewis Lung Carcinoma) cells. Tumor growth and angiogenesis was significantly enhanced in TRPV4KO mice compared to WT mice. Immunohistochemical analysis of tumor sections obtained from TRPV4KO tumors revealed the vessels were immature, i.e. large vessels with decreased pericyte coverage. Further, TRITC-dextran perfusion experiments confirmed that these vessels were leaky, as the dextran fluorescence was enhanced in the tumor tissue in TRPV4KO mice compared to WT mice. Notably, these findings suggest that TRPV4 is critical for maintaining vessel structure and integrity and that absence of TRPV4 not only increases tumor angiogenesis but also inhibits vessel maturity.

Expression and function of TRPV4 channels in normal and tumor endothelial cells. (a) Western blot showing TRPV4 protein expression in normal (NEC) and tumor-derived endothelial cells (TEC). (b) Densitometric analysis of the Western blots showing significant (p ≤ 0.05) reduction in TRPV4 expression in TEC compared to NEC. (c) Relative changes in cytosolic calcium in response to a selective TRPV4 agonist, GSK1016790A (100 nM) in Fluo-4 loaded NEC and TEC. Arrow denotes the time when the cells were stimulated with the TRPV4 agonist. (d) Quantitative analysis of cytosolic calcium influx induced by GSK1016790A in NEC and TEC. (F/F0 = ratio of normalized Fluo-4 fluorescence intensity relative to time 0). The results shown are mean ± SEM from three independent experiments. The significance was set at p ≤ 0.05 (reprinted from Adapala et al., Oncogene 2016; 35:314–22 [122])
Thus, our study has not only provided a unique role for the mechanosensitive TRPV4 channel in the tumor vasculature, but opens up an uncharted therapeutic target in vascular normalization strategies. Specifically we have demonstrated TRPV4 activation with GSK1016790A normalized tumor vasculature (increased pericyte coverage) and reduced tumor growth when given in combination with the anti-cancer drug Cisplatin, but not alone. These findings suggest that normalization of tumor vasculature by TRPV4 activation may have improved the delivery of Cisplatin and reduced tumor growth (Fig. 12.5). Thus, our work is the first to study the functional significance of TRPV4 in tumor angiogenesis in vitro and in vivo and identify a novel role for TRPV4 in the regulation of tumor vessel growth and maturity.

TRPV4 activation normalizes tumor vasculature enhancing chemotherapeutic drug delivery. The tumor vasculature is characterized by tortuous, hyper-permeable, and poorly perfused vessels surrounded by varying regions of hypoxia and normoxia. The treatment with a specific small molecular activator of TRPV4, GSK1016790A, normalizes the tumor vasculature and improves the efficient delivery of chemotherapeutic drug (Cisplatin) leading to reduced tumor growth in WT mice injected subcutaneously with LLC (Lewis Lung Carcinoma) cells (Adapala et al., Oncogene 2016; 35:314–22 [122])
Conclusion and Perspectives
The neovascularization of solid tumors create dysfunctional endothelium, generating a microenvironment in which the vasculature becomes heterogeneous and erratic blood flow ensues. This ultimately affects the delivery and efficacy of systemically administered agents. Although endothelial cells are generally heterogeneous, they are commonly regulated by many soluble, membrane-bound, and mechanical factors, including ion channels. Because most ion channels are found in the plasma membrane, they are accessible to the blood stream, making them favorable targets when vascular diseases arise. Most current therapies concentrate on targeting the cytokines involved in tumor angiogenesis. Recent pre-clinical and clinical evidence has demonstrated that anti-VEGF drugs, as well as many other direct or indirect angiogenesis inhibitors, can transiently promote the normalization of the tumor vasculature. However, growth factor mediated normalization therapies have largely been unsuccessful due to several challenges, such as drug resistance and redundancy among others. Considering these findings, targeting mechanotransduction may offer a more effective means to treating or reducing solid tumor growth. We have shown mechanosensitive ion channel TRPV4 modulates tumor endothelial function and angiogenesis, and have unraveled a TRPV4-dependent mechanotransduction mechanism in angiogenesis. These findings could lead to the development of novel growth factor-independent therapeutic targets, not only to induce vascular normalization and improve cancer therapy, but also for other angiogenic disorders such as diabetic retinopathy and age-related macular degeneration .
References
Nagy JA, et al. Why are tumour blood vessels abnormal and why is it important to know? Br J Cancer. 2009;100(6):865–9.
Fukumura D, et al. Tumor microvasculature and microenvironment: novel insights through intravital imaging in pre-clinical models. Microcirculation. 2010;17(3):206–25.
Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–6.
Nagy JA, et al. Vascular permeability factor/vascular endothelial growth factor induces lymphangiogenesis as well as angiogenesis. J Exp Med. 2002;196(11):1497–506.
Ferrara N. VEGF as a therapeutic target in cancer. Oncology. 2005;69 Suppl 3:11–6.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307.
Boucher Y, Baxter LT, Jain RK. Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: implications for therapy. Cancer Res. 1990;50(15):4478–84.
Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62.
Kim P, et al. In vivo wide-area cellular imaging by side-view endomicroscopy. Nat Methods. 2010;7(4):303–5.
Hashizume H, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156(4):1363–80.
Morikawa S, et al. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2002;160(3):985–1000.
Abramsson A, Lindblom P, Betsholtz C. Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest. 2003;112(8):1142–51.
Greenberg JI, et al. A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature. 2008;456(7223):809–13.
Jain RK. Determinants of tumor blood flow: a review. Cancer Res. 1988;48(10):2641–58.
Padera TP, et al. Pathology: cancer cells compress intratumour vessels. Nature. 2004;427(6976):695.
Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29(6 Suppl 16):15–8.
Folkman J. The role of angiogenesis in tumor growth. Semin Cancer Biol. 1992;3(2):65–71.
Bottaro DP, Liotta LA. Cancer: out of air is not out of action. Nature. 2003;423(6940):593–5.
DeClerck K, Elble RC. The role of hypoxia and acidosis in promoting metastasis and resistance to chemotherapy. Front Biosci (Landmark Ed). 2010;15:213–25.
Dudley AC. Tumor endothelial cells. Cold Spring Harb Perspect Med. 2012;2(3):a006536.
St Croix B, et al. Genes expressed in human tumor endothelium. Science. 2000;289(5482):1197–202.
Nanda A, et al. Identification of a binding partner for the endothelial cell surface proteins TEM7 and TEM7R. Cancer Res. 2004;64(23):8507–11.
Bussolati B, et al. Bifunctional role for VEGF-induced heme oxygenase-1 in vivo: induction of angiogenesis and inhibition of leukocytic infiltration. Blood. 2004;103(3):761–6.
Allport JR, Weissleder R. Murine Lewis lung carcinoma-derived endothelium expresses markers of endothelial activation and requires tumor-specific extracellular matrix in vitro. Neoplasia. 2003;5(3):205–17.
Hida K, et al. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res. 2004;64(22):8249–55.
Hida K, Klagsbrun M. A new perspective on tumor endothelial cells: unexpected chromosome and centrosome abnormalities. Cancer Res. 2005;65(7):2507–10.
Dudley AC, et al. Calcification of multipotent prostate tumor endothelium. Cancer Cell. 2008;14(3):201–11.
Ghosh K, et al. Tumor-derived endothelial cells exhibit aberrant Rho-mediated mechanosensing and abnormal angiogenesis in vitro. Proc Natl Acad Sci U S A. 2008;105(32):11305–10.
Asahara T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275(5302):964–7.
Hendrix MJ, et al. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer. 2003;3(6):411–21.
Khoury CC, Ziyadeh FN. Angiogenic factors. Contrib Nephrol. 2011;170:83–92.
Kim KJ, et al. The vascular endothelial growth factor proteins: identification of biologically relevant regions by neutralizing monoclonal antibodies. Growth Factors. 1992;7(1):53–64.
Kim KJ, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362(6423):841–4.
Saltz LB, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008;26(12):2013–9.
Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7(9):987–9.
Shojaei F, Ferrara N. Role of the microenvironment in tumor growth and in refractoriness/resistance to anti-angiogenic therapies. Drug Resist Updat. 2008;11(6):219–30.
Van der Veldt AA, et al. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: implications for scheduling of anti-angiogenic drugs. Cancer Cell. 2012;21(1):82–91.
Huang S, Ingber DE. Cell tension, matrix mechanics, and cancer development. Cancer Cell. 2005;8(3):175–6.
Suresh S. Biomechanics and biophysics of cancer cells. Acta Biomater. 2007;3(4):413–38.
Ingber DE, Folkman J. Mechanochemical switching between growth and differentiation during fibroblast growth factor-stimulated angiogenesis in vitro: role of extracellular matrix. J Cell Biol. 1989;109(1):317–30.
Gkika D, Prevarskaya N. Molecular mechanisms of TRP regulation in tumor growth and metastasis. Biochim Biophys Acta. 2009;1793(6):953–8.
O'Neil RG, Heller S. The mechanosensitive nature of TRPV channels. Pflugers Arch. 2005;451(1):193–203.
Liedtke W, Kim C. Functionality of the TRPV subfamily of TRP ion channels: add mechano-TRP and osmo-TRP to the lexicon! Cell Mol Life Sci. 2005;62(24):2985–3001.
Christensen AP, Corey DP. TRP channels in mechanosensation: direct or indirect activation? Nat Rev Neurosci. 2007;8(7):510–21.
Pedersen SF, Nilius B. Transient receptor potential channels in mechanosensing and cell volume regulation. Methods Enzymol. 2007;428:183–207.
Kwan HY, Huang Y, Yao X. TRP channels in endothelial function and dysfunction. Biochim Biophys Acta. 2007;1772(8):907–14.
Yao X, Garland CJ. Recent developments in vascular endothelial cell transient receptor potential channels. Circ Res. 2005;97(9):853–63.
Venkatachalam K, Zheng F, Gill DL. Regulation of canonical transient receptor potential (TRPC) channel function by diacylglycerol and protein kinase C. J Biol Chem. 2003;278(31):29031–40.
Singh I, et al. Galphaq-TRPC6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin. J Biol Chem. 2007;282(11):7833–43.
Cioffi DL, Stevens T. Regulation of endothelial cell barrier function by store-operated calcium entry. Microcirculation. 2006;13(8):709–23.
Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86(1):279–367.
Dhennin-Duthille I, et al. High expression of transient receptor potential channels in human breast cancer epithelial cells and tissues: correlation with pathological parameters. Cell Physiol Biochem. 2011;28(5):813–22.
Zeng B, et al. TRPC channels and their splice variants are essential for promoting human ovarian cancer cell proliferation and tumorigenesis. Curr Cancer Drug Targets. 2013;13(1):103–16.
Yang SL, et al. Transient receptor potential channel C3 contributes to the progression of human ovarian cancer. Oncogene. 2009;28(10):1320–8.
El Boustany C, et al. Capacitative calcium entry and transient receptor potential canonical 6 expression control human hepatoma cell proliferation. Hepatology. 2008;47(6):2068–77.
Thebault S, et al. Differential role of transient receptor potential channels in Ca2+ entry and proliferation of prostate cancer epithelial cells. Cancer Res. 2006;66(4):2038–47.
Beck B, et al. TRPC channels determine human keratinocyte differentiation: new insight into basal cell carcinoma. Cell Calcium. 2008;43(5):492–505.
Veliceasa D, et al. Transient potential receptor channel 4 controls thrombospondin-1 secretion and angiogenesis in renal cell carcinoma. FEBS J. 2007;274(24):6365–77.
Bomben VC, Sontheimer HW. Inhibition of transient receptor potential canonical channels impairs cytokinesis in human malignant gliomas. Cell Prolif. 2008;41(1):98–121.
Chigurupati S, et al. Receptor channel TRPC6 is a key mediator of Notch-driven glioblastoma growth and invasiveness. Cancer Res. 2010;70(1):418–27.
Cai R, et al. Blockade of TRPC6 channels induced G2/M phase arrest and suppressed growth in human gastric cancer cells. Int J Cancer. 2009;125(10):2281–7.
Jiang HN, et al. Involvement of TRPC channels in lung cancer cell differentiation and the correlation analysis in human non-small cell lung cancer. PLoS One. 2013;8(6):e67637.
Morita H, et al. Membrane stretch-induced activation of a TRPM4-like nonselective cation channel in cerebral artery myocytes. J Pharmacol Sci. 2007;103(4):417–26.
Earley S, Straub SV, Brayden JE. Protein kinase C regulates vascular myogenic tone through activation of TRPM4. Am J Physiol Heart Circ Physiol. 2007;292(6):H2613–22.
Earley S, Waldron BJ, Brayden JE. Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ Res. 2004;95(9):922–9.
Numata T, Shimizu T, Okada Y. TRPM7 is a stretch- and swelling-activated cation channel involved in volume regulation in human epithelial cells. Am J Physiol Cell Physiol. 2007;292(1):C460–7.
Owsianik G, et al. Permeation and selectivity of TRP channels. Annu Rev Physiol. 2006;68:685–717.
Suh SH, et al. ATP and nitric oxide modulate a Ca(2+)-activated non-selective cation current in macrovascular endothelial cells. Pflugers Arch. 2002;444(3):438–45.
Aarts M, et al. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115(7):863–77.
Wang J, et al. TRPM7 is required for ovarian cancer cell growth, migration and invasion. Biochem Biophys Res Commun. 2014;454(4):547–53.
Mochizuki T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272(5266):1339–42.
Kim K, et al. Polycystin 1 is required for the structural integrity of blood vessels. Proc Natl Acad Sci U S A. 2000;97(4):1731–6.
Nagata K, et al. Nociceptor and hair cell transducer properties of TRPA1, a channel for pain and hearing. J Neurosci. 2005;25(16):4052–61.
Earley S, Gonzales AL, Crnich R. Endothelium-dependent cerebral artery dilation mediated by TRPA1 and Ca2+-activated K+ channels. Circ Res. 2009;104(8):987–94.
Kwan KY, et al. TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron. 2006;50(2):277–89.
Corey DP, et al. TRPA1 is a candidate for the mechanosensitive transduction channel of vertebrate hair cells. Nature. 2004;432(7018):723–30.
Cordero-Morales JF, Gracheva EO, Julius D. Cytoplasmic ankyrin repeats of transient receptor potential A1 (TRPA1) dictate sensitivity to thermal and chemical stimuli. Proc Natl Acad Sci U S A. 2011;108(46):E1184–91.
Takahashi N, et al. TRPA1 underlies a sensing mechanism for O2. Nat Chem Biol. 2011;7(10):701–11.
Earley S. TRPA1 channels in the vasculature. Br J Pharmacol. 2012;167(1):13–22.
Schaefer EA, et al. Stimulation of the chemosensory TRPA1 cation channel by volatile toxic substances promotes cell survival of small cell lung cancer cells. Biochem Pharmacol. 2013;85(3):426–38.
Oehler B, et al. TRPA1 is functionally expressed in melanoma cells but is not critical for impaired proliferation caused by allyl isothiocyanate or cinnamaldehyde. Naunyn Schmiedebergs Arch Pharmacol. 2012;385(6):555–63.
Caterina MJ, et al. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389(6653):816–24.
Rami HK, Gunthorpe MJ. The therapeutic potential of TRPV1 (VR1) antagonists: clinical answers await. Drug Discov Today Ther Strateg. 2004;1(1):97–104.
Prescott ED, Julius D. A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science. 2003;300(5623):1284–8.
Poblete IM, et al. Anandamide elicits an acute release of nitric oxide through endothelial TRPV1 receptor activation in the rat arterial mesenteric bed. J Physiol. 2005;568(Pt 2):539–51.
Golech SA, et al. Human brain endothelium: coexpression and function of vanilloid and endocannabinoid receptors. Brain Res Mol Brain Res. 2004;132(1):87–92.
Min JK, et al. Capsaicin inhibits in vitro and in vivo angiogenesis. Cancer Res. 2004;64(2):644–51.
Caterina MJ, et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288(5464):306–13.
Davis JB, et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405(6783):183–7.
Domotor A, et al. Immunohistochemical distribution of vanilloid receptor, calcitonin-gene related peptide and substance P in gastrointestinal mucosa of patients with different gastrointestinal disorders. Inflammopharmacology. 2005;13(1-3):161–77.
Hartel M, et al. Vanilloids in pancreatic cancer: potential for chemotherapy and pain management. Gut. 2006;55(4):519–28.
Lazzeri M, et al. Transient receptor potential vanilloid type 1 (TRPV1) expression changes from normal urothelium to transitional cell carcinoma of human bladder. Eur Urol. 2005;48(4):691–8.
Sanchez MG, et al. Expression of the transient receptor potential vanilloid 1 (TRPV1) in LNCaP and PC-3 prostate cancer cells and in human prostate tissue. Eur J Pharmacol. 2005;515(1-3):20–7.
Nilius B, et al. Transient receptor potential cation channels in disease. Physiol Rev. 2007;87(1):165–217.
Vriens J, et al. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci U S A. 2004;101(1):396–401.
Liedtke WB. TRPV channels’ function in osmo- and mechanotransduction. In: Liedtke WB, Heller S, editors. TRP ion channel function in sensory transduction and cellular signaling cascades. Boca Raton, FL: CRC Press; 2007.
Nilius B, et al. TRPV4 calcium entry channel: a paradigm for gating diversity. Am J Physiol Cell Physiol. 2004;286(2):C195–205.
Owsianik G, et al. Structure-function relationship of the TRP channel superfamily. Rev Physiol Biochem Pharmacol. 2006;156:61–90.
Nilius B, Owsianik G. The transient receptor potential family of ion channels. Genome Biol. 2011;12(3):218.
Cuajungco MP, et al. PACSINs bind to the TRPV4 cation channel. PACSIN 3 modulates the subcellular localization of TRPV4. J Biol Chem. 2006;281(27):18753–62.
D’Hoedt D, et al. Stimulus-specific modulation of the cation channel TRPV4 by PACSIN 3. J Biol Chem. 2008;283(10):6272–80.
Strotmann R, Schultz G, Plant TD. Ca2+-dependent potentiation of the nonselective cation channel TRPV4 is mediated by a C-terminal calmodulin binding site. J Biol Chem. 2003;278(29):26541–9.
Suzuki M, Hirao A, Mizuno A. Microtubule-associated [corrected] protein 7 increases the membrane expression of transient receptor potential vanilloid 4 (TRPV4). J Biol Chem. 2003;278(51):51448–53.
Liedtke W, et al. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell. 2000;103(3):525–35.
Delany NS, et al. Identification and characterization of a novel human vanilloid receptor-like protein, VRL-2. Physiol Genomics. 2001;4(3):165–74.
Alvarez DF, et al. Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: a novel mechanism of acute lung injury. Circ Res. 2006;99(9):988–95.
Yin J, et al. Negative-feedback loop attenuates hydrostatic lung edema via a cGMP-dependent regulation of transient receptor potential vanilloid 4. Circ Res. 2008;102(8):966–74.
Gao X, Wu L, O'Neil RG. Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C-dependent and -independent pathways. J Biol Chem. 2003;278(29):27129–37.
Liedtke W, et al. Mammalian TRPV4 (VR-OAC) directs behavioral responses to osmotic and mechanical stimuli in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2003;100 Suppl 2:14531–6.
Kohler R, et al. Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arterioscler Thromb Vasc Biol. 2006;26(7):1495–502.
Hartmannsgruber V, et al. Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLoS One. 2007;2(9):e827.
Bubolz AH, et al. Activation of endothelial TRPV4 channels mediates flow-induced dilation in human coronary arterioles: role of Ca2+ entry and mitochondrial ROS signaling. Am J Physiol Heart Circ Physiol. 2012;302(3):H634–42.
Zhang DX, et al. Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension. 2009;53(3):532–8.
Adapala RK, et al. PKCalpha mediates acetylcholine-induced activation of TRPV4-dependent calcium influx in endothelial cells. Am J Physiol Heart Circ Physiol. 2011;301(3):H757–65.
Hamanaka K, et al. TRPV4 initiates the acute calcium-dependent permeability increase during ventilator-induced lung injury in isolated mouse lungs. Am J Physiol Lung Cell Mol Physiol. 2007;293(4):L923–32.
Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995;75(3):519–60.
Chen CS, et al. Geometric control of cell life and death. Science. 1997;276(5317):1425–8.
Kaunas R, et al. Cooperative effects of Rho and mechanical stretch on stress fiber organization. Proc Natl Acad Sci U S A. 2005;102(44):15895–900.
Naruse K, Yamada T, Sokabe M. Involvement of SA channels in orienting response of cultured endothelial cells to cyclic stretch. Am J Physiol. 1998;274(5 Pt 2):H1532–8.
Thodeti CK, et al. TRPV4 channels mediate cyclic strain-induced endothelial cell reorientation through integrin-to-integrin signaling. Circ Res. 2009;104(9):1123–30.
Matthews BD, et al. Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface beta1 integrins. Integr Biol (Camb). 2010;2(9):435–42.
Adapala RK, et al. Activation of mechanosensitive ion channel TRPV4 normalizes tumor vasculature and improves cancer therapy. Oncogene. 2016;35(3):314–22.
Paszek MJ, Weaver VM. The tension mounts: mechanics meets morphogenesis and malignancy. J Mammary Gland Biol Neoplasia. 2004;9(4):325–42.
Dvorak HF. Rous-Whipple Award Lecture. How tumors make bad blood vessels and stroma. Am J Pathol. 2003;162(6):1747–57.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Cappelli, H.C., Thoppil, R.J., Adapala, R.K., Meszaros, J.G., Paruchuri, S., Thodeti, C.K. (2016). Role of Mechanosensitive TRP Channels in Abnormal Vasculature of Tumors. In: Levitan, PhD, I., Dopico, MD, PhD, A. (eds) Vascular Ion Channels in Physiology and Disease. Springer, Cham. https://doi.org/10.1007/978-3-319-29635-7_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-29635-7_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-29633-3
Online ISBN: 978-3-319-29635-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)