
Abstract
The synthesis of complex molecular materials directly on surfaces under ultra-high vacuum is a new chemical approach attracting increasing interest. Inspired in the well-established conventional organic chemistry, an increasing number of reactions are being tested and demonstrated to work also under vacuum. Among those reactions, we find enediyne cyclizations, which open up promising new routes to create π-conjugated molecular materials that can be tailored through the choice of appropriate precursors and substrates. This chapter reviews the pioneering experiments of enediyne-based chemistry on atomically clean surfaces under ultra-high vacuum, put in relation to available knowledge from the conventional wet-chemistry analogues.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- Scanning Tunneling Microscopy
- Monomer Subunit
- Cyclized Monomer
- Bond Length Alternation
- Bond Length Variation
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
The tunable properties of molecular materials place them among the favorites for a variety of future generation devices. Beyond the readily commercialized organic electronic devices, the range of potential applications foreseen for supramolecular materials is dramatically increasing and includes functionalities and device structures with great degrees of sophistication. For example, self-assembly of well-defined molecular structures is considered among the most promising strategies to maintain the current trend of device miniaturization, for which a significant modification of the present top-down production methods will soon be required. A reliable synthesis of functional supramolecular structures from the bottom-up could pave the way toward industrial production of single-/few-molecule devices with dramatically reduced length scales. And the potential of precisely controlled molecular structures reaches even further. It would, for example, allow control over new and fascinating properties emerging in nanoscale-structured systems, which could in turn be used for optimized macroscopic devices such as sensors, filters, solar cells, or catalysts.
Along these lines, the main challenge remains to synthesize appropriate materials optimized for each particular functionality, which depending on the latter might require structures of substantial complexity. This in turn necessitates on the one hand a sufficient understanding of the structure–property relations to allow the rational design of molecular structures with the desired properties, and on the other hand the capability to build those previously designed structures. Acquisition of such understanding and control of matter at the molecular scale has been the goal of countless investigations over the last decades, which have indeed led to significant advances. Molecular self-assembly has, for example, been successfully used to create many kinds of supramolecular structures [1, 2]. Even restricting ourselves to materials on solid surfaces under ultra-high vacuum (UHV), an enormous variety of two-dimensional, one-dimensional, or zero-dimensional molecular structures with diverse compositions, symmetries, electronic, optic, or magnetic properties have been synthesized [1, 2]. However, self-assembly typically relies on weak, non-covalent intermolecular interactions such as hydrogen bonds or metal coordination. This carries some disadvantages, as is the reduced assembly’s resistance and consequently the compromised durability of a potential device, or also a strong localization of the electronic states, which limits the charge carrier mobility and thereby the structure’s efficiencies in many electronic or optical processes.
Only recently, formation of covalently bonded supramolecular structures directly on surfaces was demonstrated [3, 4]. And although the first pioneering examples still relied on the manual guidance of the reactions by means of a scanning tunneling microscope (STM), inspiring works demonstrating the self-assembled growth of covalently bonded molecular networks, tailored by appropriate design of the precursors, began appearing in 2007 [5, 6]. The consequences of this approach are far reaching. Not only is the stability of the molecular structures greatly increased and the charge transfer integral along the network enhanced, but it also allows the synthesis of complex molecular structures directly on surfaces of technological interest. Most applications require solid supports, and the transfer of complex molecular building blocks synthesized ex situ by conventional chemistry is often hampered by solubility problems or by thermal fragmentation if deposited by sublimation. Therefore, in addition to the intrinsic advantages of covalently linked structures, further compelling aspects of this so-called on-surface chemistry are that tedious transfer procedures [7] might be avoided, as well as that it can allow the occurrence of reactions and the synthesis of materials not achievable by other means [8, 9].
Among the multiple differences between conventional organic chemistry and on-surface chemistry under UHV is the absence of a traditionally crucial and tunable parameter: the solvent. However, here it is the surface which, in a somewhat related role, becomes a key player. In addition to the 2D confinement that it imposes on the reactants (or even 1D if appropriately structured surfaces are used [9–11]), it might act itself as a catalyzer [11, 12]. Understanding the role of the surfaces to rationally use it according to our needs thus becomes one of the important challenges lying ahead toward the full exploitation of on-surface chemistry’s potential.
2 Enediyne Cyclization Chemistry
In spite of the associated research having boomed in the last years, it is believed that only an insignificant part of the potential of UHV on-surface chemistry has been unveiled to date. Most efforts have been aimed at the demonstration of particular chemical reactions inspired in conventional wet chemistry [13, 14]. The positive verification of some of those reactions on surfaces even under UHV has been extremely stimulating, since it places a vast wealth of knowledge accumulated over decades in conventional organic chemistry as an enormous test bed for UHV on-surface chemistry. Among the successfully run reactions, we find the cyclization of enediynes [15].
After Bergman’s seminal report on the cyclization of enediynes upon thermal activation [16], the interest in this reaction sparked when enediynes were found to be the bioactive site of naturally occurring antibiotics [17]. The diradicals formed upon enediyne cyclization (Fig. 1) can abstract hydrogen from the DNA backbone, causing DNA cleavage and cell death. But in addition to its relevance in biochemistry, it was John and Tour who noticed the potential of the Bergman cyclization reaction for the material science community and demonstrated its use for the synthesis of conjugated aromatic polymers (such as polyphenylenes or polynaphthalene derivatives) by radical step-growth polymerization (Fig. 1) [18, 19]. However, it was soon found that in addition to the radical step growth, initially generated radicals can also attack the alkynes of still unreacted enediynes, greatly increasing the possible reaction paths. This translates into associated products including highly irregular structures with 6- and also 5-membered rings, branched chains, and unreacted alkynes (Fig. 1) [20].

Schematic representation of various possible cyclization and polymerization routes of an enediyne
Besides, in addition to the Bergman or C1–C6 cyclization, different C1–C5 and even C1–C4 cyclizations can equally occur (Fig. 1). While the two latter are generally thermodynamically unfavored [21], several factors have been shown to sway the energetic balance toward C1–C5 cyclizations [22]. This is, for example, the case if bulky alkyne terminal groups are present, causing steric congestion upon cyclization [21–23]. Electronic effects have a similarly significant influence on the cyclization, in which two mutually perpendicular arrays of π-orbitals play along with different roles. In-plane π-orbitals are sacrificed to form a σ-bond and two radicals, while out-of-plane π-orbitals evolve smoothly toward the cyclic aromatic π-system product [22, 24, 25]. The relative energies of in-plane and out-of-plane molecular orbitals cross along the cyclization. Thus, while the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO, respectively) of the reactant typically involves out-of-plane π-orbitals, in the cyclization product the frontier orbitals are normally localized in in-plane (radical) orbitals. This crossing has little importance in the thermal reactivity of neutral unexcited enediynes, but becomes relevant if the LUMO (HOMO) gets populated (depopulated) by reduction (oxidation) or photochemical excitation, often favoring C1–C5 cyclizations [22, 25, 26]. In particular, as a result of this crossing and additional aromaticity changes upon enediyne reduction, calculations show that the radical–anionic cyclization mechanism differs substantially from the thermal counterpart. Energetically, radical–anionic cyclization of reduced enediynes lowers the transition state and increases the exothermicity of both C1–C6 and C1–C5 cyclizations, but shows a significantly stronger influence on the latter [22, 25]. Also, the presence of certain catalysts has been reported to favor the C1–C5 pathway for thermal cyclizations [27]. Besides, different reactions such as alkyne homocoupling might also occur and contribute to possible dimerization or polymerization processes of yet uncyclized enediynes [12, 28, 29].
Altogether, the use of enediyne cyclization chemistry for synthetic applications is challenging due to the great number of possible reaction pathways: multiple cyclization options, each allowing both for subsequent radical step growth or radical attack of still unreacted enediynes (Fig. 1), alkyne coupling, and combinations thereof. Nevertheless, in spite of all the complexity, enediyne cyclization has become an important platform for conventional wet-chemistry-based synthesis of functional polymers [30]. Furthermore, a similarly interesting application of enediyne cyclizations is for the transformation of readily synthesized polymers. The cyclization chain reaction of poly-alkynes arranged in what can be seen as overlapping enediyne units has, for example, been proposed as a potential synthesis route to obtain graphene nanoribbons with atomically controlled structures (Fig. 2a) [31, 32].

a Example of poly-alkyne on which an initial enediyne cyclization triggers a subsequent radical cascade that may afford graphene nanoribbons upon additional cyclodehydrogenation. b C1–C5 and C1–C6 radical cyclizations, and some of the factors influencing their competing balance. c Gold(I)-catalyzed cyclization cascade by hydroamination and hydroarylations reported in Ref. [34]
All the above is referred to enediyne chemistry in solution, but is expected to apply at least to some extent also on surfaces under ultra-high vacuum. Under these new conditions, the substrate may additionally influence the chemical processes by acting as a catalyst, as a template, causing molecule-substrate charge transfer, etc. In this frame, it is interesting to remark that enediyne cyclization in principle does not require catalyzing agents. Thus, while the experiments under UHV reported to date have been performed on metal surfaces, this reaction holds promise to work also on insulators, as would be ultimately required for many technological applications. In the following, experiments performed on surfaces under UHV will be described and put in relation to our available knowledge from wet chemistry for each of these two approaches: chain reaction in poly-alkynes and radical step-growth polymerization of cyclized enediynes.
3 Chain Reactions
In molecular structures as shown in Fig. 2a, featuring coupled alkynes that form “overlapping enediynes,” an initial cyclization generates diradicals that subsequently attack the neighboring alkynes and thereby trigger a chain reaction [32]. These radical cyclization cascades differ from the analogous thermal single enediyne cyclization reactions, but the product structures still result from the balance of competing C1–C5 and C1–C6 pathways [33]. Among the factors affecting that balance we find the benzannelation of the conjugated radical reactants, steric congestion from bulky alkyne terminal groups, or strain (Fig. 2b) [33]. While the two former tend to favor C1–C5 cyclizations, the latter reverses the selectivity and favors C1–C6 cyclizations. Noteworthy, strain is particularly relevant in cascade radical cyclizations where rigid polycyclic frameworks are created.
A phenomenologically different example of chain reaction is displayed in Fig. 2c, by means of which polycyclic conjugated frameworks have been created in solution [34]. In that case, in the presence of a gold(I) catalyst, an initial hydroamination forming a 5-membered ring triggers subsequent hydroarylations with the available alkynes that form exclusively 6-membered rings, as would be desirable, for example, for graphene nanoribbon formation from longer polymeric chains. However, experiments on surfaces under UHV following this approach have not yet been reported, in contrast to radical cyclization cascades.
In particular, the cyclization cascade of 1,2-bis((2-ethynylphenyl)ethynyl)benzene on Ag(100) surfaces under UHV has been studied at the single molecule level with a powerful combination of scanning tunneling microscopy (STM) and tuning fork-based non-contact atomic force microscopy (nc-AFM), by means of which the local density of states (LDOS) and internal bond structure of the molecules can be imaged, respectively [35, 36]. The product structures resulting after annealing the precursor-decorated Ag(100) surface to temperatures above 90 °C are multiple, although close to 80 % is made up by the two majority structures displayed in Fig. 3a.

a Frequency shift nc-AFM microscopy images at constant height (together with the corresponding wireframe chemical structures) of reactant 1 before annealing and of the majority products 2 and 3 generated in yields of 51 and 28 %, respectively, after annealing the sample at T > 90 °C. b Reaction pathway and the associated energies as calculated by DFT for the transformation of 1 into 2. c Same for the transformation of 1 into 3. Adapted from Ref. [36]. Reprinted with permission from AAAS
The reactivity of the precursor can be rationalized considering it as three independent but overlapping benzannelated enediyne systems with terminal alkyne substitutions of two phenyl rings for the central enediyne, or one phenyl ring and one hydrogen atom in the terminal segments (Fig. 3). This treatment suggests three potential cyclizations along the reaction pathway. However, additional isomerization processes such as [1,2]-radical shifts and bond rotations are required to explain the reactant transformation into the majority products. Combinations of those processes leading to the products in a minimal number of steps were explored and analyzed using density functional theory (DFT) calculations displayed in Fig. 3b, c [36]. The calculations indicate that due to the exothermic character of the initial cyclizations, all barriers associated with subsequent isomerization steps remain at lower energies and are thus not rate limiting. That is, the effective transition state is found along the initial cyclization. This is in line with the experimental observations: The reactant molecules on Ag(100) are stable up to temperatures of ~90 °C, and above that threshold temperature, no intermediates on the reaction pathway to the final products is observed. Consequently, the order of the various processes following the rate limiting initial cyclizations cannot be strictly determined experimentally. However, the sequence does not change the overall reaction kinetics and thermodynamics discussed above.
Interestingly, both reaction pathways involve C1–C5 cyclizations. This can be explained by the multiple factors applying to this system that, as described above, tend to favor C1–C5 versus C1–C6 cyclizations: (i) bulky phenyl substituents at the alkyne ends, (ii) a catalytic metallic substrate, (iii) a molecular reduction (DFT calculations predict a charge transfer of 0.5 electrons from substrate to reactant), and (iv) the radical cyclization (as opposed to the initial thermal cyclization) of some of the enediyne units. And also of interest is to see that in both reaction pathways, the isomerization processes include an intramolecular recombination of the diradicals initially generated by the enediyne cyclizations. The result is an additional cyclization involving in both cases a great energetic gain (Fig. 3b, c), making it thermodynamically very favorable. Its occurrence further explains the quenching of a subsequent polymerization by radical step growth, and the consequently scarce amount of dimers or oligomers among the products.
4 Radical Step-Growth Polymerization
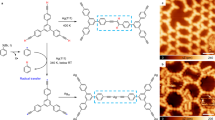
The use of enediyne cyclization to form conjugated polymers by radical step growth, as initially proposed by John and Tour [18, 19], has been recently applied with 1,6-di-2-naphthylhex-3-ene-1,5-diyne molecules on Cu(110) surfaces under UHV [15]. As opposed to the scenario described above with more complex molecules used in chain reactions, these simpler precursors lack easily accessible isomerization steps that could end up in a thermodynamically favorable cyclization by intramolecular radical recombination. Instead, the radicals remain available for step-growth polymerization, as schematically displayed in Fig. 4a. Indeed, STM measurements reveal the pristine reactants as single molecules after deposition on a cold substrate (Fig. 4b), which are then transformed into regular and uniaxially aligned molecular chains upon annealing the sample to 400 K (Fig. 4c). This temperature is thus sufficient to overcome the initial Bergman cyclization barrier, generating diradical species, and provide them enough mobility to diffuse on the surface and meet each other, setting in a radical step-growth polymerization. The discrete azimuthal chain alignment along the Cu[1-10] direction (just as the discrete azimuthal orientation of the precursors before cyclization and their preferential diffusion directions along the Cu[1-10] direction) remarks the relevant role of the substrate in the growth process. DFT-based STM simulations of the expected polymer structure show a good agreement with high-resolution experimental images (Fig. 4d) and thus support the proposed growth scenario (Fig. 4a) and product structure (Fig. 4e) [15].

a Wireframe molecular structure of the reactant and the associated reaction mechanism. b STM image of the reactant deposited on Cu(110) held at 170 K. c STM image showing the formation of one-dimensional molecular chain structures along the [1-10] direction after annealing the substrate to 400 K. d High-resolution close-up image of the molecular chain and DFT-based STM simulation (right part) of the proposed product structure. e Perspective view of the structural model showing the alternatingly tilted naphthyl groups. Adapted with permission from Ref. [15]. Copyright (2013) American Chemical Society
Another example of enediyne cyclization-based radical step-growth polymerization under UHV is that of 1,2-bis(2-ethynylphenyl)ethyne (Fig. 5a). Low-temperature (T = 4 K) STM and nc-AFM characterizations provide detailed insight into the involved chemistry and electronic properties of reactant and products [37]. nc-AFM images of the reactants deposited on Au(111) surfaces held at room temperature (Fig. 5b) display two different isomer conformations: a C 2h symmetric trans-conformation and a C 2ν symmetric cis-conformation. Annealing of the submonolayer reactant-decorated Au(111) surface at 160 °C brings about two intramolecular cyclizations per monomer. Most common is the occurrence of two C1–C5 cyclizations to yield the highly reactive 3,3′-diradical structure displayed in Fig. 5c. In turn, recombination of these resultant diradicals in a step-growth polymerization process leads to covalently linked oligo-(E)-1,1′-bi-(indenylidene) chains (Fig. 5c). More than 70 % of the material on the surface ends up forming covalently linked molecular assemblies with lengths n ≥ 3 (where n denotes the number of monomer subunits), most chains containing 5–10 monomer units and the longest chains sometimes exceeding 20 units.

a Wireframe chemical structure of the pristine reactant, intermediate product after cyclization, and final product after radical step-growth polymerization. The correspondent nc-AFM images on a Au(111) surface are shown in b (reactant), c (cyclized monomer), and d (polymer). Dark (light) arrows mark the long (short) bonds between indenyl units in the polymer (color coded accordingly in the wireframe structure above). e Polymer including non-cyclized reactants (highlighted with yellow arrows) and differently cyclized units (next to non-cyclized reactants). f Image of branched polymer chains. Adapted from Ref. [37] with permission under CC BY
However, a defect-free oligo-(E)-1,1′-bi-(indenylidene) structure as shown in Fig. 5a, d is typically retained only over segments of three to five monomers, since different monomer cyclizations also occur (e.g., C1–C6) whose product structures are equally included into the polymer chains. Examples thereof are shown in Fig. 5e. In fact, even non-cyclized monomer units are found within the molecular chains (marked with arrows in Fig. 5e). This is attributed to an attack of the radicals in cyclized species to the terminal alkyne carbon in non-cyclized precursors. While an analogue situation with the precursors described in Fig. 4 is presumably prevented by steric hindrance, this process is already known from solution-based enediyne polymerization reactions [20]. And as displayed in Fig. 1, it may cause irregular structures with different cyclization patterns, unreacted alkynes, and branched chains. An example of the latter is provided in Fig. 5f. Furthermore, cyclization of a monomer unit readily linked to a molecular chain implies a more complex “reactant” structure. In analogy to the chain reactions of 1,2-bis((2-ethynylphenyl)ethynyl)benzene on Ag(100) described in the previous section, following the exothermic cyclizations it could undergo additional isomerization reactions ending up in an intramolecular radical recombination that quenches the polymer step growth. This may indeed be an explanation to the common occurrence of irregular monomer subunits at the chain ends (Fig. 5e, f) reported in Ref. [37].
The main polymerization product oligo-(E)-1,1′-bi-(indenylidene) is an oligo-acetylene derivative with a π-conjugated carbon backbone (Fig. 5a). Bond length alternation, defined as the difference in length between long (C–C) and short (C=C) carbon–carbon bonds in a conjugated molecule, is considered as one of the phenomenological measures of aromaticity [38]. Smaller bond length alternation implies enhanced aromaticity and π-electron delocalization, as well as a smaller bandgap in extended conjugated systems. In spite of the small dimensions of such bond length variations, tuning fork-based nc-AFM measurements with carbon monoxide functionalized probes artificially enhance that difference in the images and thereby allow its visualization [39]. This is observed best in the bonds between the five-membered rings of adjacent indenyl groups (Fig. 5d), for which the bond length variation measured experimentally reaches ~50 %, while density functional theory predictions amount only to about 3 % [37]. Shorter bonds are highlighted with light gray arrows and longer ones with dark arrows (Fig. 5d), corresponding to double and single bonds, respectively (as displayed in the accordingly marked molecular structure drawing of Fig. 5a). Also, the bonds within the five-membered rings exhibit bond length modulation. In particular, the double bonds in indenyl end groups show a distinctive deviation compared to indenyl groups along the extended oligomer chain. This different geometry resembles that in cyclized monomers (Fig. 5c) and might thus be related to the interaction of a radical with the surface [40].
The bandgap of organic semiconductors is known to scale inversely proportional to the number of π-conjugated electrons both in linear [38, 41] and 2D structures [42]. Combining on-surface synthesis, which provides varied product structures, with scanning tunneling spectroscopy to probe the electronic properties of each structure independently, the same effect could be concluded from the reduced energy of the LUMO of polyphenylene oligomers with increasing length [43]. An equivalent phenomenology is observed on oligo-(E)-1,1′-bi-(indenylidene) chains, whose LUMO energy with respect to the Fermi level drops from ~1.2 to ~0.13 eV by going from a cyclized monomer (Fig. 5c) to an oligomer containing four monomer subunits [37]. Mapping of the local density of states at the LUMO energy further reveals its delocalization along the π-conjugated oligomer backbone and agrees with the distribution calculated by density functional theory. Altogether, it provides a fully coherent picture of the experimental findings on enediyne cyclization-based oligomers synthesized on Au(111) surfaces under UHV by radical step-growth polymerization. Moreover, it highlights the potential of this approach for the synthesis of fully conjugated low-bandgap derivatives of all-trans polyacetylene [37].
5 Summary and Conclusions
The use of enediyne cyclizations for on-surface chemistry under UHV has been demonstrated following two different approaches. On the one hand, cyclization-induced diradical species have been linked in a step-growth polymerization process to afford conjugated molecular chains. On the other hand, previously available alkyne chains in what can be seen as overlapping enediyne systems have been transformed via cyclization cascades into polycyclic aromatic hydrocarbons. Experiments following both approaches evidence, in line with the conventional wet-chemistry analogues, a significant complexity in the involved chemistry: multiple reaction pathways competing in a subtle balance that is in turn affected by many different parameters. Nevertheless, with a sufficient understanding of their respective effects, a rational choice of molecular precursors and substrate surfaces might convert enediyne-based chemistry into a versatile UHV synthetic route.
References
Barth, J.V., Costantini, G., Kern, K.: Engineering atomic and molecular nanostructures at surfaces. Nature 437, 671–679 (2005)
Elemans, J.A.A.W., Lei, S., de Feyter, S.: Molecular and supramolecular networks on surfaces: from two-dimensional crystal engineering to reactivity. Angew. Chem. Int. Ed. 48, 7298–7332 (2009)
Hla, S.W., Bartels, L., Meyer, G., Rieder, K.-H.: Inducing all steps of a chemical reaction with the scanning tunneling microscope tip: towards single molecule engineering. Phys. Rev. Lett. 85, 2777–2780 (2000)
Okawa, Y., Aono, M.: Nanoscale control of chain polymerization. Nature 409, 683–684 (2001)
Grill, L., Dyer, M., Lafferenz, L., Persson, M., Peters, M.V., Hecht, S.: Nano-architectures by covalent assembly of molecular building blocks. Nat. Nanotech. 2, 687–691 (2007)
Weigelt, S., Busse, C., Bombis, C., Krudsen, M.M., Gothelf, K.V., Strunkus, T., Woll, C., Dahlbom, M., Kammer, B., Laegsgaard, E., Besenbacher, F., Linderoth, T.R.: Covalent interlinking of an aldehyde and an amine on a Au(111) surface in ultrahigh vacuum. Angew. Chem. Int. Ed. 46, 9227 (2007)
Bennett, P., Pedramrazi, Z., Madani, A., Chen, Y.-C., de Oteyza, D. G., Chen, C., Fischer, F., Crommie, M., Bokor, J.: Bottom-Up Graphene Nanoribbon Field-Effect Transistors. Appl. Phys. Lett. 103, 253114–1-4 (2013)
Cai, J., Ruffieux, P., Jaafar, R., Bieri, M., Braun, T., Blankenburg, S., Muoth, M., Seitsonen, A.P., Saleh, M., Feng, X., Mullen, K., Fasel, R.: Atomically precise bottom-up fabrication of graphene nanoribbons. Nature 466, 470–473 (2010)
Zhong, D., Franke, J.-H., Podiyanachari, S.K., Blomker, T., Zhang, H., Kehr, G., Erker, G., Fuchs, H., Chi, L.: Linear alkane polymerization on a gold surface. Science 334, 213–216 (2011)
Ruffieux, P., Cai, J., Plumb, N.C., Patthey, L., Prezzi, D., Ferreti, A., Molinari, E., Feng, X., Mullen, K., Pignedoli, C.A., Fasel, R.: Electronic structure of atomically precise graphene nanoribbons. ACS Nano 6, 6930–6935 (2012)
Saywell, A., Schwarz, J., Hecht, S., Grill, L.: Polymerization on stepped surfaces: alignment of polymers and identification of catalytic sites. Angew. Chem. Int. Ed. 51, 5096–5100 (2012)
Gao, H.-Y., Wagner, H., Zhong, D., Franke, J.-H., Studer, A., Fuchs, H.: Glaser coupling at metal surfaces. Angew. Chem. Int. Ed. 52, 4024–4028 (2013)
Mendez, J., Lopez, M.F., Martin-Gago, J.A.: On-surface synthesis of cyclic organic molecules. Chem. Soc. Rev. 40, 4578–4590 (2011)
Bjork, J., Hanke, F.: Towards design rules for covalent nanostructures on metal surfaces. Chem. Eur. J. 20, 928–934 (2014)
Sun, Q., Zhang, C., Li, Z., Kong, H., Tan, Q., Hu, A., Xu, W.: On-surface formation of one-dimensional polyphenylene through bergman cyclization. J. Am. Chem. Soc. 135, 8448–8451 (2013)
Jones, R.R., Bergman, R.G.: p-Benzyne. generation as an intermediate in a thermal isomerization reaction and trapping evidence for the 1,4-Benzenediyl Structure. J. Am. Chem. Soc. 94, 660–661 (1972)
Nicolaou, K.C., Dai, W.-M.: Chemistry and biology of the enediyne anticancer antibiotics. Angew. Chem. Int. Ed. 30, 1387–1416 (1991)
John, J.A., Tour, J.M.: Synthesis of polyphenylenes and polynaphthalenes by thermolysis of enediynes and dialkynylbenzenes. J. Am. Chem. Soc. 116, 5011–5012 (1994)
John, J.A., Tour, J.M.: Synthesis of polyphenylene derivatives by thermolysis of enediynes and dialkynylaromatic monomers. Tetrahedron 53, 15515–15534 (1997)
Johnson, J.P., Bringley, D.A., Wilson, E.E., Lewis, K.D., Beck, L.W., Matzger, A.J.: Comparison of “polynaphthalenes” prepared by two mechanistically distinct routes. J. Am. Chem. Soc. 125, 14708 (2003)
Prall, M., Wittkopp, A., Schreiner, P.R.: Can fulvenes form from enediynes? A systematic high-level computational study on parent and benzannelated enediyne and enyne-allene cyclizations. J. Phys. Chem. A 105, 9265–9274 (2001)
Mohamed, R.K., Peterson, P.W., Alabugin, I.V.: Concerted reactions that produce diradicals and zwitterions: electronic, steric, conformational, and kinetic control of cycloaromatization processes. Chem. Rev. 113, 7089–7129 (2013)
Vavilala, C., Byrne, N., Kraml, C.M., Ho, D.M., Pascal Jr, R.A.: Thermal C1–C5 diradical cyclization of enediynes. J. Am. Chem. Soc. 130, 13549–13551 (2008)
Galbraith, J.M., Schreiner, P.R., Harris, N., Wei, W., Wittkopp, A., Shaik, S.: A valence bond study of the bergman cyclization: geometric features, resonance energy, and Nucleus-Independent Chemical Shift (NICS) values. Chem. Eur. J. 6, 1446–1454 (2000)
Alabugin, I.V., Manoharan, M.: Radical-anionic cyclizations of enediynes: remarkable effects of benzannelation and remote substituents on cyclorearomatization reactions. J. Am. Chem. Soc. 125, 4495–4509 (2003)
Alabugin, I.V., Kovalenko, S.V.: C1–C5 photochemical cyclization of enediynes. J. Am. Chem. Soc. 124, 9052–9053 (2002)
Lee, C.-Y., Wu, M.-J.: Synthesis of benzofulvenes by palladium-catalyzed cyclization of 1,2-dialkynylbenzenes. Eur. J. Org. Chem. 2007, 3463–3467 (2007)
Batsanov, A.S., Collings, J.C., Fairlamb, I.J.S., Holland, J.P., Howard, J.A.K., Lin, Z., Marder, T.B., Parsons, A.C., Ward, R.M., Zhu, J.: Requirement for an oxidant in Pd/Cu co-catalyzed terminal alkyne homocoupling to give symmetrical 1,4-disubstituted 1,3-diynes. J. Org. Chem. 70, 703–706 (2005)
Zhang, Y.-Q., Kepcija, N., Kleinschrodt, M., Diller, K., Fischer, S., Papageorgiou, A.C., Allegretti, F., Bjork, J., Klyatskaya, S., Klappenberger, F., Ruben, M., Barth, J.V.: Homo-coupling of terminal alkynes on a noble metal surface. Nat. Commun. 3, 1286 (2012)
Xiao, Y., Hu, A.: Bergman cyclization in polymer chemistry and material science. Macromol. Rap. Comm. 32, 1688–1698 (2011)
Byers, P.M., Alabugin, I.V.: Polyaromatic ribbons from oligo-alkynes via selective radical cascade: stitching aromatic rings with polyacetylene bridges. J. Am. Chem. Soc. 134, 9609–9614 (2012)
Alabugin, I.V., Gilmore, K., Patil, S., Manoharan, M., Kovalenko, S.V., Clark, R.J., Ghiviriga, I.: Radical cascade transformations of tris(o-aryleneethynylenes) into substituted benzo[a]indeno[2,1-c]fluorenes. J. Am. Chem. Soc. 130, 11535–11545 (2008)
Alabugin, I.V., Manoharan, M.: Thermodynamic and strain effects in the competition between 5-exo-dig and 6-endo-dig cyclizations of vinyl and aryl radicals. J. Am. Chem. Soc. 127, 12583 (2005)
Hirano, K., Inaba, Y., Takasu, K., Oishi, S., Takemoto, Y., Fujii, N., Ohno, H.: Gold(I)-catalyzed polycyclizations of polyenyne-type anilines based on hydroamination and consecutive hydroarylation cascade. J. Org. Chem. 76, 9068–9080 (2011)
Gross, L., Mohn, F., Moll, N., Liljeroth, P., Meyer, G.: The chemical structure of a molecule resolved by atomic force microscopy. Science 325, 1110–1114 (2009)
de Oteyza, D.G., Gorman, P., Chen, Y.-C., Wickenburg, S., Riss, A., Mowbray, D.J., Etkin, G., Pedramrazi, Z., Tsai, H.-Z., Rubio, A., Crommie, M.F., Fischer, F.R.: Direct imaging of covalent bond structure in single-molecule chemical reactions. Science 340, 1434–1437 (2013)
Riss, A., Wickenburg, S., Gorman, P., Tan, L.Z., Tsai, H.-Z., de Oteyza, D.G., Chen, Y.-C., Bradley, A.J., Ugeda, M.M., Etkin, G., Louie, S.G., Fischer, F.R., Crommie, M.F.: Local electronic and chemical structure of oligo-acetylene derivatives formed through radical cyclizations at a surface. Nano Lett. 14, 2251–2255 (2014)
Kertesz, M., Choi, C.H., Yang, S.: Conjugated polymers and aromaticity. Chem. Rev. 105, 3448–3481 (2005)
Gross, L., Mohn, F., Moll, N., Schuler, B., Criado, A., Guitian, E., Pena, D., Gourdon, A., Meyer, G.: Bond-Order discrimination by atomic force microscopy. Science 337, 1326–1329 (2012)
Van der Lit, J., Boneschanscher, M. P., Vanmaekelbergh, D., Ijas, M., Uppstu, A., Ervasti, M., Harju, A., Liljeroth, P., Swart, I.: Suppression of electron–vibron coupling in graphene nanoribbons contacted via a single atom. Nat. Commun. 4, 2023 (2013)
Torras, J., Casanovas, J., Aleman, C.: Reviewing extrapolation procedures of the electronic properties on the π-conjugated polymer limit. J. Phys. Chem. A 115, 7571–7583 (2012)
Gutzler, R., Perepichka, D.F.: π-electron conjugation in two dimensions. J. Am. Chem. Soc. 135, 16585–16594 (2013)
Wang, S., Wang, W., Lin, N.: Resolving band-structure evolution and defect-induced states of single conjugated oligomers by scanning tunneling microscopy and tight-binding calculations. Phys. Rev. Lett. 106, 206803 (2011)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this paper
Cite this paper
de Oteyza, D.G. (2016). Enediyne Cyclization Chemistry on Surfaces Under Ultra-High Vacuum. In: Gourdon, A. (eds) On-Surface Synthesis. Advances in Atom and Single Molecule Machines. Springer, Cham. https://doi.org/10.1007/978-3-319-26600-8_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-26600-8_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-26598-8
Online ISBN: 978-3-319-26600-8
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)