
Abstract
The Dynathor project aims at understanding the interaction of cytochrome P450 (CYP, P450) enzymes and their redox partner, cytochrome P450 reductase (CPR), in a phospholipid bilayer. Through simulation studies on the HLRS CRAY systems (initially XE6, later on XC40), we investigated the interactions of models of membrane-bound P450s (CYP51 and CYP1A1) and CPR. A model of membrane-bound T. brucei CYP51 and the human CPR was successfully built and simulated for 217.5 ns. A model of human CYP1A1 and the human CPR in a phospholipid bilayer was also built and is being simulated. These models can be used as starting points to understand the selectivity of the interactions of CYPs with CPR in the native membrane-bound forms, and thus may aid drug discovery projects.
Furthermore, we evaluated the method of Random Acceleration Molecular Dynamics (RAMD) for use in calculating relative residence times of proteins with small molecules. This would allow the computation of relative off rates for drug molecules. As a small model system for this evaluation, we use the N-terminal domain of human heat shock protein 90 (HSP90) and a set of different ligands. The obtained RAMD simulated residence times show a clear correlation with experimental values.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- Nicotinamide Adenine Dinucleotide Phosphate
- Flavin Adenine Dinucleotide
- Nicotinamide Adenine Dinucleotide Phosphate
- Cytochrome P450 Reductase
- Redox Partner
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Technical Description
1.1 NAMD Simulations
NAMD is a software package for molecular dynamics simulations, provided by the University of Illinois [1–3]. Simulations where initially performed on the HLRS Hermit, a Cray XE6 computer [4]. Starting from November 2014, the new HLRS Hornet system, a Cray XC40 [5] computer was used. On Hermit, we used NAMD version 2.9, compiled using the Cray software stack. Simulations on Hornet used the newest NAMD version 2.10b2. This version switch was required since NAMD version 2.9 did not support the Cray Aries network environment implemented on the Hornet system. The Hornet system with NAMD 2.10b2 shows an approximate 2. 4× increase in simulation performance at 1024 cores compared to the Hermit system using NAMD 2.9 (see Fig. 1). Calculations for the cytochrome P450 system were performed using 1024 cores.

Scaling of NAMD simulations. NAMD 2.9 simulations of CYP51-CPR complex in the membrane (390,422 atoms) on a Cray XE6 ( filled triangle) compared to NAMD 2.10b2 simulations on a Cray XC40 (open circle). At 1024 cores, simulations on the Cray XC40 were 2.4 times faster using NAMD 2.10b2
1.2 RAMD Simulations
Random Acceleration Molecular Dynamics (RAMD) [6, 7] uses the NAMD software package to perform molecular dynamics simulations of two molecule systems bound in a complex. This method can be used to simulate the egress of a bound ligand from a protein. The method relies on the application of random forces to guide a bound molecule out of the binding pocket. One problem of this random force application is that it disrupts simulation and requires all threads to synchronize. Therefore, so far, only a maximum of 48 cores was used for a single RAMD simulation. However, for calculations of off-rates for bound ligands, a large number of similar independent simulations needs to be carried out in the same system. These simulations can be run in parallel. Also new scripting techniques within NAMD [8] possibly will not interfere with the parallel performance. Since this would reduce the average runtime of a single NAMD run from currently several hours up to 20 h on hornet, the migration of the RAMD code would be beneficial (Fig. 2).

Scaling of RAMD simulations of HSP90 with inhibitor (41,831 atoms). RAMD is an algorithm added to the NAMD software via TCL text commands. The nature of TCL text commands disrupts the parallel performance of NAMD of a single RAMD run. Although some of these commands can be used in a parallel environment, RAMD so far does not take advantage of this. New scripting techniques within NAMD [8] might overcome this limitation. A large number of RAMD simulations are required for calculating relative off-rates for ligands
For the method evaluation we have used N-terminal domain (NTD) of human HSP90 (heat shock protein 90), for which experimental dissociation rates for a quite large set of compounds are available. We have carried out simulations for 11 ligands; four of them bound to the helical conformation and the rest bound to the loop conformation of the HSP90 binding site. For estimation of a relative residence time, 25 RAMD simulations for each ligand were performed. For evaluation of the sensitivity of ligand residence time to the magnitude of the force applied in RAMD simulations, we performed two set of simulations for each ligand using two different values of the acceleration force: 15 and 14 kcal mol-1 Å-1 (data not shown). The maximum length of each trajectory was 10 ns-5,000,000 steps, which takes about 20 h on 2 nodes. In practice, however, the most of ligands require much less time for dissociation, and thus most simulations are much shorter.
Effective RAMD residence times obtained for 11 ligands show a clear correlation with experimental values (data not shown) although the scaling factor is different for the two protein conformations simulated. The result was found to be quite robust with respect to variations of the magnitude of the force applied if the RAMD residence time was longer than hundreds of picoseconds.
2 Simulations of Membrane-Bound Complexes of P450 and CPR
2.1 Introduction
Cytochrome P450 (CYP, P450) enzymes play important roles in drug metabolism, steroid biosynthesis and xenobiotic degradation. In humans, P450s are essential for the metabolism of 70–80 % of all drugs. The catalytic function of P450s is monooxygenation, which is to add one oxygen atom to an organic bond (-RH). In the catalytic cycle of P450s, two electrons need to be transferred to the active site of P450s from their redox partners. For many P450s in eukaryotes, the redox partner is cytochrome P450 reductase (CPR). CPR consists of three domains: the FMN domain which contains the flavin mononucleotide (FMN), the FAD domain with the flavin adenine dinucleotide (FAD), and the NADP domain with the nicotinamide adenine dinucleotide phosphate (NADP). In eukaryotes, both proteins locate in the endoplasmic reticulum (ER) membrane with a trans-membrane helix anchor. The association of the globular domains of the two proteins is mainly driven by electrostatic interactions. The positively charged proximal side of P450 interacts with the negatively charged FMN domain of the CPR.
Many studies have been carried out for the soluble forms of P450s and their redox partners. The crystal structure of the complex of the P450BM3 CYP domain and its FMN domain (which, exceptionally for P450s, are in one polypeptide chain in P450BM3) gives direct evidence of how P450s may interact with CPR [9]. A model of the CYP2B4-cytochrome b5 complex was built using solid-state NMR data on CYP2B4 and the membrane-bound cytochrome b5 [10]. For the interactions of CYP2B4 with CPR, mutation studies have shown residues that affect binding. However, no crystal structure has been solved of complexes of P450s and CPR, and little is known about their interactions with the membrane or possible conformational changes of the proteins upon binding or during the catalytic cycle.
Understanding the P450-CPR interactions in the membrane is of great interest. First of all, it can help to investigate the catalytic mechanism of P450s in the native membrane-bound form. It can give insights into the association and dissociation of the two proteins in the membrane environment and how this varies with P450 sequence. Furthermore, the bound complex can be used to study ligand access and egress tunnels in P450s which can be influenced by the protein-protein interactions. Lastly, it can help to improve the prediction of drug metabolism by P450s and the design of drugs which bind to P450s.
2.2 Methods
Molecular dynamics (MD) simulations have been used to build models of different CYPs in the membrane. Our laboratory has established a systematic multiscale simulation protocol employing coarse-grain and atomic detail models, to model and simulate P450 in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer and this has been used to model human CYP2C9 and Trypanosoma brucei CYP51 in a POPC bilayer [11, 12]. Other researchers have also recently built and simulated models of P450s in bilayers including for CYP2C9 [13], CYP3A4 [14, 15], CYP19 [16] and other human drug-metabolizing P450s [17]. Sündermann et al. [18] performed a simulation of a complex of CYP2D6 and the open form of CPR in a bilayer. This complex was however built manually and the time scale of the simulation (10 ns) was too short to investigate the dynamics of the equilibrated complex in the membrane.
Our in-house software, Simulation of Diffusional Association (SDA, http://mcm.h-its.org/sda7/) has provided a useful tool for predicting the structures of protein-protein encounter complexes by using Brownian dynamics simulations [19–21]. It is particularly useful for electrostatically driven complexes [22, 23]. Our predicted structure of the P450cam-putidaredoxin complex published in 2007 [24] is in good agreement with the later published crystal structure of the complex in 2013 [25, 26].
We have tested using SDA to predict the complex of soluble forms of CYP2B4 and CPR. The complex is in good agreement with experimental data and refinement of the docked encounter complex using molecular dynamics simulation improves agreement with experimental data [9, 27]. Thus, we used SDA to predict the complexes of CYP51 with CPR and of CYP1A1 with CPR. The membrane-bound models of CYP51 and CYP1A1 have been built and simulated by using the protocol described in [11]. The P450-CPR encounter complexes defined using SDA rigid-body docking were superimposed onto the membrane-bound models of CYP51 and CYP1A1. The missing trans-membrane helix of CPR was then be inserted into the membrane and the missing linker residues modeled to connect the trans-membrane helix and the FMN domain of the CPR in the P450-CPR complexes. These models of membrane-bound complexes of P450 and CPR provided starting structures for MD simulations at HLRS. The Amber force field ff99SB is used for the protein and the Amber gaff force field for the POPC lipids and the cofactors. The systems simulated contain roughly 1 million atoms each. The procedure for this study is shown in Fig. 3.

Protocol to build and simulate membrane-bound models of CYP and CPR
2.3 Results
Using the protocol depicted in Fig. 3, we successfully built a membrane-bound model of the complex of T. brucei CYP51 and human CPR and simulated this model for 217.5 ns, which is 20 times longer than the recent simulation of a related system [18]. The starting structure and the equilibrated structure are shown in Fig. 4. In Fig. 4a, the membrane-bound T. brucei CYP51 was taken after MD simulation and the CPR was built onto it. In this model, the globular domain of CPR was relatively far away from the membrane, with the FMN domain interacting with the proximal side of CYP51 and the FAD domain extending away from the membrane. However, the FAD domain of CPR got much closer to the membrane in the simulation, although the distance between the center of mass of CPR and that of the membrane remained mostly similar during the simulation. The globular domain of CPR has a much larger distance to the membrane than that of CYP51 in the simulation. The reason can be that CPR is prohibited from getting much closer to the membrane because of its interaction with CYP51.

(a) The starting structure and (b) the equilibrated structure of CYP51 (green colored cartoons) and CPR (orange colored cartoons) in membrane (sticks view)
In the simulation of the membrane-bound complex of CYP51 and CPR, CYP51 maintained an orientation similar to the starting structure. The starting orientation of CYP51 in the membrane has been shown to be consistent with experimental data [12]. The 217.5 ns simulation of the model shows that the interaction of CYP51 and CPR is stable in the membrane-bound form.
The proposed protocol gives the possibility to build models of membrane-bound forms of different P450s and CPR. Having membrane-bound models of P450s and CPR can help to compare P450s, to understand selectivity of CPR for different P450s and thus to aid drug design. The model of the membrane-bound form of CYP51 and CPR provides a basis for understanding the behavior of both CYP51 and CPR in the complex on a lipid bilayer. This model may help to design drugs against parasitic CYP51s, taken their interaction with CPR into consideration.
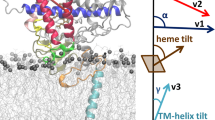
A model of the membrane-bound complex of CYP1A1 and CPR was also built using the same protocol (Fig. 5a and b). The CYP1A1-CPR membrane bound complex was converted to coarse-grained model and 12 μs of MD simulation was run. During the simulation, the CYP1A1-CPR complex was stable in the membrane. The distance between CYP1A1 and FMN binding domain of CPR reduced from 32 to 29 Å and distance between FMN and HEME co-factors reduced from 24 to 19 Å (Fig. 5c). Thus, CYP1A1 and FMN domain of CPR came closer during the course of 12 μs of coarse-grain MD simulation.

(a) The orientation of the CYP1A1-CPR complex in the membrane, (b) the positions of the HEME and FMN co-factors in the CYP1A1-CPR complex. (c) the observed parameters characterizing the arrangement of the two proteins in the bilayer during the coarse-grained CYP1A1-CPR complex simulation
The final structure obtained from coarse-grained MD simulation was converted to an all-atom model using the previously described protocol [11]. The all-atom structure of the CYP1A1-CPR complex in the membrane was then simulated for 15 ns using NAMD software. The RMSD of CYP1A1 and the FMN binding domain, showed a continuous increase during the simulation, indicating that it is not yet equilibrated. The length of the simulation will be increased to obtain a well converged structure of CYP1A1-CPR complex in membrane.
3 Future Plans
So far 900,000 CPU×h of the 4,000,000×h have been used. The remaining CPU time will primarily be used for NAMD production runs of CYP systems.
In future studies, we are interested in continuing to investigate the dynamics of the P450-CPR complex, especially in the membrane-bound form. Specifically, we will investigate how the two proteins interact with each other in the soluble form and, when the two proteins are in the membrane-bound form, how they orient in the membrane and how the membrane influences the association and dissociation of the two proteins and the structure of their complex. The studies will be carried out for complexes of three different P450s with CPR allowing for comparison of the effects of sequence differences on binding and identification of critical interactions and conformational changes. These studies will provide the basis for subsequent simulations to investigate ligand access and egress mechanism in the P450-CPR complexes. Furthermore, these studies will provide a step towards understanding how the electrons are transferred from the CPR to the P450 and how drugs are metabolized by this complex.
References
Phillips, J.C., Braun, R., Wang, W., Gumbart, J., Tajkhorshid, E., Villa, E., Chipot, C., Skeel, R.D., Kalé, L., Schulten, K.: Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 (2005)
Theoretical and U. o. I. a. U.-C. Computational Biophysics Group: NAMD 2.9 release notes. http://www.ks.uiuc.edu/Research/namd/2.9/notes.html. Online; Accessed 27 March 2015
Theoretical and U. o. I. a. U.-C. Computational Biophysics Group: NAMD 2.10b2 release notes. http://www.ks.uiuc.edu/Research/namd/2.10b2/notes.html. Online; Accessed 27 March 2015
HLRS: Cray XE6. https://wickie.hlrs.de/platforms/index.php/Cray_XE6. Online; Accessed 27 March 2015
HLRS: Cray XC40. https://wickie.hlrs.de/platforms/index.php/Cray_XC40. Online; Accessed 27 March 2015
Lüdemann, S.K., Lounnas, V., Wade, R.C.: How do substrates enter and products exit the buried active site of cytochrome P450cam? 1. Random expulsion molecular dynamics investigation of ligand access channels and mechanisms. J. Mol. Biol. 303, 797–811 (2000)
Lüdemann, S.K., Lounnas, V., Wade, R.C.: How do substrates enter and products exit the buried active site of cytochrome P450cam? 2. Steered molecular dynamics and adiabatic mapping of substrate pathways. J. Mol. Biol. 303, 813–830 (2000)
Phillips, J.C., Stone, J.E., Vandivort, K.L., Armstrong, T.G., Wozniak, J.M., Wilde, M., Schulten, K.: Petascale tcl with NAMD, VMD, and Swift/T, in Proceedings of the 1st First Workshop for High Performance Technical Computing in Dynamic Languages, HPTCDL ’14, (Piscataway, NJ, USA), pp. 6–17. IEEE Press, New York (2014)
Sevrioukova, I.F., Li, H., Zhang, H., Peterson, J.A., Poulos, T.L.: Structure of a cytochrome P450-redox partner electron-transfer complex. Proc. Natl. Acad. Sci. U.S.A. 96, 1863–1868 (1999)
Ahuja, S., Jahr, N., Im, S.-C., Vivekanandan, S., Popovych, N., Le Clair, S.V., Huang, R., Soong, R., Xu, J., Yamamoto, K., Nanga, R.P., Bridges, A., Waskell, L., Ramamoorthy, A.: A model of the membrane-bound cytochrome b5-cytochrome P450 complex from nmr and mutagenesis data. J. Biol. Chem. 288, 22080–22095 (2013)
Cojocaru, V., Balali-Mood, K., Sansom, M.S.P., Wade, R.C.: Structure and dynamics of the membrane-bound cytochrome P450 2C9. PLoS. Comput. Biol. 7, e1002152 (2011)
Yu, X., Cojocaru, V., Mustafa, G., Salo-Ahen, O.M.H., Lepesheva, G.I., Wade, R.C.: Dynamics of CYP51: implications for function and inhibitor design. J. Mol. Recognit. 28, 59–73 (2015)
Berka, K., Hendrychová, T., Anzenbacher, P., Otyepka, M.: Membrane position of ibuprofen agrees with suggested access path entrance to cytochrome P450 2C9 active site. J. Phys. Chem. A 115, 11248–11255 (2011)
Denisov, I.G., Shih, A.Y., Sligar, S.G.: Structural differences between soluble and membrane bound cytochrome P450s. J. Inorg. Biochem. 108, 150–158 (2012)
Baylon, J.L., Lenov, I.L., Sligar, S.G., Tajkhorshid, E.: Characterizing the membrane-bound state of cytochrome P450 3A4: structure, depth of insertion, and orientation. J. Am. Chem. Soc. 135, 8542–8551 (2013)
Sgrignani, J., Magistrato, A.: Influence of the membrane lipophilic environment on the structure and on the substrate access/egress routes of the human aromatase enzyme. A computational study. J. Chem Inf. Model. 52, 1595–1606 (2012)
Berka, K., Paloncýová, M., Anzenbacher, P., Otyepka, M.: Behavior of human cytochromes P450 on lipid membranes. J. Phys. Chem. B 117, 11556–11564 (2013)
Sündermann, A., Oostenbrink, C.: Molecular dynamics simulations give insight into the conformational change, complex formation, and electron transfer pathway for cytochrome P450 reductase. Protein Sci. 22, 1183–1195 (2013)
Gabdoulline, R.R., Wade, R.C.: Brownian dynamics simulation of protein-protein diffusional encounter. Methods 14, 329–341 (1998)
Gabdoulline, R.R., Wade, R.C.: On the contributions of diffusion and thermal activation to electron transfer between phormidium laminosum plastocyanin and cytochrome f: Brownian dynamics simulations with explicit modeling of nonpolar desolvation interactions and electron transfer events. J. Am. Chem. Soc. 131, 9230–9238 (2009)
Motiejunas, D., Gabdoulline, R., Wang, T., Feldman-Salit, A., Johann, T., Winn, P.J., Wade, R.C.: Protein-protein docking by simulating the process of association subject to biochemical constraints. Proteins 71, 1955–1969 (2008)
Sandikci, A., Gloge, F., Martinez, M., Mayer, M.P., Wade, R., Bukau, B., Kramer, G.: Dynamic enzyme docking to the ribosome coordinates N-terminal processing with polypeptide folding. Nat. Struct. Mol. Biol. 20, 843–850 (2013)
Pachov, G.V., Gabdoulline, R.R., Wade, R.C.: On the structure and dynamics of the complex of the nucleosome and the linker histone. Nucleic Acids Res. 39, 5255–5263 (2011)
Karyakin, A., Motiejunas, D., Wade, R.C., Jung, C.: FTIR studies of the redox partner interaction in cytochrome P450: the PDX-P450cam couple. Biochim. Biophys. Acta 1770, 420–431 (2007).
Tripathi, S., Li, H., Poulos, T.L.: Structural basis for effector control and redox partner recognition in cytochrome P450. Science 340, 1227–1230 (2013)
Hiruma, Y., Hass, M.A.S., Kikui, Y., Liu, W.-M., Ölmez, B., Skinner, S.P., Blok, A., Kloosterman, A., Koteishi, H., Löhr, F., Schwalbe, H., Nojiri, M., Ubbink, M.: The structure of the cytochrome P450cam-putidaredoxin complex determined by paramagnetic nmr spectroscopy and crystallography. J. Mol. Biol. 425, 4353–4365 (2013)
Im, S.-C., Waskell, L.: The interaction of microsomal cytochrome P450 2B4 with its redox partners, cytochrome P450 reductase and cytochrome b(5). Arch Biochem. Biophys. 507, 144–153 (2011)
Acknowledgements
We gratefully acknowledge the support of the Klaus Tschira Foundation, the German Academic Exchange Service (DAAD) for scholarships to Prajwal Nandekar and Ghulam Mustafa as well as the EU/EFPIA Innovative Medicines Initiative (IMI) Joint Undertaking, “Kinetics for Drug Discovery”, K4DD (grant no. 115366). Finally, we would like to thank the HLRS for providing CPU time for this project.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this paper
Cite this paper
Yu, X., Kokh, D.B., Nandekar, P., Mustafa, G., Richter, S., Wade, R.C. (2016). Dynathor: Dynamics of the Complex of Cytochrome P450 and Cytochrome P450 Reductase in a Phospholipid Bilayer. In: Nagel, W., Kröner, D., Resch, M. (eds) High Performance Computing in Science and Engineering ’15. Springer, Cham. https://doi.org/10.1007/978-3-319-24633-8_17
Download citation
DOI: https://doi.org/10.1007/978-3-319-24633-8_17
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-24631-4
Online ISBN: 978-3-319-24633-8
eBook Packages: Mathematics and StatisticsMathematics and Statistics (R0)