
Abstract
Chromosomal translocations frequently involve fusion of the ends of two separate DNA double-stranded breaks (DSBs) at distinct genomic locations. Recurrent chromosomal translocations are found in various cancers. Recently developed high-throughput approaches to clone genome-wide translocations that involve site-specific DSBs have provided new insights into mechanisms of chromosomal translocations. Such studies confirmed that, beyond cellular selection forces, basic mechanistic factors, including DSB frequency and persistence, as well as aspects of three dimensional genome organization, can contribute to recurrent translocations in a cell population. This review discusses our current view of the contribution of such mechanistic factors to recurrent chromosomal translocations.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Overview: Mechanisms of Recurrent Oncogenic Translocations
Chromosomal translocations (“translocations”) were observed cytogenetically over 50 years ago; since then, recurrent translocations have been identified in many types of cancers, most frequently in lymphoid and myeloid neoplasms, but also in solid tumours, such as lung and prostate cancers [1–4]. Translocations can contribute to both initiation and progression of neoplastic transformation, most frequently by leading to abnormal activation of cellular oncogenes [3]. In this regard, many translocations likely arise at very low frequency and are strongly selected during the tumourigenesis process leading to their appearance as clonal events in tumour cells. Cancer genome studies indicate that most translocations result from end-joining of two DNA double-stranded breaks (DSBs) that occur at two separate genomic locations. In this context, most joins of DSB ends from two distinct DSBs, whether on a separate chromosome to generate translocations or on the same chromosome to generate interstitial deletions or inversions, appear mechanistically related and can be considered as translocations [5].
Translocations between two genes can result in expression of hybrid fusion proteins, which generate aberrant activation of proto-oncogenes. A notable example is the BCR-ABL1 translocation between human chromosomes 9 and 22, also known as the Philadelphia chromosome, that is found in chronic myelogenous leukaemia and early B cell leukaemias [6–8]. Many other such examples for other tumour types have been elucidated [9, 10]. Translocations between different genomic sequences also can activate proto-oncogenes by deregulating their expression, often by linking them to strong cis-regulatory elements, a mechanism common in lymphoid malignancies in which translocations link strong transcriptional enhancers or super-enhancers in antigen receptor loci to cellular oncogenes [4, 9, 11]. A classic example of this type of oncogenic translocation are translocations that fuse immunoglobulin (IG) heavy chain locus (IGH) and its 3′ regulatory region, a known super-enhancer, to the MYC oncogene in Burkitt lymphomas (BL), with a variety of others having been well-characterized [2, 4].
Oncogene overexpression in tumours also can be achieved via gene amplification, the first form of genomic instability in cancer cells described at the molecular level [12]. One mechanism for oncogene amplification in cancer models involves generation of dicentric chromosomal translocations with breakpoint fusions in the vicinity of oncogenes [13–16]. In cancer cells deficient in the cellular G1 DSB checkpoint (e.g., TP53 or ataxia telangiectasia mutated (ATM) protein deficient), breakage-fusion-bridge cycles [17] (BFB) of such dicentrics can rapidly lead to oncogene amplification [18]. Although such BFB mechanisms of oncogene amplification are likely most common during solid tumour progression [15, 19, 20], they have also been observed in human B cell malignancies [21], including multiple myeloma [22]. In ATM-deficient mouse T cell lymphoma models, dicentric chromosomes that result from aberrant V(D)J recombination events at T cell receptor (TCR) δ loci (TRD) lead to BFB-generated amplification of linked sequences and to new BFB-generated DSBs that participate in translocations that may delete tumour suppressor genes [23] (see below). Inter- and intra-chromosomal translocations/deletions have been implicated in deletions of tumour suppressors that contribute to various cancers [24–27].
2 Mechanistic Factors Influence Generation of Recurrent Chromosomal Translocations
Beyond cellular selection for oncogenic translocations, general mechanistic factors of translocations can affect the propensity of two genomic sequences to translocate to each other recurrently, and can even impact oncogene choice in different malignancies [28, 29]. Many recent studies of mechanistic factors involved in promoting translocations have been done in lymphoid cells, which will be a main focus of this review.
As translocations often involve the fusion of ends from two separate DSBs, the frequency of DSBs at the two participating sites to be joined will directly influence the rate of a particular translocation. In this context, such DSB frequency will reflect both the frequency at which the participating DSBs are generated by various mechanisms and also how long they persist before being properly repaired [5] (see below). In the latter context, DSB persistence reflects the efficiency at which DSBs are repaired. Mammalian cells possess multiple DSB sensing mechanisms that are linked to DSB repair pathways that efficiently repair DSBs, often by end-joining them back together [30–32]. DSB sensing pathways, in addition to participating in repair, also activate checkpoints that either delay cell cycle progression of cells with unrepaired DSBs until they are joined or eliminate cells with persistent unjoined DSBs [30, 33] (see below). The two major pathways for DSB repair are homologous recombination (HR) [34], which is primarily involved in repair of post-replicative DSBs, and classical non-homologous DNA end-joining (C-NHEJ) which is functional throughout the cell cycle but is predominant in G1 when HR is not active [35] (discussed below). We will focus mainly on C-NHEJ due to the exclusive involvement of this repair pathway in joining programmed DSBs in lymphocytes and in suppressing their translocation [35].
For two separate DSBs to be joined to form translocations, they must also be juxtaposed (“synapsed”) at the time they are broken [5]. Thus, in a population of cells, the synapsis frequency of two regions that contain DSBs will directly influence their translocation frequency. Certain sequences are more frequently synapsed than others in the genome due to general principles of chromatin folding, as well as being involved in common processes, such as transcription [36, 37]. Beyond this, translocation of DSBs that are much less frequently synapsed in cells within a population can still occur, due to cellular heterogeneity with respect to three-dimensional (3D) spatial genome organization [38]. At a local level, synapsis of breaks that are in relatively close proximity also may occur through Brownian or Langevin motion [38–40]. Finally, active movement of DSB ends has been reported in yeast [41, 42] and in mammalian cells [43, 44], and may also contribute to synapsis.
In the past, inability to identify newly occurring translocations in normal cells, without the biases imposed by cellular or oncogenic selection, has limited analyses of mechanistic factors involved in the generation of translocations. Such limitations have been overcome by the recent development of high-throughput genome-wide translocation cloning techniques [45, 46]. In this regard, the use of rare-cutting, site-specific restriction endonucleases, such as the yeast I-SceI meganuclease or homothallic (HO) endonuclease, have provided critical tools for studying translocations by providing a method to generate initiating DSBs at one or more desired genomic locations [47]. The finding that I-SceI generated DSBs at introduced target sites in the IgH locus in B lymphocytes could substitute for endogenous mechanisms that generate IgH class switch recombination (CSR) provided the foundation for the development of genome-wide methods for studying translocation mechanisms [48]. These high-throughput methods, referred to generically here as “translocation cloning” methods, have been used to identify endogenous DSBs genome-wide based on their translocation to fixed I-SceI induced “bait” break-sites in activated B lymphocytes or G1-arrested progenitor B cells lines as well as in non-lymphoid cells [38, 39, 45, 46, 49].
Advances in custom nuclease tools that target endogenous genomic sequences, including zinc finger nucleases (ZFNs) [50], transcription activator-like effector nucleases (TALENs) [51], and Cas9:gRNAs [52], have provided additional ectopic DSB-generating approaches to study generation of translocations from DSBs at specific sites in mammalian genomes without introducing a target DSB sequence [39, 53, 54]. Recently, Cas9:gRNAs and TALENs have been used to very successfully generate bait DSBs for translocation cloning at desired endogenous sites in human cells [55], allowing tests of basic principles of translocations, modelling of recurrent oncogenic translocations, and potentially the development of improved cancer diagnostics. This approach has also provided potential benefits to genome-engineering and gene therapy fields by providing a very robust method to identify genome-wide off-target and wide-spread, low-level DSB activity of custom nucleases and to also detect collateral damage of such agents including recurrent translocations and/or deletions [55].
Below, we will further discuss the mechanistic factors outlined above and how they can contribute to recurrent translocations or recurrent classes of translocations based on insights obtained from the recently developed translocation cloning assays. We will focus this discussion largely on mechanisms revealed from studies of lymphoid cells and tumour models, but also indicate the more general relevance of the findings of these studies.
3 General Cellular DSBs Provide Translocation Substrates
Human dividing cells may undergo as many as 50 DSBs per cell cycle [56]. General DSBs can occur spontaneously or be induced by various endogenous or exogenous damaging factors. Some byproducts generated during cell metabolism, including reactive oxygen species and endogenous alkylating agents can lead to DSBs [57]. DNA replication is another source of endogenous DSBs [58]. When encountering DNA lesions or other replication barriers, DNA replication forks stall, accumulate single-stranded DNA (ssDNA) and finally collapse, resulting in DSBs [59–61]. Fragile sites, a common replication barrier, provide break-sites for many gross chromosomal rearrangements found in early-stage tumours or precancerous cells [58]. Fragile sites appear to contribute to breakpoints of recurrent translocations in acute lymphoblastic, myeloid leukaemias, or BL (See Chap. 5 by Jiang et al., “Common Chromosomal Fragile Sites and Cancer”) [62–64]. Recent studies have also identified another class of “early replicating fragile sites” in B lymphocytes that were enriched in areas of repetitive sequences and/or CpG nucleotides, some of which map near translocation breakpoints in B cell lymphomas [65]. Transcription also has been implicated in DSB generation [36, 45, 46, 66]. Transcription-associated DSBs may result from head-on collisions between DNA and RNA polymerases [67, 68], topological constraints arising from transcription induction [69], or formation of unstable DNA structures, such as R-loop and G-quadruplexes [70–72]. Transcription has also been implicated in the generation of DSBs that participate in translocations in activated B lymphocytes [45, 46] (see below).
In addition to endogenous factors, DSBs also can be generated by exposure to external agents, such as ionizing radiation (IR) and various types of chemotherapeutics [73, 74]. 1 Gray (Gy) of γ-irradiation generates about 20 DSBs in mammalian cells [75]. Translocation cloning studies from IR-treated cells confirmed that IR-derived non-specific DSBs can generate translocation substrates genome-wide [38]. Topoisomerase II inhibitors, commonly used for anticancer treatment, prevent topoisomerase II from releasing topological constraints during DNA replication or transcription, thus promoting DSBs, implicated, for example, in the emergence of therapy-related myeloid neoplasms with recurrent oncogenic translocations [76, 77].
4 Programmed DNA DSBs/Rearrangements in Lymphocytes
DSBs are necessary intermediates of the programmed rearrangements that take place during the V(D)J recombination process that assembles diverse sets of antigen receptor gene segments in developing B and T lymphocytes and the CSR process that changes the expressed IGH constant region (CH) exons in activated mature B lymphocytes [5]. These programmed DSB-based gene rearrangement processes, which might be considered programmed intra-chromosomal translocations, involve the coordinated introduction of two separate DSBs at targeted IGH locus sites followed by their joining [35, 78]. Although the joining of these DSBs is generally regulated to ensure “proper” joining within IGH, they also can be aberrantly joined to other genomic DSBs to generate oncogenic translocations [5, 79].
The B cell receptor (BCR) is composed of two pairs of identical IGH and IG light chains (IGL/IGK). The secreted forms of BCRs are known as antibodies. Similarly, TCRs are heterodimers of either αβ or γδ chains [80, 81]. The exons that encode the N-terminal antigen-binding variable region exons of IGH chains are somatically assembled in progenitor B lymphocytes from 100s of different variable (V), 13 diversity (D), and 4 joining (J) genes that lie within distinct segments of the several megabase (Mb) long variable region portion of the IgH locus [5] (Fig. 3.1). Organization of IgL, IgK and TcR loci is similar and the general aspects of cleavage and joining of IgH V, D, and J segments outlined below also applies to these loci. The lymphocyte-specific endonuclease, RAG endonuclease, comprised of the recombination activating gene 1 and 2 proteins (RAG1/RAG2) [82] initiates V(D)J recombination by introducing DSBs between appropriate pairs of V, D, or J coding segments and short conserved recombination signal sequences (RSSs) that flank them; notably, RAG does not cleave, at least efficiently, at isolated RSSs [83]. RAG cleavage generates a pair of hairpin-sealed coding ends and a pair of blunt, broken RSS ends [81, 84].

Programmed DSBs and genomic rearrangements in developing and mature B cells. (Top) Diagram of the murine IgH locus on chromosome 12 (not to scale). The IgH locus contains of hundreds of V, 13 D, and 4 J gene segments arranged in clusters as indicated. (Left) In pro-B cells, RAG cleaves synapsed V, D, and J gene segments at appropriately paired RSSs (grey triangles) which are then joined by C-NHEJ factors to generate V(D)J exons (see text for details). In the IgH locus, DJH joining occurs first followed by VH gene segment joining to the DJH segment. See text for more details. (Right) In response to antigen, mature B cells can undergo CSR to exchange their initially expressed IgH CH exons from Cμ to one of a set of downstream (from 100 to 200 kb) exons encoding Cγ3, Cγ1, Cγ2b, Cγ2a, Cε, or Cα. Each set of CH exons is preceded by a long (1–10 kb) repetitive Switch (S) sequence (ovals). Transcription through the donor Sμ and a target downstream S region promotes AID-initiated DSBs (arrow heads) which are then joined between donor and acceptor S regions to delete the intervening sequences and replace Cμ with the targeted downstream CH (See text for more details). AID can also introduce somatic mutations into the assembled V(D)J to allow affinity maturation of the BCR (See text for more details)
The D and J segments lie proximal to each other in IgH and are cut and joined first in development. Subsequent, synapsis of VH segments with the DJH complex is thought to be facilitated by physical contraction of the IgH locus [85, 86], which enhances synapsis by bringing the 100s of VH segments scattered over Mb linear distances into close enough proximity to sample the DJH complex by a form of diffusion referred to as Langevin motion [40]. The RAG complex also binds DJH regions, facilitated by particular histone modifications associated with transcription [87–90] before VH synapsis allowing formation of “recombination centres” that stabilize synapsed VH to DJH complexes once formed and which generate paired RSS cleavage [90]. Formation of DH to JH joins likely occurs similarly, but due to the close proximity of the DH and JH segments, may not require physical locus contraction. Following cleavage, RAG, with the aid of other repair factors (see below), holds coding and RSS ends in a post-cleavage synaptic complex [84], and channels their repair exclusively to the C-NHEJ pathway [5, 91], C-NHEJ directly fuses the blunt RSS ends and, along with other factors, further processes coding ends before joining them, thereby contributing to V(D)J exon coding diversity [5]. The RAG post-cleavage complex, and perhaps other factors, also contributes to directing the joining of coding ends to each other and RSS ends to each other, thereby prescribing a specific chromosomal orientation of V(D)J recombination which results in deletions or inversions depending on the orientation of the participating V, D, and J segments [5, 92].
The portion of the IgH locus downstream of the V, D, and J segments contains multiple sets of exons encoding for different CHs within an approximately 200 kb region [93, 94]. The Cμ exons, which lie closest to the V(D)J, are transcribed to yield a V(D)J Cμ transcript that encodes μ heavy chains, which activates assembly of IgL/IgK variable region exons and ultimately associate with IgL/IgK chains to form an IgM BCR resulting in “mature” B lymphocytes [95]. Antigen-dependent activation can induce mature B cells to undergo CSR to exchange Cμ exon for one of the sets of CH exons that lie downstream (Fig. 3.1). CSR involves introduction of DSBs into a donor switch (S) region just upstream of Cμ and into an acceptor S region upstream of a targeted set of downstream CH exons. Subsequently, the upstream end of the DSB in Sμ is joined to the downstream end of the DSB in the target S region to delete Cμ and other intervening sequence and juxtapose the new set of CH exons to the V(D)J exon [94]. Exchanging the CH exon, changes the effector functions of the expressed antibody. Unlike V(D)J recombination, which is completely dependent on C-NHEJ, joining of DSBs within S regions to complete CSR can occur, at somewhat reduced levels, in the absence of C-NHEJ via alternative end-joining (A-EJ) pathways [35] (see below).
CSR is initiated by the activation-induced cytidine deaminase (AID) encoded by the AICDA gene [96], which acts on ssDNA to deaminate cytidine residues within short (4 bp) target motifs of which the sequence AGCT is a canonical representative [97, 98]. S regions are very long (1–10 kb) and very rich in AID target motifs [99]. AID cytidine deamination also initiates an antigen dependent variable region diversification process termed somatic hypermutation (SHM) [100]. During CSR and SHM, AID-initiated C to U lesions are processed into DSBs and point mutations, respectively, via related processes that require activities of the normal base excision and mismatch repair pathways [97, 101]. Various mechanisms have been proposed to promote DSB versus mutational processing of AID, although these two outcomes are not totally separable [93, 100–102]. Cytidine deamination of target sequences by AID requires their transcription to both recruit AID and provide ssDNA substrates [5]. Following transcription of the GC-rich S regions, AID access to the non-template strand is promoted by the formation of stable R-loops [71, 72]. Additional details of mechanisms of AID targeting to IgH S regions and variable regions exons also have been elucidated [5, 103–105].
Currently, S region synapsis has been proposed to potentially involve diffusion (Langevin motion) due to their relatively proximal location within a 200 kb domain, with high levels of AID-initiated DSBs helping to ensure breakage of S regions while synapsed [39] (see below). Aspects of IgH locus organization, and potentially the 53BP1 (TP53BP1) DSB response factor (see below), may also help to facilitate/stabilize S region synapsis [103, 106]. Unlike RAG-initiated DSBs, S region DSBs can occur in unsynapsed S regions; however, these DSBs are usually joined internally in S regions to generate intra-S region deletions as opposed to translocation [93]. As AID, thus far, has no known downstream roles in S region synapsis, and indeed, substantial CSR can be generated by I-SceI-initiated DSBs at target sites replacing S regions [39, 48], CSR may be analogous to a targeted form of an intra-chromosomal translocation [48]. CSR must occur in a deletional, versus inversional, orientation to generate productive CSR; however, while RAG cleavage and generation of a post-cleavage complex may contribute to orientation-specific joining during V(D)J recombination, little has been reported about if and how orientation-specific joining occurs during CSR [5]. If joining is orientation specific during CSR, it must employ specialized mechanisms since, in translocation cloning assays, bait DSBs generally join equally to both ends of other DSBs across the genome in activated B lymphocytes [45].
5 Involvement of RAG- and AID-Initiated DSBs in Translocations
The potential of RAG-initiated DSBs to contribute to translocations is counteracted at several levels. RAG cleavage is restricted by the “12/23 rule” to paired RSSs with appropriate complementarity, which limits generation of “off-target” RAG-cleavage at “cryptic” RSSs across the genome [84]. RAG expression also is limited to G1-phase lymphoid cells [87], which contributes to restricting repair to C-NHEJ and also provides the G1 checkpoint to prevent replicative propagation of RAG initiated DSBs, for example via dicentric formation [13, 16, 49]. Moreover, formation of the RAG post-cleavage synaptic complex limits direct joining of coding and RSS ends and limits availability to translocate to other DSBs. Finally, the ATM DSB response complex [107] cooperates with RAG2 [108, 109] to stabilize of RAG-initiated post cleavage DSB complexes and to prevent their separation and translocation (see below). In the latter context, translocation cloning from G1-arrested ATM-deficient pro-B cell lines that induce RAG, clearly showed I-SceI or RAG-induced bait DSBs can translocate to other DSBs genome-wide, but endogenous hotspots were all provided by RAG-initiated DSBs at various Ig and TcR loci (due to the high frequency of target DSBs in these loci) and, in IR-treated cells, to DSBs genome-wide with highly preferential joining to DSBs in cis on the same chromosome due to 3D proximity influences [38] (Fig. 3.2). In this context, oncogenic translocations between V(D)J recombination-associated DSBs and other DSBs have been demonstrated to occur in the context of TP53/C-NHEJ deficient mouse pro-B cell lymphoma models [13, 16].

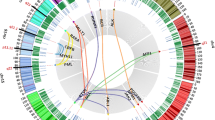
Circos plots of G1-arrested pro-B translocation cloning libraries. (a) custom circos plots (See ref. [55]) of genome-wide translocation cloning libraries from two different G1-arrested pro-B cell lines with singly integrated I-SceI substrate bait DSB sequence on either chromosome 18 (left) or chromosome 2 (right). Both RAG and I-SceI were induced to generate endogenous DSBs in the G1-arrested cells prior to genome-wide library generation from the I-SceI bait DSB. Black bars indicate I-SceI translocation junction frequency to genome-wide DSBs over 5 Mb bins on a custom log scale plot. Inner red lines link the bait DSB site to recurrently joined antigen receptor loci (e.g., show translocation hotspots). In these cells, all translocation hotspots from either chromosome 18 or chromosome 2 baits were RAG-initiated DSBs at the various endogenous antigen receptor loci. No other translocation junction regions qualified as hotspots. (b) IR treatment of the same pro-B lines to introduce frequent DSBs genome-wide (e.g., normalize DSB frequency genome-wide) decreased enrichment of antigen receptor locus translocation junctions and led the endogenous cis chromosome containing the bait sequence to become a translocation hotspot region (due to increased influence of 3D proximity when DSBs are not limiting) In these plots, all libraries are size-normalized to allow direct comparison (See ref. [55]). Chromosomes are displayed centromere to telomere in a clock-wise orientation (Data are adapted from [38]. See text, ref. [5, 38] for further details)
Translocations involving RAG-initiated DSBs at TCR loci, with translocations from TRD segments being most prominent, and DSBs near various oncogenes, including TAL1 and TAL2, LMO1 and LMO2, and MYC, are common in human T-cell acute lymphoblastic leukaemia (T-ALL) [27]. Among human B cell tumours, translocations involving IG variable region gene segments are rare in B-ALL, accounting for only about 3 % of cases [110], but are found in mature B cell neoplasms, such as the recurrent IGH/MYC translocation in endemic BL [79] or IGH/BCL2 translocation in follicular lymphomas [2]. Mouse model studies suggest some such translocations may persist through development [111] (see below) or result from RAG activity during secondary recombination events in peripheral B cells referred to as receptor editing [29, 112]. RAG can also contribute to oncogenic translocations by generating DSBs at RSS-like sequences across the genome, termed cryptic RSSs (cRSSs); the frequency of cRSSs in the human genome has been estimated at about 1 per 500 bp [113, 114]. A recent survey of a large series of translocation junctions between TCR loci and various oncogenes in human T-ALL revealed that about 25 % of such rearrangements involved cRSSs at the TCR translocation partner loci [27]. In addition, some interstitial deletions that contribute to human T-ALL oncogenesis appear to involve RAG-initiated DSBs at cRSSs in both TCR and/or non-TCR partners [27, 115]. Finally, recent studies in ATM-deficient mouse B cell lymphomas also suggested that oncogenic translocations originated from RAG cutting at IgH loci and putative cRSSs downstream of Myc [111].
AID-generated DSBs in IGH S regions during CSR have been implicated in the generation of recurrent oncogenic translocations in human mature B cell lymphomas involving IGH and AID off-targets, including the IGH/BCL6 translocation in diffuse large B-cell lymphoma [116] and IGH/MYC translocations in sporadic BL [79]. AID-initiated DSBs in V(D)J exons during SHM also have been implicated in certain oncogenic translocations in human B cell lymphomas [117]. Translocation cloning studies in activated mouse B cells clearly demonstrated that, beyond the IgH locus S region targets, AID activity also promotes lower level DSBs at dozens of other genes across the genome, referred to as AID off-targets [45, 46]. These studies also demonstrated that both IgH and off-target DSBs translocated robustly to bait I-SceI-generated DSBs in the Myc locus [45, 46, 49, 118].
IGH S regions and IG V(D)J exons likely have evolved mechanisms to recruit AID activity during CSR and SHM, such as high density AID target motifs and transcription dependent ability to generated secondary structures such as R loops for S regions [93, 119]. However, how AID is directed to off-target sites in activated B cells is still under investigation. In this context, AID is also directed to a series of off-target sites in germinal centre B cells that may contribute to SHMs and occasional DSBs that contribute to germinal centre B cell lymphomas [10, 120, 121]. Translocation cloning studies in CSR-activated B cells demonstrated high correlations between AID-dependent translocation hotspots and active transcription start sites, with translocations often clustering just downstream of active TSSs [45, 46]. However, as the vast majority of transcribed genes in activated B cells are not AID off-targets, additional factors beyond transcription per se must be involved in such AID “off-targeting” [5]. Recent studies have demonstrated that such factors include “super-enhancers” and convergent transcription [122].
6 Role of DNA End-Joining in DSB Repair and Translocations
The C-NHEJ machinery comprises four evolutionarily conserved “core” factors, Ku70 (XRCC6), Ku80 (XRCC5), XRCC4 and DNA ligase 4 (LIG4), which are essential for joining all types of DSBs via C-NHEJ [35]. Two additional C-NHEJ factors include DNA-dependent protein kinase catalytic subunit (PRKDC/DNA-PKcs) and the Artemis endonuclease (DCLRE1C), which together are important for joining DSBs in need of further processing, such as opening hairpin V(D)J coding ends [123]. The XRCC6/XRCC5 (Ku) heterodimer provides the C-NHEJ DSB recognition component, which binds DSBs to protect them from resection [124] and recruits downstream factors including PRKDC (to form the DNA-PK holoenzyme). Ku also recruits XRCC4 and LIG4, which form the ligase complex for C-NHEJ [35]. All of these factors are required for generation of V(D)J recombination coding joins; with the core factors being absolutely required for both coding and RSS joins [35, 125]. The XLF (NHEJ1) (“XRCC4 like factor”) also has been implicated in C-NHEJ based on the IR sensitivity and apparent DSB repair defects in human patients with NHEJ1 mutations [126, 127]. XLF, which interacts with the XRCC4-LIG4 complex [128–130], has been suggested to play a role in ligation of DSBs with incompatible or blunt ends [131] (see below). Deficiency of any core C-NHEJ factors dramatically increases genome instability, including translocations, in various cell types; whereas deficiency for the other C-NHEJ factors increases genomic instability but usually not as dramatically [35, 125]. In mice, deficiency for core C-NHEJ factors and DCLRE1C and PRKDC leads to severe combined immunodeficiency (“SCID”) due to inability join V(D)J recombination-associated breaks required for assembly of antigen receptor genes [5]. LIG4 hypomorphic mutations and XLF deficiencies also lead to variable immunodeficiency due to V(D)J recombination defects in human patients [132, 133].
Mice deficient for XRCC4 and LIG4 die in late embryonic development in association with severe apoptosis of newly developed neurons, along with abrogated V(D)J recombination [134, 135]. Less severe neuronal apoptosis occurs in Ku-deficient mice [136]. When XRCC4 or LIG4-deficiency are combined with TP53-deficiency, which removes the TP53-dependent G1 DSB checkpoint, neuronal apoptosis and embryonic lethality (but not V(D)J recombination) are rescued [137, 138]. Notably, TP53-deficient mice that are also deficient for any core C-NHEJ factor or for PRKDC or DCLRE1C die from progenitor B cell lymphomas that generate RAG-dependent dicentric translocations between the IgH locus and the Myc (or N-myc) loci leading to amplification of these oncogenes [9, 139]. The generation of such dicentric translocations and BFB cycles results from propagation of the RAG-generated IgH locus breaks through the cell cycle in the absence of the G1 DSB checkpoint enforced by TP53 [16]. Where examined, such C-NHEJ and TP53 double-deficient mice also develop medulloblastomas in situ and, indeed, conditional inactivation of XRCC4 in developing neurons of TP53 deficient mice leads to inevitable medulloblastomas with highly recurrent translocations and gene amplifications which include N-myc or Myc amplifications [140, 141]. Why C-NHEJ is required for neural development and protection from medulloblastomas with recurrent translocations is not yet known. Notably, XLF plus TP53 double deficient mice do not generally succumb to B lineage lymphomas, reflecting lack of absolute requirement for XLF in V(D)J recombination in an otherwise normal background (see below); but they do develop medulloblastomas, reflecting the requirement for XLF in general DSB repair by C-NHEJ [142].
The frequency of translocations that form in various types of core C-NHEJ deficient cells [141, 143–145] and the recurrent translocations that occur in core C-NHEJ-deficient tumours revealed that chromosomal translocations can be catalyzed by A-EJ pathways [16, 35]. In this regard, C-NHEJ-deficient mammalian cells join DSBs in plasmid-based assays by A-EJ pathway [146, 147]. Likewise, while V(D)J recombination absolutely requires C-NHEJ, CSR can occur at up to 50 % normal levels in the absence of core C-NHEJ factors [148, 149], or even the absence of both Ku70 plus LIG4—which eliminates both DSB recognition and joining components of C-NHEJ [148]. The latter studies definitively prove the existence of relatively robust A-EJ pathways in mammalian cells that are completely distinct from C-NHEJ. A number of known DNA repair factors have been implicated in A-EJ pathways (reviewed by [35]).
CSR junctions in the different types of C-NHEJ deficient cells were essentially totally (e.g., XRCC4 or LiG4-deficiency), or substantially (e.g., Ku-deficiency), mediated by short micro-homologies (MHs) [148, 149]. Similarly, oncogenic translocation junctions found in nine independent XRCC4- or LIG4- plus TP53-deficient pro-B lymphomas were MH-mediated [16]. In this regard, short MHs are found in many translocations genome-wide in activated mouse B cells [45], human cancer genomes [150], and tumour translocation junctions [151]. However, A-EJ can also generate substantial levels of blunt junctions in various contexts, perhaps dependent on the DSB ends presented for joining [152, 153]. Moreover, C-NHEJ frequently uses short MHs [154, 155]. Therefore, A-EJ, which could represent several different pathways [35], cannot be categorized unequivocally as MH-mediated. It should also be noted that, while A-EJ may, indeed, be a translocation prone pathway [156], its predominant contributions to translocations in the absence of C-NHEJ also may be contributed to increased levels of unrepaired substrate DSB ends for translocations [32]. Finally, C-NHEJ may contribute to translocations in C-NHEJ proficient cells (e.g., [157]); although the relative contribution of A-EJ remains to be determined.
7 The ATM DNA Damage Response Pathway and Its Multiple Roles in Suppressing Translocations
Ataxia telangiectasia (AT), a syndrome characterized by neurodegeneration, immunodeficiency, sensitivity to ionizing irradiation, and cancer susceptibility is associated with mutations in the ATM gene [158]. In response to DSBs, ATM, a serine/threonine kinase, activates a downstream DNA damage response (DDR) pathway that includes series of chromatin bound factors that regulate cell cycle progression at the G1 checkpoint and contribute directly to DSB repair by C-NHEJ [5, 31]. A key ATM substrate is the TP53 tumour suppressor, a transcription factor that directly activates the G1/S cell cycle checkpoint to arrest cells for DSB repair or that triggers apoptosis to eliminate cells with persistent DSBs [159]. ATM DDR substrates include the H2AX (H2AFX) histone variant, MDC1, and 53BP1, which assemble into large macromolecular complexes, called “foci”, that can spread in chromatin over several hundred kb or more on either side of DSBs [160, 161]. The DDR also employs additional downstream factors that facilitate repair pathway choice and provide additional chromatin modifications that promote DSB repair (reviewed by [34, 162–165]).
The ATM DDR has been implicated in contributing directly to C-NHEJ of DSBs, potentially by tethering DSB ends and, thereby, contributing to appropriate re-joining by C-NHEJ [166, 167]. In this regard, ATM deficiency has long been known to lead to genomic instability and recurrent translocations, particularly in lymphoid cells and tumours [168, 169]. Such translocations are likely facilitated by the dual effects of ATM deficiency on C-NHEJ (e.g. during V(D)J recombination) and abrogation of G1 DSB checkpoint [78], analogous to combined C-NHEJ and TP53 deficiency. Recent translocation cloning studies have confirmed the increased levels of genome-wide translocations from DSBs in the Myc gene in ATM-deficient activated mouse B cells relative to wild-type B cells [49] (Fig. 3.3).

Circos plots of stimulated primary B cell high throughput genome-wide translocation sequencing libraries. Translocation libraries generated with a bait I-SceI break-site in intron 1 of Myc gene on chromosome 15 from CSR-activated primary B cells that either do not express AID (left panel) or do express AID (right panel). Blue and red lines link the Myc I-SceI bait DSBs to cryptic I-SceI-generated translocation hotspots genome-wide and red lines link bait DSBs to AID-dependent hotspots genome-wide. Chromosomes are displayed centromere-to-telomere in a clock-wise orientation (Data are adapted from [49]. See text and ref. [49] or further details)
Like ATM deficiency, H2AX deficiency in various cell types leads to marked increases in genomic instability, increased chromosomal translocations [170, 171] and, in the absence of TP53, progenitor and mature B cell lymphomas with complex Myc translocations (involving IgH) and amplifications [172–174]. ATM or H2AX deficiency also moderately impairs CSR (decreasing levels to about 50 % or less of normal [5, 160]) accompanied by accumulation of substantial levels of AID-dependent IgH locus chromosome breaks and translocations [171, 174]. Notably, however, 53BP1 deficiency, while not dramatically increasing genomic instability in most tested cell types other than CSR-activated B cells, nearly abrogates CSR [175, 176]. Yet, 53BP1 deficiency leads to similar level of AID-dependent IgH breaks and translocations as observed in the context of ATM- or H2AX deficiency, which together with other findings suggest a specialized role for 53BP1 in CSR that may involve S region synapsis, end-protection, or other yet to be identified functions [103, 125].
Deficiency for ATM also has moderate effects on V(D)J recombination that have been attributed to destabilization of the post-cleavage synaptic complex, allowing some RSS or coding ends to escape and participate in translocations [78]. Correspondingly, high-throughput translocation libraries from ATM-deficient pro-B cell lines revealed the major translocation hotspots from various bait DSBs in different chromosomal locations to be the various Ig and TcR loci which are RAG-targets in these cells [38] (Fig. 3.2). Despite the impact on CSR, C-NHEJ and genomic stability in activated B cells, deficiencies for downstream ATM substrates H2AX and 53BP1 have little or no obvious effect on V(D)J recombination [170, 176, 177]. The relatively modest impact of deficiencies of ATM DDR factors on V(D)J recombination results from functional redundancy between these factors with the small XLF factor [178]. In this regard, despite the C-NHEJ role implied by the phenotype of XLF-deficient human patients and their cells, XLF deficiency in mice does not markedly impact V(D)J recombination in developing lymphocytes, despite leading to more general genomic instability and IR sensitivity [142]. However, combined deficiency for XLF and ATM, H2AX or 53BP1 leads to an essentially complete block in V(D)J recombination, along with more general DSB repair and CSR defects that indicate a nearly complete loss of C-NHEJ [178–180] (reviewed by [125]). Thus, in the absence of XLF, ATM and downstream DDR factors are required for C-NHEJ and vice versa, raising the possibility that variations in the expression of XLF in different tissues or individuals could contribute to differential manifestations of ATM deficiency. The nature of this functional redundancy is still being studied [125].
In humans, germline or somatic mutations in ATM have been associated with development of both B and T cell lymphomas [181, 182]. However, ATM deficiency in mice predisposes only to thymic lymphomas, but not B cell lymphomas [168]. ATM-deficient T cell lymphomas nearly universally have complex translocations involving the Trd (Tcrd) locus on chromosome 14 [23]. Notably, TRD translocations are the most common oncogenic translocation in human T-ALLs [27]. In mouse T cell lymphomas, the translocations involve formation of dicentric chromosomes downstream of RAG-induced Trd DSBs and subsequent amplification of chromosome 14 sequences along with potential Trd translocation-mediated deletion of a tumour suppressor on chromosome 12 [23]. Notably, TP53-deficient mice that harbour a homozygous germline mutation that leads to a C-terminal truncation in the RAG2 protein develop T cell lymphomas with essentially identical Trd-based translocations as observed in ATM deficient T cell lymphomas [109]. In this case, the RAG2 truncation is speculated to destabilize the post-cleavage V(D)J recombination complex similar to ATM deficiency [109].
Recently, several mouse models have been generated that develop peripheral mature B cell lymphomas in the context of ATM-deficiency [111]. These peripheral B cell lymphomas routinely harbour amplified Myc genes that result from RAG-initiated dicentric translocations between the IgH JH locus and sequences downstream of Myc [111] (see above). How RAG-initiated DSBs, which occur in progenitor B cells, could contribute to translocations and amplifications in mature B cells was an intriguing question. In this regard, prior studies suggested that RAG-generated breaks on chromosome 12 could be generated frequently due to the V(D)J joining defect associated with ATM deficiency and that the resulting telomere-deleted portions of chromosome 12 (IgH is near the telomere) could persist through development into mature B cells due to the G1 checkpoint defect associated with ATM deficiency [183]. Translocation cloning studies further revealed that such RAG-initiated DSBs in progenitor B cells are developmentally propagated into mature B cells in the form of dicentric chromosomes. These dicentrics then undergo BFB cycles in ATM-deficient mature B cells to generate new DSBs in a large region of chromosome 12 downstream of the IgH locus that robustly translocate to DSBs near the Myc gene and undergo BFB amplification of Myc [49]. In the latter context, these ATM-deficient mouse mature B cell lymphomas share similar mechanisms of Myc amplification to mouse pro-B cell lymphomas deficient for both C-NHEJ and TP53 [13, 16].
8 Three-Dimensional Genome Organization and Translocations
Our current understanding of genome organization is derived from early cytogenetic studies [184] and more recent chromosome conformation capture (3C)-based methods [38, 185, 186]. In interphase nuclei, chromosomes are non-randomly organized and each chromosome fills a nuclear space or territory. At the 1–10 Mb scale, active and inactive regions exist in separate compartments and conform to a fractal globule capable of dynamic local compaction across the length of the chromosome [37]. Smaller topologically associated domains (TADs) of approximately 1 Mb (“1 Mb domains”) exist within these compartments and comprise the majority of specific chromosomal contacts [185, 187, 188].
Translocations require DSBs at two independent sites and also require the two sites to be synapsed at the time they are broken. Various studies in yeast indicate increased chromatin mobility of sequences containing DSBs [42, 189]. Likewise, chromosomes with eroded telomeres, equivalent to DSBs, display 53BP1-dependent movement [43] and recent live cell tracking in mammalian systems which simultaneously follow interchromosomal I-SceI DSBs from many cells displayed non-directional saltatory motion with increased pairing of inter-chromosomal DSBs over time [44]. Thus, increased movements of DSBs may contribute to their synapsis. In the absence of enforced movements, the frequency of a translocation can, in simplistic terms, be considered proportional to the frequency of un-joined DSBs at site 1 times the frequency of DSBs at site 2 times the frequency at which these DSBs are synapsed: (DSBfreq1) × (DSBfreq2) × (Synapsisfreq) [5]. These principles apply to spatially proximal and distal sites, and both in the context of developmentally programmed events such as V(D)J recombination and CSR or through spontaneous illicit joining to genome-wide DSBs.
Translocation cloning studies have clearly demonstrated that highly frequent DSBs can drive recurrent translocations irrespective of their relative average position in the genome [38, 39, 45, 46, 49, 118]. This phenomenon derives from the finding that spatial heterogeneity in 3D genome organization allows most genomic sites to be proximal in some cells in a population [38]. Thus, highly-frequent DSBs can multiplicatively dominate the translocation frequency equation by greatly increasing the chance that two more rarely synapsed sites will be broken in cells in which they are synapsed [5, 38], allowing translocations across compartments that, on average, would be considered distal. In this regard, translocation cloning studies on G1-arrested ATM-deficient pro-B cell lines revealed that DSBs from eight independent I-SceI DSB bait sites on various chromosomes translocated recurrently to five different antigen receptor loci on different chromosomes (40 pairs of loci); thus, recurrent detection of dominant antigen receptor locus DSBs translocated to dominant I-SceI DSBs independent of chromosomal location due to 3D genome heterogeneity in the cell population (Fig. 3.2a) [38]. This explanation can also explain the dominance of AID hotspot DSBs in defining the translocation landscape of CSR activated B cells independent of chromosomal location (Fig. 3.3) [45, 46, 49].
In cases where particular DSBs are not dominant, synapsis frequency can play a much more dominant role in driving translocations. In experimental conditions, DSBs across the genome of ATM-deficient pro-B cell lines were normalized by treating cells with 5Gy of IR to induce, on average, 100 DSBs per cell. In such cases, antigen receptor locus DSBs were no longer so dominant and factors that increase synapsis frequency of two sequences became more influential (Fig. 3.2b) [38]. In accord with Hi-C mapping studies, such factors include placement in active versus inactive chromatin, associating with similarly-sized chromosomes, (more prominently) residing on the same chromosome in cis (demonstrated by SNP mapping), and (most prominently) lying within Mb domains in cis on a chromosome [5, 38]. A most striking feature of the greatly increased probability of sequences on the same chromosome lying proximal to each other was the finding that IR treatment of G1-arrested pro-B cells led the length of the cis-chromosome harbouring a bait DSB to become a major hotspot region for translocation of bait DSBs (Fig. 3.2b) [38].
Within a cis-chromosome, DSBs within Mb domains of a bait DSB have the highest frequency of translocation to the bait DSB [5, 38]. This phenomenon is thought to be due, at least in part, to sequences within such domains having a greater probability of being synapsed via Brownian (Langevin) motion [5, 38, 39]. In this regard, I-SceI and/or Cas9:gRNA DSBs separated by 100 kb translocated to each other within the IgH locus and within the Myc locus in B cells, T cells or fibroblasts at frequencies high enough to support substantial IgH CSR [39]. These findings suggest CSR may have evolved to employ the high frequency synapsis of sequences in Mb domains, as opposed to or in addition to more specialized synapsis mechanisms, with the high frequency of AID-initiated DSBs to helping to drive physiological levels of CSR [5, 39]. Such mechanisms have also been implicated in synapsis of V, D, and J segments during V(D)J recombination [40] and may contribute to recurrent interstitial deletions found in T-ALLs and other cancers (e.g., [27, 190]).
References
Gainor JF, Shaw AT (2013) Novel targets in non-small cell lung cancer: ROS1 and RET fusions. Oncologist 18(7):865–875
Kuppers R (2005) Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer 5(4):251–262
Mitelman F, Johansson B, Mertens F (2007) The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 7(4):233–245
Nambiar M, Kari V, Raghavan SC (2008) Chromosomal translocations in cancer. Biochim Biophys Acta 1786(2):139–152
Alt FW et al (2013) Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 152(3):417–429
Groffen J et al (1984) Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell 36(1):93–99
Lugo TG et al (1990) Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 247(4946):1079–1082
Nowell P, Hungerford D (1960) A minute chromosome in human chronic granulocytic leukemia [abstract]. Science 132:1497
Gostissa M, Alt FW, Chiarle R (2011) Mechanisms that promote and suppress chromosomal translocations in lymphocytes. Annu Rev Immunol 29:319–350
Robbiani DF, Nussenzweig MC (2013) Chromosome translocation, B cell lymphoma, and activation-induced cytidine deaminase. Annu Rev Pathol 8:79–103
Hnisz D et al (2013) Super-enhancers in the control of cell identity and disease. Cell 155(4):934–947
Alt FW et al (1978) Selective multiplication of dihydrofolate reductase genes in methotrexate-resistant variants of cultured murine cells. J Biol Chem 253(5):1357–1370
Difilippantonio MJ et al (2002) Evidence for replicative repair of DNA double-strand breaks leading to oncogenic translocation and gene amplification. J Exp Med 196(4):469–480
Papenhausen PR, Griffin S, Tepperberg J (2005) Oncogene amplification in transforming myelodysplasia. Exp Mol Pathol 79(2):168–175
Sharpless NE et al (2001) Impaired nonhomologous end-joining provokes soft tissue sarcomas harboring chromosomal translocations, amplifications, and deletions. Mol Cell 8(6):1187–1196
Zhu C et al (2002) Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell 109(7):811–821
McClintock B (1941) The stability of broken ends of chromosomes in Zea mays. Genetics 26(2):234–282
Murnane JP (2012) Telomere dysfunction and chromosome instability. Mutat Res 730(1-2):28–36
Albertson DG et al (2003) Chromosome aberrations in solid tumors. Nat Genet 34(4):369–376
Rikova K et al (2007) Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131(6):1190–1203
Albertson DG (2006) Gene amplification in cancer. Trends Genet 22(8):447–455
Munshi NC, Avet-Loiseau H (2011) Genomics in multiple myeloma. Clin Cancer Res 17(6):1234–1242
Zha S et al (2010) ATM-deficient thymic lymphoma is associated with aberrant tcrd rearrangement and gene amplification. J Exp Med 207(7):1369–1380
Burkhardt L et al (2013) CHD1 is a 5q21 tumor suppressor required for ERG rearrangement in prostate cancer. Cancer Res 73(9):2795–2805
Gutierrez A et al (2011) The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood 118(15):4169–4173
Krohn A et al (2014) Heterogeneity and chronology of PTEN deletion and ERG fusion in prostate cancer. Mod Pathol 27(12):1612–1620
Larmonie NS et al (2013) Breakpoint sites disclose the role of the V(D)J recombination machinery in the formation of T-cell receptor (TCR) and non-TCR associated aberrations in T-cell acute lymphoblastic leukemia. Haematologica 98(8):1173–1184
Gostissa M et al (2009) Chromosomal location targets different MYC family gene members for oncogenic translocations. Proc Natl Acad Sci U S A 106(7):2265–2270
Wang JH et al (2009) Mechanisms promoting translocations in editing and switching peripheral B cells. Nature 460(7252):231–236
Deriano L, Roth DB (2013) Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu Rev Genet 47:433–455
Shiloh Y, Ziv Y (2013) The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14(4):197–210
Zhang Y et al (2010) The role of mechanistic factors in promoting chromosomal translocations found in lymphoid and other cancers. Adv Immunol 106:93–133
Symington LS, Gautier J (2011) Double-strand break end resection and repair pathway choice. Annu Rev Genet 45:247–271
Chapman JR, Taylor MR, Boulton SJ (2012) Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 47(4):497–510
Boboila C, Alt FW, Schwer B (2012) Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol 116:1–49
Kim N, Jinks-Robertson S (2012) Transcription as a source of genome instability. Nat Rev Genet 13(3):204–214
Lieberman-Aiden E et al (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326(5950):289–293
Zhang Y et al (2012) Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell 148(5):908–921
Gostissa M et al (2014) IgH class switching exploits a general property of two DNA breaks to be joined in cis over long chromosomal distances. Proc Natl Acad Sci U S A 111(7):2644–2649
Lucas JS et al (2014) 3D trajectories adopted by coding and regulatory DNA elements: first-passage times for genomic interactions. Cell 158(2):339–352
Dion V, Gasser SM (2013) Chromatin movement in the maintenance of genome stability. Cell 152(6):1355–1364
Mine-Hattab J, Rothstein R (2012) Increased chromosome mobility facilitates homology search during recombination. Nat Cell Biol 14(5):510–517
Dimitrova N et al (2008) 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 456(7221):524–528
Roukos V et al (2013) Spatial dynamics of chromosome translocations in living cells. Science 341(6146):660–664
Chiarle R et al (2011) Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 147(1):107–119
Klein IA et al (2011) Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell 147(1):95–106
Haber JE (2006) Transpositions and translocations induced by site-specific double-strand breaks in budding yeast. DNA Repair (Amst) 5(9–10):998–1009
Zarrin AA et al (2007) Antibody class switching mediated by yeast endonuclease-generated DNA breaks. Science 315(5810):377–381
Hu J et al (2014) Developmental propagation of V(D)J recombination-associated DNA breaks and translocations in mature B cells via dicentric chromosomes. Proc Natl Acad Sci U S A 111(28):10269–10274
Kim YG, Cha J, Chandrasegaran S (1996) Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A 93(3):1156–1160
Christian M et al (2010) Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186(2):757–761
Jinek M et al (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096):816–821
Brunet E et al (2009) Chromosomal translocations induced at specified loci in human stem cells. Proc Natl Acad Sci U S A 106(26):10620–10625
Piganeau M et al (2013) Cancer translocations in human cells induced by zinc finger and TALE nucleases. Genome Res 23(7):1182–1193
Frock RL et al (2015) Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol 33(2):179–86
Vilenchik MM, Knudson AG (2003) Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci U S A 100(22):12871–12876
De Bont R, van Larebeke N (2004) Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis 19(3):169–185
Aguilera A, Garcia-Muse T (2013) Causes of genome instability. Annu Rev Genet 47:1–32
Branzei D, Foiani M (2010) Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 11(3):208–219
Hu J et al (2012) The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell 149(6):1221–1232
Lambert S, Carr AM (2013) Impediments to replication fork movement: stabilisation, reactivation and genome instability. Chromosoma 122(1–2):33–45
Ferber MJ et al (2004) Positioning of cervical carcinoma and Burkitt lymphoma translocation breakpoints with respect to the human papillomavirus integration cluster in FRA8C at 8q24.13. Cancer Genet Cytogenet 154(1):1–9
Morelli C et al (2002) Cloning and characterization of the common fragile site FRA6F harboring a replicative senescence gene and frequently deleted in human tumors. Oncogene 21(47):7266–7276
Sinclair PB et al (2004) A fluorescence in situ hybridization map of 6q deletions in acute lymphocytic leukemia: identification and analysis of a candidate tumor suppressor gene. Cancer Res 64(12):4089–4098
Barlow JH et al (2013) Identification of early replicating fragile sites that contribute to genome instability. Cell 152(3):620–632
Fong YW, Cattoglio C, Tjian R (2013) The intertwined roles of transcription and repair proteins. Mol Cell 52(3):291–302
Helmrich A, Ballarino M, Tora L (2011) Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell 44(6):966–977
Prado F, Aguilera A (2005) Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J 24(6):1267–1276
Haffner MC et al (2011) Transcription-induced DNA double strand breaks: both oncogenic force and potential therapeutic target? Clin Cancer Res 17(12):3858–3864
Li X, Manley JL (2005) Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 122(3):365–378
Shinkura R et al (2003) The influence of transcriptional orientation on endogenous switch region function. Nat Immunol 4(5):435–441
Yu K et al (2003) R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol 4(5):442–451
Curtin NJ (2012) DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer 12(12):801–817
Mehta A, Haber JE (2014) Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol 6(9):a016428
Asaithamby A, Chen DJ (2009) Cellular responses to DNA double-strand breaks after low-dose gamma-irradiation. Nucleic Acids Res 37(12):3912–3923
Joannides M, Grimwade D (2010) Molecular biology of therapy-related leukaemias. Clin Transl Oncol 12(1):8–14
Joannides M et al (2011) Molecular pathogenesis of secondary acute promyelocytic leukemia. Mediterr J Hematol Infect Dis 3(1), e2011045
Helmink BA, Sleckman BP (2012) The response to and repair of RAG-mediated DNA double-strand breaks. Annu Rev Immunol 30:175–202
Janz S (2006) Myc translocations in B cell and plasma cell neoplasms. DNA Repair (Amst) 5(9–10):1213–1224
Krangel MS (2009) Mechanics of T cell receptor gene rearrangement. Curr Opin Immunol 21(2):133–139
Nishana M, Raghavan SC (2012) Role of recombination activating genes in the generation of antigen receptor diversity and beyond. Immunology 137(4):271–281
Schatz DG, Baltimore D (2004) Uncovering the V(D)J recombinase. Cell 116(2 Suppl):S103–S106, 2 p following S106
Fugmann SD et al (2000) The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu Rev Immunol 18:495–527
Schatz DG, Swanson PC (2011) V(D)J recombination: mechanisms of initiation. Annu Rev Genet 45:167–202
Bossen C, Mansson R, Murre C (2012) Chromatin topology and the regulation of antigen receptor assembly. Annu Rev Immunol 30:337–356
Hewitt SL, Chaumeil J, Skok JA (2010) Chromosome dynamics and the regulation of V(D)J recombination. Immunol Rev 237(1):43–54
Desiderio S (2010) Temporal and spatial regulatory functions of the V(D)J recombinase. Semin Immunol 22(6):362–369
Ji Y et al (2010) The in vivo pattern of binding of RAG1 and RAG2 to antigen receptor loci. Cell 141(3):419–431
Matthews AG, Oettinger MA (2009) RAG: a recombinase diversified. Nat Immunol 10(8):817–821
Schatz DG, Ji Y (2011) Recombination centres and the orchestration of V(D)J recombination. Nat Rev Immunol 11(4):251–263
Taccioli GE et al (1993) Impairment of V(D)J recombination in double-strand break repair mutants. Science 260(5105):207–210
Alt FW, Baltimore D (1982) Joining of immunoglobulin heavy chain gene segments: implications from a chromosome with evidence of three D-JH fusions. Proc Natl Acad Sci U S A 79(13):4118–4122
Chaudhuri J et al (2007) Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol 94:157–214
Honjo T, Kinoshita K, Muramatsu M (2002) Molecular mechanism of class switch recombination: linkage with somatic hypermutation. Annu Rev Immunol 20:165–196
Jung D et al (2006) Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu Rev Immunol 24:541–570
Muramatsu M et al (2000) Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102(5):553–563
Peled JU et al (2008) The biochemistry of somatic hypermutation. Annu Rev Immunol 26:481–511
Rogozin IB, Diaz M (2004) Cutting edge: DGYW/WRCH is a better predictor of mutability at G:C bases in Ig hypermutation than the widely accepted RGYW/WRCY motif and probably reflects a two-step activation-induced cytidine deaminase-triggered process. J Immunol 172(6):3382–3384
Hackney JA et al (2009) DNA targets of AID evolutionary link between antibody somatic hypermutation and class switch recombination. Adv Immunol 101:163–189
Storb U (2014) Why does somatic hypermutation by AID require transcription of its target genes? Adv Immunol 122:253–277
Di Noia JM, Neuberger MS (2007) Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem 76:1–22
Stavnezer J, Guikema JE, Schrader CE (2008) Mechanism and regulation of class switch recombination. Annu Rev Immunol 26:261–292
Daniel JA, Nussenzweig A (2013) The AID-induced DNA damage response in chromatin. Mol Cell 50(3):309–321
Keim C et al (2013) Regulation of AID, the B-cell genome mutator. Genes Dev 27(1):1–17
Matthews AJ et al (2014) Regulation of immunoglobulin class-switch recombination: choreography of noncoding transcription, targeted DNA deamination, and long-range DNA repair. Adv Immunol 122:1–57
Wuerffel R et al (2007) S-S synapsis during class switch recombination is promoted by distantly located transcriptional elements and activation-induced deaminase. Immunity 27(5):711–722
Bednarski JJ, Sleckman BP (2012) Lymphocyte development: integration of DNA damage response signaling. Adv Immunol 116:175–204
Chaumeil J et al (2013) Higher-order looping and nuclear organization of Tcra facilitate targeted rag cleavage and regulated rearrangement in recombination centers. Cell Rep 3(2):359–370
Deriano L et al (2011) The RAG2 C terminus suppresses genomic instability and lymphomagenesis. Nature 471(7336):119–123
Dyer MJ et al (2010) Immunoglobulin heavy chain locus chromosomal translocations in B-cell precursor acute lymphoblastic leukemia: rare clinical curios or potent genetic drivers? Blood 115(8):1490–1499
Tepsuporn S et al (2014) Mechanisms that can promote peripheral B-cell lymphoma in ATM-deficient mice. Cancer Immunol Res 2(9):857–866
Wesemann DR et al (2013) Microbial colonization influences early B-lineage development in the gut lamina propria. Nature 501(7465):112–115
Merelli I et al (2010) RSSsite: a reference database and prediction tool for the identification of cryptic recombination signal sequences in human and murine genomes. Nucleic Acids Res 38(Web Server issue):W262–W267
Tsai AG, Lieber MR (2010) Mechanisms of chromosomal rearrangement in the human genome. BMC Genomics 11(Suppl 1):S1
Mendes RD et al (2014) PTEN microdeletions in T-cell acute lymphoblastic leukemia are caused by illegitimate RAG-mediated recombination events. Blood 124(4):567–578
Pasqualucci L et al (2001) Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature 412(6844):341–346
Liu M et al (2008) Two levels of protection for the B cell genome during somatic hypermutation. Nature 451(7180):841–845
Hakim O et al (2012) DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature 484(7392):69–74
Pavri R, Nussenzweig MC (2011) AID targeting in antibody diversity. Adv Immunol 110:1–26
Liu M, Schatz DG (2009) Balancing AID and DNA repair during somatic hypermutation. Trends Immunol 30(4):173–181
Pasqualucci L et al (2008) AID is required for germinal center-derived lymphomagenesis. Nat Genet 40(1):108–112
Meng F-L et al (2014) Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell 159(7):1538–1548
Lieber MR (2010) The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79:181–211
Sun J et al (2012) Human Ku70/80 protein blocks exonuclease 1-mediated DNA resection in the presence of human Mre11 or Mre11/Rad50 protein complex. J Biol Chem 287(7):4936–4945
Kumar V, Alt FW, Oksenych V (2014) Functional overlaps between XLF and the ATM-dependent DNA double strand break response. DNA Repair (Amst) 16:11–22
Buck D et al (2006) Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell 124(2):287–299
Du L et al (2012) Cernunnos influences human immunoglobulin class switch recombination and may be associated with B cell lymphomagenesis. J Exp Med 209(2):291–305
Ahnesorg P, Smith P, Jackson SP (2006) XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 124(2):301–313
Lu H et al (2007) Length-dependent binding of human XLF to DNA and stimulation of XRCC4.DNA ligase IV activity. J Biol Chem 282(15):11155–11162
Mahaney BL et al (2013) XRCC4 and XLF form long helical protein filaments suitable for DNA end protection and alignment to facilitate DNA double strand break repair. Biochem Cell Biol 91(1):31–41
Tsai CJ, Kim SA, Chu G (2007) Cernunnos/XLF promotes the ligation of mismatched and noncohesive DNA ends. Proc Natl Acad Sci U S A 104(19):7851–7856
de Villartay JP (2009) V(D)J recombination deficiencies. Adv Exp Med Biol 650:46–58
van der Burg M et al (2009) A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J Clin Invest 119(1):91–98
Frank KM et al (1998) Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature 396(6707):173–177
Gao Y et al (1998) A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell 95(7):891–902
Gu Y et al (2000) Defective embryonic neurogenesis in Ku-deficient but not DNA-dependent protein kinase catalytic subunit-deficient mice. Proc Natl Acad Sci U S A 97(6):2668–2673
Frank KM et al (2000) DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell 5(6):993–1002
Gao Y et al (2000) Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature 404(6780):897–900
Rooney S, Chaudhuri J, Alt FW (2004) The role of the non-homologous end-joining pathway in lymphocyte development. Immunol Rev 200(1):115–131
Frappart PO et al (2009) Recurrent genomic alterations characterize medulloblastoma arising from DNA double-strand break repair deficiency. Proc Natl Acad Sci U S A 106(6):1880–1885
Yan CT et al (2006) XRCC4 suppresses medulloblastomas with recurrent translocations in p53-deficient mice. Proc Natl Acad Sci U S A 103(19):7378–7383
Li G et al (2008) Lymphocyte-specific compensation for XLF/cernunnos end-joining functions in V(D)J recombination. Mol Cell 31(5):631–640
Boboila C et al (2010) Alternative end-joining catalyzes robust IgH locus deletions and translocations in the combined absence of ligase 4 and Ku70. Proc Natl Acad Sci U S A 107(7):3034–3039
Ferguson DO et al (2000) The interplay between nonhomologous end-joining and cell cycle checkpoint factors in development, genomic stability, and tumorigenesis. Cold Spring Harb Symp Quant Biol 65:395–403
Wang JH et al (2008) Oncogenic transformation in the absence of Xrcc4 targets peripheral B cells that have undergone editing and switching. J Exp Med 205(13):3079–3090
Boulton SJ, Jackson SP (1996) Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J 15(18):5093–5103
Kabotyanski EB et al (1998) Double-strand break repair in Ku86- and XRCC4-deficient cells. Nucleic Acids Res 26(23):5333–5342
Boboila C et al (2010) Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. J Exp Med 207(2):417–427
Yan CT et al (2007) IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 449(7161):478–482
Stephens PJ et al (2009) Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 462(7276):1005–1010
Zhang Y, Rowley JD (2006) Chromatin structural elements and chromosomal translocations in leukemia. DNA Repair (Amst) 5(9–10):1282–1297
Grabarz A et al (2012) Initiation of DNA double strand break repair: signaling and single-stranded resection dictate the choice between homologous recombination, non-homologous end-joining and alternative end-joining. Am J Cancer Res 2(3):249–268
Guirouilh-Barbat J et al (2007) Defects in XRCC4 and KU80 differentially affect the joining of distal nonhomologous ends. Proc Natl Acad Sci U S A 104(52):20902–20907
Komori T, Sugiyama H (1993) N sequences, P nucleotides and short sequence homologies at junctional sites in VH to VHDJH and VHDJH to JH joining. Mol Immunol 30(16):1393–1398
Stavnezer J et al (2010) Mapping of switch recombination junctions, a tool for studying DNA repair pathways during immunoglobulin class switching. Adv Immunol 108:45–109
Simsek D, Jasin M (2010) Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat Struct Mol Biol 17(4):410–416
Ghezraoui H et al (2014) Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol Cell 55(6):829–842
Perlman S, Becker-Catania S, Gatti RA (2003) Ataxia-telangiectasia: diagnosis and treatment. Semin Pediatr Neurol 10(3):173–182
Choi M et al (2012) Attractor landscape analysis reveals feedback loops in the p53 network that control the cellular response to DNA damage. Sci Signal 5(251):ra83
Nussenzweig A, Nussenzweig MC (2010) Origin of chromosomal translocations in lymphoid cancer. Cell 141(1):27–38
Scully R, Xie A (2013) Double strand break repair functions of histone H2AX. Mutat Res 750(1–2):5–14
Al-Hakim A et al (2010) The ubiquitous role of ubiquitin in the DNA damage response. DNA Repair (Amst) 9(12):1229–1240
Panier S, Boulton SJ (2014) Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 15(1):7–18
Sirbu BM, Cortez D (2013) DNA damage response: three levels of DNA repair regulation. Cold Spring Harb Perspect Biol 5(8):a012724
Zimmermann M, de Lange T (2014) 53BP1: pro choice in DNA repair. Trends Cell Biol 24(2):108–117
Bassing CH, Alt FW (2004) The cellular response to general and programmed DNA double strand breaks. DNA Repair (Amst) 3(8–9):781–796
Franco S, Alt FW, Manis JP (2006) Pathways that suppress programmed DNA breaks from progressing to chromosomal breaks and translocations. DNA Repair (Amst) 5(9-10):1030–1041
Lavin MF (2008) Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol 9(10):759–769
Xu Y et al (1996) Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev 10(19):2411–2422
Bassing CH et al (2002) Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci U S A 99(12):8173–8178
Franco S et al (2006) H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell 21(2):201–214
Bassing CH et al (2003) Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell 114(3):359–370
Celeste A et al (2003) H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 114(3):371–383
Ramiro AR et al (2006) Role of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature 440(7080):105–109
Manis JP et al (2004) 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol 5(5):481–487
Ward IM et al (2004) 53BP1 is required for class switch recombination. J Cell Biol 165(4):459–464
Celeste A et al (2002) Genomic instability in mice lacking histone H2AX. Science 296(5569):922–927
Zha S et al (2011) ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature 469(7329):250–254
Liu X et al (2012) Overlapping functions between XLF repair protein and 53BP1 DNA damage response factor in end joining and lymphocyte development. Proc Natl Acad Sci U S A 109(10):3903–3908
Oksenych V et al (2012) Functional redundancy between repair factor XLF and damage response mediator 53BP1 in V(D)J recombination and DNA repair. Proc Natl Acad Sci U S A 109(7):2455–2460
Gumy-Pause F, Wacker P, Sappino AP (2004) ATM gene and lymphoid malignancies. Leukemia 18(2):238–242
McKinnon PJ (2012) ATM and the molecular pathogenesis of ataxia telangiectasia. Annu Rev Pathol 7:303–321
Callen E et al (2007) ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. Cell 130(1):63–75
Roix JJ et al (2003) Spatial proximity of translocation-prone gene loci in human lymphomas. Nat Genet 34(3):287–291
Dixon JR et al (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485(7398):376–380
Nagano T et al (2013) Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502(7469):59–64
Dekker J, Marti-Renom MA, Mirny LA (2013) Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet 14(6):390–403
Gibcus JH, Dekker J (2013) The hierarchy of the 3D genome. Mol Cell 49(5):773–782
Dion V et al (2012) Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat Cell Biol 14(5):502–509
Onozawa M, Aplan PD (2012) Illegitimate V(D)J recombination involving nonantigen receptor loci in lymphoid malignancy. Genes Chromosom Cancer 51(6):525–535
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Frock, R.L., Hu, J., Alt, F.W. (2015). Mechanisms of Recurrent Chromosomal Translocations. In: Rowley, J., Le Beau, M., Rabbitts, T. (eds) Chromosomal Translocations and Genome Rearrangements in Cancer. Springer, Cham. https://doi.org/10.1007/978-3-319-19983-2_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-19983-2_3
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-19982-5
Online ISBN: 978-3-319-19983-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)