
Abstract
Dermatomyositis (DM) is a systemic autoimmune disease characterized by inflammation of multiple organs, most commonly the skin, muscle and lungs. This disease poses a challenge to clinicians because of its rarity, diverse clinical presentations, and variable organ involvement. Depending on the clinical manifestations, patients with DM can present first to either rheumatologists, dermatologists, or neurologists, among other specialists. In this chapter we review the manifestations of dermatomyositis and associated differential diagnosis by organ system. Rheumatologists and dermatologists approach this disorder with unique perspectives, both of which are often necessary for optimal care of the patient. The purpose of this chapter is to highlight the vital contributions that each specialty can make to patient care in this complex disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
FormalPara Key Points-
Dermatomyositis (DM) is a systemic autoimmune disease affecting the skin, muscle, and lungs, and is associated with a malignancy in 10–20% of cases.
-
Interstitial lung disease (ILD) is a major source of morbidity and mortality in DM, with increased risk conferred by the presence of anti-synthetase and anti-MDA5 antibodies.
-
Malignancy screening with computed tomography scans of the chest, abdomen and pelvis may be of benefit to detect occult cancers in patients with DM that may be missed on routine age-appropriate screening.
-
Multidisciplinary collaboration between rheumatology and dermatology, among other specialties, is important to assess all potentially involved organs and select an appropriate treatment plan.
-
Treatment of cutaneous DM can be challenging and discordant with treatment for the muscle disease. Multiple agents may be necessary to achieve complete remission; the risks and benefits each agent should be considered carefully given the potentially prolonged treatment course.
Interdisciplinary Introduction
Dermatomyositis (DM) is a systemic autoimmune disease characterized by inflammation of multiple organs, most commonly the skin, muscle and lungs. This disease poses a challenge to clinicians because of its rarity, diverse clinical presentations, and variable organ involvement. Depending on the clinical manifestations, patients with DM can present first to either rheumatologists, dermatologists, or neurologists, among other specialists.
A timely diagnosis is imperative, not only to prevent internal organ damage from the disease itself, but also to initiate appropriate malignancy screening, given the increased risk of cancer around the time of first symptoms [1, 2]. In this chapter we review the manifestations of DM and associated differential diagnosis by organ system. However, making the diagnosis of DM necessitates consideration of the complete clinical context in which the patient presents, including the history of present illness, contributory past medical history, review of systems, physical examination, laboratory analysis, biopsy results, and imaging or electromyographic studies.
Another important goal in DM is to select therapeutic agents that target manifestations in multiple organs to gain control of the disease while minimizing the risks to the patient. Patients with DM are therefore best served by multidisciplinary collaboration. Rheumatologists and dermatologists approach this disorder with unique perspectives, both of which are often necessary for optimal care of the patient. The purpose of this chapter is to highlight the vital contributions that each specialty can make to patient care in this complex disease.
Epidemiology and Risk Factors
Age of onset in DM has a bimodal distribution, occurring in two peaks, one at 5–14 years and the other at 45–64 years of life. The female to male ratio is 2–3:1. There are insufficient epidemiologic data, however, to determine the true incidence and prevalence of DM. Regional variation and differences in case ascertainment methods have complicated efforts to do so [3, 4].
One study based on data from Olmstead County, Minnesota, estimated population age- and sex-adjusted prevalence at 21.42 per 100,000 persons (95% CI, 13.07–29.77) and incidence at 9.63 per million per decade (95% CI 6.09–13.17) [5]. Another estimate, based on review of hospital discharge diagnosis codes in Spain from 1997–2004, found a lower annual incidence, estimated at 4.9 cases/million/year (95% CI, 4.7–5.2). Lower still, the annual incidence determined in a review of one million records from a health insurance database between 2005–2009 was 0.7 cases per 100,000 person-years (95% CI, 0.5–1.0) [6].
The regional variation noted in DM incidence and prevalence may relate in part to geographic differences in risk factors for the disease. For example, intensity of ultraviolet radiation may influence the development and modulate the expression of DM. In the Northern Hemisphere, the relative incidence of DM as compared to polymyositis displays a latitudinal gradient, with the greatest incidence of DM in Athens, Greece and lowest incidence in Reykjavik, Iceland [7]. This finding was replicated in another study across 14 different countries, which found the highest relative proportion of DM in Guatemala and the lowest in Glasgow, Scotland [8]. These authors concluded that surface ultraviolet radiation was the major geoclimatic factor associated with the relative proportion of DM. Similarly, in the United States, Love et al. found a positive association between the annual ultraviolet index in seven U.S. regions and the proportion of patients with DM, along with the relative frequency of anti-Mi-2 autoantibodies in women [9].
On the other hand, Marcelo Petri et al. found a significant difference in the prevalence of anti-Mi-2 antibodies in Mexico City (26/44 DM patients, or 59%) versus Guadalajara (0/17 DM patients), cities that have comparable surface UV radiation, suggesting that additional genetic or environmental factors determine the autoimmune phenotype [10]. Another study found that the prevalence of juvenile DM patients with anti-NXP2 antibodies was inversely correlated with surface UV exposure, suggesting that, at least for some DM patients, UV is not an epidemiologic risk factor [11].
Classification
DM is currently classified as an idiopathic, inflammatory myopathy. In epidemiologic studies, it is often grouped with other inflammatory myopathies, including polymyositis. In 1975, Bohan and Peter empirically defined diagnostic and classification criteria for DM and polymyositis [12, 13]. They divided DM into four groups: idiopathic DM, juvenile DM, DM associated with cancer, and DM associated with other connective tissue diseases.
Since that time, efforts have been made to subclassify patients. The term “amyopathic dermatomyositis” was coined by Carl Pearson in 1979 to describe patients with the classic cutaneous manifestations of DM but minimal to no evidence of muscle involvement [14]. In 1991, Euwer and Sontheimer proposed the designation of “clinically amyopathic dermatomyositis” (CADM) to describe patients with the hallmark skin findings of DM but no clinical evidence of myopathy on physical examination or muscle enzyme analysis for at least 6 months after disease onset [15].
In 2002, Sontheimer proposed cutaneous criteria for establishing a diagnosis of CADM. These included three major criteria and 14 minor criteria. The major criteria were as follows: the pathognomonic heliotrope sign (violaceous erythema on the upper eyelids), Gottron’s papules (papules overlying the metacarpophalangeal and interphalangeal joints) and Gottron’s sign (erythema overlying the knees, elbows, or interphalangeal joints) (Table 4.1) [16]. The presence of 2 major criteria, or one 1 major criterion and 2 minor criteria, in addition to skin biopsy showing histopathologic changes consistent with DM, was required to establish a diagnosis [16]. Although these criteria have not been formally validated, they are often cited in studies as inclusion criteria for CADM patients.
We value the CADM classification criteria for formally recognizing the significant subset of roughly 20% [5] of DM patients who do not have overt muscle disease and would therefore otherwise be excluded from a clinical diagnosis of DM as well as from clinical trials and translational studies for DM patients [17]. However, existing data do not support the concept that CADM patients uniformly differ from classic DM patients in any other clinical or pathologic manner. These CADM patients have similar skin manifestations (both clinically and histologically), as well as an increased risk for interstitial lung disease (ILD) and internal malignancy. Any differences that do exist between these two subgroups may be largely accounted for by differences in autoantibody profile (Table 4.2) and not simply the presence or absence of clinical myositis.
Other subclassification schemes have been proposed based on serologies. In 1991, Love et al. suggested that myositis-specific antibodies may define groups of patients who share certain clinical features [18]. Approximately 80% of DM patients will have a detectable myositis-specific antibody, including transcriptional intermediary factor 1-gamma (TIF1-γ), nuclear matrix protein 2 (NXP2), melanoma differentiation-associated gene 5 (MDA5), small ubiquitin-like modifier activating enzyme (SAE), Mi-2, Jo-1 and the other anti-synthetase antibodies. These myositis-specific autoantibodies have been associated with distinct clinical subsets and appear to be useful in the diagnosis and classification of DM (Table 4.2). Dr. Manabu Fujimoto used results from Japanese studies to create an autoantibody-based classification of DM [19]. With improved phenotyping of the myositis-specific antibodies with respect to disease features and clinical course as well as increasing availability of testing for myositis-specific antibodies, this classification method may add value to existing definitions by facilitating improved prognostication, targeted screening and potentially tailored therapy.
Clinical Presentation
Skin Disease
Classic Features
A careful history will elicit common features of skin disease in DM. In some patients, onset of disease is associated with a recent history of significant UV exposure. Patients may also describe sensitivity to sunlight. Pruritus is typical and further questioning often reveals a subjective dysesthetic component to the itch, often described as a sensation of skin tightness, burning, or crawling. This sensation is especially common on the scalp. Patients complain of swelling of the eyelids, which is frequently misdiagnosed as allergic contact dermatitis or angioedema. Additionally, patients will describe the eruption to be chronic, relapsing, and progressive.
On physical examination, skin changes in DM are distributed on archetypal regions on the body (Table 4.1). Of note, many of these are not necessarily in areas of UV exposure (so-called “photodistributed”). In order to improve the sensitivity of the examination, proper patient positioning and exam room lighting are critical. Overhead lighting tends to cast shadows over the brow, nose, and chin, which may conceal faint erythema or telangiectasias on the body surfaces inferiorly. Also, we find that bright direct lighting often obscures the subtle color changes seen in DM skin. Examination with natural lighting is recommended whenever possible.
The distinction between disease activity and damage in DM skin is critical for clinical decision-making, so that immunosuppressive treatments are not erroneously utilized for skin damage. In addition to itch, cutaneous disease activity is characterized by violaceous erythema, induration (papules or plaques), scale, or ulceration. Epidermal and vascular damage, by contrast, may be evident on examination as telangiectasias, atrophy, and dyspigmentation.
Although erythema is often an important sign of activity, it may also represent damage, and thus care must be taken not to escalate therapy based solely on the presence of erythema. Telangiectasias , for example, cause erythema but are a sign of damage. Livedo reticularis, a vascular phenomenon associated with DM, may likewise be confused with active erythema in DM. Careful identification of the netlike pattern of livedo and presence on photoprotected surfaces may help avoid this confusion. Skin damage due to DM may also be reflected in reticulated patches, but these are more brown, post-inflammatory hyperpigmented patches in areas of prior disease activity.
When substantial inflammation has been present, patients may present with a distinctive and pathognomonic pattern comprised of reticulated, sometimes atrophic, white macules, adjacent to erythema and/or telangiectasias, which we call “red on white” (Fig. 4.1a–c). The scalp and the skin along the bitemporal hairline (Fig. 4.2) are frequent sites of involvement, though this pattern does not necessarily occur only in sun-exposed areas. It is becoming increasingly clear that many of these red on white patches do not necessarily represent permanent damage, as these lesions may slowly resolve with time, even when atrophy is present. However, this morphology can be a useful diagnostic clue, as it does not seem to be associated with other connective tissue diseases, such as cutaneous lupus, but is more specific to DM.

Pathognomonic “red on white” pattern of reticulated, sometimes atrophic, white macules adjacent to erythema and/or telangiectasias, seen on the right upper back (a), central chest (b) and right lateral upper arm (c)

“Red on white” plaques confluent over the frontal hairline and hair-bearing scalp
Longstanding disease activity, typically in sun-exposed areas, results in more significant damage, characterized by atrophy, hypopigmentation, hyperpigmentation and telangiectasias, a constellation of findings that is termed poikiloderma. Poikiloderma is a late manifestation in DM and is not diagnostically specific, as it may result from many other acquired and congenital diseases, including cutaneous lupus, chronic actinic damage (poikiloderma of Civatte), poikilodermatous mycosis fungoides (poikiloderma vasculare atrophicans), borrelia infection (acrodermatitis chronic atrophicans), chronic radiation dermatitis, and graft versus host disease.
Two important signs have been proposed to be pathognomonic for DM. First, violaceous to pink papules over the dorsal proximal interphalangeal and metacarpophalangeal joints are termed Gottron’s papules (Fig. 4.3). These may display the same range of features seen elsewhere on DM skin, including poikiloderma, atrophy, hypopigmentation, hyperkeratosis or ulceration. Second, Gottron’s sign is characterized by symmetric, macular, violaceous erythema over the interphalangeal joints, olecranon processes (Fig. 4.4), patellas, and medial malleoli.

Gottron’s papules : violaceous papules overlying the dorsal proximal interphalangeal and metacarpophalangeal joints

Gottron’s sign : symmetric red patches on the elbows
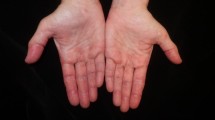
Other characteristic hand findings in DM include hyperkeratosis and fissuring along the lateral second and third digits (Fig. 4.5), which may be subtle; the rough texture is often evident only with palpation. In patients with anti-synthetase antibodies, digital hyperkeratosis and fissuring is often more extensive and usually also affects the palmar fingers and fingertips (so-called “mechanic’s hands”).

Lateral digit hyperkeratosis: pink papules with rough white scale on the bilateral lateral second digits
Involvement of the scalp with erythema, fine scale, and pruritus is one of the most ubiquitous cutaneous manifestations in DM. Scalp pruritus may be severe, have a burning or dysesthetic quality and significantly reduce the patient’s quality of life. Subtle erythema may be perceptible on the vertex scalp, along the hair part, or on the borders of the hairline, even when the remainder of the cutaneous disease is quiescent.
The periorbital skin is frequently involved. Patients may present with violaceous patches on the upper eyelids (the heliotrope sign), frequently with associated edema that may be minimal to severe. In addition, erythema of the lateral canthi, medial canthi and adjacent nasal sidewalls is common (Fig. 4.6a, b). The rest of the face may have diffuse erythema or may be uninvolved.

(a, b) : Red and violaceous macules on the lateral canthi, medial canthi and nasal sidewalls, commonly seen in association with the heliotrope sign
Areas of involvement on the trunk may include the upper back, posterior neck, posterior shoulders (shawl sign), and posterior upper arms. Confluent violaceous erythema on the sun-exposed areas of the lower anterior neck and anterior chest is termed the V-neck sign. In addition, linear patches or urticarial plaques (flagellate erythema), possibly due to excoriation or imprinting from clothing or bed sheets, may be present on the back or upper chest. Biopsy of flagellate erythema shows typical histopathology of DM [20].
We have frequently observed reticulated, violaceous patches on the lateral areas of the flanks and lower back. Violaceous erythema and poikiloderma may also affect the lateral hips and lateral thighs (Holster sign). This finding, consisting of small (1 mm), violaceous, folliculocentric macules or less likely papules, may be confused with the more common condition keratosis pilaris. However, the violaceous color and typically macular nature of the eruption, as well as the distribution typically not involving areas typical for keratosis pilaris (e.g., upper, outer arms) helps to delineate this finding to DM.
The oral mucosa may also be involved in DM. Red on white patches may be observed, particularly on the hard palate and surrounding gingival mucosa. (Fig. 4.7a). When this occurs in a distinctly oval pattern at the junction of the hard and soft palate at the midline, it is termed the “ovoid palatal patch” (Fig. 4.7b). This latter finding appears to occur most frequently in the subset of DM patients with anti-transcriptional intermediary factor 1 gamma (TIF1-γ) antibodies [21]. Biopsies from these lesions demonstrate interface mucositis, consistent with typical findings in DM. In our experience, activity of these mucosal changes seems to mirror the activity of the cutaneous disease: the hard palate changes appear with mild disease activity and fade late in the course as definitive control of the cutaneous disease is achieved. Oral manifestations of DM may be confused with the oral findings seen in discoid lupus or lichen planus, but their consistent localization to the center of the hard palate may aid in the diagnosis of DM when other cutaneous features are non-diagnostic.

“Red on white” reticulated patch (a) and ovoid palatal patch (b) seen on the posterior hard palate
The nailfolds are another classic site of involvement for DM. Nailfold capillary changes provide a window into the disease’s hallmark microangiopathy. When pronounced and easily visualized with the naked eye, these nailfold capillary changes can be highly suggestive of DM over other connective tissue disorders. The classic findings include, red, edematous, often tender, proximal nailfolds. Capillary loops are ramified and dilated, with intervening pale to white avascular areas characterized by capillary dropout, as well as cuticular hemorrhages and elongated, ragged cuticles (Fig. 4.8a, b). These changes are a sign of ongoing cutaneous disease activity [22], though persistently ramified capillaries may represent damage in longstanding DM [23].

(a, b) Dilated capillary loops, cuticular hemorrhages, and intervening yellow to white avascular areas, with elongated and ragged cuticles
The vasculopathy that plays an important role in cutaneous DM may sometimes become clinically prominent, causing ulceration, Degos-like lesions, and livedo reticularis. Degos-like lesions are most common on the dorsal fingers and are characterized by a depressed, porcelain-white papule with a rim of bright red erythema. The clinical significance of these lesions in DM is unknown. Ulceration may be present in 30% of patients and typically affects the skin over the extensor joint surfaces, the digital pulp, or the periungal skin [24]. In our U.S. cohort, ulceration was associated with anti-MDA5 antibodies, although it may be noted in other contexts as well. DM patients with anti-MDA5 antibodies display a more severe vasculopathic phenotype.
Calcinosis of the dermis, subcutaneous tissue, fascia, or muscle is a late manifestation of DM, typically involving the trunk, proximal extremities, or areas of previous disease activity. The prevalence of calcinosis is 20% in adult DM [25] and up to 40% in juvenile DM [26]. Calcinosis also occurs more rapidly after disease onset in juvenile DM compared with adult DM (2.9 years vs. 7.9 years, respectively) [27]. In juvenile DM, risk factors for development of calcinosis include longer disease duration, younger age of disease onset, sustained disease activity, and internal organ involvement [28, 29]. Calcinosis is most frequent on the proximal extremities, buttocks and trunk in DM, an important distinguishing factor from the calcinosis seen in systemic sclerosis, which typically affects the digits and elbows [27]. In both juvenile and adult DM, the presence of anti-nuclear matrix protein 2 (NXP2) antibodies are associated with an increased risk of calcinosis [30, 31]. Calcinosis is also commonly seen in the anti-MDA5 subset (especially those patients with longstanding disease) [31], which is associated with known vasculopathy. In adults with DM, fingertip ulceration has been associated with calcinosis [31], suggesting that vascular insufficiency or damage may be involved in the pathogenesis of calcinosis.
Panniculitis may occur in DM and typically affects the buttocks, trunk, and proximal extremities. It may progress to calcinosis and/or lipoatrophy [32]. Histopathology shows a lobular panniculitis, but lipomembranous change as seen in lupus panniculitis may be present, and septal thickening may be seen, as in deep morphea. Panniculitis appears to be more common among the anti-MDA5 DM group.
Non-scarring alopecia may occur in DM, either secondary to scalp inflammation or due to telogen effluvium. This manifestation is particularly common in the anti-MDA5 group (Fig. 4.9a, b). In this group, alopecia closely mirrors the cutaneous disease activity.

(a, b) Non-scarring alopecia , seen in DM patients with anti-MDA5 antibodies
Rare Presentations of Cutaneous DM
There is a subset of DM patients with overlapping features of both psoriasis and DM. Their skin disease may show psoriasiform, well-demarcated, thick plaques over the MCP and PIP joints, elbows and knees, along with dilated nailfold capillaries. Skin biopsies reveal both epidermal hyperplasia and interface dermatitis. Some affected patients have a history of psoriasis, and it is unclear whether these psoriasiform lesions represent concomitant psoriasis or a psoriasiform manifestation of DM. DM and psoriasis share similar interferon gene signatures, which could at least partly explain this presentation[33, 34].
Other rare presentations of DM include subcutaneous edema in the distal extremities and generalized edema, both of which may portend more severe muscle inflammation or aggressive disease [35, 36]. Lastly, DM may rarely present with erythroderma, in which 90% or more of the body surface is involved with confluent erythema.
Cutaneous Signs of Interstitial Lung Disease
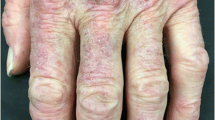
Hyperkeratosis and fissuring along the ulnar aspect of the thumb and radial aspect of the index and middle fingers were first described as mechanic’s hands by Stahl et al. in 1979, in a series of eight patients with inflammatory myopathies [37]. In 1991, Love et al. found that myositis patients with anti-synthetase antibodies were more likely to have mechanic’s hands, ILD, fever, and arthritis [18]. In 2012, Sato et al. noted an increased prevalence of ILD among DM patients with mechanic’s hands (7/9 patients, 78%) as compared to those without mechanic’s hands (12/30 patients, 40%) [38], suggesting that mechanic’s hands may be a cutaneous clue to the presence of ILD. We have observed that additional features of mechanic’s hands in anti-synthetase antibody positive DM patients include hyperkeratosis and fissuring of the distal fingertips and palmar fingers.
Japanese case series have suggested that cutaneous ulceration is associated with lung disease in DM [38,39,41]. In our U.S. cohort, we did not find an association between ulceration and ILD [24]. However, in the presence of anti-MDA5 antibodies, cutaneous ulceration was associated with a markedly increased odds of having ILD (OR 35.19, 95% CI 3.55–3.49, p = 0.0024) [24]. Thus, the significance of ulceration in DM may depend upon the autoantibody status of the patient. Anti-MDA5 antibodies are more commonly seen in Japanese DM patients, and this may explain the data associating ulceration with lung disease in the patients from Japan. In addition, in the anti-MDA5 patients, we have found a correlation between the severity of the ulceration and the severity of ILD (unpublished data). Worsening cutaneous ulceration in a patient with anti-MDA5 DM, therefore, may be a cutaneous sign of worsening ILD.
Because anti-MDA5 antibodies are strongly associated with lung disease, other cutaneous features associated with this serologic group should also raise suspicion for underlying lung inflammation. These include the frankly eroded papules (“inverse Gottron’s papules”) on the palmar fingers that are virtually pathognomonic for patients with anti-MDA5 antibodies (Fig. 4.10). Patients with these antibodies may also have violaceous reticular erythema over the palmar surfaces and digital pulps. Severe alopecia may also be a sign of anti-MDA5 antibodies.

Eroded papules on the palmar fingers overlying the interphalangeal joints in a DM patient with anti-MDA5 antibodies
Cutaneous Signs of Internal Malignancy
Cutaneous necrosis and cutaneous small vessel vasculitis have been reported to be associated with paraneoplastic DM [42, 43]. However, necrosis causing ulceration should raise suspicion for other conditions, such as ILD, as reviewed above. Additionally, vasculitis has been reported in cases not associated with malignancy [43,44,45,46,48]. Acquired ichthyosis, manifesting as a paraneoplastic dermatosis, has been described in association with malignancy and DM [48,49,51].
Histopathology
Biopsy of involved skin in DM classically shows an interface dermatitis, basement membrane thickening, epidermal atrophy, perivascular lymphocytic infiltrate, increased dermal mucin and vascular ectasia. However, many of these features are often subtle or absent, including the interface dermatitis. Smith et al. reviewed 40 DM skin biopsies and noted that when interface dermatitis was absent (20% of cases), increased dermal mucin was always present [52]. Magro et al. described supervening dermal sclerosis in DM as a sign of more severe endothelial damage and potentially a more severe disease course [53].
Routine histopathology cannot reliably be used to distinguish cutaneous lupus from DM. Likewise, direct immunofluorescence is not always reliable. The presence of immunoreactants along the dermoepidermal junction, a finding often observed in sun-exposed lesional skin in acute cutaneous lupus, has been variably reported in lesional skin in DM. Black et al. found that 65% (19 of 29) of lesional biopsies from DM patients demonstrated positive IgM, IgG or C3 at the basement membrane[54]. Magro et al. suggested a more stringent definition of the DIF findings seen in lupus (sometimes called the lupus band), requiring either a continuous, moderately intense band of IgM and/or presence of IgG (interrupted or continuous) at the dermoepidermal junction [55]; using this definition, DM skin, unlike that from lupus patients, virtually never displayed a positive result. Using these criteria, DIF may be a useful test in distinguishing acute cutaneous lupus from DM. However, it is important to note that DIF may also be negative in lupus patients.
Building on the observations of Mascaró Jr. et al. [56], Magro et al. suggest that the presence of membrane attack complex (C5b-C9) deposits around the dermoepidermal junction and vessels is a characteristic finding in DM. When coupled with the absence of direct immunofluorescence findings seen in lupus, this finding yielded a sensitivity for the diagnosis of DM of 93.5% and a specificity of 78.3% [55]. The absence of lupus direct immunofluorescence findings alone or positive C5b-C9 deposition alone yielded specificities of 64.5% and 78.6%, respectively [55]. The utility of immunofluorescence testing in the diagnosis of DM warrants further evaluation.
Differential Diagnosis
A common differential diagnosis for the cutaneous findings of DM includes cutaneous lupus, psoriasis, acne rosacea, phototoxic or photoallergic drug eruption, atopic dermatitis, and mycosis fungoides.
As compared to cutaneous lupus erythematosus (CLE), the erythema is more violaceous in color in DM than in CLE and has a different distribution, affecting the extensor joint surfaces. Erythema over the dorsal fingers may be seen in acute or subacute cutaneous lupus, but it is typically more prominent over the hair-bearing interphalangeal skin, with relative sparing of the PIP joints, although there are many instances bearing exception to this rule. Periungual erythema and nailfold capillary changes can be seen in both CLE and DM, including dilation, hemorrhage and dropout; however, in our experience, severe capillary changes are more common in DM. Although marked periorbital edema and erythema has been described in discoid lupus and can mimic the heliotrope rash, these findings tend to be unilateral and affect the lower eyelids [56,57,59].
The extensor surfaces and the scalp may be involved in both psoriasis and DM; however, the heliotrope, V-neck and shawl signs should be absent in psoriasis. Psoriasis also tends to present with more abundant, thick, silvery to whitish scale than is seen in DM. Scalp tightness and dysesthesia are typically absent in psoriasis. As a caveat, as reviewed, DM and psoriasis may present as an overlap syndrome, including clinical and histologic features of both psoriasis and DM.
Facial involvement in erythematotelangiectatic rosacea is most prominent on the mid-cheeks, chin and glabella, and it tends to spare the upper eyelids, factors that can distinguish it from DM. In neurogenic rosacea, which is a rare variant, there may be intense burning symptoms out of proportion to examination findings. These symptoms may resemble the dysesthesia seen in DM [60, 61]; however, facial disease in DM tends not to be highly symptomatic.
Photoprotected sites are not typically involved phototoxic or photoallergic eruptions, whereas these sites may be involved in DM, e.g., the holster sign on the lateral hips and Gottron’s sign on the knees. Nailfold capillary changes are absent in phototoxic or photoallergic eruptions. When a photoeruption becomes chronic, as in chronic actinic dermatitis, the skin becomes lichenified, whereas the chronically involved skin in DM tends to become atrophied.
Periorbital and scalp erythema, edema, and scale may be present in atopic dermatitis, but the eczematous plaques should also involve flexural areas. Additionally, the eruption in atopic dermatitis becomes more lichenified with time, and nailfold capillary changes are absent.
Although DM patients may have puffy fingers and nailfold capillary changes similar to those seen in systemic sclerosis, facial involvement with microstomia is not observed in DM. The dyspigmentation on the face and trunk seen in systemic sclerosis is accompanied by cutaneous sclerosis, which is absent in DM.
Poikilodermatous mycosis fungoides (previously named poikiloderma vasculare atrophicans and parapsoriasis variegata) is a variant of mycosis fungoides presenting with large, violaceous plaques characterized by hyperpigmentation, hypopigmentation, atrophy and telangiectasias, which may be clinically indistinguishable from those of DM. In this variant of MF, however, and in contrast to DM, the majority of the body surface area of the trunk, buttocks and flexural surfaces is involved.
Muscle Disease
Classic Presentation
Myopathy in DM typically presents as symmetrical proximal muscle weakness. Muscle symptoms may occur before, after, or at the same time as cutaneous manifestations [62]. As discussed, approximately 20% of DM patients are classified as clinically amyopathic, such that even with mild muscle enzyme elevations or abnormalities in an electromyogram, magnetic resonance imaging (MRI), or muscle histopathology, there are no signs of objective weakness on physical examination. Those DM patients that do develop weakness often do so within the first year of symptom onset although weakness can present many years after disease onset [63]. The temporal course of myositis is generally acute or subacute, and progressive.
Patients most often experience weakness of the extensor muscles surrounding the shoulder and pelvic girdles and of proximal limbs. Patients with shoulder and upper extremity weakness may experience difficulty washing their hair or reaching for items in overhead cupboards. Quadriceps and gluteal muscle weakness may manifest as difficulty in rising from a seated position, climbing stairs, or stepping onto curbs. Distal muscle weakness in the hands, manifesting as difficulty opening jars or holding onto objects, typically occurs late in the disease, although patients with the anti-NXP2 antibody may develop distal disease early in the course of DM. With neck flexor muscle involvement, patients may have difficulty raising the head off the table while laying supine. Along with neck weakness, patients experiencing difficulty swallowing liquids and/or solids or having dysphonia may portend poor prognosis. Patients with DM may also describe global symptoms of myopathy, such as fatigue or decreased exercise capacity.
Muscle involvement may also result in symptoms other than classic weakness. Involvement of respiratory muscles of the chest wall or diaphragm may lead to respiratory insufficiency and occasionally respiratory failure. Patients may note a hoarse or raspy voice (dysphonia) due to cricoarytenoid muscle involvement, which occurs in up to 40% of DM patients [64]. Dysphagia may occur in 20% to 50% of cases, due to weak pharyngeal musculature and thus an inability to propel food in the pharyngeal phase of swallowing [65]. Dysphagia is often experienced by the patient as having to “think about swallowing” solids, choking on liquids, or a feeling of not being able to clear their throat. The dermatologist must not only ask patients about weakness and myalgias but also about these bulbar symptoms such as dysphagia or dysphonia, as they can be important clues of muscle activity, and, if severe, can portend need for hospitalization or more aggressive care. Significant bulbar symptoms, especially in cases with cranial nerve involvement, should alert the clinician to consider an overlap with myasethenia gravis, which can occur concomitantly with DM. Approximately 30% of patients will complain of muscle pain with or without muscle weakness [66].
Early in the course of myositis, serum muscle enzymes (i.e., creatine kinase [CK], aldolase, lactate dehydrogenase [LDH], aspartate aminotransferase [AST], and alanine aminotransferase [ALT]) are sensitive biomarkers of muscle inflammation. However, in the mid- to late course of myositis, their sensitivities decrease. CK may be elevated as high as over 100 times above the upper limit of normal; alternative benign causes of myositis, by contrast, which include strenuous exercise, viral illness, and muscle trauma, typically result in CK elevations less than 5 times the upper limit of normal. African-Americans and muscular individuals may have baseline CK levels above the reference laboratory range, usually less than three times the upper limit [67].
Histopathology
Classic cutaneous findings along with weakness and muscle enzyme elevation are sufficient for making a clinical diagnosis of DM and for treating. When performed, biopsy of involved muscle in DM typically demonstrates perifascicular atrophy, degenerating and regenerating myofibers, endothelial cell swelling and capillary necrosis, and membrane attack complex deposition in the endomysial capillary walls [68]. The perifascicular pathology has been proposed to result from the destruction of capillaries populating this region, which could result in localized hypoxia and subsequent myofiber injury. An inflammatory infiltrate is present, consisting of CD4+ T cells [69], plasmacytoid dendritic cells secreting interferon alpha [70], B cells, macrophages, and plasma cells.
If muscle biopsy is performed at the time of acute presentation, features of concomitant rhabdomyolysis with overwhelming necrosis may obscure the primary underlying pathologic process. Nonetheless, a muscle biopsy may still be warranted at this time if immunosuppressive therapy is to be initiated. Prednisone and other immunomodulatory therapies will decrease the yield of the muscle biopsy, resulting in a false negative due to the presence of patchy muscle inflammation. If a muscle biopsy is necessary to confirm the diagnosis of DM, we recommend it be performed within 2 weeks of the initiation of immunomodulatory therapy.
Electromyography
In some cases, electromyography (EMG) may be a helpful adjunct in identifying inflammatory myopathy. EMG can suggest the category of disease (i.e., neuropathic vs myopathic) and will identify patterns of abnormalities to allow for further characterization within each category.
On EMG, DM patients demonstrate the classic triad of small amplitude, short duration, polyphasic motor unit potentials; fibrillations and positive sharp waves; and complex repetitive discharges [71]. Similar patterns may be noted in patients with other inflammatory myopathies, such as polymyositis [72].
Early in the course of disease, EMG detects myositis in 70–90% of DM patients. Later in the course, the sensitivity of EMG in detecting myositis decreases. A potential explanation for decreased sensitivity of EMG (as well as muscle enzymes) over time is that longstanding myositis may result in perifascicular muscle atrophy and fibrosis, leading to less dramatic results, even while inflammation persists.
Imaging
MRI may be useful to assess for signs of myositis when muscle enzymes and EMG studies are inconclusive. MRI can also be used to direct the site of a diagnostic biopsy [73] or, in some cases, assess clinical response to treatment [74]. MRI provides a detailed view of the muscle anatomy, allowing for localization and discrimination of pathologic processes, e.g., edema, inflammation, fibrosis, calcifications or atrophy. On short tau inversion recovery (STIR) sequencing, in which normal muscle is dark and inflamed muscle is bright, an increased signal intensity is noted within muscles affected by inflammation, necrosis, and/or degeneration [75]. Yoshida et al. performed MRI studies on 14 newly diagnosed DM patients and noted that on STIR sequences, fasciitis was the predominant finding in the first 2 months after symptom onset. They also analyzed en bloc muscle biopsies from each patient and found histopathologic evidence of fasciitis in 12 of 14 patients, suggesting that the fascial microvasculature may be a primary site of involvement [76]. In T1-weighted MRI images, in which fat is bright and normal muscle is dark, chronic muscle damage may be identified as fatty replacement of skeletal muscle [77].
Differential Diagnosis
The differential diagnosis for weakness is broad. The presence of characteristic cutaneous findings of DM obviously help to point away from other causes. However, in the case of a patient with a photodistributed rash and muscle weakness, the possibility that they are two unrelated conditions must be considered by the clinician before assuming a diagnosis of DM. In cases when the cutaneous eruption is subtle or specific features are in question, other causes of myositis may be considered. It is also essential to distinguish between myopathic and nonmyopathic causes of weakness.
Nonmyopathic etiologies of weakness include other disorders of the motor unit [67] as well as global causes, including chronic pain and chronic fatigue syndromes [78]. Neuromuscular diseases such as myasthenia gravis have been described as co-existing with DM [79]. The presence of ptosis, diplopia, fatiguability, and bulbar symptoms should raise concern for myasthenia gravis. Neuromyelitis optica (Devic’s disease), another neuromuscular disease, has been described in a juvenile and adult patient with DM [80, 81].
Myopathic causes may be hereditary and may include channelopathies and muscular dystrophies. Acquired causes of myopathy include autoimmune (DM, polymyositis, inclusion body myositis, immune-mediated necrotizing myopathy, other connective tissue disease associated), toxic (drug-induced) or metabolic myopathies (thyroid and adrenal dysfunction) [67].
Systemic lupus erythematosus (SLE) often presents with a photodistributed eruption, and myositis is present in 4% to 16% of SLE patients [81,82,83,84,86]. Moreover, 50% of SLE patients may complain of myalgias [87]. Symptoms of myositis in these patients, as in DM, frequently include fatigue and proximal muscle weakness [82]. CK levels are elevated in the majority of cases, and electromyographic studies show signs of myositis [88]. Muscle biopsy shows a nonspecific perivascular and perimysial infiltrate of inflammatory cells, without invasion of non-necrotic fibers and type II muscle fiber atrophy [89].
Polymyositis lacks a well-defined clinical phenotype. These patients may present with proximal muscle weakness, elevated muscle enzymes, and myopathic changes on EMG; however, the cutaneous manifestations of DM are, by definition, absent [90]. Muscle biopsy is essential to confirm the diagnosis of polymyositis. Classic histopathologic features include an endomysial inflammatory infiltrate consisting predominantly of CD8+ T cells, as well as muscle fiber necrosis and regeneration [20].
Anti-synthetase antibody syndrome , characterized by fever, arthritis, myositis, ILD, mechanic’s hands, Raynaud’s phenomenon and the presence of an anti-synthetase autoantibody, may be seen in both DM and polymyositis. Specific histopathologic findings in muscle biopsies of anti-synthetase antibody syndrome have been identified [91], while the ultrastructural findings of myonuclear actin aggregation and intranuclear rod formation have been found to have 81% sensitivity and 100% specificity for anti-synthetase syndrome-myositis [92].
Inclusion body myositis typically affects men older than 40 years and has an insidious onset, preferentially affecting the finger flexors (causing difficulty with fine motor movements) and the quadriceps [93]. Muscle biopsy shows a mixed infiltrate of CD8+ T cells and monocytes surrounding non-necrotic myofibers, rimmed vacuoles and amyloid and p62 inclusions within myofibers [94].
Up to 30% of patients with systemic sclerosis have a myopathy [94,95,97]. The subset of systemic sclerosis patients with anti-PM-Scl antibodies may have an inflammatory myositis in roughly 50% of cases [98]. Systemic sclerosis patients typically lack classic cutaneous manifestations of DM, however. There are case reports of sclerodermatomyositis [99, 100], however, described in patients with systemic sclerosis and myositis who have anti-PM-Scl antibodies.
Trichinosis is a meat-borne parasitic disease caused by ingestion of roundworm larvae from the Trichinella species, usually found in undercooked pork. Fifteen cases were confirmed in the U.S in 2012 [101]. It manifests with an initial enteral phase with diarrhea and abdominal pain within 1 week of larvae ingestion. In the systemic phase, beginning 1–6 weeks following ingestion, patients develop eosinophilic myositis, characterized by fever, myalgias and periorbital edema [102]. Muscle pain is typically present in the nuchal muscles, masseters, and upper and lower extremities. No cutaneous manifestations are observed. Diagnosis may be made with serum anti-Trichinella antibodies or by a muscle biopsy [103]. The severity of symptoms may range from mild to death from myocarditis or meningoencephalitis, depending on the number of larvae ingested and the host immune response. Treatment for the systemic phase is with albendazole and systemic corticosteroids [101].
Numerous medications have been implicated in drug-induced myopathies, including anti-depressants, antipsychotics, anti-retrovirals, anticonvulsants, colchicine and statins [67]. Corticosteroids are the most common cause. Glucocorticoids induce atrophy of type II (predominantly type IIb, fast-twitch) fibers. The clinical presentation may be acute, within 4 weeks of administration of high-dose fluorinated glucocorticoids, such as dexamethasone or triamcinolone. Onset may also be insidious, over weeks to months. Risk factors include older age, malignancy, and poor nutritional status. The pelvic girdle and proximal leg muscles are more commonly affected than the shoulders and arms [104]. Muscle enzymes are normal. EMG testing may be normal early in the course but may show myopathic changes in late stages, such small-amplitude polyphasic action potentials without spontaneous activity upon needle insertion. Muscle biopsy provides the most definitive diagnosis of corticosteroid myopathy, with histology demonstrating nonspecific atrophy of type IIb muscle fibers, absence of inflammatory infiltrates, and variations in fiber size with centrally-placed nuclei [105].
Although myalgias occur in 10% of statin users [106], statin myopathy occurs in 5 in 100,000 persons and is characterized by elevated CK levels greater than 10 times normal [107]. Rhabdomyolysis is a rare, severe form of statin myopathy with an incidence of 0.44 cases per 10,000 person-years, characterized by massive myonecrosis potentially leading to renal failure and death [108]. Risk factors for statin myopathy include higher doses, the particular statins fluvastatin and pravastatin [109], the DRB1*11:01 allele [110], SLCO1B1 gene variants [111], obesity, older age, hypothyroidism, and preexisting liver disease [112]. Statin myopathy is self-limited, with resolution of symptoms seen in an average of 2 months following discontinuation of the drug culprit [113].
Immune-mediated necrotizing myopathy from anti-HMG-CoA reductase antibodies may also present with symmetric proximal muscle weakness in a patient on a statin. However, 25% of patients with this statin-associated immune-mediated necrotizing myopathy will not have a history of statin exposure [114]. The average duration of statin exposure prior to symptom onset is 3 years (range 2 months to 10 years) [115]. Laboratory evaluation reveals highly elevated CK levels (mean 10,000 IU/L) and anti-HMG-CoA reductase antibodies [116]. An irritant myopathy may be evident on EMG; MRI may show muscle edema [116]. Muscle biopsy, which is not diagnostically specific, shows prominent necrosis with minimal inflammatory cell infiltrate. Autoantibody testing is a more direct method of confirming the diagnosis [117]. Myopathic symptoms do not resolve with drug discontinuation.
An important caveat in the differential diagnosis of DM is that hepatic inflammation from a variety of causes, including nonalcoholic steatohepatitis, drug-induced hepatitis, and infectious hepatitis, may cause elevations in any of the muscle enzymes, particularly AST, ALT, and/or aldolase. Therefore, if a DM patient develops such elevations in muscle enzymes without clinical evidence of weakness, it is prudent to exclude hepatic injury. Initial investigation includes obtaining serum gamma glutamyl transferase and alkaline phosphatase as well as a hepatic ultrasound. Further work-up, including anti-smooth muscle antibody, anti-liver/kidney microsomal antibody-1 antibody, and anti-mitochondrial antibody, may be helpful in diagnosing concomitant autoimmune hepatitis, which has been previously described in patients with DM [118, 119].
It is important to consider that continued weakness despite adequate treatment or that is disconnected from improvement in skin activity may be a sign of other myopathy related to ongoing corticosteroid use, hydroxychloroquine-related myopathy, de-conditioning or post-inflmmatoary myopathy.
Systemic Disease
Pulmonary Manifestations
Pulmonary manifestations in DM include ILD, pulmonary hypertension, and aspiration pneumonia. Drug-induced pneumonitis due to agents such as methotrexate is also an important consideration. Prevalence estimates for methotrexate-induced pneumonitis in patients treated for rheumatologic indications range from 0.5% to 1%[120, 121].
Interstitial Lung Disease
Interstitial lung disease (ILD) is a leading cause of morbidity and mortality in DM [122]. ILD affects between 15% and 50% of patients with DM, depending upon the population studied and the autoantibody distribution [122,123,124,126]. Large case series have suggested that 75–86% of patients who have an anti-synthetase antibody will develop ILD [127, 128]. Similarly, DM patients with anti-MDA5 antibodies have a marked increase in risk for developing ILD, with 50–100% of these patients developing this manifestation[24, 129]. Rapidly-progressive ILD (RP-ILD) is an aggressive form of ILD that responds poorly to immunosuppressive therapies, having a 6-month survival rate of approximately 40% [130]. RP-ILD affects 40–60% of patients with anti-MDA5 antibodies [129, 130,131,133]. Serum ferritin is often highly elevated (>500 mg/dl) in anti-MDA5 DM patients [134] and may serve as a useful biomarker in assessing severity and the clinical response of ILD [135, 136].
Symptom onset in ILD is often insidious. Patients present with dry cough, decreased exercise capacity, or dyspnea with relatively minor exertion. Less commonly, acute onset of shortness of breath, hypoxia and respiratory failure may occur. While there are no formal guidelines for ILD screening, it is prudent to obtain baseline pulmonary function tests (PFTs) with diffusion capacity, and then repeat screening annually for at least the first 3–5 years after diagnosis. Screening PFTs may be performed more frequently if new pulmonary symptoms develop.
Diffusion capacity of carbon monoxide (DLCO) and forced vital capacity (FVC) are the two most informative parameters in evaluating for ILD: both are reduced below 80% of the predicted value [137]. In systemic sclerosis patients, who may also develop ILD, DLCO correlates better with disease severity, as assessed by high resolution computed tomography (HRCT) scans, than do lung volumes or other spirometric values [138]. Additionally, in idiopathic pulmonary fibrosis, an absolute change of 10% in FVC or 15% in DLCO signifies disease progression and an increased risk of mortality, and these parameters are reasonably applicable to ILD associated with connective tissue diseases [139]. The significance of isolated reductions in FVC of less than 5% or in DLCO of less than 10% are difficult to interpret in the absence of a clear trend over time. This degree of change may be influenced by patient effort and/or intrinsic variability of the test.
Exertional oxygen desaturation on the 6-minute walk test provides a global assessment of cardiopulmonary function and exercise performance. However, it may not be the most accurate assessment of lung function in patients with CTDs due to confounding factors, such as deconditioning, myopathy, arthritis, respiratory muscle weakness and pulmonary hypertension [140].
When there is a concern for ILD on based on pulmonary function testing or 6-minute walk testing, HRCT scan of the chest should be the next step in evaluation. HCRT of the chest may also be useful in detecting subclinical fibrosis prior to symptom onset, which is noted in up to 65% of patients with polymyositis and DM [141]. The most common pulmonary radiographic and histologic pattern in DM is nonspecific interstitial pneumonia (NSIP), which was reported in 81.8% (18/22 cases) of DM patients in one series [142]. In this study by Douglas et al. there were also two cases of diffuse alveolar damage (9%), one case of usual interstitial pneumonia (UIP, 4.5%) and one case of cryptogenic organizing pneumonia (OP, 4.5%) [142]. Radiographically, basilar and peripheral ground glass opacities and subpleural sparing characterize NSIP. By contrast, UIP is characterized by basilar and peripheral honeycombing and subpleural involvement [139]. More than one pattern may exist in a single patient. Patients with the NSIP and OP patterns experience better responses to systemic corticosteroids than those with UIP [143].
Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PAH) is a rare manifestation in DM. Symptoms may include increased fatigue, shortness of breath, dyspnea on exertion, palpitations, chest pain, edema, lightheadedness, and, rarely, pre-syncopal or syncopal episodes. A loud pulmonic component of the second heart sound may be audible at the left second intercostal space, corresponding to elevated pressure in the pulmonary arteries and delayed closure of the pulmonic valve [144]. Electrocardiogram may show right axis deviation due to right ventricular (RV) hypertrophy. Pulmonary function testing revealing a disproportionately low DLCO compared to a relatively normal FVC should prompt further screening for PAH. In systemic sclerosis patients, a DLCO of <55% and a normal FVC or an FVC/DLCO ratio of greater than 1.6 were highly associated with PAH [121].
Echocardiographic findings suggestive of pulmonary hypertension include a RV systolic pressure of >40 mmHg, RV enlargement, maximum tricuspid regurgitant velocity > 3.0 m/s, and presence of a pericardial effusion [145]. Tricuspid annular plane systolic excursion (TAPSE, a measure of RV contractility) < 1.7 cm signifies worse RV function and has been associated with higher mortality in systemic sclerosis patients [146]. Transthoracic echocardiogram has a sensitivity of only 82% and specificity of 69% in detecting pulmonary hypertension and cannot be used to differentiate patients with PAH from those with left heart disease or ILD-associated pulmonary hypertension [145]. The gold standard for the diagnosis of PAH is right heart catheterization showing a mean pulmonary artery pressure of ≥25 mmHg at rest and an end-expiratory pulmonary artery wedge pressure (PAWP) ≤15 mm Hg [147].
Cardiac Manifestations
DM-Specific Cardiac Involvement
Cardiac involvement is increasingly recognized as an important clinical feature in DM. It is typically subclinical but can present with electrocardiographic abnormalities, most commonly including ST-T segment changes and conduction abnormalities in 12.5–56.7% and 25–38.5%, respectively, of DM patients [148]. Echocardiographic findings include ventricular hypertrophy and left ventricular diastolic dysfunction in 8–15% and 42%, respectively [149]. Myocarditis may lead to myocardial fibrosis, ventricular dysfunction and thus cardiomyopathy. Rosenbohm et al. screened 11 DM patients with cardiac MRI and found that 54% (6 of 11) displayed evidence of late gadolinium enhancement, consistent with myocardial inflammation [150]. Cardiac troponin I may be a useful biomarker in detecting subclinical cardiac muscle involvement [151], and this topic warrants further inquiry.
DM patients with anti-MDA5 antibodies may be at higher risk for cardiac involvement [152, 153]. We currently follow three anti-MDA5 DM patients with cardiomyopathy; a cardiac muscle biopsy from one of these patients revealed endomyocardial fibrosis. Further studies in this patient subgroup with cardiac MRI and/or biopsies are necessary to determine the true frequency and pathogenesis of cardiac involvement in these patients.
Coronary Artery Disease
DM patients are at increased risk of developing coronary artery disease (CAD) compared with the general population. The etiology for the accelerated atherosclerosis is likely multifactorial. Important factors include the chronic inflammatory state, endothelial dysfunction, hypercoagulability [154], and prolonged exposure to prednisone in the context of traditional cardiovascular disease risk factors [155], such as hypertension, hyperglycemia, and dyslipidemia [156].
A meta-analysis of four large epidemiologic studies of patients with idiopathic inflammatory myopathies showed an increased frequency of cardiovascular events compared to controls with a risk ratio of 2.26 (95% confidence interval 1.02–4.92) [157]. A U.S. retrospective case-control study of 50,322 hospitalization records of DM patients found that 20% of these hospitalizations were associated with a concurrent atherosclerotic cardiovascular diagnosis or procedure. Additionally, DM patients with CAD were twice as likely to die during hospitalization compared to age and sex-matched controls with CAD, and compared to DM patients without CAD [158].
In a prospective case-control study in Taiwan in which 907 DM patients and 4535 age and sex-matched controls were followed over 2 years, 14 patients with DM (1.5%) vs. 18 controls (0.4%) sustained acute myocardial infarctions. After adjustment for cardiovascular risk factors, the hazard ratio for an acute myocardial infarction among patients with DM was 3.37 (95% confidence interval 1.67–6.8, p = 0.0007) [159]. In this study, 46 DM patients (5.1%) and 133 controls (2.9%) experienced an ischemic cerebrovascular event, resulting in an adjusted hazard ratio of 1.78 (95% confidence interval 1.29–2.49, p = 0.0028) [159].
These studies underscore the importance of recognizing the increased CAD risk in DM patients. Cardiovascular risk factors should be addressed and corticosteroid exposure should be minimized while the underlying inflammation is controlled with corticosteroid-sparing therapies.
Thromboembolic Disease
In a large retrospective study of 355 DM patients and 443 polymyositis patients, an increased risk of deep vein thrombosis (DVT) was found among patients with DM (hazard ratio 9.40 [95% CI 2.88 to 30.68]) and polymyositis (hazard ratio 6.16 [95% CI 2.50 to 13.92])as compared to age- and sex-matched controls [111]. In this study, risk of pulmonary embolism (PE) was significantly increased among polymyositis patients (hazard ratio 9.42 [95% CI 4.59 to 18.70]), but this risk did not reach statistical significance in the DM cohort (hazard ratio 4.70 [95% CI 0.85 to 25.98]) [160].
Similarly, a trend towards increased risk of venous thromboembolism (VTE) was noted in a Spanish cohort of 87 DM patients in whom 6 developed the event (6.8%)[161]. In large epidemiologic studies combing DM and polymyositis patients from Sweden [162] and the United Kingdom [163], the risk of PE was increased (standardized incidence ratio 16.44 [95% CI 11.57–22.69]) compared to the baseline population risk. Thus, while the risk of DVT appears to be elevated in DM, the risk of VTE or PE needs further clarification.
Joint Manifestations
Arthritis is reported in 30–40% of DM patients [163,164,166]. Although arthritis frequently presents concurrently with the initial presentation of myositis, flares of arthritis affect only 50% of patients during disease relapses [167].
Arthritis and arthralgias are more common among DM patients with anti-MDA5 and anti-synthetase antibodies. Hall et al. found 9 of 11 (81.8%) anti-MDA5 patients exhibited an inflammatory arthritis, as compared to 40 of 149 (26.7%, p < 0.001) non-MDA5 DM patients [168]. DM patients with anti-synthetase antibodies, most commonly Jo-1, may also demonstrate non-erosive arthritis in up to 93% of cases [167]. In these patients, the non-erosive arthritis may occur in the setting of “anti-synthetase syndrome” which consists of fever, arthritis, myositis, ILD, mechanic’s hands and/or Raynaud’s phenomenon, as reviewed above.
Symmetric non-erosive polyarthropathy is the most common arthritis presentation in patients with anti-MDA5 and anti-synthetase antibodies [77, 168]. However, erosive changes have been reported, more commonly in the anti-synthetase antibody subset [168,169,170,172]. In these instances, DM patients with arthritis as the presenting symptom may be misdiagnosed with rheumatoid arthritis or a rheumatoid arthritis overlap disease [173].
In general, the arthritis in DM is mild to moderate in severity and typically involves the small joints of the hands (including the wrists and metacarpophalangeal and proximal interphalangeal joints) as well as the shoulders, elbows, and ankles [167]. Signs and symptoms of inflammatory arthritis include joint swelling, morning stiffness for 30 minutes or longer, or joint pain that improves with activity.
On examination, one must first determine if the pain is articular in origin, as opposed to involving the periarticular soft tissue such as tendons, ligaments or bursae. Asking the patient to point and localize the exact area of pain may be helpful. Key points in the rheumatologic exam include inspection for deformity, swelling, and muscle wasting. Examination for synovitis, indicative of active joint inflammation, includes palpation of the small joints of the hands as well as elbows, shoulders, knees and symptomatic joints, evaluating for warmth, range of motion, swelling or palpable fluid, and tenderness. True articular pathology will limit both active and passive range of motion, in contrast to tendonitis, in which pain will be elicited with active range of motion alone. Inflammatory markers (erythrocyte sedimentation rate, C-reactive protein) may or may not be elevated.
The differential diagnosis for joint pain in DM patients includes osteoarthritis, rheumatoid arthritis, polymyalgia rheumatica, infectious arthritis, crystalline arthropathies and chronic pain syndromes.
In osteoarthritis, morning stiffness may last only one to several minutes, and the pain is often worse with activity. Joint swelling is typically absent.
Rheumatoid arthritis is typically associated with elevated acute phase reactants in addition to elevated titers of rheumatoid factor and antibodies to cyclic citrullinated peptide. Rash and myositis are rare.
Polymyalgia rheumatica is characterized by morning stiffness in the shoulder and hip girdles in patients over the age of 50 years. Characteristically, it is associated with an elevated sedimentation rate. Although weakness may be reported, examination will demonstrate normal muscle strength [174].
Infectious arthritis should be considered in any immunosuppressed host presenting with acute onset of monoarticular joint pain and swelling. Fever, tachycardia, malaise, and/or leukocytosis may or may not be present. If the suspicion is high for infectious arthritis, urgent referral for arthrocentesis and evaluation for septic arthritis is necessary.
Crystalline arthropathies include gout and pseudogout or chondrocalcinosis. These entities typically present with acute onset of mono- or polyarticular arthritis with crystals (monosodium urate in gout and calcium pyrophosphate crystals in pseudogout) observed upon arthrocentesis of the involved joints.
Chronic pain syndrome is often seen in combination with rheumatic conditions. Pain is diffuse, and there are multiple tender points, typically over myofascial points instead of over joints.
Patients with joint disease having synovitis / inflammation may benefit from being on hydroxychloroquine and may warrant the choice of methorexate as initial immunosuppressive therapy.
Renal Manifestations
Direct renal involvement in DM is rare and has not been well described. However, renal complications in DM may be more common than previously thought. These complications occur via three broad mechanisms: (1) rhabdomyolysis, (2) presence of an associated glomerulopathy, and (3) drug-induced nephrotoxicity.
In cases of acute fulminant myositis, which typically occurs at disease onset, rhabdomyolysis may result in myoglobin-induced acute tubular necrosis (183,184).
Multiple types of nephropathies have been reported in association with DM, the most common (~50%) of which is immune complex-mediated glomerulonephritis [175]. Other reported glomerulopathies in association with DM include IgA nephropathy [175,176,178], membranous nephropathy [178,179,181], diffuse proliferative glomerulonephritis [182], and anti-neutrophil cytoplasmic antibody (ANCA)-associated crescentic glomerulonephritis [183, 184].
A recent French retrospective cohort study by Couvrat-Desvergnes et al. found that among 96 DM patients, 21 (22%) had evidence of chronic kidney disease (CKD) [175]. In 40% of these patients, nephrotoxicity was thought to be medication-induced. Five of the DM patients underwent renal biopsy, which revealed vascular lesions in two patients, minimal change disease in two patients, and focal segmental glomerulosclerosis and vascular lesions in one patient. Risk factors for development of CKD were presence of cardiovascular risk factors such as hypertension and diabetes (HR 16.56, 95% CI 2.56–107.16, p = 0.0032), previous episode of acute kidney injury (HR 15.09, 95% CI 6.19–36.79, p < 0.0001) and age at myositis onset (HR per year of increased age 1.05, 95% CI 1.02–1.08, p = 0.0016). Female sex was protective (HR 0.4, 95% CI 0.18–0.89, p = 0.0024).
Further study is needed to validate these associations in DM patients and to characterize the mechanism of renal injury. Nonetheless, it is important to be aware of the potential for renal involvement in DM patients. It is prudent to carefully monitor renal function, particularly early in the disease course, especially in patients with severe myositis, cardiovascular risk factors, previous episodes of acute kidney injury, or exposure to nephrotoxic medications.
Rare Manifestations
Macrophage activation syndrome (MAS) , also known as acquired hemophagocytic lymphohistiocytosis (HLH), is a severe and potentially fatal complication or presenting syndrome in DM. MAS is a state of dysregulated immune hyperactivation of macrophages that can lead to multisystem organ failure and death. Clinical features include prolonged high fever, lymphadenopathy, and hepatosplenomegaly, with laboratory findings of hyperferritinemia (>500 mg/dl), cytopenias (hemoglobin <9 g/dl, platelets <100,000/mm3, neutrophils <1000/mm3), hypertriglyceridemia (>265 mg/dl), hemophagocytosis in the bone marrow, spleen or lymph nodes, low natural killer cell activity, and elevation of soluble CD25 (> 400 U/l) [185].
Cancer
DM is associated with an internal malignancy in 10–20% of cases [2]. A meta-analysis by Olazagasti et al. analyzed seven population-based and three hospital-based DM cohorts with follow-up of 3.7 to 10.4 years. They found a standardized incidence ratio of 4.79 for cancer development during follow-up (95% confidence interval 3.71–5.87) [1]. Buchbinder et al. retrospectively reviewed 537 patients with biopsy-proven idiopathic inflammatory myopathy. DM was diagnosed in 85 of these cases over an average follow-up period of 5.3 years. A malignancy was observed in 32 (42%) patients, and the risk of cancer diagnosis was greatest within the first 3 years after diagnosis of DM [186].
Although cancer types appear to vary based on population studied, the most common malignancies associated with DM include breast, lung, ovarian, prostate, colorectal, gastric, and pancreatic cancers, as well as non-Hodgkin lymphoma [186,187,189]. Nasopharyngeal cancer is more common among Southeast Asians [190].
The autoantibodies anti-TIF1-γ and possibly anti-NXP2 are associated with an increased risk of cancer in DM. NXP2 has a role in activating p53 and inducing cellular senescence [191, 192] while TIF1-γ interacts with Smad2/3 in embryonic stem cells to modulate transcriptional elongation and tissue differentiation [192]. In collaboration with researchers at Johns Hopkins, we found that anti-NXP2 and anti-TIF1-γ antibodies were observed in 37 of 213 DM patients (17%) and 82 of 213 (38%), respectively. A cancer was detected in 14% (29/213) of these DM patients. Among the 20 patients with cancer, 24 (83%) had antibodies to either NXP2 or TIF1-γ [193]. In addition, anti-NXP2 antibodies were disproportionately represented among male DM patients having cancer (7 of 9 patients, 78%). Similarly, Ichimura et al. reviewed 457 cases of DM and found that seven patients (1.6%) had anti-NXP2 antibodies [191]. Of those, three (43%) had associated malignancies, all of whom were male. Trallero-Araguas et al. meta-analyzed six studies that included 312 adult DM patients and found a 27-fold higher odds of developing cancer-associated myositis (95% CI 6.59–112.82) among anti-TIF1-γ positive DM patients [194]. Of note, however, at least in the U.S. population, most DM patients with these antibodies still do not harbor a malignancy.
Regardless of autoantibody status, the frequency of cancer-associated DM increases in patients over age 60 years [195]. Other proposed risk factors include male sex, presence of constitutional symptoms [196], highly elevated erythrocyte sedimentation rate, and cutaneous necrosis (208).
There are no existing guidelines for cancer screening in patients with newly diagnosed DM. In a case series of 33 DM patients, 13 of whom had coexisting malignancies, initial routine cancer screening failed to discover 4 malignancies (30%) [196]. In collaboration with the University of Louisville, we have retrospectively examined cancer screening practices at our two institutions in a cohort of 400 DM patients [197]. In this cohort, 16 patients harbored an unknown internal malignancy at the time of DM diagnosis but had no symptoms or signs of cancer based on physical examination and routine blood testing (blood counts, chemistry and urinalysis). Blind testing with CT scan, colonoscopy, mammogram, and prostate-specific antigen evaluation revealed the cancer in these patients. Blind screening may therefore be of benefit in detecting malignancy in at least a proportion of DM patients. Identifying the most appropriate screening tests, the timing and frequency of these screenings, and the population subset most appropriate for these screenings is a high priority.
In addition to a complete history and physical examination, routine age-appropriate cancer screening studies (colonoscopy, mammogram, prostate exam) and relevant screening bloodwork (complete blood count, renal and liver function tests) as well as a urinalysis are indicated at the time of DM diagnosis. The role of other blood tests, including erythrocyte sedimentation rate, C-reactive protein, serum cancer markers, and serum and urine protein immunofixation electrophoresis, is currently not established. Our practice is to perform screening CT scans of the chest, abdomen and pelvis, and to consider screening ultrasound of the thyroid gland and transvaginal ultrasound of the ovaries.
The necessity for annual re-screening for malignancy is even less clear. We recommend that re-screening be considered in clinically high-risk patients whose disease is difficult to control or those who have experienced a substantial disease flare after a sustained quiescent period. Of note, treatment of the associated malignancy may result in disease remission.
Principles of Management
Overview
Appropriate management of DM hinges on ascertaining a comprehensive understanding of the involved organ systems. ILD and underlying malignancies are the leading causes of disease-related death in DM and thus should be prioritized in treatment. Establishing collaborative relationships with co-managing providers (rheumatologist, dermatologist, cardiologist, pulmonologist) is essential to monitor disease activity and optimize therapeutic strategies when multiple organ systems are involved.
Many DM patients ultimately enter a long-term remission, often induced by prolonged immunomodulation and/or immunosuppression [198]. Rarely, DM may spontaneously remit without therapy [199]. In our experience, sustaining immunosuppression or immunomodulation for at least 9–12 months after remission is achieved may decrease the likelihood of relapse.
The evidence for medical therapies in DM is derived largely from single-center, retrospective case series, case reports, and expert opinion. Only 15 randomized clinical trials have been performed on DM treatment, including the Rituximab in Myositis (RIM) trial, which represents the largest clinical trial to date, with 76 adult and 48 juvenile DM patients. Gordon et al. identified 14 additional randomized clinical trials in a Cochrane Review from 2012[200]. Ten of these trials, the largest of which included 62 patients, were analyzed in that review. As such, therapeutic strategies are currently informed mostly by expert opinion [199,200,201,204].
Treatment of the skin disease in DM is challenging. In a single patient, multiple systemic immunomodulatory and immunosuppressive agents must often be combined to achieve control. Moreover, skin disease and muscle disease often have discordant response to treatment[205]. In some patients, recalcitrant skin disease may be active for years after remission is achieved for myopathy. Given the chronicity, and in some cases the recalcitrance, of the skin disease, it is worthwhile to weigh the long-term toxicity of the prescribed therapy against the achieved or potential cutaneous benefit. Commonly used systemic agents for cutaneous DM are discussed below, along with other management strategies.
Behavioral Change
Photoprotection is an essential first step in management of the cutaneous disease in DM. However, it is important to note that up to 60% of DM patients are only minimally photosensitive, and as few as 20% report disease exacerbation after UV exposure [205, 206]. Despite this observation, it is prudent to counsel patients on UV protection. Patients should be advised to use broad-spectrum sunscreens that protect against both UVA and UVB radiation, wear sun-protective clothing, avoid exposure during hours of high UV intensity (10 a.m. to 4 p.m.), and seek shade whenever possible.
Physical Medicine and Rehabilitation
Strength training has been shown to improve muscle strength and function in patients with DM[205,206,209], while aerobic exercise has been shown to improve endurance [210, 211]. In a randomized clinical trial of patients with active disease, a home strength training program was shown to be safe [212] but conferred no benefit in strength or disease control over the control group, who performed range of motion exercises [213]. We advise all patients to enroll in a physical therapy program soon after diagnosis to prevent injury and to maintain mobility and muscle strength.
Topical Agents
Topical Corticosteroids
Topical corticosteroids play a supportive role in suppressing cutaneous inflammation in DM and can provide temporary relief, but they are unlikely to fully control even mild cutaneous disease activity.
For the scalp and body, class I or class II topical corticosteroids, applied twice daily, are typically necessary to ameliorate pruritus, erythema and scale. They can be applied under occlusion with plastic wrap to increase potency, a technique that is particularly helpful for painful nail bed disease or hyperkeratotic plaques on the elbows and knees.
For the face and intertriginous zones, low potency (class VI) topical corticosteroidsare preferred to minimize the risk of atrophy and hypopigmentation. When necessary, class I or class II corticosteroids may be applied to the face or intertriginous area for brief intervals (i.e., 2 weeks) followed by similar periods (i.e., 2 weeks) off therapy.
Topical Calcineurin Inhibitors
Topical calcineurin inhibitors (TCIs), such as tacrolimus 0.1% ointment, have shown modest efficacy in several cases of cutaneous DM [212,213,214,217]. One study in 6 patients with cutaneous DM using a split face design with vehicle applied on the contralateral face showed no detectable benefit of tacrolimus ointment 0.1% after 2 months [218]. Pimecrolimus 1% cream used twice daily for 4–6 months was reported to significantly improve cutaneous disease in DM in two patients who were concomitantly treated with hydroxychloroquine, prednisone and methotrexate [219].
There are no studies comparing the efficacy of TCIs to topical corticosteroids in DM. However, unlike potent topical corticosteroids, TCIs may be used safely on the face without concern for atrophy or hypopigmentation. As such, TCIs may represent valuable topical corticosteroid-sparing agents in DM owing to their favorable side effect profile.
Transient application site reactions are the most common adverse effects of TCIs, manifesting as a warm or burning sensation in up to 58% of patients [220]. This burning is likely due to local induction of release of neuropeptides, such as substance P, from sensory nerve endings [221]. The burning sensation typically lasts 10–15 minutes after application and subsides after 3–7 days of regular use [222].
Systemic Medications
Antimalarials
Antimalarials are often used as first-line agents for cutaneous DM. Retrospective studies suggest that improvement is seen in approximately 30–50% of cutaneous DM patients on antimalarials[223]. In our experience, however, this improvement is generally mild. In addition, up to 30% of DM patients may experience a drug eruption (morbilliform or lichenoid) in the days or weeks after initiating hydroxychloroquine [224]. Antimalarials are therefore a reasonable choice for management of cutaneous DM in mild cases where the patient prefers to avoid immunosuppression and is comfortable waiting 4–6 months for a detectable response. The addition of quinacrine to hydroxychloroquine or chloroquine may result in higher efficacy than single-agent therapy [225]. Hydroxychloroquine may also be useful for treating mild symptoms of inflammatory arthritis, and chloroquine was noted to ameliorate arthritis in DM in one case report [226].
Methotrexate
Methotrexate is effective in significantly reducing cutaneous disease severity in 50–100% of DM patients [225,226,227,230]. In combination with prednisone, it also represents first-line treatment for myositis; doses of 20–25 mg per week are typically necessary to control muscle inflammation [164, 231]. Methotrexate is also a preferable treatment choice when concomitant arthritis is present [232].
It is important to recognize that elevation of serum transaminases should not preclude the use of methotrexate in a patient with active myositis, as these elevations might be related to the muscle disease. Switching from oral to subcutaneous or intramuscular administration may improve gastrointestinal tolerability [231] and efficacy [228] as compared with oral administration. Splitting the methotrexate dose into two, each 12 hours apart, also improved bioavailability. Patients should be counseled regarding oligospermia, teratogenicity, hepatotoxicity and bone marrow suppression with methotrexate. The cutaneous side effects include mucositis and hair shedding, which are mitigated by increasing folic acid supplementation. When ILD is present, it is prudent to avoid methotrexate due to its potential to induce acute pneumonitis and pulmonary fibrosis [233, 234].
Systemic Corticosteroids
For cutaneous DM, systemic corticosteroids are undesirable agents as monotherapy, as they usually elicit only partial responses, even at moderate to high doses, and require long-term administration. The predictable toxicities associated with prolonged high-dose corticosteroids outweigh the potential cutaneous benefits.
In myositis, by contrast, systemic corticosteroids are considered first-line, though formal, controlled studies regarding dose or tapering regimens have not been performed [235]. Complete clinical responses in muscle inflammation with prednisone monotherapy at doses greater than 0.5 mg/kg/day have been achieved in 27% [236] to 87% [237] of DM patients [238]. Oral or intra-articular corticosteroids are typically highly effective for the arthritis associated with DM [232].
The addition of corticosteroid-sparing agents may improve control of myositis and extramuscular inflammation, but their critical role is to minimize toxicities of oral corticosteroids, including the risk for corticosteroid-induced myopathy [239]. At our institution, a corticosteroid-sparing agent is started simultaneously or soon after oral corticosteroids. Although oral corticosteroids are used as empiric therapy for ILD associated with DM [127], response rates with monotherapy may be as low as 50% [238,239,240,243], and thus the addition or substitution of a corticosteroid-sparing agent is usually required.
Mycophenolate Mofetil
Mycophenolate mofetil (MMF) has been shown to be effective in reducing cutaneous disease severity [244, 245] and myositis in DM at doses of 2–3 grams divided daily [244,245,248]. It is considered the first line oral agent when ILD is present [247,248,249,250,253].
A significant proportion (20%) of patients will experience nausea or diarrhea on MMF at 2 g daily [254]. When gastrointestinal side effects are dose-limiting, switching to enteric-coated mycophenolate sodium is an option for maintaining the current dosing with improved tolerability[255].
Methotrexate and mycophenolate may be combined when either alone is insufficient to achieve disease control. Patients receiving combination therapy may require increased monitoring for bone marrow suppression, infection and neoplasia.
Intravenous Immunoglobulin
Intravenous immunoglobulin (IVIG) appears to be the single most effective agent for cutaneous DM, with up to 70–80% of patients achieving an almost complete or complete response (272,273). IVIG is also effective for myositis [256, 257]. The first randomized placebo-controlled crossover trial of IVIG in DM, including 15 patients in 1993 [258], showed a significant improvement in muscle strength in 9/12 (75%) patients and dramatic improvements in skin disease based on clinical photographs in 8/12 (67%) patients who received IVIG.
The standard dosing regimen is 2 grams/kg divided over 2–3 days, infused monthly. The therapeutic effect may be perceived as early as 2 weeks, but it may not be fully evident until 3–4 months of treatment. Because IVIG works relatively quickly in decreasing muscle inflammation, along with high dose corticosteroids, we often use this agent in hospitalized patients who are rapidly declining or are acutely ill with dysphagia or respiratory muscle involvement. In circumstances in which immunosuppression is relatively contraindicated, such as in patients with a history of malignancy or recurrent infection, IVIG offers a safe and effective means to gain disease control.
There are case reports of IVIG ameliorating ILD in DM [259], polymyositis [260], and systemic sclerosis [261, 262]. Further study is needed to clarify the benefit of IVIG in ILD. There is also interest in transitioning to subcutaneous immunoglobulin due to ease of dosing, particularly to maintain disease control after remission is achieved with IVIG[261,262,265].
IVIG is generally well-tolerated, though up to 56% of patients may experience headache, which can be severe and debilitating. The rate of infusion, the total dose [266] and the formulation of IVIG [267] may influence the occurrence of headache. Aseptic meningitis is a rare adverse effect of IVIG, manifesting as fever, headache, photophobia, meningismus, and neutrophilic pleocytosis or eosinophilia in the cerebrospinal fluid [268, 269]. It occurs 24–48 hours after infusion and generally resolves spontaneously without sequelae in 2–7 days. Acute kidney injury is another rare complication of IVIG; risk factors include pre-existing renal insufficiency, concomitant administration of nephrotoxic medications or sucrose preparations, and dehydration [269]. Anaphylaxis is also rare but may occur in patients with primary IgA deficiency; checking serum IgA levels prior to infusion is therefore recommended. There is no evidence that having low but detectable immunoglobulin levels confers any increased risk for anaphylaxis [266]. Finally, the risk for thrombotic complications with IVIG must be considered in patients with concomitant hypercoagulable states.
Rituximab
Rituximab has shown mixed results for cutaneous DM [270, 271], and the current evidence does not support its use for cutaneous disease alone. With regard to myositis, the RIM trial randomized 200 patients (76 with polymyositis, 76 DM, 48 juvenile DM) to either rituximab at week 0 (early) or week 8 (late), with the primary endpoint of time to disease improvement. Although there was no significant difference in the time to improvement, 83% of refractory adult and juvenile myositis patients ultimately met criteria for disease improvement [272]. A shorter time to improvement was seen in patients with anti-synthetase antibodies, namely anti-Jo-1 (hazard ratio 3.08, p < 0.01) and anti-Mi-2 (hazard ratio 2.5 p < 0.01) [273].
Rituximab has been reported to be successful in retrospective studies for the treatment of ILD. Andersson et al. published their experience with 112 patients with anti-synthetase syndrome-related ILD: of the 24 patients with severe ILD who received rituximab, 24% had improvement in FVC and 17% had improvement in diffusion capacity at 1 year [274]. The benefit was most pronounced in the 7 patients with disease duration of less than 12 months and those with acute onset of ILD. Another retrospective cohort study found that 4 of 5 patients with DM or polymyositis-associated ILD exhibited 18% improvement in FVC and 22% in diffusion capacity 9–12 months after receiving rituximab [275].
Rituximab dosing in DM typically follows the rheumatoid arthritis protocol of 1000 mg administered intravenously at day 0 and day 14 [271]. Infectious complications are the most frequent [276] serious adverse effects in DM patients, with rare reports of progressive multifocal leukoencephalopathy [274,275,278].
Cyclosporine
Cyclosporine , a calcineurin inhibitor, binds cyclophilin, inhibiting interleukin-2 production and T cell activation. The evidence for cyclosporine in cutaneous DM is limited to case reports [279], but it has been found to be a rapidly acting and effective agent for myositis [280]. A randomized clinical trial of 36 patients with DM (n = 20) or polymyositis (n = 16) comparing cyclosporine (3–3.5 mg/kg/day) with methotrexate (7.5-15 mg weekly) in addition to oral corticosteroids found comparable decreases in CK and improvements in strength between the groups at 6 months [281].
Cyclosporine is utilized in the setting of severe ILD as rescue therapy [241, 282] and has also been shown to improve overall survival in retrospective studies [283]. When used in combination with intravenous cyclophosphamide and pulse dose methylprednisolone, cyclosporine has been found beneficial for rapidly progressive ILD in anti-MDA5 DM patients in Asia [284]. Cyclosporine is dosed based on ideal body weight and has improved bioavailability in its hydrophilic microemulsion [285].
Careful monitoring of renal function and blood pressure is needed for the duration of treatment with cyclosporine; nephrotoxicity risk is highest at doses above 3 mg/kg/day [286, 287]. Cyclosporine may also induce hirsutism, gingival hyperplasia, and increased LDL cholesterol and triglyceride levels.
Tofacitinib
Tofacitinib is an oral Janus kinase (JAK)-1/3 inhibitor approved for use in rheumatoid arthritis, psoriatic arthritis, and ulcerative colitis. It suppresses interferon signaling [288], which is upregulated in DM [289], and it has therefore been investigated as an off-label treatment for this condition.
In a 2016 case series of three patients with refractory cutaneous DM, all subjects demonstrated improvement in the validated Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI) activity score as well as in pruritus [290]. Similarly, in a 2019 case series, four patients with DM refractory to immunosuppressive and immunomodulatory therapy experienced significant improvement in cutaneous and extracutaneous manifestations when treated with tofacitinib [291].
Adverse effects of tofacitinib include infection, nasopharyngitis, myocardial infarction, stroke, cancer, blood clots, and death. Laboratory monitoring is recommended due to risk of lymphocytopenia, neutropenia, anemia, elevated liver function tests, and increased serum cholesterol.
Tacrolimus
Tacrolimus , another calcineurin inhibitor, binds FK binding protein and is 100 times more potent a T-cell inhibitor than is cyclosporine [292]. There are case reports of improvement in cutaneous DM with tacrolimus [293], most often for patients with juvenile DM [294, 295]. Tacrolimus has been found to be beneficial in treatment of myositis and may allow for accelerated corticosteroid tapering [293]. However, this agent is typically reserved for refractory ILD, with several retrospective reports supporting its efficacy [294,295,298], particularly in the anti-synthetase antibody group [299]. Tacrolimus has also shown prolonged survival benefit for treatment of ILD compared with prednisone alone [300].
As with cyclosporine , due to a low therapeutic index and high inter-patient variability in pharmacodynamics, close monitoring of renal function is necessary during tacrolimus therapy to avoid nephrotoxicity. In addition, tacrolimus has been associated with GI upset and hypomagnesemia [301].
Nonsteroidal Anti-inflammatory Drugs
Nonsteroidal anti-inflammatory drugs (NSAIDs) have been reported to be useful as initial therapy for arthritis associated with juvenile DM [232]. NSAIDs are typically not effective as monotherapy for moderate or severe arthritis [226] but may be useful adjunctive agents in cases where arthritis becomes more symptomatic as oral corticosteroids are tapered. Cycloxygenase-2 selective NSAIDs such as celecoxib or meloxicam are preferred due to better gastrointestinal tolerability [302, 303], particularly if the patient is on concomitant oral corticosteroids.
Azathioprine
Azathioprine has not been assessed specifically in cutaneous DM. In combined studies of DM and polymyositis, azathioprine has shown efficacy in improving myositis, in up to 75% of cases [302,303,306], as well as overall survival [307]. The first randomized controlled trial of 28 patients with polymyositis or DM compared prednisolone in combination with azathioprine 2.5 mg/kg/day versus prednisolone in combination with methotrexate 15 mg weekly and found no difference in efficacy for myositis [25]. The second randomized controlled crossover trial of 30 patients with polymyositis or DM showed improved response in the group receiving combination oral methotrexate and azathioprine (8/15, 53%) as compared to the group receiving methotrexate alone (3/15, 20%) [280].
We occasionally combine low dose azathioprine with methotrexate when myositis is persistent with methotrexate alone. Azathioprine is commonly used as maintenance therapy in the treatment of ILD associated with idiopathic inflammatory myopathies [127, 142], typically following induction with cyclophosphamide [308, 309].
Azathioprine has comparable tolerability to methotrexate, with risks of bone marrow suppression and gastrointestinal upset [306]. Initially, thiopurine methyltransferase (TPMT) levels should be checked to avoid severe bone marrow toxicity. Additionally, a rare systemic hypersensitivity reaction, manifesting with fever, myalgia, nausea, vomiting, hypotension and shock, has been reported; it may occur in the first 4 weeks of therapy [310] and resolves with drug discontinuation.
Leflunomide
Leflunomide inhibits pyrimidine synthesis, leukocyte adhesion to vascular endothelium, and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [311], resulting in immunosuppressive and anti-inflammatory effects. Although leflunomide is used primarily to treat rheumatoid arthritis, there are 4 reported cases of its use to treat DM, with evidence of improvement in both skin and muscle disease at 20 mg daily [312, 313]. We find it useful particularly in the setting of arthritis in DM, or as a corticosteroid-sparing agent for both skin and muscle disease in patients with intolerance or poor response to methotrexate or MMF.
The most commonly reported side effectsof leflunomide are gastrointestinal, with diarrhea, nausea and abdominal pain [314]seen in 10–25% of patients. Less common side effects include alopecia, hypertension and hepatotoxicity [315, 316]. Leflunomide may induce acute interstitial pneumonitis in patients with inflammatory arthritis [315,316,319] and therefore may not be preferable in patients with ILD.
Dapsone
Dapsone is a sulfone antibiotic that inhibits neutrophil function and complement activation [320]. Its potential for benefit in cutaneous DM has been noted in several case reports [319,320,323]. In one case report [323], there was concomitant improvement in skin and muscle disease on dapsone 75 mg daily. There is no evidence for the use of dapsone in ILD.
Quantitative glucose-6-phosphate dehydrogenase (G6PD) levels should be checked prior to initiation to prevent methemoglobinemia and severe hemolysis. Common side effects include a dose-dependent hemolysis and gastrointestinal upset [324]. Rarely, hepatotoxicity, agranulocytosis and aplastic anemia have been reported, justifying serial blood monitoring [325].
Thalidomide
Thalidomide is a glutamic acid derivative that inhibits expression of tumor necrosis factor alpha (TNFα) adhesion molecules on neutrophils, TNFα synthesis, neutrophil phagocytosis, and angiogenesis [326]. We noted one case report describing its benefit in cutaneous DM, with 60% improvement [327]. Teratogenicity and increased risk of thrombosis [328] preclude its use in many patients. As many as 25% of patients may develop a dose-dependent sensory peripheral neuropathy [17, 329].
Cyclophosphamide
Skin disease rarely warrants treatment with cytotoxic agents, except perhaps when there is evidence of progressive cutaneous vasculitis [45]. The alkylating agent cyclophosphamide may be used, however, for refractory or rapidly progressive myositis. Most commonly in DM, cyclophosphamide is indicated for the treatment of ILD [45, 309, 328,329,330,331,332,333,334,335,338].
Although a randomized controlled trial demonstrated efficacy over placebo in the treatment of systemic sclerosis-associated ILD [339], the supportive evidence for the treatment of ILD associated with inflammatory myopathies derives primarily from case reports. In a randomized trial of 10 DM patients with rapidly progressive ILD, Kameda et al. compared a three-drug combination regimen (prednisolone 0.5 mg/kg/day, intravenous cyclophosphamide 10–30 mg/kg every 3–4 weeks, and cyclosporine 2–4 mg/kg/day) with dual agent therapy consisting of corticosteroids plus either agent alone [299]. Yamasaki et al. treated 14 DM patients with refractory ILD with intravenous cyclophosphamide 300–800 mg/m2 every 4 weeks and observed significant improvements in HRCT, PFTs, and dyspnea [45].
Cyclophosphamide may produce a host of immediate toxicities, including nausea and vomiting, alopecia, myelosuppression, hemorrhagic cystitis and, rarely, interstitial pneumonitis [340]. Long-term toxicities include malignancy (skin, bladder and hematologic) [341], infertility and gonadal failure [342]. Vigilant monitoring with serial blood tests and urinalyses is essential.
Special Cases: Calcinosis Cutis and Ulceration
Calcinosis is the Achilles heel for DM patients and clinicians alike. Surgical excision for localized lesions remains the definitive therapy [312]. Data are lacking to guide medical therapy. Multiple agents have been proposed, including anti-inflammatories and calcium and phosphate modulators, but no single agent is reliably effective [26]. IVIG has been reported to be effective in some cases, especially in juvenile DM, [313, 343, 344] but not others [345]. Bisphosphonates have been cited as effective for calcinosis in juvenile DM, but controlled studies are needed [346, 347]. The editor (AG) of this textbook has observed softening and size reductions of plaques and nodules of calcinosis with both intralesional and intravenous sodium thiosulfate (personal communication). Lastly, a study of risk factors for calcinosis identified that digital ulcers were present in 50% of DM patients with digital ulcers vs. 9% without digital ulcers (p < .001), suggesting a common underlying vascular mechanism [31]; given these data, it is plausible that long-term vasodilatory therapy may be effective for the prevention and treatment of calcinosis.
Cutaneous ulceration in anti-MDA5 DM patients can also be challenging to treat. We have found coexisting thrombophilias in several of our patients with this autoantibody. Treatment of any underlying hypercoagulable state may be instrumental in ulcer healing. We speculate that the pathophysiology of ulceration in anti-MDA5 DM is a vasculopathy with endothelial damage and microvascular occlusion. The most common sites of ulceration are the extensor surfaces over the joints or on the digits [24]. We have achieved resolution of chronic ulceration by using potent vasodilators, such as phosphodiesterase inhibitors like sildenafil 20–40 mg three times daily.
Summary
Dermatomyositis (DM) is a systemic autoimmune disease that commonly manifests with inflammation of the skin, muscle and lungs. Patients are at increased risk of malignancy at disease onset and should undergo cancer screening. ILD is another important cause of morbidity and mortality in DM. Collaboration between rheumatology and dermatology, among other disciplines, is essential to ensure appropriate assessment of all possible involved organs and treatment monitoring.
Skin and muscle disease often respond at different rates and require different treatments. Patients may require multiple agents to achieve remission, and the risks and benefits of such treatment must be weighed carefully given the frequent need for long-term treatment.
References
Olazagasti JM, et al. Cancer risk in dermatomyositis: a meta-analysis of cohort studies. Am J Clin Dermatol. 2015;16(2):89–98.
Madan V, et al. Defining cancer risk in dermatomyositis. Clin Exp Dermatol. 2009;34:451–5.
Meyer A, et al. Incidence and prevalence of inflammatory myopathies: a systematic review. Rheumatology (Oxford). 2015;54(1):50–63.
Bernatsky S, et al. Estimating the prevalence of polymyositis and dermatomyositis from administrative data: age, sex and regional differences. Ann Rheum Dis. 2009;68:1192–6.
Bendewald MJ, et al. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146(1):26–30.
See LC, et al. Sex-and age-specific incidence of autoimmune rheumatic diseases in the Chinese population: a Taiwan population-based study. Semin Arthritis Rheum. 2013;43(3):381–6.
Hengstman GJ, et al. The relative prevalence of dermatomyositis and polymyositis in Europe exhibits a latitudinal gradient. Ann Rheum Dis. 2000;59:141–2.
Okada S, et al. Global surface ultraviolet radiation intensity may modulate the clinical and immunologic expression of autoimmune muscle disease. Arthritis Rheum. 2003;48(8):2285–93.
Love LA, et al. Ultraviolet radiation intensity predicts the relative distribution of dermatomyositis and anti-Mi-2 autoantibodies in women. Arthritis Rheum. 2009;60:2499–504.
Petri MH, et al. Implications in the difference of anti-Mi-2 and-p155/140 autoantibody prevalence in two dermatomyositis cohorts from Mexico City and Guadalajara. Arthritis Res Ther. 2013;15(2):R48.
Shah M, et al. Brief report ultraviolet radiation exposure is associated with clinical and autoantibody phenotypes in juvenile myositis. Arthritis Rheum. 2013;65(7):1934–41.
Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975;292(8 SRC):403–7.
Bohan A, et al. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344–7.
Pearson C. Arthritis and allied conditions: a textbook of rheumatology, Polymyositis and dermatomyositis. 9th ed. Philadelphia: Lea & Febiger; 1972.
Euwer RL, Sontheimer RD. Amyopathic dermatomyositis (dermatomyositis sine myositis). Presentation of six new cases and review of the literature. J Am Acad Dermatol. 1991;24:959–66.
Sontheimer RD. Dermatomyositis: an overview of recent progress with emphasis on dermatologic aspects. Dermatol Clin. 2002;20(3):387–408.
Bastuji-Garin S, et al. Incidence and risk factors for thalidomide neuropathy: a prospective study of 135 dermatologic patients. J Invest Dermatol. 2002;119:1020–6.
Love LA, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore). 1991;70:360–74.
Schultz HY, et al. From pathogenesis, epidemiology, and genetics to definitions, diagnosis, and treatments of cutaneous lupus erythematosus and dermatomyositis: a report from the 3rd International Conference on Cutaneous Lupus Erythematosus (ICCLE). J Dermatol. 2015;135(1):7–12.
Nousari HC, et al. Centripetal flagellate erythema: a cutaneous manifestation associated with dermatomyositis. J Rheumatol. 1999;26(3):692–5.
Bernet L, et al. JAMA Dermatology. 2016;152(9):1049–51. PMID: 27224238.
Smith RL, et al. Skin involvement in juvenile dermatomyositis is associated with loss of end row nailfold capillary loops. J Rheumatol. 2004;31(8):1644–9.
Manfredi A, et al. Nailfold capillaroscopic changes in dermatomyositis and polymyositis. Clin Rheumatol. 2015;34(2):279–84.
Narang NS, et al. Cutaneous ulceration in dermatomyositis: association with anti-melanoma differentiation-associated gene 5 antibodies and interstitial lung disease. Arthritis Care Res (Hoboken). 2015;67(5):667–72.
Miller J, et al. Randomised double blind controlled trial of methotrexate and steroids compared with azathioprine and steroids in the treatment of idiopathic inflammatory myopathy. J Neurol Sci. 2002;199:S53.
Hoeltzel MF, et al. The presentation, assessment, pathogenesis, and treatment of calcinosis in juvenile dermatomyositis. Curr Rheumatol Rep. 2014;16:467.
Balin SJ, et al. Calcinosis cutis occurring in association with autoimmune connective tissue disease: the Mayo Clinic experience with 78 patients. Arch Dermatol. 2012;148:455–62.
Sallum AM, et al. Risk factors associated with calcinosis of juvenile dermatomyositis. J Pediatr. 2008;84(1):68–74.
Sanner H, et al. Cumulative organ damage and prognostic factors in juvenile dermatomyositis: a cross-sectional study median 16.8 years after symptom onset. Rheumatology (Oxford). 2009;48(12):1541–7.
Gunawardena H, et al. Autoantibodies to a 140-kd protein in juvenile dermatomyositis are associated with calcinosis. Arthritis Rheum. 2009;60:1807–14.
Valenzuela A, et al. Identification of clinical features and autoantibodies associated with calcinosis in dermatomyositis. JAMA Dermatol. 2014;150(7):724–9.
Hansen CB, Callen JP. Connective tissue panniculitis: lupus panniculitis, dermatomyositis, morphea/scleroderma. Dermatol Ther. 2010;23:341–9.
Sarin KY, et al. Molecular profiling to diagnose a case of atypical dermatomyositis. J Invest Dermatol. 2013;133(12):2796–9.
Wong D, et al. Interferon and biologic signatures in dermatomyositis skin: specificity and heterogeneity across diseases. PLoS One. 2012;7:e29161.
Milisenda JC, et al. Dermatomyositis presenting with severe subcutaneous edema: five additional cases and review of the literature. Semin Arthritis Rheum. 2014;44(2):228–33.
Tu J, McLean-Tooke A, Junckerstorff R. Increasing recognition of dermatomyositis with subcutaneous edema-is this a poorer prognostic marker? Dermatol Online J. 2014;20(1):21244.
Taggart AJ, et al. Anti Jo-1 myositis. 'Mechanic's hands' and interstitial lung disease. Ulster Med. 2002;71(1):68–71.
Sato Y, et al. Clinical characterization of dermatomyositis associated with mechanic's hands. J Dermatol. 2012;39(12):1093–5.
Ishigaki K, et al. Skin ulcer is a predictive and prognostic factor of acute or subacute interstitial lung disease in dermatomyositis. Rheumatol Int. 2013;33:2381–9.
Ideura G, et al. Interstitial lung disease associated with amyopathic dermatomyositis: review of 18 cases. Respir Med. 2007;101:1406–11.
Mimori A, et al. Dermatomyositis and cutaneous necrosis: report of five cases. Mod Rheumatol. 2000;10(2):117–20.
Di Rollo D, et al. Cancer risk in dermatomyositis: a systematic review of the literature. G Ital Dermatol Venereol. 2014;149(5):525–37.
Lu X, et al. Factors predicting malignancy in patients with polymyositis and dermatomyostis: a systematic review and meta-analysis. PLoS One. 2014;9:e94128.
Ueda-Hayakawa I, et al. Cutaneous necrotizing vasculitis in a patient with dermatomyositis positive for anti-PL-7 antibody. Eur J Dermatol. 2013;23(6):889–90.
Tsujimura S, Saito K, Tanaka Y. Complete resolution of dermatomyositis with refractory cutaneous vasculitis by intravenous cyclophosphamide pulse therapy. Intern Med. 2008;47(21):1935–40.
Kawakami T, et al. Histopathological evidence of small-vessel vasculitis within the skin and lungs associated with interstitial pneumonia in an adult patient with dermatomyositis. Clin Exp Dermatol. 2008;33(4):415–7.
Kadoya A, et al. Cutaneous vasculitis in a patient with dermatomyositis without muscle involvement. Intern Med. 1994;33:809–12.
Martini A, et al. Recurrent juvenile dermatomyositis and cutaneous necrotizing arteritis with molecular mimicry between streptococcal type 5 M protein and human skeletal myosin. J Pediatr. 1992;121(5 Pt 1):739–42.
Inuzuka M, et al. Acquired ichthyosis associated with dermatomyositis in a patient with hepatocellular carcinoma. Br J Dermatol. 2001;144:416–7.
Roselino AM, et al. Dermatomyositis and acquired ichthyosis as paraneoplastic manifestations of ovarian tumor. Int J Dermatol. 1997;36(8):611–4.
Urrutia S, et al. Sanchez acquired ichthyosis associated with dermatomyositis. J Am Acad Dermatol. 1987;16(3 Pt 1):627–9.
Smith ES, et al. Dermatomyositis: a clinicopathological study of 40 patients. Am J Dermatopathol. 2009;31(1):61–7.
Magro CM, et al. Cutaneous lesions of dermatomyositis with supervening fibrosis. J Cutan Pathol. 2008;35(1):31–9.
Jones SA, Black MM. The value of direct immunofluorescence as a diagnostic aid in dermatomyositis-a study of 35 cases. Clin Exp Dermatol. 1997;22(2):77–81.
Magro CM, Crowson AN. The immunofluorescent profile of dermatomyositis: a comparative study with lupus erythematosus. J Cutan Pathol. 1997;24(9):543–52.
Mascaro JMJ, et al. Membrane attack complex deposits in cutaneous lesions of dermatomyositis. Arch Dermatol. 1995;131(12):1386–92.
Serarslan G, Atik E, Sarikaya G. Periorbital edema and erythema: an unusual localization of DLE in a patient with psoriasis. J Dermatol. 2011;38(5):486–8.
Braun RP, et al. Periorbital oedema and erythema as a manifestation of discoid lupus erythematosus. Dermatology. 2002;205:194–7.
Cyran S, Douglass MC, Silverstein JL. Chronic cutaneous lupus erythematosus presenting as periorbital edema and erythema. J Am Acad Dermatol. 1992;26(2 Pt 2):334–8.
Parkins GJ, Maan A, Dawn G. Neurogenic rosacea: an uncommon and poorly recognized entity? Clin Exp Dermatol. 2015;40:930.
Scharschmidt TC, et al. Neurogenic rosacea: a distinct clinical subtype requiring a modified approach to treatment. Arch Dermatol. 2011;147(1):123–6.
Rockerbie NR, et al. Cutaneous changes of dermatomyositis precede muscle weakness. J Am Acad Dermatol. 1989;20(4):629–32.
Cosnes A, et al. Dermatomyositis without muscle weakness. Longterm followup of 12 patients without systemic corticosteroids. Arch Dermatol. 1995;131:1381–5.
Pachman LM, et al. Juvenile dermatomyositis at diagnosis: clinical characteristics of 79 children. J Rheumatol. 1998;25(6):1198–204.
Parodi A, et al. De La dermatomyositis in 132 patients with different clinical subtypes: cutaneous signs, constitutional symptoms and circulating antibodies. Acta Derm Venereol. 2002;82(1):48–51.
Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971–82.
Barohn RJ, Dimachkie MM, Jackson CE. A pattern recognition approach to patients with a suspected myopathy. Neurol Clin. 2014;32:569–93.
Kissel JT, Mendell JR, Rammohan KW. Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med. 1986;314(6):329–34.
Arahata K, Engel AG. Monoclonal antibody analysis of mononuclear cells in myopathies I Quantitation of subsets according to diagnosis and sites of accumulation and demonstration and counts of muscle fibers invaded by T cells. Ann Neurol. 1984;16:193–208.
Greenberg SA, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol. 2005;57(5):664–78.
Bohan A, et al. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56:255–86.
Briani C, et al. Update on idiopathic inflammatory myopathies. Autoimmunity. 2006;39:161–70.
Del Grande F, et al. Magnetic resonance imaging of inflammatory myopathies. Top Magn Reson Imaging. 2011;22(2):39–43.
Curiel RV, Jones R, Brindle K. Magnetic resonance imaging of the idiopathic inflammatory myopathies: structural and clinical aspects. Ann N Y Acad Sci. 2009;1154:101–14.
Adams EM, et al. The idiopathic inflammatory myopathies: spectrum of MR imaging findings. Radiographics. 1995;15:563–74.
Yoshida K, et al. Fasciitis as a common lesion of dermatomyositis, demonstrated early after disease onset by en bloc biopsy combined with magnetic resonance imaging. Arthritis Rheum. 2010;62(12):3751–9.
Mammen AL. Autoimmune myopathies: autoantibodies, phenotypes and pathogenesis. Nat Rev Neurol. 2011;7(6):343–54.
Smith BW, McCarthy JC, Dawley CA. Suspect myopathy? Take this approach to the work-up. J Fam Pract. 2014;63(11):631–8.
Paik JJ, Corse AM, Mammen AL. The co-existence of myasthenia gravis in patients with myositis: a case series. Semin Arthritis Rheum. 2014;43(6):792–6.
Fraga MM, et al. Devic's disease in an adolescent girl with juvenile dermatomyositis. Rev Bras Reumatol. 2015;S0482-5004:00020-0.
Delman D, et al. Dermatomyositis as a presentation of neuromyelitis optica spectrum disorder. J Neuroimmunol. 2015;278:108–11.
Tsokos GC, Moutsopoulos HM, Steinberg AD. Muscle involvement in systemic lupus erythematosus. JAMA. 1981;246(7):766–8.
Jacobsen S, et al. A multicentre study of 513 Danish patients with systemic lupus erythematosus II. Disease manifestations and analyses of clinical subsets. Clin Rheumatol. 1998;17:468–77.
Foote RA, Kimbrough SM, Stevens JC. Lupus myositis. Muscle Nerve. 1982;5(1):65–8.
Garton MJ, Isenberg DA. Clinical features of lupus myositis versus idiopathic myositis: a review of 30 cases. Br J Rheumatol. 1997;36(10):1067–74.
Font J, et al. Clusters of clinical and immunologic features in systemic lupus erythematosus: analysis of 600 patients from a single center. Semin Arthritis Rheum. 2004;33(4):217–30.
Isenber DA, Snaith ML. Muscle disease in systemic lupus erythematosus: a study of its nature, frequency and cause. J Rheumatol. 1981;8(6):917–24.
Maazoun F, et al. Systemic lupus erythematosusmyositis overlap syndrome: report of 6 cases. Clin Pract. 2011;1(4):e89.
Jakati S, et al. SLE myopathy: a clinicopathological study. Int J Rheum Dis. 2015;18:886.
Milisenda JC, Selva-O'Callaghan A, Grau JM. The diagnosis and classification of polymyositis. Autoimmun Rev. 2014;48-49:118–21.
Mahler M, Miller FW, Fritzler MJ. Idiopathic inflammatory myopathies and the anti-synthetase syndrome: a comprehensive review. Autoimmun Rev. 2014;13:367–71.
Stenzel W, et al. Nuclear actin aggregation is a hallmark of anti-synthetase syndrome-induced dysimmune myopathy. Neurology. 2015;84(13):1346–54.
Griggs RC, et al. Inclusion body myositis and myopathies. Ann Neurol. 1995;38(5):705–13.
Brady S, et al. A retrospective cohort study identifying the principal pathological features useful in the diagnosis of inclusion body myositis. BMJ Open. 2014;4:e004552.
Hong J, et al. Muscle disease in systemic sclerosis is associated with an increased risk for cardiac involvement. Dent Abstr. 2014;721.
Paik JJ, et al. Myopathy in scleroderma, its identification, prevalence, and treatment: lessons learned from cohort studies. Curr Opin Rheumatol. 2014;26(2):124–30.
Muangchan C, et al. The 15% rule in scleroderma: the frequency of severe organ complications in systemic sclerosis. A systematic review. J Rheumatol. 2013;40(9):1545–56.
Koschik RWN, et al. Anti-PM-Scl antibody in patients with systemic sclerosis. Clin Exp Rheumatol. 2012;30:S12–6.
Toll A, et al. Sclerodermatomyositis associated with severe arthritis. Dermatol Online J. 2004;10(2):18.
Garcia-Patos V, et al. Childhood sclerodermatomyositis: report of a case with the anti-PM/Scl antibody and mechanic's hands. Br J Dermatol. 1996;135:613–6.
Wilson NO, et al. Trichinellosis surveillance-United States, 2008-2012. MMWR Surveill Summ. 2015;1:2008–12.
Kociecka W. Trichinellosis: human disease, diagnosis and treatment. Vet Parasitol. 2000;93(3–4):365–83.
Neghina R, Neghina AM, Marincu I. Reviews on trichinellosis (III): cardiovascular involvement. Foodborne Pathog Dis. 2011;8(8):853–60.
Gupta A, Gupta Y. Glucocorticoid-induced myopathy: pathophysiology, diagnosis, and treatment. Indian J Endocrinol Metab. 2013;17:913–6.
Pereira RM, Freire de Carvalho J. Glucocorticoid-induced myopathy. Joint Bone Spine. 2011;78(1):41–4.
Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol. 2006;97:52C–60.
Bruckert E, et al. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients–the PRIMO study. Cardiovasc Drugs Ther. 2005;19(6):403–14.
Graham DJ, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA. 2004;292:2585–90.
Molokhia M, et al. Statin induced myopathy and myalgia: time trend analysis and comparison of risk associated with statin class from. PLoS One. 2008;3(6):1991–2006.
Mammen AL, et al. Increased frequency of DRB1*11:01 in anti-hydroxymethylglutaryl-coenzyme a reductase-associated autoimmune myopathy. Arthritis Care Res Hoboken. 2012;64(8):1233–7.
Group, S.C, et al. SLCO1B1 variants and statin-induced myopathy–a genomewide study. N Engl J Med. 2008;359(8):789–99.
Mammen AL. Toxic myopathies. Continuum (Minneap Min), 2013. 19(6 Muscle Disease): p. 1634–49.
Hansen KE, et al. Outcomes in 45 patients with statin-associated myopathy. Arch Intern Med. 2005;165:2671–6.
Mammen AL, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme a reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum. 2011;63(3):713–21.
Needham M, et al. Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul Disord. 2007;17(2):194–200.
Christopher-Stine L, et al. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. 2010;62:2757–66.
Nance JR, Mammen AL. Diagnostic evaluation of rhabdomyolysis. Muscle Nerve. 2015;51(6):793–810.
Pamfil C, et al. Primary biliary cirrhosis-autoimmune hepatitis overlap syndrome associated with dermatomyositis, autoimmune thyroiditis and antiphospholipid syndrome. J Gastrointestin Liver Dis. 2015;24(1):101–4.
Noda S, et al. A case of dermatomyositis with "liver disease associated with rheumatoid diseases" positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29(8):941–3.
Saravanan V, Kelly C. Drug-related pulmonary problems in patients with rheumatoid arthritis. Rheumatology (Oxford). 2006;45(7):787–9.
Sathi N, et al. How common is methotrexate pneumonitis? A large prospective study investigates. Clin Rheumatol. 2012;31(1):79–83.
Marie I, et al. Short-term and long-term outcomes of interstitial lung disease in polymyositis and dermatomyositis: a series of 107 patients. Arthritis Rheum. 2011;63(11):3439–47.
Kang EH, et al. Interstitial lung disease in patients with polymyositis, dermatomyositis and amyopathic dermatomyositis. Rheumatology (Oxford). 2005;44:1282–6.
Fiorentino D, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. Acad Dermatol. 2011;65:25–34.
Sun Y, et al. Interstitial lung disease in clinically amyopathic dermatomyositis (CADM) patients: a retrospective study of 41 Chinese Han patients. Rheumatol Int. 2013;33(5):1295–302.
Mukae H, et al. Clinical differences between interstitial lung disease associated with clinically amyopathic dermatomyositis and classic dermatomyositis. Chest. 2009;136(5):1341–7.
Connors GR, et al. Interstitial lung disease associated with the idiopathic inflammatory myopathies: what progress has been made in the past 35 years? Chest. 2010;138:1464–74.
Richards TJ, et al. Characterization and peripheral blood biomarker assessment of anti-Jo-1 antibody-positive interstitial lung disease. Arthritis Rheum. 2009;60(7):2183–92.
Cao H, et al. Clinical manifestations of dermatomyositis and clinically amyopathic dermatomyositis patients with positive expression of anti-melanoma differentiation-associated gene 5 antibody. Arthritis Care Res Hoboken. 2012;64:1602–10.
Chen Z, et al. Utility of anti-melanoma differentiation-associated gene 5 antibody measurement in identifying patients with dermatomyositis and a high risk for developing rapidly progressive interstitial lung disease: a review of the literature and a meta-analysis. Arthritis Care Res Hoboken. 2013;65:1316–24.
Hamaguchi Y, et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Arch Dermatol. 2011;147:391–8.
Gono T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713–9.
Labrador-Horrillo M, et al. Anti-MDA5 antibodies in a large Mediterranean population of adults with dermatomyositis. J Immunol Res. 2014;2014:290797.
Horai Y, et al. Serum interferon-alpha is a useful biomarker in patients with anti-melanoma differentiation-associated gene 5 (MDA5) antibody-positive dermatomyositis. Mod Rheumatol. 2015;25:85–9.
Gono T, et al. Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatology (Oxford). 2012;51:1563–70.
Mimori T, Nakashima R, Hosono Y. Interstitial lung disease in myositis: clinical subsets, biomarkers, and treatment. Curr Rheumatol Rep. 2012;14(3):264–74.
Martinez FJ, Flaherty K. Pulmonary function testing in idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006;3(4):315–21.
Wells AU, et al. Fibrosing alveolitis in systemic sclerosis: indices of lung function in relation to extent of disease on computed tomography. Arthritis Rheum. 1997;40(7):1229–36.
Raghu G, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824.
Silva JA, et al. Da Safety of low dose glucocorticoid treatment in rheumatoid arthritis: published evidence and prospective trial data. Ann Rheum Dis. 2006;65:285–93.
Fathi M, et al. Interstitial lung disease, a common manifestation of newly diagnosed polymyositis and dermatomyositis. Ann Rheum Dis. 2004;63(3):297–301.
Douglas WW, et al. Polymyositis-dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med. 2001;164(7):1182–5.
Johkoh T. Nonspecific interstitial pneumonia and usual interstitial pneumonia: is differentiation possible by high-resolution computed tomography? Semin Ultrasound CT MR. 2014;35(1):24–8.
Feiner J, et al. Chapter 23, in The second heart sound. In: Clinical methods the history physical and laboratory examinations. 3rd ed. Boston: Butterworths; 1990.
Zhang RF, et al. Diagnostic value of transthoracic Doppler echocardiography in pulmonary hypertension: a meta-analysis. Am J Hypertens. 2010;23(12):1261–4.
Mathai SC, et al. Tricuspid annular plane systolic excursion is a robust outcome measure in systemic sclerosis-associated pulmonary arterial hypertension. J Rheumatol. 2011;38(11):2410–8.
Hoeper MM, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25):D42–50.
Zhang L, et al. Cardiac involvement in adult polymyositis or dermatomyositis: a systematic review. Clin Cardiol. 2012;35(11):686–91.
Gonzalez-Lopez L, et al. Cardiac manifestations in dermato-polymyositis. Clin Exp Rheumatol. 1996;14(4):373–9.
Rosenbohm A, et al. Early diagnosis of cardiac involvement in idiopathic inflammatory myopathy by cardiac magnetic resonance tomography. J Neurol. 2015;262(4):949–56.
Hughes M, et al. Cardiac troponin testing in idiopathic inflammatory myopathies and systemic sclerosis-spectrum disorders: biomarkers to distinguish between primary cardiac involvement and low-grade skeletal muscle disease activity. Ann Rheum Dis. 2015;74(5):795–8.
Pau-Charles I, et al. Anti-MDA5 positive clinically amyopathic dermatomyositis presenting with severe cardiomyopathy. J Eur Acad Dermatol Venereol. 2014;28(8):1097–102.
Sakurai N, et al. Anti-CADM-140 antibody-positive juvenile dermatomyositis with rapidly progressive interstitial lung disease and cardiac involvement. J Rheumatol. 2011;38(5):963–4.
Esmon CT. The interactions between inflammation and coagulation. Br J Haematol. 2005;131(4):417–30.
Limaye VS, et al. Idiopathic inflammatory myositis is associated with a high incidence of hypertension and diabetes mellitus. Int J Rheum Dis. 2010;13(2):132–7.
Durante A, Bronzato S. The increased cardiovascular risk in patients affected by autoimmune diseases: review of the various manifestations. J Clin Med Res. 2015;7(6):379–84.
Ungprasert P, et al. Risk of coronary artery disease in patients with idiopathic inflammatory myopathies: a systematic review and meta-analysis of observational studies. Semin Arthritis Rheum. 2014;44(1):63–7.
Linos E, et al. Atherosclerotic cardiovascular disease and dermatomyositis: an analysis of the Nationwide Inpatient Sample survey. Arthritis Res Ther. 2013;15(1):R7.
Lai YT, et al. Dermatomyositis is associated with an increased risk of cardiovascular and cerebrovascular events: a Taiwanese population-based longitudinal follow-up study. Br J Dermatol. 2013;168(5):1054–9.
Carruthers EC, et al. Risk of deep venous thrombosis and pulmonary embolism in individuals with polymyositis and dermatomyositis: a general population-based study. Ann Rheum Dis. 2014;75:110.
Selva-O'Callaghan A, et al. Venous thromboembolism in patients with dermatomyositis and polymyositis. Clin Exp Rheumatol. 2011;29(5):846–9.
Zoller B, et al. Risk of pulmonary embolism in patients with autoimmune disorders: a nationwide follow-up study from Sweden. Lancet. 2012;379(9812):244–9.
Ramagopalan SV, et al. Risk of venous thromboembolism in people admitted to hospital with selected immune-mediated diseases: record-linkage study. BMC Med. 2011;9:1–7015.
Koh ET, et al. Adult onset polymyositis/dermatomyositis: clinical and laboratory features and treatment response in 75 patients. Ann Rheum Dis. 1993;52(12):857–61.
Citera G, et al. Maldonado joint involvement in polymyositis/dermatomyositis. Clin Rheumatol. 1994;13(1):70–4.
Porkodi R, et al. Clinical spectrum of inflammatory myositis in South India-a ten year study. J Assoc Physicians India. 2002;50:1255–8.
Klein M, et al. Arthritis in idiopathic inflammatory myopathy: clinical features and autoantibody associations. J Rheumatol. 2014;41(6):1133–9.
Hall JC, et al. Anti-melanoma differentiation-associated protein 5-associated dermatomyositis: expanding the clinical spectrum. Arthritis Care Res Hoboken. 2013;65(8):1307–15.
Wasko MC, et al. Dermatomyositis with erosive arthropathy: association with the anti-PL-7 antibody. J Rheumatol. 1999;26(12):2693–4.
Oddis CV, et al. Subluxing arthropathy associated with the anti-Jo-1 antibody in polymyositis/dermatomyositis. Arthritis Rheum. 1990;33(11):1640–5.
Schumacher HR, et al. Articular manifestations of polymyositis and dermatomyositis. Am J Med. 1979;67(2):287–92.
Levin J, Werth VP. Skin disorders with arthritis. Best Pract Res Clin Rheumatol. 2006;20(4):809–26.
Mumm GE, McKown KM, Bell CL. Antisynthetase syndrome presenting as rheumatoid-like polyarthritis. J Rheumatol. 2010;16(7):307–12.
Nesher G. Polymyalgia rheumatica-diagnosis and classification. J Autoimmun. 2014;48–49:76.
Couvrat-Desvergnes G, et al. The spectrum of renal involvement in patients with inflammatory myopathies. Medicine (Baltimore). 2014;93(1):33–41.
Yen TH, et al. Unexpected IgA nephropathy during the treatment of a young woman with idiopathic dermatomyositis: case report and review of the literature. J Nephrol. 2003;16(1):148–53.
Barros TB, et al. IgA nephropathy and polymyositis: a rare association. Rev Bras Reumatol. 2014;54(3 SRC):231–3.
Civilibal M, et al. Selcuk nephropathy associated with juvenile dermatomyositis. Pediatr Nephrol. 2009;24(10):2073–5.
Akashi Y, et al. Dermatomyositis associated with membranous nephropathy in a 43-year-old female. Am J Nephrol. 2002;22(4):385–8.
Makino H, et al. Membranous nephropathy developing during the course of dermatomyositis. J Rheumatol. 1994;21(7):1377–8.
Rose JD. Membranous glomerulonephritis, dermatomyositis, and bronchial carcinoma. Br Med J. 1979;2(6191):641.
Xie Q, et al. Diffuse proliferative glomerulonephritis associated with dermatomyositis with nephrotic syndrome. Rheumatol Int. 2010;30(6):821–5.
Kawai H, et al. Myeloperoxidase-antineutrophil cytoplasmic antibody-related crescentic glomerulonephritis after treatment for clinically amyopathic dermatomyositis: a coincidental combination or not? Clin Exp Nephrol. 2011;15(4):577–81.
Yuste C, et al. Overlap between dermatomyositis and ANCA vasculitides. Clin Kidney J. 2014;7(1):59–61.
Henter JI, et al. HLH-: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–31.
Buchbinder R, et al. Incidence of malignant disease in biopsy-proven inflammatory myopathy. A population based cohort study. Ann Intern Med. 2001;134(12):1087–95.
Hill CL, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357(9250):96–100.
Chow WH, et al. Cancer risk following polymyositis and dermatomyositis: a nationwide cohort study in Denmark. Cancer Causes Control. 1995;6(1):9–13.
Sigurgeirsson B, et al. Risk of cancer in patients with dermatomyositis or polymyositis. A population based study. N Engl J Med. 1992;326(6):363–7.
Huang YL, et al. Malignancies associated with dermatomyositis and polymyositis in Taiwan: a nationwide population-based study. Br J Dermatol. 2009;161(4):854–60.
Ichimura Y, et al. Anti-NXP2 autoantibodies in adult patients with idiopathic inflammatory myopathies: possible association with malignancy. Ann Rheum Dis. 2012;71(5):710–3.
Cammas F, et al. TRIM involvement in transcriptional regulation. Adv Exp Med Biol. 2012;770:59–76.
Fiorentino DF, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1gamma. Arthritis Rheum. 2013;65(11):2954–62.
Trallero-Araguas E, et al. Usefulness of anti-p155 autoantibody for diagnosing cancer-associated dermatomyositis: a systematic review and meta-analysis. Arthritis Rheum. 2012;64(2):523–32.
Shah AA, Casciola-Rosen L, Rosen A. Review: cancer-induced autoimmunity in the rheumatic diseases. Arthritis Rheum. 2015;67(2):317–26.
Sparsa A, et al. Routine vs extensive malignancy search for adult dermatomyositis and polymyositis: a study of 40 patients. Arch Dermatol. 2002;138(7):885–90.
Leatham H, et al. Medicine (Baltimore). 2018;97(2):e9639. PMID: 29480875.
Kim S, et al. Complete and sustained remission of juvenile dermatomyositis resulting from aggressive treatment. Arthritis Rheum. 2009;60(6):1825–30.
Jordan JR, et al. Spontaneously remitting dermatomyositis. Acad Dermatol Venereol. 2004;18(4):495–8.
Gordon PA, et al. Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database Syst Rev. 2012;(8):CD003643.
Femia AN, Vleugels RA, Callen JP. Cutaneous dermatomyositis: an updated review of treatment options and internal associations. Am J Clin Dermatol. 2013;14(4):291–313.
Lam C, Vleugels RA. Management of cutaneous dermatomyositis. Dermatol Ther. 2012;25(2):112–34.
Strowd LC, Jorizzo JL. Review of dermatomyositis: establishing the diagnosis and treatment algorithm. J Dermatolog Treat. 2013;24(6):418–21.
Jorizzo JL, Vleugels RA. Dermatomyositis. In: Bolognia J, Jorizzo JL, Schaefer JV, editors. Dermatology. 3rd ed. Elsevier: Edinburgh; 2012.
Dourmishev L, Meffert H, Piazena H. Dermatomyositis: comparative studies of cutaneous photosensitivity in lupus erythematosus and normal subjects. Photodermatol Photoimmunol Photomed. 2004;20(5 SRC):230–4.
Cheong WK, et al. Cutaneous photosensitivity in dermatomyositis. Br J Dermatol. 1994;131(2):205–8.
Mattar MA, et al. Safety and possible effects of low-intensity resistance training associated with partial blood flow restriction in polymyositis and dermatomyositis. Arthritis Res Ther. 2014;16(5):473–014.
Alexanderson H, et al. Benefits of intensive resistance training in patients with chronic polymyositis or dermatomyositis. Arthritis Rheum. 2007;57:768–77.
Wiesinger GF, et al. Improvement of physical fitness and muscle strength in polymyositis/dermatomyositis patients by a training programme. Br J Rheumatol. 1998;37(2):196–200.
Riisager M, et al. Aerobic training in persons who have recovered from juvenile dermatomyositis. Neuromuscul Disord. 2013;23(12):962–8.
Omori CH, et al. Exercise training in juvenile dermatomyositis. Arthritis Care Res (Hoboken). 2012;64(8):1186–94.
Alexanderson H, et al. The safety of a resistive home exercise program in patients with recent onset active polymyositis or dermatomyositis. Scandinavica. 2000;29:295–301.
Alexanderson H, et al. Resistive home exercise in patients with recent-onset polymyositis and dermatomyositis-a randomized controlled single-blinded study with a 2-year followup. J Rheumatol. 2014;41:1124–32.
Peyrot I, et al. Topical tacrolimus and resistant skin lesions of dermatomyositis. Rev Med Interne. 2006;27(10 SRC):730–5.
Lampropoulos CE, D'Cruz DP. Topical tacrolimus treatment in a patient with dermatomyositis. Ann Rheum Dis. 2005;64(9):1376–7.
Hollar CB, Jorizzo JL. Topical tacrolimus 0.1% ointment for refractory skin disease in dermatomyositis a pilot study. J Dermatolog Treat. 2004;15(1):35–9.
Yoshimasu T, et al. Topical FK506 (tacrolimus) therapy for facial erythematous lesions of cutaneous lupus erythematosus and dermatomyositis. Eur J Dermatol. 2002;12(1):50–2.
Garcia-Doval I, Cruces M. Topical tacrolimus in cutaneous lesions of dermatomyositis: lack of effect in side-by-side comparison in five patients. Dermatology. 2004;209(3):247–8.
Kim JE, et al. Successful treatment of cutaneous lesions of dermatomyositis with topical pimecrolimus. Ann Dermatol. 2011;23(3):348–51.
Soter NA, et al. Tacrolimus ointment for the treatment of atopic dermatitis in adult patients: part II, safety. J Am Acad Dermatol. 2001;44(1):S39–46.
Pereira U, et al. Mechanisms of the sensory effects of tacrolimus on the skin. Br J Dermatol. 2010;163(1):70–7.
Rustin MH. The safety of tacrolimus ointment for the treatment of atopic dermatitis: a review. Br J Dermatol. 2007;157(5):861–73.
Woo TY, et al. Cutaneous lesions of dermatomyositis are improved by hydroxychloroquine. J Am Acad Dermatol. 1984;10(4):592–600.
Pelle MT, Callen JP. Adverse cutaneous reactions to hydroxychloroquine are more common in patients with dermatomyositis than in patients with cutaneous lupus erythematosus. Arch Dermatol. 2002;138(9):1231–3.
Ang GC, Werth VP. Combination antimalarials in the treatment of cutaneous dermatomyositis: a retrospective study. Arch Dermatol. 2005;141:855–9.
Climent-Albaladejo A, et al. Dermatomyositis sine myositis and antisynthetase syndrome. Joint Bone Spine. 2002;69:72–5.
Zieglschmid-Adams ME, et al. Treatment of dermatomyositis with methotrexate. J Am Acad Dermatol. 1995;32(5 Pt 1):754–7.
Kasteler JS, Callen JP. Low-dose methotrexate administered weekly is an effective corticosteroid-sparing agent for the treatment of the cutaneous manifestations of dermatomyositis. J Acad Dermatol. 1997;36:67–71.
Hornung T, et al. Efficacy of low-dose methotrexate in the treatment of dermatomyositis skin lesions. Clin Exp Dermatol. 2012;37:139–42.
Click JW, et al. Methotrexate for the treatment of cutaneous dermatomyositis. Acad Dermatol. 2013;68:1043–5.
Metzger AL, et al. Polymyositis and dermatomyositis: combined methotrexate and corticosteroid therapy. Ann Intern Med. 1974;81(2):182–9.
Tse S, et al. The arthritis of inflammatory childhood myositis syndromes. J Rheumatol. 2001;28(1):192–7.
Camus P, et al. Interstitial lung disease induced by drugs and radiation. Respiration. 2004;71:301–26.
Kim YJ, Song M, Ryu JC. Mechanisms underlying methotrexate-induced pulmonary toxicity. Expert Opin Drug Saf. 2009;8:451–8.
Iorizzo L Jr, Jorizzo JL. The treatment and prognosis of dermatomyositis: an updated review. J Am Acad Dermatol. 2008;59(1):99–112.
Chwalinska-Sadowska H, Maldykowa H. Polymyositis-dermatomyositis: 25 years of follow-up of 50 patients disease course, treatment, prognostic factors. Mater Med Pol. 1990;22(3 SRC):213–8.
Troyanov Y, et al. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore). 2005;84(4):231–49.
Marie I, et al. Polymyositis and dermatomyositis: short term and longterm outcome, and predictive factors of prognosis. J Rheumatol. 2001;28(10):2230–7.
Dalakas MC. Inflammatory myopathies: management of steroid resistance. Curr Opin Neurol. 2011;24(5):457–62.
Schwarz MI, et al. Interstitial lung disease in polymyositis and dermatomyositis: analysis of six cases and review of the literature. Medicine (Baltimore). 1976;55(1):89–104.
Nawata Y, et al. Corticosteroid resistant interstitial pneumonitis in dermatomyositis/polymyositis: prediction and treatment with cyclosporine. J Rheumatol. 1999;26(7):1527–33.
Schnabel A, et al. Interstitial lung disease in polymyositis and dermatomyositis: clinical course and response to treatment. Semin Arthritis Rheum. 2003;32(5):273–84.
Dickey BF, Myers AR. Pulmonary disease in polymyositis/dermatomyositis. Semin Arthritis Rheum. 1984;14:60–76.
Edge JC, et al. Mycophenolate mofetil as an effective corticosteroid-sparing therapy for recalcitrant dermatomyositis. Arch Dermatol. 2006;142:65–9.
Gelber AC, et al. Mycophenolate mofetil in the treatment of severe skin manifestations of dermatomyositis: a series of 4 cases. J Rheumatol. 2000;27:1542–5.
Majithia V, Harisdangkul V. Mycophenolate mofetil (CellCept): an alternative therapy for autoimmune inflammatory myopathy. Rheumatology (Oxford). 2005;44(3):386–9.
Pisoni CN, et al. Mycophenolate mofetil treatment in resistant myositis. Rheumatology (Oxford). 2007;46(3):516–8.
Rowin J, et al. Mycophenolate mofetil in dermatomyositis: is it safe? Neurology. 2006;66(8):1245–7.
Tsuchiya H, et al. Mycophenolate mofetil therapy for rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis. Mod Rheumatol. 2014;24(4):694–6.
Cozzani E, et al. Amyopathic dermatomyositis with lung involvement responsive to mycophenolate mofetil. Immunopharmacol Immunotoxicol. 2013;35:687–92.
Morganroth PA, Kreider ME, Werth VP. Mycophenolate mofetil for interstitial lung disease in dermatomyositis. Arthritis Care Res Hoboken. 2010;62(10):1496–501.
Mira-Avendano IC, et al. A retrospective review of clinical features and treatment outcomes in steroid-resistant interstitial lung disease from polymyositis/dermatomyositis. Respir Med. 2013;107(6):890–6.
Saketkoo LA, Espinoza LR. Experience of mycophenolate mofetil in 10 patients with autoimmune-related interstitial lung disease demonstrates promising effects. Am J Med Sci. 2009;337(5):329–35.
Kitchin JE, et al. Rediscovering mycophenolic acid: a review of its mechanism, side effects, and potential uses. J Acad Dermatol. 1997;37:445–9.
Sterneck M, et al. Improvement in gastrointestinal and health-related quality of life outcomes after conversion from mycophenolate mofetil to enteric-coated mycophenolate sodium in liver transplant recipients. Transplant Proc. 2014;46(1):234–40.
Cherin P, et al. Intravenous immunoglobulin for polymyositis and dermatomyositis. Lancet. 1990;116:336.
Cherin P, et al. Efficacy of intravenous gammaglobulin therapy in chronic refractory polymyositis and dermatomyositis: an open study with 20 adult patients. Am J Med. 1991;91:162–8.
Dalakas MC, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med. 1993;329:1993–2000.
Suzuki Y, et al. Intravenous immunoglobulin therapy for refractory interstitial lung disease associated with polymyositis/dermatomyositis. Lung. 2009;187(3):201–6.
Bakewell CJ, Raghu G. Polymyositis associated with severe interstitial lung disease: remission after three doses of IV immunoglobulin. Chest. 2011;139:441–3.
Mauhin W, et al. Improvement in lung fibrosis using intravenous immunoglobulin in systemic sclerosis with myositis. Scand J Rheumatol. 2014;43(2):170–1.
Cantarini L, et al. Intravenous immunoglobulins (IVIG) in systemic sclerosis: a challenging yet promising future. Immunol Res. 2015;61:326–37.
Danieli MG, et al. Subcutaneous immunoglobulin in polymyositis and dermatomyositis: a novel application. Autoimmun Rev. 2011;10:144–9.
Danieli MG, et al. Open-label study on treatment with 20% subcutaneous IgG administration in polymyositis and dermatomyositis. Clin Rheumatol. 2014;33:531–6.
Schleinitz N, et al. Subcutaneous immunoglobulin administration: an alternative to intravenous infusion as adjuvant treatment for dermatomyositis? Clin Rheumatol. 2008;27(8):1067–8.
Caress JB, Kennedy BL, Eickman KD. Safety of intravenous immunoglobulin treatment. Expert Opin Drug Saf. 2010;9:971–9.
Feldmeyer L, et al. Not all intravenous immunoglobulin preparations are equally well tolerated. Acta Derm Venereol. 2010;90:494–7.
Sekul EA, Cupler EJ, Dalakas MC. Aseptic meningitis associated with high-dose intravenous immunoglobulin therapy: frequency and risk factors. Ann Intern Med. 1994;121(4):259–62.
Obando I, et al. Aseptic meningitis due to administration of intravenous immunoglobulin with an unusually high number of leukocytes in cerebrospinal fluid. Pediatr Emerg Care. 2002;18(6):429–32.
Dinh HV, et al. Rituximab for the treatment of the skin manifestations of dermatomyositis: a report of 3 cases. Acad Dermatol. 2007;56:148–53.
Chung L, Genovese MC, Fiorentino DF. A pilot trial of rituximab in the treatment of patients with dermatomyositis. Arch Dermatol. 2007;143:763–7.
Oddis CV, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum. 2013;65(2):314–24.
Aggarwal R, et al. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis Rheum. 2014;66:740–9.
Andersson H, et al. Long-term experience with rituximab in anti-synthetase syndrome-related interstitial lung disease. Rheumatology (Oxford). 2015;54:1420.
Keir GJ, et al. Severe interstitial lung disease in connective tissue disease: rituximab as rescue therapy. Eur Respir J. 2012;40:641–8.
Marie I, et al. Infectious complications in polymyositis and dermatomyositis: a series of 279 patients. Semin Arthritis Rheum. 2011;41(1):48–60.
Belhassen-Garcia M, et al. Atypical progressive multifocal leukoencephalopathy in a patient with antisynthetase syndrome. Intern Med. 2015;54:519–24.
Vulliemoz S, et al. Favourable outcome of progressive multifocal leucoencephalopathy in two patients with dermatomyositis. J Neurol Neurosurg Psychiatry. 2006;77(9):1079–82.
Mehregan DR, Su WP. Cyclosporine treatment for dermatomyositis/polymyositis. Curtis. 1993;51(1):59–61.
Villalba L, et al. Treatment of refractory myositis: a randomized crossover study of two new cytotoxic regimens. Arthritis Rheum. 1998;41(3):392–9.
Vencovsky J, et al. Cyclosporine a versus methotrexate in the treatment of polymyositis and dermatomyositis. Scand J Rheumatol. 2000;29(2):95–102.
Maeda K, et al. Cyclosporine treatment for polymyositis/dermatomyositis: is it possible to rescue the deteriorating cases with interstitial pneumonitis? Scand J Rheumatol. 1997;26:24–9.
Takada K, Nagasaka K, Miyasaka N. Polymyositis/dermatomyositis and interstitial lung disease: a new therapeutic approach with T-cell-specific immunosuppressants. Autoimmunity. 2005;38(5):383–92.
Kameda H, et al. Combination therapy with corticosteroids, cyclosporin a, and intravenous pulse cyclophosphamide for acute/subacute interstitial pneumonia in patients with dermatomyositis. J Rheumatol. 2005;32:1719–26.
Nagai K, et al. Therapeutic drug monitoring of cyclosporine microemulsion in interstitial pneumonia with dermatomyositis. Mod Rheumatol. 2011;21(1):32–6.
Ryan C, Amor KT, Menter A. The use of cyclosporine in dermatology: part II. J Am Acad Dermatol. 2010;63(6):949–72.
Amor KT, et al. The use of cyclosporine in dermatology: part I. Acad Dermatol. 2010;63:925–46, quiz 9478
Rosengren S, et al. The JAK inhibitor CP-690,550 (tofacitinib) inhibits TNF-induced chemokine expression in fibroblast-like synoviocytes: autocrine role of type I interferon. Ann Rheum Dis. 2012;71(3):440–7.
Kao L, Chung L, Fiorentino DF. Pathogenesis of dermatomyositis: role of cytokines and interferon. Curr Rheumatol Rep. 2011;13(3):225–32.
Kurtzman DJ, et al. Tofacitinib citrate for refractory cutaneous dermatomyositis: an alternative treatment. JAMA Dermatol. 2016;152(8):944–5.
Moghadam-Kia S, et al. Management of refractory cutaneous dermatomyositis: potential role of Janus kinase inhibition with tofacitinib. Rheumatology (Oxford). 2019;58:1011.
Dumont FJ. FK506, an immunosuppressant targeting calcineurin function. Curr Med Chem. 2000;7:731–48.
Shimojima Y, et al. Coadministration of tacrolimus with corticosteroid accelerates recovery in refractory patients with polymyositis/dermatomyositis: a retrospective study. BMC Musculoskelet Disord. 2012;13:228.
Hassan J, van der Net JJ, van Royen-Kerkhof A. Treatment of refractory juvenile dermatomyositis with tacrolimus. Clin Rheumatol. 2008;27:1469–71.
Nalda A, et al. Martin Modesto Arnal Boronat Barcelo efficacy of tacrolimus (FK-506) in the treatment of recalcitrant juvenile dermatomyositis: study of 6 cases. Med Clin (Barc). 2006;127(18):697–701.
Kurita T, et al. The efficacy of tacrolimus in patients with interstitial lung diseases complicated with polymyositis or dermatomyositis. Rheumatology (Oxford). 2015;54:39–44.
Yokoyama Y, et al. Corticosteroid-sparing effect of tacrolimus in the initial treatment of dermatomyositis and polymyositis. Mod Rheumatol. 2015;30:1–5.
Rigby AL, Plit M, Glanville AR. Tacrolimus rescue therapy for severe respiratory failure in the anti-synthetase syndrome. Respirol Case Rep. 2014;2(2):70–2.
Wilkes MR, et al. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum. 2005;52(8):2439–46.
Gordon P, Gooptu B. Tacrolimus in idiopathic inflammatory myopathy-associated interstitial lung disease: defining roles and responders. Rheumatology (Oxford). 2015;54:3–4.
Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol. 2009;4(2):481–508.
McCormack PL. Celecoxib: a review of its use for symptomatic relief in the treatment of osteoarthritis, rheumatoid arthritis and ankylosing spondylitis. Drugs. 2011;71(18):2457–89.
Mallen SR, Essex MN, Zhang R. Gastrointestinal tolerability of NSAIDs in elderly patients: a pooled analysis of 21 randomized clinical trials with celecoxib and nonselective NSAIDs. Curr Med Res Opin. 2011;27(7):1359–66.
Ramirez G, et al. Adult-onset polymyositis-dermatomyositis: description of 25 patients with emphasis on treatment. Semin Arthritis Rheum. 1990;20(2):114–20.
de Vere Hollingworth P, et al. Intensive immunosuppression versus prednisolone in the treatment of connective tissue diseases. Ann Rheum Dis. 1982;41:557–62.
Joffe MM, et al. Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med. 1993;94:379–87.
Yu KH, et al. Survival analysis of patients with dermatomyositis and polymyositis: analysis of 192 Chinese cases. Clin Rheumatol. 2011;30(12):1595–601.
Hoyles RK, et al. A multicenter, prospective, randomized, double-blind, placebo-controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis Rheum. 2006;54:3962–70.
Mok CC, C.H. To, Szeto ML. Successful treatment of dermatomyositis-related rapidly progressive interstitial pneumonitis with sequential oral cyclophosphamide and azathioprine. Scand J Rheumatol. 2003;32(3):181–3.
Bidinger JJ, et al. The cutaneous and systemic manifestations of azathioprine hypersensitivity syndrome. Acad Dermatol. 2011;65:184–91.
Sangle VS, Sangle SR, D'Cruz DP. Leflunomide as a remission-maintaining therapy in difficult-to-treat dermatomyositis. Ann Rheum Dis. 2008;67(5):723.
Welborn MC, et al. Juvenile dermatomyositis: of calcinosis cutis of the elbow and review of the literature. J Pediatr Orthop. 2014;35:e43.
Schanz S, Ulmer A, Fierlbeck G. Response of dystrophic calcification to intravenous immunoglobulin. Arch Dermatol. 2008;144(5):585–7.
Hewitson PJ, et al. Leflunomide and rheumatoid arthritis: a systematic review of effectiveness, safety and cost implications. Pharm Ther. 2000;25:295–302.
Alcorn N, Saunders S, Madhok R. Benefit-risk assessment of leflunomide: an appraisal of leflunomide in rheumatoid arthritis 10 years after licensing. Drug Saf. 2009;32(12):1123–34.
Osiri M, et al. Leflunomide for treating rheumatoid arthritis. Cochrane Database Syst Rev. 2017;(1):CD002047.
Roubille C, Haraoui B. Interstitial lung diseases induced or exacerbated by DMARDS and biologic agents in rheumatoid arthritis: a systematic literature review. Semin Arthritis Rheum. 2014;43(5):613–26.
Inokuma S. Leflunomide-induced interstitial pneumonitis might be a representative of disease-modifying antirheumatic drug-induced lung injury. Expert Opin Drug Saf. 2011;10:603–11.
Lee MA, Hutchinson DG. HRCT-proven leflunomide pneumonitis in a patient with psoriatic arthritis and normal lung function tests and chest radiography. Rheumatology (Oxford). 2010;49:1206–7.
Coleman MD. Dapsone: modes of action, toxicity and possible strategies for increasing patient tolerance. Br J Dermatol. 1993;129:507–13.
Kawachi Y, et al. Pruritic poikilodermatous eruption associated with dermatomyositis: successful treatment with dapsone. Eur J Dermatol. 2012;22:289–90.
Cohen JB, And J. Cutaneous involvement of dermatomyositis can respond to Dapsone therapy. Int J Dermatol. 2002;41:182–4.
Konohana A, Kawashima J. Successful treatment of dermatomyositis with dapsone. Clin Exp Dermatol. 1994;19:367.
Mok CC, Lau CS, Wong RW. Toxicities of dapsone in the treatment of cutaneous manifestations of rheumatic diseases. J Rheumatol. 1998;25(6):1246–7.
Wolverton SE, Remlinger K. Suggested guidelines for patient monitoring: hepatic and hematologic toxicity attributable to systemic dermatologic drugs. Dermatol Clin. 2007;25(2):195–205.
Lenardo TM, Calabrese LH. The role of thalidomide in the treatment of rheumatic disease. J Rheumatol. 2000;6:19–26.
Flores-Suarez LF, et al. Drug-induced amyopathic dermatomyositis. Rheumatology. 2002;8:50–4.
Piette JC, Sbai A, Frances C. Warning: thalidomide-related thrombotic risk potentially concerns patients with lupus. Lupus. 2002;11(2):67–70.
Gaspari A. Thalidomide neurotoxicity in dermatological patients: the next "STEP". J Invest Dermatol. 2002;119(5):987–8.
Watanabe R, et al. Successful multi-target therapy using corticosteroid, tacrolimus, cyclophosphamide, and rituximab for rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis. Mod Rheumatol. 2015;20:1–6.
Hildebrand BAJ, Arroyo R. Evolution of clinically amyopathic dermatomyositis despite aggressive immunosuppression with cyclophosphamide and prednisone. Rheumatology. 2010;16:143–5.
Murota H, et al. Successful treatment with regimen of intravenous gamma globulin and cyclophosphamide for dermatomyositis accompanied by interstitial pneumonia, opportunistic infection and steroid psychosis. Allergol Int. 2006;55(2):199–202.
Tanaka F, et al. Successful combined therapy of cyclophosphamide and cyclosporine for acute exacerbated interstitial pneumonia associated with dermatomyositis. Intern Med. 2000;39(5):428–30.
Takashi S, et al. Amyopathic dermatomyositis with interstitial pneumonia: effective treatment with cyclophosphamide pulse therapy. Nihon Kokyuki Gakkai Zasshi. 1999;37(8):647–51.
Yoshida T, et al. Pulse intravenous cyclophosphamide treatment for steroid-resistant interstitial pneumonitis associated with polymyositis. Intern Med. 1999;38(9):733–8.
Shinohara T, et al. Rapidly progressive interstitial lung disease associated with dermatomyositis responding to intravenous cyclophosphamide pulse therapy. Intern Med. 1997;36(7):519–23.
Takizawa H, Ito K. Cyclophosphamide pulse therapy for rapidly progressive interstitial pneumonia in dermatomyositis. A new possibility for rescue? Intern Med. 1997;36(7):448–9.
Yamasaki Y, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford). 2007;46(1):124–30.
Tashkin DP, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354(25):2655–66.
Fraiser LH, Kanekal S, Kehrer JP. Cyclophosphamide toxicity. Characterising and avoiding the problem. Drugs. 1991;42:781–95.
Radis CD, et al. Effects of cyclophosphamide on the development of malignancy and on long-term survival of patients with rheumatoid arthritis. A 20year followup study. Arthritis Rheum. 1995;38(8):1120–7.
Kenney LB, et al. High risk of infertility and long term gonadal damage in males treated with high dose cyclophosphamide for sarcoma during childhood. Cancer. 2001;91:613–21.
Shahani L. Refractory calcinosis in a patient with dermatomyositis: response to intravenous immune globulin. BMJ Case Rep. 2012;2012:bcr2012006629.
Penate Y, et al. Calcinosis cutis associated with amyopathic dermatomyositis: response to intravenous immunoglobulin. J Am Acad Dermatol. 2009;60(6):1076–7.
Kalajian AH, Perryman JH, Callen JP. Intravenous immunoglobulin therapy for dystrophic calcinosis cutis: unreliable in our hands. Arch Dermatol. 2009;145(3):334.
Palaniappan P, Lionel AP, Kumar S. Successful treatment of calcinosis cutis in juvenile dermatomyositis with pamidronate. J Clin Rheumatol. 2014;20(8):454–5.
Tayfur AC, et al. Bisphosphonates in juvenile dermatomyositis with dystrophic calcinosis. Mod Rheumatol. 2015;24:1–6.
Casciola-Rosen L, Mammen AL. Myositis autoantibodies. Curr Opin Rheumatol. 2012;24:602–8.
Tansley SL, McHugh NJ. Myositis specific and associated autoantibodies in the diagnosis and management of juvenile and adult idiopathic inflammatory myopathies. Curr Rheumatol Rep. 2014;16(12):464.
Yamasaki Y, et al. Unusually high frequency of autoantibodies to PL-7 associated with milder muscle disease in Japanese patients with polymyositis/dermatomyositis. Arthritis Rheum. 2006;54(6):2004–9.
Targoff IN, Arnett FC. Clinical manifestations in patients with antibody to PL-12 antigen (alanyl-tRNA synthetase). Am J Med. 1990;88(3):241–51.
Hirakata M, et al. Clinical and immunogenetic features of patients with autoantibodies to asparaginyl-transfer RNA synthetase. Arthritis Rheum. 2007;56:1295–303.
Sato S, Kuwana M, Hirakata M. Clinical characteristics of Japanese patients with anti-OJ (anti-isoleucyl-tRNA synthetase) autoantibodies. Rheumatology (Oxford). 2007;46(5):842–5.
Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology (Oxford). 2009;48:607–12.
Komura K, et al. Prevalence and clinical characteristics of anti-Mi-2 antibodies in Japanese patients with dermatomyositis. J Dermatolog Sci. 2005;40(3):215–7.
Targoff IN, Reichlin M. The association between Mi-2 antibodies and dermatomyositis. Arthritis Rheum. 1985;28:796–803.
Sato S, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60:2193–200.
Koga T, et al. The diagnostic utility of anti-melanoma differentiation-associated gene 5 antibody testing for predicting the prognosis of Japanese patients with DM. Rheumatology (Oxford). 2012;51:1278–84.
Ceribelli A, et al. Anti-MJ/NXP-2 autoantibody specificity in a cohort of adult Italian patients with polymyositis/dermatomyositis. Arthritis Res Ther. 2012;R97:14.
Tarricone E, et al. Anti-SAE antibodies in autoimmune myositis: identification by unlabelled protein immunoprecipitation in an Italian patient cohort. J Immunol Methods. 2012;384:128–34.
Fujimoto M, et al. Autoantibodies to small ubiquitin-like modifier activating enzymes in Japanese patients with dermatomyositis: comparison with a cohort. Ann Rheum Dis. 2013;72:151–3.
Bodoki L, et al. Four dermatomyositis-specific autoantibodies-anti-TIF1gamma, anti-NXP2, anti-SAE and anti-MDA5-in adult and juvenile patients with idiopathic inflammatory myopathies in a Hungarian cohort. Autoimmun Rev. 2014;13:1211–9.
Muro Y, Sugiura K, Akiyama M. Low prevalence of anti-small ubiquitin-like modifier activating enzyme antibodies in dermatomyositis patients. Autoimmunity. 2013;46:279–84.
Betteridge Z, et al. Identification of a novel autoantibody directed against small ubiquitin-like modifier activating enzyme in dermatomyositis. Arthritis Rheum. 2007;56:3132–7.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Lewis, M., Chung, L., Fiorentino, D. (2022). Dermatomyositis. In: Garg, A., Merola, J.F., Fitzpatrick, L. (eds) Interdisciplinary Approaches to Overlap Disorders in Dermatology & Rheumatology. Springer, Cham. https://doi.org/10.1007/978-3-319-18446-3_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-18446-3_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-18445-6
Online ISBN: 978-3-319-18446-3
eBook Packages: MedicineMedicine (R0)