
Abstract
This article covers aspects of the on-surface synthesis of large molecular systems, in particular metal complexes, organometallic compounds, and organic molecules. It is shown that this approach is especially useful if the desired molecular species are too large or thermally unstable for vapor deposition, or when their synthesis in solution is not possible. Another advantage is the possible formation of well-ordered two-dimensional network structures with a hybrid covalent/van der Waals bonding scheme. The first part of the article focuses on the on-surface synthesis of metalloporphyrins, metallocorroles, and metallophthalocyanines by direct metalation of the respective ligand molecules with coadsorbed metal atoms. The underlying metalation reaction proceeds with high yields and without by-products except hydrogen, which readily desorbs. The second part is devoted to the on-surface synthesis of hydrocarbon macrocycles by means of the surface Ullmann reaction and also discusses the organometallic reaction intermediates with C–Cu–C bonds.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Prefixes for porphyrin and phthalocyanine ligands: 2H or 2D = freebase, M = metal complex
1 Introduction
The synthesis of metal complexes, organic molecules, and organometallic compounds at the solid/vacuum interface is a promising approach for surface modification and functionalization. It is an important alternative to the direct vapor deposition of the required molecules or metal complexes and has substantial advantages especially in the following cases:
-
1.
The substance cannot be synthesized as a bulk material and then (vapor) deposited onto a surface, either because no bulk synthesis is available or because the substance is too labile for vapor deposition. The former issue typically arises in the case of large conjugated hydrocarbons, where a solution-based synthesis often requires that long alkyl chains are attached to the parent structure in order to achieve sufficient solubility [1–3]. The corresponding naked parent structures have only been accessible by on-surface synthesis [4]. In addition, catalytic and template effects exerted by the surface can help driving the reaction in the desired direction, especially in the case of planar molecular structures.
-
2.
The on-surface synthesis of covalently bonded two-dimensional networks is often hampered by large defect concentrations. This issue results from the irreversible character of the bond formation, which prevents the healing of initially formed structural defects. An alternative strategy uses the on-surface synthesis of large molecular moieties, which are still sufficiently mobile to self-assemble and to segregated defect structures. The resulting van der Waals network can then be fused by covalent bonds in a subsequent step.
In this contribution, both cases will be addressed and illustrated by examples from the recent literature. The first part focuses on the surface-confined synthesis of large metal complexes based on porphyrins, corroles, and phthalocyanines. Monolayers and multilayers of these complexes can be synthesized by the reaction of the metal-free macrocycle ligands with coadsorbed metal atoms (or metal atoms from the substrate), sometimes followed by the attachment of another ligand on the metal center. Mechanistic details of this in situ redox reaction and properties of the resulting complexes will be discussed. The second part deals with reactions of bromoarenes on Cu surfaces. Temperature-dependent studies show that the C–Br bonds dissociate already at low temperatures and are replaced by C–Cu–C bonds, which lead to the formation of linear, cyclic, or two-dimensional organometallic oligomers and polymers. A prominent example for a molecule for which no solution-based synthesis is available, but which can be made by on-surface synthesis, is hyperbenzene, a hexagonal octadecaphenylen, which is formed by an Ullmann-type reaction from six dibromo-m-terphenyl molecules. As a room temperature stable intermediate, the corresponding organometallic macrocycle with 6 C–Cu–C bridges was found, accompanied by other cyclic and chain-like organometallic oligomers.
2 On-Surface Synthesis of Porphyrin, Phthalocyanine, and Corrole Complexes
Formation of coordinative bonds between adsorbed ligand molecules and coadsorbed (or substrate) metal atoms has frequently been used to synthesize two-dimensional metal-organic coordination networks [5–8]. With a similar approach, molecular complexes can be synthesized on surfaces, as has been shown for various porphyrins and phthalocyanines. Their parent structures are displayed in Fig. 1.

Porphin, corrole, and phthalocyanine, the parent structures of the respective classes of molecules. Corrole differs from porphin only by the absence of one meso-carbon atom in the macrocycle (position marked in red); the hydrogen atom from this meso-position (formally) goes to one of the N atoms, resulting in three pyrrolic nitrogens (–NH–) in the corrole
Porphyrins and their metal complexes are widespread in nature as the active centers of many enzymes and other functional biomolecules, such as hemoproteins [9, 10] or chlorophylls [11]. Porphyrin derivatives also find widespread application in science and technology, such as heterogeneous catalysis [12, 13], electrocatalysis [14], photocatalysis [15], sensor systems [16–18], organic electronics [19, 20], and various types of light-harvesting systems [21, 22]. Porphyrins have also been employed in the medical context as sensitizers in photodynamic therapy for cancer treatment [23–26].
Phthalocyanines are not found in nature, but can efficiently be synthesized from phthalonitriles in the presence of metals [27]. They are produced on an industrial scale and find their main applications as pigments in printing inks, paints, plastics, and other mass products, including color filters for LCD and TFT displays [27]. They are also used as oxidation catalysts [28], in organic semiconductors in thin-film transistors [29], organic light-emitting diodes [30], and molecular organic photovoltaics [31–33], in liquid crystalline materials [34, 35], as photoactive element in photocopies and laser printers [32, 36], and as photosensitizers in dye solar cells [37] and for photodynamic cancer therapy [38, 39].
Porphyrins and phthalocyanines possess a rich and diverse coordination chemistry, which arises from the large variety of complexes which they form with most stable elements in the periodic table, except nitrogen, the halogens, and the rare gases (Figs. 2 and 3) [40]. A similar rich coordination chemistry was observed at solid/vacuum interfaces [41], although there are characteristic mechanistic differences, which partly result from the presence or absence of a solvent: In solution, stabilization by solvatation favors ionic reactants, products, and intermediates, especially in polar solvents, while in the absence of solvatation, reactions preferentially proceed via neutral species. This is well known for gas-phase reactions, where atoms and radicals dominate the reaction mechanism, but also holds true for many surface reactions. It also applies to the formation of metalloporphyrins and metallophthalocyanines by direct metalation of the ligand. In solution, the metalation reaction proceeds as a replacement of two protons by a metal ion, i.e., as an ion exchange [42]. In contrast, the corresponding surface reaction follows a redox mechanism, resulting in the oxidation of a metal atom to its dication and the related reduction of the protons to H2 (Fig. 2) [43–45]. Besides the coordination reactions at the center of the macrocycle and the coordination of axial ligands to the metal center, porphyrins with suitable substituents can also form coordinative bonds at the molecule’s periphery [7].

Metalation of a porphyrin (here: porphin, 2HP) in the adsorbed state

All elements shown in this periodic system form porphyrin and phthalocyanine complexes. To date, on-surface metalation in UHV was achieved only with the elements in the gray-shaded boxes
2.1 Synthesis of Complexes Using Pre-deposited Ligand Molecules
Adsorbed metalloporphyrins and phthalocyanines at the solid/vacuum interface can be synthesized by reaction between the pre-adsorbed ligand molecules and post-deposited metal atoms. This type of reaction was first reported for Co and meso-tetraphenylporphyrin (2HTPP) [43, 46], Zn and 2HTPP [43, 47], Fe and meso-tetrapyridylporphyrin (2HTPyP) [48], as well as Fe and 2HTPP [49]. In all these experiments, the metals were vapor deposited onto monolayers of the ligands on an inert metal surface. The metal atoms reacted with the porphyrin molecules as is shown in Fig. 2.
The reaction proceeds at room temperature for V [50], Cr [51], Mn [52], Fe, Co [43], and Ni [53], but requires elevated temperatures for Ti [54], Zn [43], and Rh [52], as will be discussed in Sect. 2.3. A periodic table with all elements that form metal complexes with porphyrins and those for which on-surface metalation was achieved (gray-shaded boxes) is shown in Fig. 3. As can be seen, there are plenty of opportunities for the further exploration of on-surface metalation.
The reaction progress was monitored mainly with XPS, STM, and NEXAFS. Representative N 1s XPS spectra for the metalation of a 2HTPP monolayer on Ag(111) with post-deposited Fe atoms are displayed in Fig. 4 [55]. The XP spectrum of a 2HTPP monolayer shows two components: one for the iminic nitrogen (–N=) at lower binding energy (398.2 eV) and another for pyrrolic nitrogen (–NH–) at higher binding energy (400.1 eV) [56]. Upon deposition of Fe atoms, the two 2HTPP related peaks are increasingly replaced by a single peak at 398.7 eV, which indicates the formation of iron(II) meso-tetraphenylporphyrin (FeTPP), because the four nitrogen atoms are chemically equivalent in the complex.

On-surface synthesis of iron(II) meso-tetraphenylporphyrin (FeTPP) by metalation of meso-tetraphenylporphyrin (2HTPP) with vapor-deposited Fe. Left, N 1s XP spectra for increasing dosages of Fe (from top to bottom). The STM images (right) show the 2HTPP monolayer after deposition of 0.012, 0.024, 0.036, and 0.048 ML Fe, resulting in metalation degrees of 29, 60, 73, and 89 %, respectively. The indicated Fe coverages are relative to the number of surface Ag atoms. The stoichiometric Fe coverage would be 0.037 ML (one Fe atom per one 2HTPP molecule). Adapted with permission from Ref. [55], © (2008) American Chemical Society (left). Adapted with permission from Ref. [49] (right), © (2007) John Wiley and Sons (right)
The reaction progress can also be monitored by STM, if there is sufficient contrast between the unmetalated and the metalated species. Figure 4b shows STM images of a 2HTPP monolayer on Ag(111) during metalation with Fe [49]. The first image was taken after the deposition of 0.012 ML Fe, which led to the metalation of 29 % of the molecules in the monolayer. The FeTPP complexes appeared bright because of the large contribution of iron d-orbitals near E F [57]. The subsequent images show the monolayer after additional Fe deposition steps, resulting in the metalation of 60, 73, and 89 % of the molecules. Typical yields were 70–90 % relative to the deposited amount of metal atoms. Excessive metal deposition onto the porphyrin monolayer led to the formation of metal clusters, which preferentially formed at substrate step edges. For deposition of Fe onto 2HTPP/Ag(111), the Fe clusters were reported to sit between the Ag surface and the FeTPP layer formed by metalation [58]. Similar observations were made for the metalation of 2HTPP with Ni on Au(111) [53].
Up to now, there is no evidence for a significant influence of the peripheral substituents at the porphyrin core on the metalation reaction with post-deposited metal atoms (but see Sect. 2.2 for substitutent effects on the metalation with pre-deposited or substrate metal atoms). Metalation of 2HTPP [49, 55], 2HTPyP [48], and 2HOEP [59] with Fe on Ag(111) led to very similar results; all these reactions were found to take place at room temperature, if the metal was deposited onto the porphyrin layer.
Metalation with post-deposited metal atoms was also used as part of a two-step on-surface synthesis of porphyrin complexes with an additional axial ligand: (NH3)ZnTPP was made on Ag(111) by metalation of 2HTPP with Zn followed by coordination of NH3 at the Zn ion [47]. With a similar approach, the complex (NO)FeTPP was synthesized by successive reaction of Fe and nitric oxide (NO) with a 2HTPP monolayer on Ag(111) [60, 61].
Metalation of porphyrin monolayers is not limited to metallic substrates, but can also be performed on oxides. On a TiO2(110)-(1×1) surface, 2HTPP (sub)monolayers were metalated with post-deposited Ni atoms to from NiTPP. The yield was found to be lower than in the case of metalation on metal surfaces, reaching only 60 % even with a threefold Ni excess [62].
Phthalocyanines undergo similar metalation reactions as porphyrins. 2HPc monolayers have been metalated with Fe [58, 63] and V [50] on Ag(111). The yields were similar to those found for porphyrin metalation. Another example is the metalation of 2HPc with Fe on thin Pb films on Si(111), which was dependent on the thickness of the Pb film (cf. Sect. 2.3) [64].
Corroles, which differ from porphyrins by the lack of one of the four meso-carbon atoms, have also been shown to undergo metalation. Since the corrole macrocycle contains three pyrrolic nitrogen atoms (–NH–), the metal atom can be oxidized to its +III state, as was shown for the metalation of 2,3,8,12,17,18-hexaethyl-7,13-dimethyl-corrole with Co on Ag(111) [65].
Instead of vapor-deposited metal atoms, metal carbonyls can be used for metalation. Ru3(CO)12 was reported to metalate a 2HTPP derivative on Ag(111). (The 2HTPP derivative was formed by surface-assisted partial dehydrogenation.) After adsorption of the carbonyl at room temperature, the reaction was induced by heating to 550 K [66].
Double-decker complexes containing a metal ion in a formal +IV oxidation state sandwiched between two porphyrins or phthalocyanine ligands are formed by metals with large atomic radii and preference for higher oxidation states, such as rare earth metals. Complexes of this structure were produced by exposing 2HTPP multilayers on Ag(111) to a beam of Ce atoms. After heating to 500 K, which induces the metalation reaction and ensures desorption of excessive 2HTPP, Ce(TPP)2 double-decker complexes were observed by LT-STM. Additional bright features in the STM images were interpreted as Ce2(TPP)3 triple-decker complexes [67]. If only (sub)monolayers of 2HTPP are available, Ce deposition leads to the formation of CeTPP complexes. Due to its size, the Ce ion sits outside the porphyrin plane, but it is not clear whether it points toward or away from the surface [68].
If the metalation reaction does not proceed spontaneously, it can be induced by manipulation with an STM tip, as was demonstrated by Sperl et al. with the metalation of a single phthalocyanine (2HPc) molecule on a Ag(111) surface at 7 K, resulting in the formation of AgPc [69]. The first step of the forced metalation consisted in the dehydrogenation of the pyrrolic nitrogen atoms (–NH–) by applying voltage pulses with the STM tip (Fig. 5). In the second step, the Ag-coated W tip was approached to the molecular center at low bias voltage. At a certain distance, a sudden change of the tunneling current indicated the transfer of an Ag atom from the tip to the ligand. The identity of the synthesized AgPc was established by comparison with the tunneling spectra with those of directly deposited AgPc. Note that spontaneous formation of silver(II) phthalocyanine (AgPc) (or any Ag porphyrin) on Ag(111) by reaction with substrate atoms has not been observed as yet. Due to its large ion radius, the Ag center is located outside the molecular plane of the Pc ligand. After the STM-induced synthesis, the Ag ion points to the vacuum side (Ag-up position). By applying a voltage pulse, the ion can be pushed downward to a position between the substrate surface and molecular plane of the Pc ligand (Ag-down position). A similar conversion was reported for SnPc on Ag(111). SnPc molecules in direct contact to the Ag surface can only be switched from a Sn-up to a Sn-down position. If the molecules sit on a decoupling layer of other SnPc molecules, reversible switching in both directions was possible [70].

Synthesis of silver(II) phthalocyanine (AgPc) by forced metalation of phthalocyanine (2HPc) on Ag(111) with a Ag atom from the STM tip. a, b Gas-phase structures of 2HPc and AgPc. c–j Constant-current STM images of 2HPc and derived molecules, along with molecular models (Insets). c–e Tautomerization of 2HPc induced by voltage pulses of 1.6 V. f, g STM images of HPc obtained after removal of one pyrrolic H atom from 2HPc by a pulse of 3.0 V. Hopping of the remaining pyrrolic H atom was induced by pulses of 2.5 V. h Pc molecule fabricated by pulsing the voltage to 3.5 V. i After approach of the Ag tip to the center of the Pc, MPc-up (MPc↑) is formed. j A voltage pulse of 3.0 V leads to an interconversion of MPc into MPc-down (MPc↓). By comparison with the image of AgPc on Ag(111) prepared by sublimation from a heated crucible (k), the molecule in (j) was identified as AgPc, with the Ag atom between the surface and the molecular plane (AgPc↓). Adapted with permission from Ref. [69], © (2011) John Wiley and Sons
2.2 Synthesis of Complexes Using Post-deposited Ligand Molecules
Synthesis of porphyrin and phthalocyanine complexes is also possible by applying the reverse order of deposition, as was first demonstrated for a Zn/Ag(111) substrate, onto which 2HTPP was deposited. Subsequent heating to 550 K induced reaction to ZnTPP, as was shown by XPS [71]. A similar reaction was observed between 2HTPP and Fe/Ag(111): Initial vapor deposition of small amounts of Fe onto Ag(111) led to the formation cluster at monatomic step edges (Fig. 6a). In the next step, 2HTPP was deposited, which formed an ordered monolayer on the terraces (Fig. 6b). At room temperature, the Fe and 2HTPP coexisted without reaction. Upon heating to 550 K, however, the clusters dissolved and the Fe atoms reacted with the 2HTPP molecules on the terraces to form FeTPP. The fact the bright protrusions of FeTPP are randomly distributed over the terraces (and not clustered around the original positions of the Fe clusters at the step edges) indicates that the reactants are sufficiently mobile at this temperature to allow multiple diffusion events before the actual metalation reaction occurs [55]. Similar results were obtained for the metalation of 2HTPP on Ni/Au(111) [53].

Synthesis of FeTPP from 2HTPP on Fe/Ag(111). a STM image taken after deposition of 0.025 ML Fe onto Ag(111) at room temperature, showing Fe clusters at the step edges of a Ag(111) surface. b Coexistence of Fe clusters and a 2HTPP monolayer at the terraces after subsequent deposition of a 2HTPP monolayer. c After heating to 550 K, terraces individually color-coded. The bright spots in (c) indicate FeTPP formed by metalation of 2HTPP. Adapted with permission from Ref. [55], © (2008) American Chemical Society
At higher coverages of pre-deposited Fe, metalation at room temperature is possible. This has been shown for thin films of γ-Fe(110) (15 ML) on Cu(110). Vapor deposition of 1 ML 2HTPP onto this Fe layer led to immediate formation of FeTPP. A similar reactivity was found for 2HTPP on Ni(111): Deposition of 1 ML 2HTPP onto this substrate held at room temperature resulted in complete metalation to NiTPP [52]. 2HOEP deposited onto Ni islands on Cu(111) gave also rise to metalation, whereas no metalation was observed for 2HTPP on the same substrate [72].
The finding of room temperature metalation on Fe or Ni films and single-crystal surfaces also explains why elevated temperatures are necessary for the metalation with submonolayers of Fe or Ni on inert surfaces such as Ag(111) or Au(111): Apparently, not the metalation reaction itself is rate-limiting, but rather the supply of metal atoms by 2D evaporation of the metal clusters. At room temperature, the 2D vapor pressure of the clusters is too low for efficient metalation.
Metalation by substrate metal atoms, sometimes termed “self-metalation,” has also been studied on Cu surfaces [73–80]. The reaction of 2HP and 2HTPP with a Cu(111) surface was studied in great detail by Diller et al. [73, 74] using LT-STM, XPS, and NEXAFS. A 2HP submonolayer was found to undergo complete metalation upon heating to 423 K, as was deduced by STM. Detailed XPS studies of a 2HP bilayer revealed that complete reaction of both layers to CuP occured already between 373 and 393 K [73]. A 2HTPP monolayer on Cu(111) was shown to react to CuTPP starting at 420 K [74]. Ditze et al. used the slow reaction of 2HTPP with Cu on Cu(111) at 400 K to determine the reaction activation energy (cf. Sect. 2.3 below) [78]. For 2HTBPP on Cu(111), it was found that 57 % of a monolayer was metalated after heating to 350 K for 3.5 h. In contrast, substantial metalation of a 2HPc monolayer on Cu(111) occurred already below room temperature [81].
Reaction of 2HTPP with substrate Cu atoms was reported to be facilitated by the presence of chemisorbed oxygen atoms. On an oxygen-(\(\sqrt 2 \times \sqrt 2\))R45° reconstructed Cu(001) surface, metalation of 2HTPP was found to be complete already at 285 K, whereas 450 K was required on the bare Cu(001) surface [75]. Interestingly, the oxygen-containing porphyrin 2HPPIX was reported to undergo metalation on Cu(100) and Cu(110) surfaces at room temperature. It is not clear whether the oxygen of the molecule plays a role in this reaction as well or whether the high adatom density on the Cu(100) and Cu(110) surfaces was decisive, as was suspected by the authors [76].
The metalation of 2HTPP on Cu(111) was found to be strongly dependent on the 2HTPP coverage [82]. While disordered 2HTPP submonolayers on Cu(111) reacted only slowly to CuTPP, an increased reaction rate was observed for an ordered supramolecules “checkerboard” structure, which was formed at higher coverages [83].
2.3 Mechanism, Energetics, and Kinetics of Direct Metalation
In contrast to metalation in solution, where a metal ion replaces two protons at the pyrrolic N atoms of the porphyrin core, direct metalation with metal atoms is a redox reaction. In the course of this reaction, the metal atom is oxidized to its dication and hydrogen is released as H2. Thus, this reaction formally resembles the dissolution of a metal in a Brønsted acid [43].
Gas-phase DFT calculations of the reaction between a porphin molecule (2HP) and various metal atoms (Fe, Co, Ni, Zn, Cu) were reported by Shubina et al. [43, 44]. Figure 7 shows the computed lowest energy profile of the reaction between 2HP and Zn, together with selected minimum and transition state structures.

a Schematic energy diagram of the metalation of porphin (2HP) with Zn, according to gas-phase DFT calculations. Energies are given in kJ/mol, bond lengths in Å. The pyrrolic hydrogen atoms are marked in red. b Calculated activation energies for the gas-phase metalation of 2HP with as a function of the atomic number. Values were taken from Ref. [43]. Adapted with permission from Ref. [43], © (2007) American Chemical Society
As can be seen in the diagram, the metalation reaction starts with the coordination of the neutral metal atom by the intact porphin molecule. This initial step is exothermic and leads to a deformation of the ligand, such that the pyrrolic hydrogen atoms are bent out of the molecular plane. The resulting complex closely resembles the sitting-atop (SAT) complex postulated for metalation in solution [84, 85]. In the following two elementary steps, which have substantial activation barriers in the case of Zn, the pyrrolic hydrogen atoms are transferred to the coordinated Zn atom, from which they desorb as H2. As a result, the Zn atom is formally oxidized to its dication, while the porphin ligand acquires two negative charges. In this example (Zn + 2HP), the rate-limiting step is the first H transfer, which has a computed barrier of ΔE ≠ = 136 kJ/mol or ΔG ≠ = 137 kJ/mol [43]. The fact that ΔE ≠ and ΔG ≠ are almost identical indicates that the activation entropy ΔS ≠ is very small. This is consistent with an intramolecular H transfer step in a porphyrin molecule [86].
The release of H2 provides a convenient method for the experimental determination of the overall reaction barrier. For this purpose, N-deuterated 2DTPP was reacted with Zn, while the evolution of D2 was monitored with mass spectrometry. The resulting experimental ΔG ≠ value of 134 kJ/mol was found to agree well with theory [43, 44].
The height of the rate-limiting barrier depends strongly on the type of the metal center. While no barrier was computed for Fe, the activation energy was predicted to increase in the order Fe < Co < Ni < Cu < Zn, as shown in Fig. 7b. According to these barriers, one should expect metalation with Fe, Co, and Ni to occur below room temperature, whereas elevated temperatures should be required for Cu and Zn. This was confirmed by various experimental studies, including Refs. [43, 46, 52, 53, 55].
Experimental observations of the initial (SAT-like) complex provide further evidence for the proposed reaction mechanism as shown in Fig. 7. SAT complexes were first observed at room temperature by XPS for Zn-2HTPP on Ag(111) [43] and Cu-2HTBrPP on Cu(111) [87]. A Cu-2HTPyP SAT complex and its transition to CuTPyP were observed by STM after the deposition of Cu onto a 2HTPyP submonolayer on Au(111) [88].
A separate determination of the activation energy and the pre-exponential factor of porphyrin metalation with substrate atoms was achieved with isothermal rate measurements using STM [78]. 2HTPP and CuTPP were discriminated and quantified by their different mobilities, due to which only 2HTPP molecules appeared as individual moieties in the STM images, whereas the more mobile CuTPP gave rise to blurry areas. This approach yielded an activation energy of 143 ± 12 kJ/mol and a pre-exponential factor of 10(15±1.6) s−1. At higher coverages, a reduced barrier for the metalation was observed [82]. For comparison, DFT values for the activation energy, computed for the corresponding gas-phase reaction of 2HP with Cu, range from 100 to 155 kJ/mol, depending on the level of theory and the applied basis sets [43, 88]. The measured frequency factor is remarkably high, indicating a large positive activation entropy. This result points toward a dissociation reaction such as the final release of H2, rather than an intramolecular H transfer, as the rate-limiting step. This could indicate an active participation of the substrate surface in the metalation mechanism, as was also suggested on the basis of metalation experiments with N-deuterated tetraphenylporphyrin (2DTPP) on Cu(111) [89]. In addition, the metalation of adsorbed corroles may also be surface-mediated, because the third pyrrolic H atom cannot easily desorb as molecular hydrogen by the mechanism in Fig. 7a, and desorption of atomic H is energetically unfavorable. Therefore, it is likely that at least the third H atom migrates to the substrate surface and recombines there with another H atom before desorption as H2 [65]. Furthermore, for the metalation of monolayers, substantial influences of the substrate’s electronic structure have been observed. Song et al. studied the metalation of a 2HPc monolayer on Pb islands deposited on Si(111) [64]. The Pb islands had different thicknesses ranging from 9 to 19 monolayers. Due to the electron confinement between the semiconductor bandgap and the vacuum barrier, the electronic structure of the Pb film was found to be thickness dependent with an odd–even transition at 17 ML (Fig. 8) [90–96]. After deposition of substoichiometric amounts of Fe onto the 2HPc monolayer, the amounts of FePc on the islands of different heights were determined by STM. The yield of FePc was found to have a one-to-one correspondence to the local density of states at the Fermi level (LDOS(E F)), as shown in Fig. 8 [64]. Considering these findings of substantial surface influences, one may question the relevance of the (gas-phase) mechanism shown in Fig. 7a [43, 44]. However, it was shown that the metalation reaction also proceeds when metal atoms are deposited onto thick porphyrin multilayers, i.e., in the absence of a surface that could participate in the reaction [55] (cf. Sect. 2.5). This reaction most likely follows the mechanism shown in Fig. 7a.

Metalation of a 2HPc monolayer with Fe on Pb islands of different thickness on Si(111). a STM image showing different areal densities of the FePc product on 11 ML and 12 ML Pb films. The white dotted line indicates the boundary between the 11 and 12 ML Pb islands. b and c Magnified STM images from the marked regions in (a) depicting the thickness-dependent metalation. d Thickness-dependent FePc density at different Fe coverage. Each data point was obtained by averaging over ∼1300 molecules. e Experimental LDOS(E F ) as a function of Pb film thickness. There is an obvious one-to-one correspondence between the FePc areal density and LDOS (E F ). Adapted with permission from Ref. [64], © (2010) American Chemical Society
Coadsorption of other species can also influence the rate of the metalation reaction. This has been shown for metalation of 2HTPP on oxygen-covered Cu(001) [75]. As already briefly mentioned in Sect. 2.2, the reaction rate is dramatically enhanced by the presence of chemisorbed atomic oxygen, such that the reaction proceeds already at 285 K, compared to a minimum of 450 K on the bare Cu(001) surface. This was attributed to the fact that not H2, but H2O is formed as the second reaction product besides CuTPP. This reduces the energy of the final state by ~73 kJ/mol and likely also reduces the corresponding activation barrier. This hypothesis is supported by the loss of surface oxygen observed with XPS. As a possible mechanism, the initial incorporation of a Cu–O species into the porphyrin ring was proposed, which then reacts to H2O and the Cu porphyrin [75].
DFT calculations of the reaction between 2HTPP and Ni(111) were reported by Goldoni et al. [52]. In particular, an initial state configuration where a Ni adatom remains on a Ni(111) surface far from the adsorbed molecule was compared with a final state, in which the Ni atom has reacted with 2HTPP to form NiTPP and H2. This process was found to be exothermic by 86 kJ/mol. In contrast, when the reacting Ni atom was not an adatom, but was removed from a terrace site creating a vacancy, the process was endothermic by 67 kJ/mol. The importance of adatom availability was also thoroughly discussed by Diller et al. for metalation with substrate atoms on Cu surfaces [74].
2.4 Metalation and Synthesis of Two-Dimensional Networks
The metalation reaction can be accompanied by the formation of two-dimensional networks with covalent, organometallic, or coordination bonds, if the periphery of the ligand molecules possesses suitable functionalities for reaction with metal atoms and neighboring molecules. A prototypical example is the reaction of 2HTPyP with Cu atoms. In the absence of Cu, submonolayers of 2HTPyP on Au(111) were found to form densely packed, ordered islands. Deposition of Cu led to the formation of a two-dimensional metal-organic coordination network (2D-MOCN) with a square unit cell. In this structure, Cu atoms occupied the bridging positions between two pyridyl groups of neighboring molecules, forming linear N–Cu–N links. Additional Cu atoms coordinated to the centers of the porphyrin molecules. At room temperature, the reaction between Cu and the porphyrin core reached only the stage of the initial SAT complex, which means that only Cu(0) is present in the structure. Heating to 450 K induced completion of the metalation reaction, by which the Cu in the porphyrin centers was oxidized to Cu(II), according to XPS. The bridging Cu atom between the pyridyl groups maintained their position and zero oxidation state. As a result, a mixed-valence network with a regular arrangement of Cu(0) and Cu(II) centers was obtained [88]. Deposition of Fe instead of Cu onto a 2HTPyP submonolayer on Au(111) and annealing resulted in the formation a coordination network, in which each four pyridyl groups of neighboring molecules were connected by a vertically oriented pair of Fe atoms, besides metalation of some of the porphyrin centers [97]. It is also possible to have different metal species in the center and in the bridging position, either by linking adsorbed metalloporphyrins with another metal species or by sequential deposition of metal atoms onto the adsorbed freebase porphyrin. The latter has been demonstrated for 5,10,15,20-tetra[(4-cyanophenyl)phen-4-yl]porphyrin (2HTPCN), which forms a close-packed assembly on Ag(111). Vapor deposition of Gd onto this assembly results in the formation of a metal–organic coordination network with a square unit cell. In this network, each Gd atom links four cyano groups, whereas no Gd is found in the centers of the porphyrin molecules. Subsequent deposition of Co results in the metalation of the porphyrin centers [98].
On more reactive surfaces, the dehydrogenation of the porphyrin is not limited to the pyrrolic N–H bonds, as in the case of the metalation reaction; instead, also C–H bonds at the periphery can be dissociated. This can eventually lead to the formation of metal-organic frameworks with C–M–C bonds and covalent frameworks with C–C bonds. This type of reaction has been observed for 2HTMP of Cu(110) upon annealing to 423–473 K. Besides the metalation of the porphyrin core, the molecules formed oligomers by C–C bond formation between the peripheral mesityl groups [99]. Not only the peripheral substituent, but also the porphyrin core itself can undergo dehydrogenation, as was reported for 2HP and 2HDPP on Cu(110). Annealing submonolayers to temperatures up to 670 K led to metalation of the porphyrin core and to the formation of 2D organometallic networks, in which the dehydrogenation porphyrin cores were linked by C–Cu–C bonds [100, 101].
A related approach is the on-surface synthesis on phthalocyanine derivatives by tetramerization of phthalonitriles, as was first proposed by Abel et al. [102]. Coadsorption of 1,2,4,5-tetracyanobenzene (TCNB) with Fe atoms on Au(111) at room temperature resulted in structures that were attributed to individual Fe(II)-octacyanophthalocyanine (FePc(CN)8) molecules and covalently linked FePc-polymers, depending on the ratio between Fe and TCNB. However, later work suggested that these structures did not represent phthalocyanines, but rather 2D metal-organic coordination networks (2D-MOCNs), in which the intact TCNB molecules coordinated to Fe with their CN groups [103–105]. Koudia and Abel [106] later modified this approach and succeeded in the on-surface synthesis of Mn(II)-octacyanophthalocyanine (MnPc(CN)8) by codeposition of TCNB and Mn atoms on Ag(111). At room temperature, similar 2D-MOCNs as in the case of the Fe-TCNB system were obtained. Post-annealing at 415 K induced the cyclotetramerization reaction resulting in the formation of MnPc(CN)8. Polymerization of the MnPc(CN)8 molecules by reaction of the peripheral cyano group and formation of a 2D covalent MnPc network was achieved by annealing to 615 K [106]. Similar codeposition experiments with Mn and TCNB on Ag(111) were reported by Piantek et al. [104], who obtained MnPc(CN)8 molecules after annealing to 423 K. The on-surface synthesis of FePc(CN)8 was eventually achieved by annealing coadsorbed Fe and TCNB on Au(111) to 550 K [105].
2.5 Synthesis of Complexes with Ligand Multilayers and at Bulk Interfaces
Metal complexes of porphyrins and phthalocyanines can also be obtained by vapor deposition of metal atoms onto multilayers of the organic ligands or by reaction of the multilayers with the underlying metal substrate. A related approach is the codeposition of metal and porphyrin, which also leads to the respective metal complexes.
Multilayer metalation was first reported by Buchner et al., who vapor deposited Fe onto a 2HTPP multilayer on Ag(111) (Fig. 9). The reaction was not complete, i.e., not all deposited Fe atoms reacted, despite an excess of unreacted 2HTPP [55]. Similar experiments were reported by Di Santo et al. for the metalation of thin multilayers (~4 layers) of 2HTPP on Ag(111). This thin multilayer was almost completely metalated, as was shown by XPS. In addition, the thereby produced FeTPP multilayer was removed by thermal desorption, leaving a FeTPP monolayer on the surface [107]. 2HOEP multilayers (~4 layers) on Ag(111) were successfully metalated by the same approach [59]. Metalation of 2HTPP multilayers with vapor-deposited Ce atoms and subsequent desorption of excessive 2HTPP was used by Ecija et al. for the on-surface synthesis of Ce(TPP)2 double-decker and Ce2(TPP)3 triple-decker complexes [67]. The codeposition of Er and 2HTPP was used for the synthesis of clean ErTPP multilayers of up to 60 nm thickness. Composition and electronic structure of the ErTPP film were studied by XPS and UPS [108].

Multilayer metalation. a Vapor deposition of the metal onto the organic layer: N 1s XP spectra of the metalation of 2HTPP multilayers (ca. 20 layers) with vapor-deposited Fe atoms at room temperature. (A) 2HTPP multilayer. (B)–(D) After vapor deposition of Fe as indicated. The central peak in the spectra (B)–(D) represents the newly formed FeTPP, while the two outer peaks stem from residual 2HTPP. b Vapor deposition of the organic multilayer onto the metal: N 1s XP spectra taken after deposition of 2HPc onto Cu(111) at 300 K with an initial coverage of 4 monolayers and after heating to the indicated temperatures. Adapted with permission from Refs. [55, 81], © (2008) and (2014) American Chemical Society
Metalation of multilayers by reaction with a single-crystalline metal substrate has been observed for 2HTPP, 2HP, and 2HPc on Cu(111). Diller et al. reported XPS data which show that a thin 2HTPP multilayer reacts completely with Cu atoms from the substrate, forming CuTPP, upon annealing at 420 K [74]. Similar results were reported for a 2HP bilayer [73].
Reaction of a multilayer with substrate atoms raises the question how the reactants come into contact. There are two principal possibilities: diffusion of the metal into the organic phase or exchange within the organic phase, such that all molecules in the multilayer get into contact with the metal surface. This question was addressed by Chen et al., who investigated the metalation of 2HPc multilayers on Cu(111) with XPS (Fig. 9b) and TPD/TPR [81]. TPD/TPR performed after deposition of a 2HPc bilayer on Cu(111) led to desorption of CuPc around 550 K, while XPS showed that a CuPc monolayer remained on the surface. Likewise, it was found that a NiPc monolayer did not desorb at temperatures up to 650 K (instead, decomposition occurs above that temperature, according to XPS). In contrast, when a bilayer of 2HPc was deposited onto 0.8 monolayers of NiPc on Cu(111), NiPc was found to desorb along with CuPc and residual 2HPc. A similar experiment with a NiPc bilayer yielded a similar result. This experiment was explained with an exchange of molecules between the first layer and the higher layers, bringing 2HPc into contact with the Cu surface and NiPc into higher layers, from which it can desorb. Therefore, a possible mechanism for multilayer metalation was shown to be the exchange of molecules between the first layer and the higher layers [81]. However, the findings by Chen et al. could also be explained by an exchange of metal centers between the NiPc and Cu(111) surface, as was shown for another system by Doyle et al. [109].
2.6 Synthesis of Porphyrin Complexes by Metalation at Solid–Liquid Interfaces
Porphyrins adsorbed at solid–liquid interfaces can also undergo metalation. Hai et al. studied this process at an electrochemical interface using the cationic porphyrin 2HTMPyP4+ on Cu(100) in a solution containing HCl and 2HTMPyP4+ tosylate. Cu2+ ions formed during anodic oxidation of the Cu electrode were incorporated into the adsorbed porphyrin molecules, resulting in the formation of Cu(II)TMPyP4+. This was concluded from cyclic voltammetry (CV) in combination with electrochemical STM (EC-STM) and UV–vis measurements. The mechanism of the metalations is apparently based on an ion exchange process: Similar to the homogeneous reaction in solution, but unlike the metalation reaction at the solid–vacuum interface, the two pyrrolic protons are replaced by one Cu(II) ion [110].
Metalation of porphyrins adsorbed at the metal/liquid interface has also been observed by surface-enhanced resonance Raman scattering (SERRS). For 2HTMPyP4+ adsorbed on Ag colloids, it was found that 2HTMPyP4+ exists only under acidic conditions, while metalation to Ag(II)TMPyP4+ occurred in neutral and alkaline environments. On mixed Ag–Cu colloids with up to 5 at.% Cu, all adsorbates were converted to Cu(II)TMPyP4+ [111]. The metalation kinetics of 2HTMPyP4+ on Ag colloids was investigated by time-dependent SERRS and was found to be highly dependent on the presence of anions in the system [112, 113]. In another study, Zn(II) ions adsorbed on silica gel reacted rapidly with solutions of 2HTPP to ZnTPP [114].
2.7 Synthesis of Complexes with Axial Ligands
The on-surface metalation of porphyrins and phthalocyanines described in the previous sections results in adsorbed complexes with a vacant axial coordination site, to which a ligand can be attached in a subsequent step. Such two-step syntheses have rarely been performed; notable examples are the synthesis of (NH3)ZnTPP from 2HTPP, Zn, and NH3 successively adsorbed on a Ag(111) surface [47]. The same approach was used for the synthesis of (NO)FeTPP on the same substrate [61]. In both cases, it would not have been possible to vapor deposit the intact complexes because of their insufficient thermal stability. In other work, small molecules were coordinated to directly deposited metalloporphyrins and metallophthalocyanines, again to produce complexes which would have been thermally too labile for direct vapor deposition. Examples include (CO)MTPP and cis-(CO)2MTPP with M=Co, Fe [115, 116], (CO)FePc [117, 118], (CO)MnPc [119], (DABCO)ZnTBPP [120], (H)MPc with M=Fe, Mn [121, 122], (NH3)FePc [117, 123, 124], (NH3)MnPc [125], (NH3)NiTPP [126], (NO)CoOEP [127], (NO)CoTPP [60, 61, 115, 125, 128–134], (NO)FeOEP [135], (NO)FePc [117, 118, 124, 136], (NO)FeTPP [61, 115, 125], (NO)MnTPP [125], (pyridine)FePc [117], and (O)TiTPP [54]. In the case of (N)MnPc, thermal activation of N2 was necessary [137].
The attachment of the axial ligand can influence the bond between the coordinated metal center and the substrate (surface trans effect [61]), and the magnetic properties of the complex (surface spin trans effect [138]). As an illustrative example, the coordination of NO and CO to CoTPP and FeTPP on an Ag(111) surface will be compared in the following. NO formed exclusively the mononitrosyl species (NO)MTPP (M=Fe, Co) and had a large impact on the geometric and electronic structure of the porphyrins. In particular, the M–Ag(111) interaction (M=Fe, Co) was suppressed by the NO ligand, resulting in changes of the electronic structure in the valence region [60, 61]. In contrast, CO was found to form both monocarbonyl and cis-dicarbonyl species, while leaving the properties of the metalloporphyrin almost unaffected [115]. The carbonyl species were generated by dosing CO at very low temperatures of 6–20 K. Transfer of the CO molecules between neighboring CoTPP molecules by LT-STM manipulation was demonstrated [115, 116]. In contrast to NO, CO did not suppress the valence states induced by the interaction of the Fe and Co centers with the Ag surface, as was shown by STS [115]. STM images of the cis-dicarbonyl species (CO)2FeTPP and (CO)2CoTPP revealed a relatively large distance between the two CO molecules, compared to dicarbonyls formed with metal adatoms. This was explained with a bonding geometry in which the CO ligands occupy bridge positions between the metal center and a porphyrin N atom (Fig. 10) [116]. Overall, CO appears to have a weaker influence on the metal centers than NO.

Synthesis of (CO)xCoTPP on Ag(111), STM images. a Conformational adaptation with saddle-shape distortion of CoTPP adsorbed on Ag(111). b CoTPP monolayer array on Ag(111) after exposure to CO, comprising CoTPP, (CO)CoTPP (dashed circle), and (CO)2CoTPP (solid circle). c Top-view model of CoTPP overlaid on an STM image. d, e Top-view topography of undecorated CoTPP (d) and dicarbonyl (e) species. The corresponding difference image (f) emphasizes the relatively large distance of ~5.3 Å between the CO-related maxima. Adapted with permission from Ref. [116], © (2011) Nature Publishing Group
3 On-Surface Synthesis of Organometallic and Covalent Nanostructures
3.1 The Surface Ullmann Reaction
In contrast to the wide range of C–C bond formation reactions available in solution-based organic chemistry, only few reactions suitable for C–C bond formation at solid surfaces have been identified so far. The most prominent example is the Ullmann reaction between bromo- and iodoarenes on metal surfaces. Originally performed in solution with solid Cu metal as a reactant [139] and widely used in organic synthesis [140, 141], this heterogeneous reaction can also be used for C–C bond formation on well-defined metal surfaces in ultra-high vacuum (UHV) [142]. To date, detailed insight into the mechanism of this surface Ullmann reaction has been obtained by a combination with spectroscopic and microscopic techniques [143–147], complemented by theoretical investigations [148, 149].
3.2 Hierarchical Molecular Approach Toward a Hydrocarbon-Based Two-Dimensional Network
The surface Ullmann reaction has been used for the on-surface synthesis of large π-conjugated hydrocarbon macrocycles consisting of phenyl rings, i.e., cyclo-oliphenylenes. This type of model systems will be discussed here in more detail for two reasons: First, the naked parent structures of these macrocycles are only available by on-surface synthesis. Previous solution-based syntheses required the presence of large solubility enhancing alkyl substituents. As an example, Hensel and Schlüter reported a hexagonal cyclotetraicosa-phenylene macrocycle containing 24 phenyl rings and, for enhanced solubility, 12 hexyl side chains [3]. Similar compromises were necessary for the synthesis of other large, shape-persistent macrocycles [1]. In addition, even if it was possible to make the parent structures of these macrocycles in solution, they would be too large for undecomposed vapor deposition in UHV.
Second, the macrocycles made by on-surface synthesis are suitable for the fabrication of porous networks with a combined covalent and van der Waals linkage. Compared to the conventional all-covalent linkage, this approach has distinct advantages: As illustrated in Fig. 11, the on-surface synthesis of porous graphene-like structures requires careful kinetic control of the reaction conditions, which include the usage of appropriate precursors. In most cases, the reaction still results in structures with very high defect concentrations. This is rather unfortunate, because the reactions need to be performed under conditions at which the C–C bond formation is not reversible (e.g., at moderate temperatures). Otherwise, the thermodynamic equilibrium structure will be formed, which is regular graphene. Considering this fundamental issue, it appears questionable whether the direct synthesis of two-dimensional hydrocarbon nanostructures is possible at all with this approach. Therefore, alternative approaches should be developed.

Kinetic versus thermodynamic control on the example of the on-surface synthesis of (porous) graphene structures. The global minimum of the potential energy surface is usually graphene, which is formed under conditions where the C–C bond formation is reversible. In contrast, the porous graphene derivatives represent only local minima of the potential energy surface. Their synthesis therefore requires kinetic reaction control and suitable precursors. Initially formed defects cannot be healed by annealing, because this would lead to the formation of (unwanted) regular graphene
The problem of defect formation and a possible solution are illustrated in Fig. 12: Molecular precursors that undergo C–C bond formation in two dimensions form covalent networks, which are usually defect-rich (left). The irreversible nature of the C–C bond (under the required experimental conditions) makes these defects permanent. Another hierarchical strategy is shown in the right column of Fig. 12. The precursors used in this approach can only link in one dimension and form large molecular network subunits (with the formation of oligomer or polymer chains as a possible side reaction). If these subunits are still sufficiently mobile on the surface, they can self-assemble to form ordered arrays. Since this process is reversible, defects can heal and unwanted structural elements (shown in red) are able to segregate. If necessary, additional covalent linkage of the now well-ordered subunits can be achieved in an additional reaction step.

Network formation exclusively based on covalent linkage results in defect-rich structures which cannot be improved by annealing (left). In contrast, the hybrid approach shown in the right column starts with the on-surface synthesis of mobile molecular subunits, which are mobile and can form ordered supramolecular structures by reversible self-assembly. “Wrong” structural elements (red) are expelled from the lattice
3.3 Formation of Hyperbenzene and Organometallic Intermediates
An example for the strategy outlined in Sect. 3.2 is the on-surface synthesis of hyperbenzene C108H72, a hexagonal hydrocarbon macrocycle consisting of 18 phenyl rings, using the surface Ullmann reaction [4]. The reaction proceeds most likely via an intermediate state with C–Cu–C bonds [145, 150], which are stable at and well above room temperature.
3.3.1 Organometallic Intermediates with C–Cu–C Bonds
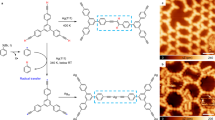
Deposition of the bromoarene precursor molecule, 4,4″-dibromo-m-terphenyl (DMTP), onto Cu(111) at room temperature resulted in the dissociation of the C–Br bonds (which occurs already below 240 K on this surface [146]) and the subsequent formation of C–Cu–C bonds between neighboring molecular m-terphenyl (MTP) fragments [150]. This process led to the formation of organometallic (MTP-Cu) n zigzag chains (as shown schematically in Fig. 13a) and cyclic (MTP-Cu) n (n = 6, 8, 14, 16, 18, 22) structures [150]. STM images of these chains are shown in Fig. 14. Deposition at elevated temperatures (440 K) yielded large islands consisting of the (MTP-Cu) n zigzgag chains (Fig. 14d) [150]. The chains have preferential orientations relative to the high-symmetry directions of the Cu(111) substrate (see Fig. 14a). The lattice constant along the chains is 26.5 Å, which is larger than the value expected for direct C–C linkage (21.8 Å) and confirms that the MTP fragment is linked by Cu atoms (in line with related work, in which dibromo-p-terphenyl was used [145]). The apparent height in STM varies along the chains, and the terphenyl units (located at the bends, large maxima in Fig. 14g) can be distinguished from the C–Cu–C bridges (at the straight parts of the chain, small maxima in Fig. 14g). Residual Br atoms sit between the chains or form separate islands with a (√3 × √3)R30° structure and a hexagonal unit cell [151]. The C–Cu bond was found to be remarkably stable, requiring temperatures above 440 K for the release of the Cu atom and formation of the C–C bond [4, 147, 150, 151].

Surface-assisted Ullmann coupling reaction of 4,4″-dibromo-m-terphenyl (DMTP) on Cu(111), resulting in the formation of a organometallic polymers at 300 K and b hyperbenzene molecules at 550 K. Adapted with permission from Ref. [4], © (2013) John Wiley & Sons

STM study of the reaction of dibromo-m-terphenyl (DMTP) on Cu(111) at 300 K and formation of organometallic oligomers and polymers with C–Cu–C bonds: a Constant-current STM image after deposition of DMTP onto Cu(111) at 300 K at low coverage. The islands consist of chains of the organometallic polymer (MTP-Cu) n , which are oriented at angles of ±5° relative to the [−110] (and equivalent) directions, as marked with green arrows. b Higher resolution image of the shaded area in (a) with overlaid molecular model and unit cell. c (MTP-Cu) n polymers at higher precursor coverage. Inset Magnified view of a cyclic hexamer (MTP-Cu)6 superimposed with molecular model. d After deposition of DMTP onto Cu(111) held at 440 K, which leads to larger (MTP-Cu) n islands. e Side view of a structure from periodic DFT calculations, illustrating the non-planar geometry of the (MTP-Cu) n chains. f DFT-calculated STM image for the tunneling parameters in (b). g Apparent height profile along the blue zigzag line in (b) from left to right. Adapted with permission from Ref. [4], © (2013) John Wiley & Sons
3.3.2 On-Surface Synthesis of Hyperbenzene Molecules
Hyperbenzene was synthesized according to the scheme in Fig. 13b by deposition of DMTP onto a hot Cu(111) surface at 550 K [4]. Besides hyperbenzene, which was found to account for ~70 % of the reaction products, oligophenylene chains (13 %) and other oligophenylene structures (17 %) were observed as side products (Fig. 15) [4]. Synthesis by heating the organometallic precursor phase from 300 K to temperatures above 500 K also induced the formation of C–C bonds, but the yield of well-defined products (and especially hyperbenzene) was considerably lower, most likely because the islands of the organometallic (MTP-Cu) n chains (as can be seen in Fig. 14d) are too dense for the formation of hyperbenzene.

Hyperbenzene: a Overview constant-current STM image obtained at 300 K after deposition of 4,4″-dibromo-m-terphenyl (DMTP) onto Cu(111) held at 550 K. The resulting hexagonal rings (hyperbenzene) show a preferential orientation relative to the high-symmetry directions of the substrate. b Magnified view of a small section with oligophenylene chains. c Magnified view of a hyperbenzene island with several defects and inclusions in the central cavities. Molecular models and a unit cell are overlaid. d DFT-calculated STM images with overlaid model of the relaxed computed structure. e Apparent height profile along the perimeter of a hyperbenzene molecule, as marked by the blue hexagon in the inset. Note that the height axis is identical to that in Fig. 14g for direct comparison. Adapted with permission from Ref. [4], © (2013) John Wiley & Sons
In line with the scheme in Fig. 12 (right column), the hyperbenzene molecules self-assemble to form a hexagonal lattice with the unit cell dimensions displayed in Fig. 15c. Products with other structures (such as chain fragments) are not embedded in the hyperbenzene islands, but segregate to the periphery (Fig. 15a).
The distance between the edges of two adjacent hyperbenzene molecules of 9.3 Å is almost identical to the distance between the coordination polymer chains in Fig. 14, indicating that the assembly is mainly driven by van der Waals interactions and that there are no C–C bonds between the rings. The variation of the apparent height along the ring is small compared to the case of the coordination oligomers (compare the height profile in Fig. 15e with Fig. 14g).
4 Summary
In this chapter, it has been shown that structurally complex molecular coordination compounds, organometallic compounds, and conjugated hydrocarbons can be synthesized on metal surfaces from suitable molecular precursors. Some of the reactions proceed with high yields and lead to by-products that desorb under the reaction conditions, while in other cases the synthesis is hamped by formation of multiple products, which remain adsorbed. Two examples have been discussed in more detail.
Adsorbed porphyrins, corroles, and phthalocyanines react with coadsorbed metal atoms such as Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ru, and Ce, resulting in the formation of the respective metal complexes. In the course of this reaction, porphyrins and phthalocyanines oxidize metal atoms to their +II ions, while corroles cause oxidation to the +III ions. The metal atoms can be provided by the substrate, vapor deposited before or after the deposition of the molecules, or delivered as carbonyls. The reaction follows a two-step mechanism, which starts with the initial coordination of the neutral metal atom by the intact freebase macrocyle, and includes two hydrogen transfer steps and is completed by with the release of H2 from the oxidized metal center. The activation barriers vary strongly with the type of the metal center. Participation of the surface in the reaction has been observed in some cases. In the resulting metal complexes, one of the two axial coordination sites at the metal center points away from the surface and can bind one or more additional ligands, which can affect the electronic and magnetic properties of the metal center. Multilayers can be metalated in the same way by the deposition of metals onto the organic phase.
The surface Ullmann reaction can be employed for the on-surface synthesis of large organic molecules, as has been shown on the example of hyperbenzene, a hexagonal macrocycle consisting of 18 phenyl rings, formed at a Cu(111) surface by covalent linking of six meta-terphenyl fragments. The macrocycles undergo self-assembly to form a honeycomb network. Extension to other and larger macrocycles as well as functionalized species appears feasible. Subsequent covalent linking of the honeycomb network could provide a possible way to well-ordered covalent networks with large pores.
Abbreviations
- CV:
-
Cyclic voltammetry
- DFT:
-
Density functional theory
- EC-STM:
-
Electrochemical scanning tunneling microscopy
- LDOS:
-
Local density of states
- LT-STM:
-
Low-temperature scanning tunneling microscopy
- MOCN:
-
Metal-organic coordination network
- NEXAFS:
-
Near-edge X-ray absorption fine structure
- RT:
-
Room temperature
- SERRS:
-
Surface-enhanced resonance Raman scattering
- STM:
-
Scanning tunneling microscopy
- STS:
-
Scanning tunneling spectroscopy
- TPD:
-
Temperature programmed desorption
- TPR:
-
Temperature programmed reaction
- UHV:
-
Ultra-high vacuum
- UPS:
-
Ultraviolet photoelectron spectroscopy
- XPS:
-
X-ray photoelectron spectroscopy
- DABCO:
-
1,4-diazabicyclo[2.2.2]octane
- DMTP:
-
4,4″-dibromo-m-terphenyl
- DPP:
-
5,15-diphenylporphyrin
- MTP:
-
meta-terphenyl biradical
- OEP:
-
2,3,7,8,12,13,17,18-octaethylporphyrin
- P:
-
Porphin
- Pc:
-
Phthalocyanine
- PPIX:
-
Protoporphyrin IX (3,7,12,17-tetramethyl-8,13-divinyl-2,18-porphindipropionic acid)
- TBPP:
-
5,10,15,20-tetrakis-(3,5-di-tert-butyl)-phenylporphyrin
- TBrPP:
-
5,10,15,20-tetrakis(4-bromophenyl)porphyrin
- TCNB:
-
1,2,4,5-tetracyanobenzene
- TMP:
-
Meso-tetramesitylporphyrin (5,10,15,20-tetrakis(2,4,6-trimethyl)phenylporphyrin)
- TMPA:
-
5,10,15,20-tetrakis[4-(trimethylammonio)phenyl]-porphyrin
- TMPyP4+ :
-
5,10,15,20-tetra(N-methyl-4-pyridinium)porphyrin
- TPCN:
-
5,10,15,20-tetra[(4-cyanophenyl)phen-4-yl]porphyrin
- TPP:
-
5,10,15,20-tetraphenylporphyrin
- TPyP:
-
5,10,15,20-tetra(4-pyridyl)porphyrin
References
Grave, C., Schlüter, A.D.: Shape-persistent, nano-sized macrocycles. Eur. J. Org. Chem. 18, 3075–3098 (2002)
Hensel, V., Lutzow, K., Jacob, J., Gessler, K., Saenger, W., Schlüter, A.D.: Repetitive construction of macrocyclic oligophenylenes. Angew. Chem.-Int. Edit. 36, 2654–2656 (1997)
Hensel, V., Schlüter, A.D.: A cyclotetraicosaphenylene. Chem.-Eur. J. 5, 421–429 (1999)
Fan, Q.T., Wang, C.C., Han, Y., Zhu, J.F., Hieringer, W., Kuttner, J., Hilt, G., Gottfried, J.M.: Surface-assisted organic synthesis of hyperbenzene nanotroughs. Angew. Chem.-Int. Edit. 52, 4668–4672 (2013)
Barth, J.V.: Molecular architectonic on metal surfaces. Annu. Rev. Phys. Chem. 58, 375–407 (2007)
Barth, J.V.: Fresh perspectives for surface coordination chemistry. Surf. Sci. 603, 1533–1541 (2009)
Lin, N., Stepanow, S., Ruben, M., Barth, J.V.: Surface-confined supramolecular coordination chemistry. In: Broekmann, P., Dotz, K.H., Schalley, C.A. (eds.) Templates in Chemistry III, Topics in Current Chemistry, pp. 1–44 (2009)
Stepanow, S., Lin, N., Barth, J.V.: Modular assembly of low-dimensional coordination architectures on metal surfaces. J. Phys.-Condens. Matter 20, 184002 (2008)
Aldridge, W.N., De Matteis, P.: Heme and Hemoproteins. Springer, Berlin (1977)
Oohora, K., Hayashi, T.: Hemoprotein-based supramolecular assembling systems. Curr. Op. Chem. Biol. 19, 154–161 (2014)
Milgrom, L.R.: The Colors of Life. Oxford University Press, Oxford (1997)
Mochida, I., Suetsugu, K., Fujitsu, H., Takeshita, K.: Enhanced catalytic activity of cobalt tetraphenylporphyrin on titanium dioxide by evacuation at elevated temperatures for intensifying the complex support interaction. J. Phys. Chem. 87, 1524–1529 (1983)
Rezaeifard, A., Jafarpour, M.: The catalytic efficiency of Fe-porphyrins supported on multi-walled carbon nanotubes in the heterogeneous oxidation of hydrocarbons and sulfides in water. Catal. Sci. Technol. 4, 1960–1969 (2014)
Qiao, J.L., Liu, Y.Y., Hong, F., Zhang, J.J.: A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 43, 631–675 (2014)
Wang, C., Li, J., Mele, G., Duan, M.Y., Lu, X.F., Palmisano, L., Vasapollo, G., Zhang, F.X.: The photocatalytic activity of novel, substituted porphyrin/TiO2-based composites. Dyes Pigm. 84, 183–189 (2010)
Takulapalli, B.R., Laws, G.M., Liddell, P.A., Andreasson, J., Erno, Z., Gust, D., Thornton, T.J.: Electrical detection of amine ligation to a metalloporphyrin via a hybrid SOI-MOSFET. J. Am. Chem. Soc. 130, 2226–2233 (2008)
Jiang, S., Cheng, R., Wang, X., Xue, T., Liu, Y., Nel, A., Huang, Y., Duan, X.F.: Real-time electrical detection of nitric oxide in biological systems with sub-nanomolar sensitivity. Nat. Commun. 4, 2225 (2013)
Dedigama, A., Angelo, M., Torrione, P., Kim, T.-H., Wolter, S., Lampert, W., Atewologun, A., Edirisoorya, M., Collins, L., Kuech, T.F., Losurdo, M., Bruno, G., Brown, A.: Hemin-functionalized InAs-based high sensitivity room temperature NO gas sensors. J. Phys. Chem. C 116, 826–833 (2012)
Klauk, H.: Organic Electronics—Materials, Manufacturing and Applications. Wiley-VCH (2006)
Tuffy, B.: Porphyrin Materials for Organic Light Emitting Diodes: A Route to Phosphorescent Emission. Lambert Academic Publishing (2011)
Imahori, H.: Giant multiporphyrin arrays as artificial light-harvesting antennas. J. Phys. Chem. B 108, 6130–6143 (2004)
Li, L.-L., Diau, E.W.-G.: Porphyrin-sensitized solar cells. Chem. Soc. Rev. 42, 291–304 (2013)
Ackroyd, R., Kelty, C., Brown, N., Reed, M.: The history of photodetection and photodynamic therapy. Photochem. Photobiol. 74, 656–669 (2001)
Nyman, E.S., Hynninen, P.H.: Research advances in the use of tetrapyrrolic photosensitizers for photodynamic therapy. J. Photochem. Photobiol. B Biology 73, 1–28 (2004)
O’Connor, A.E., Gallagher, W.M., Byrne, A.T.: Porphyrin and Nonporphyrin photosensitizers in oncology: preclinical and clinical advances in photodynamic therapy. Photochem. Photobiol. 85, 1053–1074 (2009)
Middelburg, T.A., de Vijlder, H.C., de Bruijn, H.S., van der Ploeg-van den Heuvel, A., Neumann, H.A.M., de Haas, E.R.M., Robinson, D.J.: Topical photodynamic therapy using different porphyrin precursors leads to differences in vascular photosensitization and vascular damage in normal mouse skin. Photochem. Photobiol. 90, 896–902 (2014)
Erk, P., Hengelsberg, H.: Phthalocyanine dyes and pigments. In: Kadish, K.M., Guilard, R., Smith, K.M. (eds.) The Porphyrin Handbook, vol. 19, pp. 105–150. Academic Press (2003)
Sorokin, A.B.: Phthalocyanine metal complexes in catalysis. Chem. Rev. 113, 8152–8191 (2013)
Dimitrakopoulos, C.D., Malenfant, P.R.L.: Organic thin film transistors for large area electronics. Adv. Mater. 14, 99–117 (2002)
Blochwitz, J., Pfeiffer, M., Fritz, T., Leo, K.: Low voltage organic light emitting diodes featuring doped phthalocyanine as hole transport material. Appl. Phys. Lett. 73, 729–731 (1998)
Bottari, G., de la Torre, G., Guldi, D.M., Torres, T.: Covalent and noncovalent phthalocyanine-carbon nanostructure systems: synthesis, photoinduced electron transfer, and application to molecular photovoltaics. Chem. Rev. 110, 6768–6816 (2010)
Hains, A.W., Liang, Z.Q., Woodhouse, M.A., Gregg, B.A.: Molecular semiconductors in organic photovoltaic cells. Chem. Rev. 110, 6689–6735 (2010)
Cao, W.R., Xue, J.G.: Recent progress in organic photovoltaics: device architecture and optical design. Energy Environ. Sci. 7, 2123–2144 (2014)
Ayhan, M.M., Durmus, M., Gurek, A.G.: Synthesis, photophysical and photochemical studies of novel liquid crystalline phthalocyanines. J. Porph. Phthalocyanines 13, 722–738 (2009)
Basova, T.V., Parkhomenko, R.G., Igumenov, I.K., Hassan, A., Durmus, M., Gurek, A.G., Ahsen, V.: Composites of liquid crystalline nickel phthalocyanine with gold nanoparticles: liquid crystalline behaviour and optical properties. Dyes Pigm. 111, 58–63 (2014)
Gregory, P.: Industrial applications of phthalocyanines. J. Porph. Phthalocyanines 4, 432–437 (2000)
Ince, M., Yum, J.H., Kim, Y., Mathew, S., Gratzel, M., Torres, T., Nazeeruddin, M.K.: Molecular engineering of phthalocyanine sensitizers for dye-sensitized solar cells. J. Phys. Chem. C 118, 17166–17170 (2014)
Bonnett, R.: Photosensitizers of the porphyrin and phthalocyanine series for photodynamic therapy. Chem. Soc. Rev. 24, 19–33 (1995)
Taquet, J.P., Frochot, C., Manneville, V., Barberi-Heyob, M.: Phthalocyanines covalently bound to biomolecules for a targeted photodynamic therapy. Curr. Med. Chem. 14, 1673–1687 (2007)
Buchler, J.W.: Static coordination chemistry of metalloporphyrins. In: Smith, K.M (ed.) Porphyrins and Metalloporphyrins, pp. 157–232. Elsevier (1975)
Gottfried, J.M., Marbach, H.: Surface-confined coordination chemistry with porphyrins and phthalocyanines: aspects of formation, electronic structure, and reactivity. Z. Phys. Chem. 223, 53–74 (2009)
Hambright, P.: Dynamic coordination chemistry of metalloporphyrins. In: Smith, K.M. (ed.) Porphyrins and Metalloporphyrins, pp. 234–278. Elsevier (1975)
Shubina, T.E., Marbach, H., Flechtner, K., Kretschmann, A., Jux, N., Buchner, F., Steinrück, H.P., Clark, T., Gottfried, J.M.: Principle and mechanism of direct porphyrin metalation: joint experimental and theoretical investigation. J. Am. Chem. Soc. 129, 9476–9483 (2007)
Shubina, T.E.: Computational studies on properties, formation, and complexation of M(II) porphyrins. In: VanEldik, R., Harvey, J. (ed.) Advances in Inorganic Chemistry: Theoretical and Computational Inorganic Chemistry, vol. 62, pp. 261–299 (2010)
Panighel, M., Santo, G.D., Caputo, M., Lal, C., Taleatu, B., Goldoni, A.: Review of 2H-tetraphenylporphyrins metalation in ultra-high vacuum on metal surfaces. J. Phys. Conf. Series 470, 012012 (2013)
Gottfried, J.M., Flechtner, K., Kretschmann, A., Lukasczyk, T., Steinrück, H.P.: Direct synthesis of a metalloporphyrin complex on a surface. J. Am. Chem. Soc. 128, 5644–5645 (2006)
Flechtner, K., Kretschmann, A., Bradshaw, L.R., Walz, M.M., Steinrück, H.P., Gottfried, J.M.: Surface-confined two-step synthesis of the complex (ammine)(meso-tetraphenylporphyrinato)-zinc(II) on Ag(111). J. Phys. Chem. C 111, 5821–5824 (2007)
Auwärter, W., Weber-Bargioni, A., Brink, S., Riemann, A., Schiffrin, A., Ruben, M., Barth, J.V.: Controlled metalation of self-assembled porphyrin nanoarrays in two dimensions. ChemPhysChem 8, 250–254 (2007)
Buchner, F., Schwald, V., Comanici, K., Steinrück, H.-P., Marbach, H.: Microscopic evidence of the metalation of a free-base porphyrin monolayer with iron. ChemPhysChem 8, 241–243 (2007)
Eguchi, K., Nakagawa, T., Takagi, Y., Yokoyama, T.: Direct synthesis of vanadium phthalocyanine and its electronic and magnetic states in monolayers and multilayers on Ag(111). J. Phys. Chem. C 119, 9805–9815 (2015)
Schouteden, K., Ivanova, T., Li, Z., Iancu, V., Janssens, E., Van Haesendonck, C.: Probing magnetism in 2D molecular networks after in situ metalation by transition metal atoms. J. Phys. Chem. Lett. 6, 1048–1052 (2015)
Goldoni, A., Pignedoli, C.A., Di Santo, G., Castellarin-Cudia, C., Magnano, E., Bondino, F., Verdini, A., Passerone, D.: Room temperature metalation of 2H-TPP monolayer on iron and nickel surfaces by picking up substrate metal atoms. ACS Nano 6, 10800–10807 (2012)
Chen, M., Feng, X.F., Zhang, L., Ju, H.X., Xu, Q., Zhu, J.F., Gottfried, J.M., Ibrahim, K., Qian, H.J., Wang, J.O.: Direct synthesis of nickel(II) tetraphenylporphyrin and its interaction with a Au(111) surface: a comprehensive study. J. Phys. Chem. C 114, 9908–9916 (2010)
Duncan, D.A., Deimel, P.S., Wiengarten, A., Han, R., Acres, R.G., Auwarter, W., Feulner, P., Papageorgiou, A.C., Allegretti, F., Barth, J.V.: Immobilised molecular catalysts and the role of the supporting metal substrate. Chem. Commun. 51, 9483–9486 (2015)
Buchner, F., Flechtner, K., Bai, Y., Zillner, E., Kellner, I., Steinrück, H.P., Marbach, H., Gottfried, J.M.: Coordination of iron atoms by tetraphenylporphyrin monolayers and multilayers on Ag(111) and formation of iron-tetraphenylporphyrin. J. Phys. Chem. C 112, 15458–15465 (2008)
Khandelwal, S.C., Roebber, J.L.: Photoelectron spectra of tetraphenylporphine and some metallotetraphenylporphyrins. Chem. Phys. Lett. 34, 355–359 (1975)
Lu, X., Hipps, K.W.: Scanning tunneling microscopy of metal phthalocyanines: d(6) and d(8) cases. J. Phys. Chem. B 101, 5391–5396 (1997)
Buchner, F., Kellner, I., Steinrück, H.-P., Marbach, H.: Modification of the growth of iron on Ag(111) by predeposited organic monolayers. Z. Phys. Chem. 223, 131–144 (2009)
Borghetti, P., Di Santo, G., Castellarin-Cudia, C., Fanetti, M., Sangaletti, L., Magnano, E., Bondino, F., Goldoni, A.: Adsorption geometry, conformation, and electronic structure of 2H-octaethylporphyrin on Ag(111) and Fe metalation in ultra high vacuum. J. Chem. Phys. 138, 144702 (2013)
Flechtner, K., Kretschmann, A., Steinrück, H.P., Gottfried, J.M.: NO-induced reversible switching of the electronic interaction between a porphyrin-coordinated cobalt ion and a silver surface. J. Am. Chem. Soc. 129, 12110–12111 (2007)
Hieringer, W., Flechtner, K., Kretschmann, A., Seufert, K., Auwärter, W., Barth, J.V., Görling, A., Steinrück, H.P., Gottfried, J.M.: The surface trans effect: influence of axial ligands on the surface chemical bonds of adsorbed metalloporphyrins. J. Am. Chem. Soc. 133, 6206–6222 (2011)
Wang, C., Fan, Q., Hu, S., Ju, H., Feng, X., Han, Y., Pan, H., Zhu, J., Gottfried, J.M.: Coordination reaction between tetraphenylporphyrin and nickel on a TiO2(110) surface. Chem. Commun. 50, 8291–8294 (2014)
Bai, Y., Buchner, F., Wendahl, M.T., Kellner, I., Bayer, A., Steinrück, H.-P., Marbach, H., Gottfried, J.M.: Direct metalation of a phthalocyanine monolayer on Ag(111) with coadsorbed iron atoms. J. Phys. Chem. C 112, 6087–6092 (2008)
Song, C.L., Wang, Y.L., Ning, Y.X., Jia, J.F., Chen, X., Sun, B., Zhang, P., Xue, Q.K., Ma, X.C.: Tailoring phthalocyanine metalation reaction by quantum size effect. J. Am. Chem. Soc. 132, 1456–1457 (2010)
Chen, M., Schmid, M., Schweyen, P., Bröring, M., Gottfried, J.M.: Oxidation state tuning in ligand-metal surface reaction: formation of Co(III)-Corrole on Ag(111). submitted
Papageorgiou, A.C., Fischer, S., Oh, S.C., Saglam, O., Reichert, J., Wiengarten, A., Seufert, K., Vijayaraghavan, S., Ecija, D., Auwärter, W., Allegretti, F., Acres, R.G., Prince, K.C., Diller, K., Klappenberger, F., Barth, J.V.: Self-terminating protocol for an interfacial complexation reaction in vacuo by metal-organic chemical vapor deposition. ACS Nano 7, 4520–4526 (2013)
Ecija, D., Auwärter, W., Vijayaraghavan, S., Seufert, K., Bischoff, F., Tashiro, K., Barth, J.V.: Assembly and manipulation of rotatable cerium porphyrinato sandwich complexes on a surface. Angew. Chem.-Int. Edit. 50, 3872–3877 (2011)
Weber-Bargioni, A., Reichert, J., Seitsonen, A.P., Auwärter, W., Schiffrin, A., Barth, J.V.: Interaction of cerium atoms with surface-anchored porphyrin molecules. J. Phys. Chem. C 112, 3453–3455 (2008)
Sperl, A., Kröger, J., Berndt, R.: Controlled metalation of a single adsorbed phthalocyanine. Angew. Chem.-Int. Edit. 50, 5294–5297 (2011)
Wang, Y.F., Kröger, J., Berndt, R., Hofer, W.A.: Pushing and pulling a sn ion through an adsorbed phthalocyanine molecule. J. Am. Chem. Soc. 131, 3639–3643 (2009)
Kretschmann, A., Walz, M.-M., Flechtner, K., Steinrück, H.-P., Gottfried, J.M.: Tetraphenylporphyrin picks up zinc atoms from a silver surface. Chem. Commun., 568–570 (2007)
Ditze, S., Röckert, M., Buchner, F., Zillner, E., Stark, M., Steinrück, H.P., Marbach, H.: Towards the engineering of molecular nanostructures: local anchoring and functionalization of porphyrins on model-templates. Nanotechnology 24, 115305 (2013)
Diller, K., Klappenberger, F., Allegretti, F., Papageorgiou, A.C., Fischer, S., Wiengarten, A., Joshi, S., Seufert, K., Ecija, D., Auwärter, W., Barth, J.V.: Investigating the molecule-substrate interaction of prototypic tetrapyrrole compounds: adsorption and self-metalation of porphine on Cu(111). J. Chem. Phys. 138, 154710 (2013)
Diller, K., Klappenberger, F., Marschall, M., Hermann, K., Nefedov, A., Wöll, C., Barth, J.V.: Self-metalation of 2H-tetraphenylporphyrin on Cu(111): an x-ray spectroscopy study. J. Chem. Phys. 136, 014705 (2012)
Nowakowski, J., Wäckerlin, C., Girovsky, J., Siewert, D., Jung, T.A., Ballav, N.: Porphyrin metalation providing an example of a redox reaction facilitated by a surface reconstruction. Chem. Commun. 49, 2347–2349 (2013)
Gonzalez-Moreno, R., Sanchez-Sanchez, C., Trelka, M., Otero, R., Cossaro, A., Verdini, A., Floreano, L., Ruiz-Bermejo, M., Garcia-Lekue, A., Martin-Gago, J.A., Rogero, C.: Following the metalation process of protoporphyrin IX with metal substrate atoms at room temperature. J. Phys. Chem. C 115, 6849–6854 (2011)
Xiao, J., Ditze, S., Chen, M., Buchner, F., Stark, M., Drost, M., Steinrück, H.-P., Gottfried, J.M., Marbach, H.: Temperature-dependent chemical and structural transformations from 2H-tetraphenylporphyrin to Copper(II)-tetraphenylporphyrin on Cu(111). J. Phys. Chem. C 116, 12275–12282 (2012)
Ditze, S., Stark, M., Drost, M., Buchner, F., Steinrück, H.-P., Marbach, H.: Activation energy for the self-metalation reaction of 2H-tetraphenylporphyrin on Cu(111). Angew. Chem.-Int. Edit. 51, 10898–10901 (2012)
Bürker, C., Franco-Cañellas, A., Broch, K., Lee, T.L., Gerlach, A., Schreiber, F.: Self-metalation of 2H-tetraphenylporphyrin on Cu(111) studied with XSW: influence of the central metal atom on the adsorption distance. J. Phys. Chem. C 118, 13659–13666 (2014)
Stark, M., Ditze, S., Lepper, M., Zhang, L., Schlott, H., Buchner, F., Röckert, M., Chen, M., Lytken, O., Steinrück, H.P., Marbach, H.: Massive conformational changes during thermally induced self-metalation of 2H-tetrakis-(3,5-di-tert-butyl)-phenylporphyrin on Cu(111). Chem. Commun. 50, 10225–10228 (2014)
Chen, M., Röckert, M., Xiao, J., Drescher, H.-J., Steinrück, H.-P., Lytken, O., Gottfried, J.M.: Coordination reactions and layer exchange processes at a buried metal-organic interface. J. Phys. Chem. C 118, 8501–8507 (2014)
Röckert, M., Ditze, S., Stark, M., Xiao, J., Steinrück, H.P., Marbach, H., Lytken, O.: Abrupt coverage-induced enhancement of the self-metalation of tetraphenylporphyrin with Cu(111). J. Phys. Chem. C 118, 1661–1667 (2014)
Röckert, M., Franke, M., Tariq, Q., Ditze, S., Stark, M., Uffinger, P., Wechsler, D., Singh, U., Xiao, J., Marbach, H., Steinrück, H.P., Lytken, O.: Coverage- and temperature-dependent metalation and dehydrogenation of tetraphenylporphyrin on Cu(111). Chem.-Eur. J. 20, 8948–8953 (2014)
Fleischer, E.B., Wang, J.H.: The detection of a type of reaction intermediate in the combination of metal ions with porphyrins. J. Am. Chem. Soc. 82, 3498–3502 (1960)
De Luca, G., Romeo, A., Scolaro, L.M., Ricciardi, G., Rosa, A.: Sitting-atop metallo-porphyrin complexes: experimental and theoretical investigations on such elusive species. Inorg. Chem. 48, 8493–8507 (2009)
Braun, J., Schlabach, M., Wehrle, B., Kocher, M., Vogel, E., Limbach, H.H.: NMR-study of the tautomerism of porphyrin including the kinetic HH/HD/DD isotope effect in the liquid and the solid state. J. Am. Chem. Soc. 116, 6593–6604 (1994)
Doyle, C.M., Krasnikov, S.A., Sergeeva, N.N., Preobrajenski, A.B., Vinogradov, N.A., Sergeeva, Y.N., Senge, M.O., Cafolla, A.A.: Evidence for the formation of an intermediate complex in the direct metalation of tetra(4-bromophenyl)-porphyrin on the Cu(111) surface. Chem. Commun. 47, 12134–12136 (2011)
Li, Y., Xiao, J., Shubina, T.E., Chen, M., Shi, Z., Schmid, M., Steinrück, H.-P., Gottfried, J.M., Lin, N.: Coordination and metalation bifunctionality of Cu with 5,10,15,20-tetra(4-pyridyl)porphyrin: toward a mixed-valence two-dimensional coordination network. J. Am. Chem. Soc. 134, 6401–6408 (2012)
Röckert, M., Franke, M., Tariq, Q., Lungerich, D., Jux, N., Stark, M., Kaftan, A., Ditze, S., Marbach, H., Laurin, M., Libuda, J., Steinrück, H.-P., Lytken, O.: Insights in reaction mechanistics: isotopic exchange during the metalation of deuterated tetraphenyl-21,23D-porphyrin on Cu(111). J. Phys. Chem. C 118, 26729–26736 (2014)
Aballe, L., Barinov, A., Locatelli, A., Heun, S., Kiskinova, M.: Tuning surface reactivity via electron quantum confinement. Phys. Rev. Lett. 93, 196103 (2004)
Chiang, T.C.: Photoemission studies of quantum well states in thin films. Surf. Sci. Rep. 39, 181–235 (2000)
Czoschke, P., Hong, H., Basile, L., Chiang, T.C.: Quantum size effects in the surface energy of Pb/Si(111) film nanostructures studied by surface x-ray diffraction and model calculations. Phys. Rev. B 72, 075402 (2005)
Czoschke, P., Hong, H.W., Basile, L., Chiang, T.C.: Quantum beating patterns observed in the energetics of Pb film nanostructures. Phys. Rev. Lett. 93, 036103 (2004)
Danese, A.G., Curti, F.G., Bartynski, R.A.: Quantum size effect induced modification of the chemisorption properties of thin metal films. Phys. Rev. B 70, 165420 (2004)
Ma, X., Jiang, P., Qi, Y., Jia, J., Yang, Y., Duan, W., Li, W.-X., Bao, X., Zhang, S.B., Xue, Q.-K.: Experimental observation of quantum oscillation of surface chemical reactivities. Proc. Natl. Acad. Sci. U.S.A. 104, 9204–9208 (2007)
Zhang, Y.F., Jia, J.F., Han, T.Z., Tang, Z., Shen, Q.T., Guo, Y., Qiu, Z.Q., Xue, Q.K.: Band structure and oscillatory electron–phonon coupling of Pb thin films determined by atomic-layer-resolved quantum-well states. Phys. Rev. Lett. 95, 096802 (2005)
Lin, T., Kuang, G., Wang, W., Lin, N.: Two-Dimensional lattice of out-of-plane dinuclear iron centers exhibiting kondo resonance. ACS Nano 8, 8310–8316 (2014)
Urgel, J.I., Ecija, D., Auwärter, W., Stassen, D., Bonifazi, D., Barth, J.V.: Orthogonal insertion of lanthanide and transition-metal atoms in metal-organic networks on surfaces. Angew. Chem.-Int. Edit. 54, 6163–6167 (2015)
In’t Veld, M., Iavicoli, P., Haq, S., Amabilino, D. B., Raval, R.: Unique intermolecular reaction of simple porphyrins at a metal surface gives covalent nanostructures. Chem. Commun., 1536–1538 (2008)
Haq, S., Hanke, F., Dyer, M.S., Persson, M., Iavicoli, P., Amabilino, D.B., Raval, R.: Clean coupling of unfunctionalized porphyrins at surfaces to give highly oriented organometallic oligomers. J. Am. Chem. Soc. 133, 12031–12039 (2011)
Hanke, F., Haq, S., Raval, R., Persson, M.: Heat-to-connect: surface commensurability directs organometallic one-dimensional self-assembly. ACS Nano 5, 9093–9103 (2011)
Abel, M., Clair, S., Ourdjini, O., Mossoyan, M., Porte, L.: Single layer of polymeric Fe-phthalocyanine: an organometallic sheet on metal and thin insulating film. J. Am. Chem. Soc. 133, 1203–1205 (2011)
Kezilebieke, S., Amokrane, A., Boero, M., Clair, S., Abel, M., Bucher, J.-P.: Steric and electronic selectivity in the synthesis of Fe-1,2,4,5-tetracyanobenzene (TCNB) complexes on Au(111): from topological confinement to bond formation. Nano Res. 7, 888–897 (2014)
Piantek, M., Serrate, D., Moro-Lagares, M., Algarabel, P., Pascual, J.I., Ibarra, M.R.: Manganese phthalocyanine derivatives synthesized by on-surface cyclotetramerization. J. Phys. Chem. C 118, 17895–17899 (2014)
Kezilebieke, S., Amokrane, A., Abel, M., Bucher, J.-P.: Hierarchy of chemical bonding in the synthesis of Fe-phthalocyanine on metal surfaces: a local spectroscopy approach. J. Phys. Chem. Lett. 5, 3175–3182 (2014)
Koudia, M., Abel, M.: Step-by-step on-surface synthesis: from manganese phthalocyanines to their polymeric form. Chem. Commun. 50, 8565–8567 (2014)
Di Santo, G., Castellarin-Cudia, C., Fanetti, M., Taleatu, B., Borghetti, P., Sangaletti, L., Floreano, L., Magnano, E., Bondino, F., Goldoni, A.: Conformational adaptation and electronic structure of 2H-tetraphenylporphyrin on Ag(111) during Fe metalation. J. Phys. Chem. C 115, 4155–4162 (2011)
Nardi, M., Verucchi, R., Tubino, R., Iannotta, S.: Activation and control of organolanthanide synthesis by supersonic molecular beams: erbium-porphyrin test case. Phys. Rev. B 79, 125404 (2009)
Doyle, C.M., Cunniffe, J.P., Krasnikov, S.A., Preobrajenski, A.B., Li, Z.S., Sergeeva, N.N., Senge, M.O., Cafolla, A.A.: Ni–Cu ion exchange observed for Ni(II)-porphyrins on Cu(111). Chem. Commun. 50, 3447–3449 (2014)
Hai, N.T.M., Furukawa, S., Vosch, T., De Feyter, S., Broekmann, P., Wandelt, K.: Electrochemical reactions at a porphyrin-copper interface. Phys. Chem. Chem. Phys. 11, 5422–5430 (2009)
Itoh, K., Sugii, T., Kim, M.: Surface-enhanced resonance Raman-scattering study on tetrakis(4-N-methylpyridinium)porphine adsorbed on mixed silver and copper colloids. J. Phys. Chem. 92, 1568–1571 (1988)
Prochazka, M., Hanzlikova, J., Stepanek, J., Baumruk, V.: Metalation of positively charged water soluble mesoporphyrins studied via time-resolved SERRS spectroscopy. J. Mol. Struct. 410, 77–79 (1997)
Hanzlikova, J., Prochazka, M., Stepanek, J., Bok, J., Baumruk, V., Anzenbacher, P.: Metalation of 5,10,15,20-tetrakis(1-methyl-4-pyridyl)porphyrin in silver colloids studied via time dependence of surface-enhanced resonance Raman spectra. J. Raman Spectr. 29, 575–584 (1998)
Tsukahara, S., Suzuki, N.: Characteristics of complexation reaction of tetraphenylporphine with zinc(II) at solid–liquid interface. Inorg. Chim. Acta 245, 105–108 (1996)
Seufert, K., Auwärter, W., Barth, J.V.: Discriminative response of surface-confined metalloporphyrin molecules to carbon and nitrogen monoxide. J. Am. Chem. Soc. 132, 18141–18146 (2010)
Seufert, K., Bocquet, M.L., Auwärter, W., Weber-Bargioni, A., Reichert, J., Lorente, N., Barth, J.V.: Cis-dicarbonyl binding at cobalt and iron porphyrins with saddle-shape conformation. Nat. Chem. 3, 114–119 (2011)
Isvoranu, C., Wang, B., Schulte, K., Ataman, E., Knudsen, J., Andersen, J.N., Bocquet, M.L., Schnadt, J.: Tuning the spin state of iron phthalocyanine by ligand adsorption. J. Phys.-Condens. Matter 22, 472001 (2010)
Isvoranu, C., Wang, B., Ataman, E., Knudsen, J., Schulte, K., Andersen, J.N., Bocquet, M.L., Schnadt, J.: Comparison of the carbonyl and nitrosyl complexes formed by adsorption of CO and NO on mono layers of iron phthalocyanine on Au(111). J. Phys. Chem. C 115, 24718–24727 (2011)
Strozecka, A., Soriano, M., Pascual, J.I., Palacios, J.J.: Reversible change of the spin state in a manganese phthalocyanine by coordination of CO molecule. Phys. Rev. Lett. 109, 147202 (2012)
Williams, F.J., Vaughan, O.P.H., Knox, K.J., Bampos, N., Lambert, R.M.: First observation of capping/uncapping by a ligand of a Zn porphyrin adsorbed on Ag(100). Chem. Commun., 1688–1689 (2004)
Liu, L.W., Yang, K., Xiao, W.D., Jiang, Y.H., Song, B.Q., Du, S.X., Gao, H.J.: Selective adsorption of metal-phthalocyanine on Au(111) surface with hydrogen atoms. Appl. Phys. Lett. 103, 023110 (2013)
Liu, L.W., Yang, K., Jiang, Y.H., Song, B.Q., Xiao, W.D., Li, L.F., Zhou, H.T., Wang, Y.L., Du, S.X., Ouyang, M., Hofer, W.A., Neto, A.H.C., Gao, H.J.: Reversible single spin control of individual magnetic molecule by hydrogen atom adsorption. Sci. Rep. 3, 1210 (2013)
Isvoranu, C., Wang, B., Ataman, E., Schulte, K., Knudsen, J., Andersen, J.N., Bocquet, M.L., Schnadt, J.: Ammonia adsorption on iron phthalocyanine on Au(111): influence on adsorbate-substrate coupling and molecular spin. J. Chem. Phys. 134, 114710 (2011)
Tran, N.L., Kummel, A.C.: A density functional theory study on the binding of NO onto FePc films. J. Chem. Phys. 127, 214701 (2007)
Wäckerlin, C., Tarafder, K., Siewert, D., Girovsky, J., Hahlen, T., Iacovita, C., Kleibert, A., Nolting, F., Jung, T.A., Oppeneer, P.M., Ballav, N.: On-surface coordination chemistry of planar molecular spin systems: novel magnetochemical effects induced by axial ligands. Chem. Sci. 3, 3154–3160 (2012)
Wäckerlin, C., Tarafder, K., Girovsky, J., Nowakowski, J., Hahlen, T., Shchyrba, A., Siewert, D., Kleibert, A., Nolting, F., Oppeneer, P.M., Jung, T.A., Ballav, N.: Ammonia coordination introducing a magnetic moment in an on-surface low-spin porphyrin. Angew. Chem.-Int. Edit. 52, 4568–4571 (2013)
Hermanns, C.F., Bernien, M., Kruger, A., Miguel, J., Kuch, W.: Switching the electronic properties of Co-octaethylporphyrin molecules on oxygen-covered Ni films by NO adsorption. J. Phys.-Condens. Matter 24, 394008 (2012)
Buchner, F., Seufert, K., Auwärter, W., Heim, D., Barth, J.V., Flechtner, K., Gottfried, J.M., Steinrück, H.P., Marbach, H.: NO-induced reorganization of porphyrin arrays. ACS Nano 3, 1789–1794 (2009)
Burema, S.R., Seufert, K., Auwärter, W., Barth, J.V., Bocquet, M.L.: Probing nitrosyl ligation of surface-confined metalloporphyrins by inelastic electron tunneling spectroscopy. ACS Nano 7, 5273–5281 (2013)
Kim, H., Chang, Y.H., Lee, S.H., Lim, S., Noh, S.K., Kim, Y.H., Kahng, S.J.: Visualizing tilted binding and precession of diatomic NO adsorbed to Co-porphyrin on Au(111) using scanning tunneling microscopy. Chem. Sci. 5, 2224–2229 (2014)
Wäckerlin, C., Chylarecka, D., Kleibert, A., Müller, K., Iacovita, C., Nolting, F., Jung, T.A., Ballav, N.: Controlling spins in adsorbed molecules by a chemical switch. Nat. Commun. 1, 61 (2010)
Wäckerlin, C., Siewert, D., Jung, T.A., Ballav, N.: On-surface coordination chemistry: direct imaging of the conformational freedom of an axial ligand at room temperature. Phys. Chem. Chem. Phys. 15, 16510–16514 (2013)
Kim, H., Chang, Y.H., Lee, S.H., Kim, Y.H., Kahng, S.J.: Switching and sensing spin states of Co-porphyrin in bimolecular reactions on Au(111) using scanning tunneling microscopy. ACS Nano 7, 9312–9317 (2013)
Domke, K.F., Pettinger, B.: In situ discrimination between axially complexed and ligand-free Co porphyrin on Au(111) with tip-enhanced Raman spectroscopy. ChemPhysChem 10, 1794–1798 (2009)
Miguel, J., Hermanns, C.F., Bernien, M., Kruger, A., Kuch, W.: Reversible manipulation of the magnetic coupling of single molecular spins in Fe-porphyrins to a ferromagnetic substrate. J. Phys. Chem. Lett. 2, 1455–1459 (2011)
Bishop, S.R., Tran, N.L., Poon, G.C., Kummel, A.C.: Dynamics of analyte binding onto a metallophthalocyanine: NO/FePc. J. Chem. Phys. 127, 214702 (2007)
Zhang, J.L., Wang, Z., Zhong, J.Q., Yuan, K.D., Shen, Q., Xu, L.L., Niu, T.C., Gu, C.D., Wright, C.A., Tadich, A., Qi, D., Li, H.X., Wu, K., Xu, G.Q., Li, Z., Chen, W.: Single-molecule imaging of activated nitrogen adsorption on individual manganese phthalocyanine. Nano Lett. 15, 3181–3188 (2015)
Ballav, N., Wäckerlin, C., Siewert, D., Oppeneer, P.M., Jung, T.A.: Emergence of on-surface magnetochemistry. J. Phys. Chem. Lett. 4, 2303–2311 (2013)
Ullmann, F., Meyer, G.M., Loewenthal, O., Gilli, E.: Ueber symmetrische biphenylderivate. Justus Liebigs Ann. Chem. 332, 38–81 (1904)
Fanta, P.E.: Ullmann synthesis of biaryls 1945–1963. Chem. Rev. 64, 613–632 (1964)
Fanta, P.E.: Ullmann synthesis of biaryls. Synthesis, 9–21 (1974)
Xi, M., Bent, B.E.: Mechanisms of the Ullmann coupling reaction in adsorbed monolayers. J. Am. Chem. Soc. 115, 7426–7433 (1993)
Blake, M.M., Nanayakkara, S.U., Claridge, S.A., Fernandez-Torres, L.C., Sykes, E.C.H., Weiss, P.S.: Identifying reactive intermediates in the Ullmann coupling reaction by scanning tunneling microscopy and spectroscopy. J. Phys. Chem. A 113, 13167–13172 (2009)
Lewis, E.A., Murphy, C.J., Liriano, M.L., Sykes, E.C.H.: Atomic-scale insight into the formation, mobility and reaction of Ullmann coupling intermediates. Chem. Commun. 50, 1006–1008 (2014)
Wang, W.H., Shi, X.Q., Wang, S.Y., Van Hove, M.A., Lin, N.: Single-molecule resolution of an organometallic intermediate in a surface-supported Ullmann coupling reaction. J. Am. Chem. Soc. 133, 13264–13267 (2011)
Chen, M., Xiao, J., Steinrück, H.-P., Wang, S., Wang, W., Lin, N., Hieringer, W., Gottfried, J.M.: Combined photoemission and scanning tunneling microscopy study of the surface-assisted Ullmann coupling reaction. J. Phys. Chem. C 118, 6820–6830 (2014)
Di Giovannantonio, M., El Garah, M., Lipton-Duffin, J., Meunier, V., Cardenas, L., Fagot Revurat, Y., Cossaro, A., Verdini, A., Perepichka, D.F., Rosei, F., Contini, G.: Insight into organometallic intermediate and its evolution to covalent bonding in surface-confined Ullmann polymerization. ACS Nano 7, 8190–8198 (2013)
Nguyen, M.T., Pignedoli, C.A., Passerone, D.: An ab initio insight into the Cu(111)-mediated Ullmann reaction. Phys. Chem. Chem. Phys. 13, 154–160 (2011)
Björk, J., Hanke, F., Stafström, S.: Mechanisms of Halogen-based covalent self-assembly on metal surfaces. J. Am. Chem. Soc. 135, 5768–5775 (2013)
Fan, Q., Wang, C., Han, Y., Zhu, J., Kuttner, J., Hilt, G., Gottfried, J.M.: Surface-assisted formation, assembly, and dynamics of planar organometallic macrocycles and zigzag shaped polymer chains with C–Cu–C bonds. ACS Nano 8, 709–718 (2014)
Fan, Q., Wang, C., Liu, L., Han, Y., Zhao, J., Zhu, J., Kuttner, J., Hilt, G., Gottfried, J.M.: Covalent, organometallic, and halogen-bonded nanomeshes from tetrabromo-terphenyl by surface-assisted synthesis on Cu(111). J. Phys. Chem. C 118, 13018–13025 (2014)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this paper
Cite this paper
Michael Gottfried, J. (2016). Molecular On-Surface Synthesis: Metal Complexes, Organic Molecules, and Organometallic Compounds. In: Gourdon, A. (eds) On-Surface Synthesis. Advances in Atom and Single Molecule Machines. Springer, Cham. https://doi.org/10.1007/978-3-319-26600-8_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-26600-8_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-26598-8
Online ISBN: 978-3-319-26600-8
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)