
Abstract
Cancer relapse or recurrence has been the greatest challenge in the treatment of this life threatening disease, which occurs due to resistance of cancer cells to drug or radiation therapy. Most often this resistance is developed during treatment, which makes it even more complicated, leading to the failure of chemo or radiation therapy in the majority of cases. To circumvent these problems associated with conventional therapies, newer strategies were adopted like targeted therapy using monoclonal antibodies, immunotoxins and antibody-drug conjugates. However, targeted therapy also showed failure in many in vitro and in vivo studies that was again attributed to the emergence of resistant cells. Here, we discuss the various factors and cellular mechanisms responsible for resistance against conventional therapies and targeted approaches like recombinant immunotoxins. Cancer stem cells (CSC’s) were identified as the major reason for resistance and their role in cancer relapse has been proved convincingly in recent studies. Hence, resistance mechanisms involved in CSC’s have been elaborated. We also summarize the strategies being adopted currently to overcome resistance and different means of targeting resistant cancer stem cells that could be used in the future.
Sithambaram Devilakshmi and Jayaprakasam Madhumathi contributed equally for the manuscript and should be considered first authors equally
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
2.1 Introduction
Cancer is the most devastating disease that confers threat to human health. The widely used conventional treatment for all types of cancers is chemo- and radiation therapies. However, the emerging resistance towards various chemotherapeutic drugs remains a great challenge in cancer treatment. Transcriptional misregulation of genes and accumulation of mutations commonly result in gaining resistance to the panel of chemotherapeutic drugs referred as Multi Drug Resistance (MDR). MDR can be defined as “a state of resilience against structurally and mechanistically unrelated drugs”. MDR is the principal mechanism by which many cancers develop resistance to chemotherapeutic drugs, leading to the failure of treatment in patients with a variety of blood cancers and solid tumors, including breast, ovarian, lung, and lower gastrointestinal tract cancers [1–3].
To combat chemotherapy resistance newer strategies were developed like targeted approaches which include immunotoxins, monoclonal antibodies, antibody fragments (ScFv, Fab), antibody-drug conjugates etc. However, even targeted therapy faces challenges due to the emergence of resistance by more complex mechanisms. Here, we describe the various factors involved in cancer resistance with a focus on resistance against immunotoxins, the role of cancer stem cells in resistance and strategies to overcome resistance.
2.2 Factors Responsible for Cancer Resistance
The hallmark of a cancer cell is the increased genomic instability and higher mutation rates. Drug resistant tumour is most often acquired with characteristic features like gene mutations, gene amplification, or epigenetic changes that influence the drug metabolism , or export of drugs from cells. The daughter cells from cancer cells acquire changes (mutations) at a high rate. Acquiring gene mutation is a common characteristic feature of cancer cells . Normal cells can be targeted by drug treatment. However, a mutated cell with a modified gene may have a function, which can no longer be the drug target [4]. Mutations in any of the genes involved in metabolic pathways that are essential for cell growth, survival, maintenance of cellular homeostasis and cell division may result in resistance.
Other than mutations, the expression of drug transport proteins and the tumor niche play a major role in resistance. The tumor niche is a microenvironment which consists of diverse populations of malignant cells, with variable degree of resistance, few of them being highly resistant [5]. Chemotherapy kills drug-sensitive cells, but a small population called “side population” survives, which then drives the cancer relapse. As the tumor begins to grow again, chemotherapy may fail to eliminate resistant populations that culminate into a poor prognostic recurrence of disease or even death. These side populations survive, proliferate at a higher rate and finally rebuild the tumor microenvironment .
2.2.1 Gene Mutations in Signalling Pathways
A20 is a protein encoded by the gene tumor necrosis factor alpha-induced protein 3 (TNFAIP3). It regulates the canonical NF-κB activation and also acts as the cell’s autocrine inhibitory molecule. It interacts with NF-κB upstream signalling components which keeps the pathway activation under control [6]. Recent reports suggested that NF-κB-dependent A20 exerts cell-type specific anti- or pro-apoptotic functions. Increased A20 expression in few solid human tumors likely contributes to both carcinogenesis and response to chemotherapy . However, a current approach of analysing a unique molecular signature of each tumor holds promise for a personalized chemotherapeutic regimen comprising specific A20-targeting agents i.e., both inhibitors and enhancers [7].
Ibrutinib, an FDA approved drug, targets and inhibits the Bruton tyrosine kinase (BTK), which is found in increased levels in several types of B-cell malignancies, including mantle cell lymphoma (MCL) [8]. However, about one-third of these patients do not respond to the treatment and those that respond also become resistant to the drug [9–11]. It was identified later that there was a patient-relapse–specific mutation, C481S, in patients who initially had a durable response to ibrutinib but then showed disease progression. The mutation resulted in increased BTK activation which leads to AKT activation, further driven by the cell-cycle regulator CDK4 [12, 13].
FLT3 is a cytokine receptor belonging to the receptor tyrosine kinase class III family. Mutations in the FLT3 gene linked to a poor prognosis in acute myeloid leukemia (AML) . Early trials of FLT3 inhibitors gained resistance to treatment and the development of a new FLT3 inhibitor AC220 showed promising activity in patients with highly resistant leukemia [14].
Glucocorticoid resistance is a major cause of therapeutic failure in T cell acute lymphoblastic leukemia (T-ALL). AKT1 impairs glucocorticoid-induced gene expression by direct phosphorylation of NR3C1 at position S134 and blocking glucocorticoid-induced NR3C1 translocation to the nucleus. Conversely, pharmacologic inhibition of AKT with MK2206 effectively restores glucocorticoid-induced NR3C1 translocation to the nucleus, increases the response of T-ALL cells to glucocorticoid therapy and effectively reverses glucocorticoid resistance in vitro and in vivo [10].
2.2.2 Drug Transporters
ATP-binding cassette (ABC) transporters play a major role in drug resistance. Chemotherapeutic drugs are rapidly effluxed out by this family of transporters which are localized in the plasma membrane of resistant cells. Three well studied transporters involved in multi-drug resistance are—P-glycoprotein (P-gp, MDR1, ABCB1), multidrug resistance protein 1 (MRP1, ABCC1), and breast cancer resistance protein (BCRP, ABCG2).
P-glycoprotein had shown strong evidence in support of its role in pleiotropic drug resistance in 1982, when it was shown that DNA from resistant cell lines transformed in non-resistant cells conferred resistance which also correlated with protein expression [15]. The gene for P-glycoprotein, called MDR-1, was cloned in 1985, and the protein’s putative function was postulated on the basis of sequence homologies with bacterial hemolysin transport protein as an energy-dependent pump that expels small molecules from inside the cells [16, 17]. Recent work suggests that in non-small cell lung cancer cells: there is a correlation between MRP and mutant p53 expression, suggesting that it can be used as a prognostic marker.
ABCG2 is the mitoxantrone resistance gene also known as breast cancer resistance protein (BCRP), or ABC transporter in placenta (ABC-P). Mutant ABCG2 protein is an ideal candidate for human stem cell protection and for use as a selectable marker in gene therapy [18].
2.2.3 Tumor Microenvironment and Accessibility
Hypoxia and accumulation of HIF-1 alpha in solid tumor tissues are associated with resistance to chemotherapy , radiotherapy and immunotherapy [19]. Activated HIF-1 induces the expression of vascular endothelial growth factor (VEGF) in cancer. Increase in VEGF levels promotes tumor metastasis by angiogenesis [20]. Anti-angiogenic therapy using humanized VEGF antibody and VEGF receptor tyrosine kinase inhibitors have been shown to have promising effect in solid cancer therapy.
Another important, but little studied, cause of drug resistance is the accessibility of drugs to tumor tissue. Since the diluted concentration of drug that reaches the target site, which is much less than the potential lethal concentration, it may trigger resistance in cancer cells [21].
2.3 Resistance to Immunotoxins
Targeting cancer cells by inducing apoptosis is one of the earliest approaches to control cancer. However, cancer cells bypass apoptosis by activating alternate survival pathways and/or by blocking/inactivating apoptotic pathways. Another strategy by which resistant cells evade apoptosis is through the efflux of drugs by membrane transporters by expressing multi-drug resistant genes.
Immunotoxins bind to specific cell surface receptor and are internalized into endocytic vesicles. After priming, it is then translocated to the cytosol, and inhibiting protein synthesis by acting upon ADP-ribosylation of elongation factor 2 in the cytosol. Resistance can be induced by interference at any of these steps like down regulation of receptor or poor binding of immunotoxins, degradation of internalized toxins and failure of ADP-ribosylation of EF-2 by an escape mechanism in resistant cells (Table 2.1).
Gemtuzumab ozogamicin (GO) is a conjugate of monoclonal antibody targeting CD33 and the toxic drug calicheamicin . The poor expression of CD33 or the uptake of GO was correlated with resistance in AML cell lines [22]. Similarly, higher expression of HER2 was associated with response to T-DMI, which is a conjugate of anti-HER2 antibody, trastuzumab and toxic drug moiety DMI, a derivative of maytansine, in cancer cells [23].
2.3.1 Dysfunctional Apoptotic Pathways
Anti-apoptotic factors downstream of DNA damage play a major role in gaining resistance against GO in AML. The AML cell line KG1a displayed resistance to GO due to defect in activation of the pro-apoptotic proteins Bak and Bax [24]. Activation of caspase-3 signaling was observed in HL60 and NB4 AML cells but not in GO-exposed KG1a AML cells. Bcl-2 family of anti-apoptotic proteins were reported to be involved in GO resistance in few studies. HL-60 cells with stable overexpression of Bcl-2 or Bcl-XL were reported to be resistant to GO [25]. Despite inhibition of protein synthesis by PE-based immunotoxins, cell death was not significant due to apoptosis via pro-survival proteins like Bcl-2 family. ABT-737, a BH3-only mimetic that inhibits Bcl-2 protein could restore sensitivity in resistant cell lines like DLD1 by neutralizing Bcl-2, Bcl-xl, and Bcl-w [26].
2.3.2 ABC Transporters
Multidrug resistance protein1 (MRP1 or ABCC1), is overexpressed in 7–30 % AML cases and was associated with resistance to GO. Expression of P-gp in AML blast cells were correlated with resistance by treatment with GO in phase II clinical trials. MDR1-Pgp and MRP1 efflux systems were reported to be engaged by CalCγ1 in the resistant HL60 cell line but only MDR1-Pgp over-expression could abrogate drug cytotoxicity in MDR cells [27].
The expression of multi drug resistance proteins P-gp, MRP1 and MRP2 was characterized in CD33 + cell lines and AML samples. The MRP inhibitor MK-571 showed cytotoxicity to GO in MRP-positive NB4 and HL-60 cells and the Pgp inhibitor cyclosporine (CSA) increased susceptibility to GO in P-gp-positive/MRP-positive TF1 cells [28]. In a case study, it was reported that MRP activity in all patient samples and 17 out of 23 patients showed Pgp activity and further 12 pgp positive samples were found sensitive after treatment with CSA. This proves the important role of multi-drug resistance genes in gaining cancer resistance.
Gelonin is a RIP from the seeds of Gelonium multiflorum used in the immunotoxin. The IT HuM195-gelonin that consists of a humanized mAb specific for CD33 conjugated to a recombinant gelonin toxin. P-gp was reported to be involved in resistance to HuM195-gelonin immunotoxin [25]. Resistance to HuM195-gelonin and to free rGelonin was also reported in The human leukemic cell lines HL60 and K562 by mediating multi-drug resistance through over expression of the P-glycoprotein (P-gp). Inhibiting the function of P-gp was shown to reverse resistance to IT. However, they showed that the same cells were sensitive to other protein synthesis inhibitors like cycloheximide, saponin, and Pseudomonas exotoxin A [29] .
2.3.3 Lysosomal Degradation
Lysosomes are involved in degrading internalized exogenous macromolecules which include active immunotoxin in the cytosol. Weldon et al. [30] observed that PE-based immunotoxin B3(dsFv)-PE38 targeting CD22 was susceptible to the lysosomal degradation pathway in CD22-positive human Burkitt lymphoma cell lines. Resistance to gelonin-based IT was suggested to be mediated by increased lysosomal degradation [29]. Misfolded proteins are cleared by the ER and translocated to the proteasome in the cytosol for degradation by retrotranslocation via a pathway known as ER associated degradation (ERAD). IT is known to exert its activity by translocation from the ER to the cytosol. AB toxins were suggested to act by mimicking misfolded proteins and entering the ERAD pathway to the cytosol [31]. However, this will also result in the majority of proteins being degraded through this pathway which is one of the reasons for resistance.
Resistance of myeloid cells to ricin A-chain IT CD33, p67-7·dgA, was attributed to fast and efficient lysosomal degradation. The IT was found to bind to HL60 cells but was not capable of killing whereas anti-TfR immunotoxin could kill the cells [32].
2.3.4 Other Factors
In a recent study, Moxetumomab pasudotox (HA22), an anti-CD22 Fv fused to Pseudomonas exotoxin A, showed resistance in KOPN-8 cells due to methylation of the CpG island in the DPH1 promoter that was overcome by the methylation inhibitor 5-Azacytidine [33]. Earlier Wei et al. [34] showed that HA22 resistant cell lines had low levels of DPH4 expression which in turn prevents diphthamide biosynthesis in ALL. The CpG island in the promoter region of DPH4 gene was hypermethylated in resistant cells which was reversed when incubated with 5-azacytidine.
The PE conjugated IT targeting CD22 was found to be effective in drug resistant hairy cell leukemia but not in acute lymphoblastic leukemia. In a recent work, Wei et al. [35] claimed that resistance was due to the failure of the immunotoxin to ADP-ribosylate and inactivate EF2 in the HA22-resistant lymphoma cell line. They showed that this was, in turn, owing to the deletion of the diphthamide synthesis gene WDR85, which results in the modification of diphthamide in EF2 and, thus, cannot be inactivated by the immunotoxin.
The PI3K/AKT signaling pathway is actively involved in cell growth, survival and apoptosis mediated by AKT phosphorylation. The activation of the AKT signaling pathway has been correlated with failure to therapy in AML. Recently, Rosen et al., [36] noted the association of AKT signaling with GO resistance in vitro using the single cell network profiling (SCNP) assays with the AKT inhibitor, MK-2206, in AML samples.
2.4 Cancer Stem Cells and Resistance
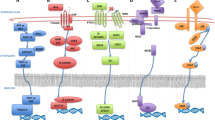
The involvement of cancer stem cells (CSC’s) in tumor recurrence and invasion has been a long debated concept. Several studies have proved the presence of cancer stem cells in drug resistant cancers convincingly (Fig. 2.1). In a recent report by Ding et al., [37], CSC’s have been proven to be involved in trastuzumab resistance in mammary carcinoma cells. Cojoc et al., [38] have extensively reviewed the mechanisms of resistance in cancer stem cells.

Mechanisms involved in cancer stem cell resistance. 1 Small molecule drugs are effluxed by the membrane transporters P-gp/MDR1, MRP1 and BCRP/ABCG2. 2 Some drugs are detoxified by the enzyme ALDH. ROS generated during radiation therapy are scavenged by enzymes like Glutathione S transferase, Peroxidase and Catalase. 3 DNA damage induced by radiation therapy or drugs is repaired by HMG proteins and the cell cycle checkpoint mechanisms ATM-Chk2 and ATR-Chk1 are activated. 4 Anti-apoptotic and developmental signaling pathways needed for survival like Notch, Wnt and Hedgehog pathways are activated. 5 Intracellular digestion by autophagy is activated by fusion of autophagosomes with endosomes and lysosomes. Targeted therapy using monoclonal antibodies and immunotoxins lead to specific binding to cell surface markers but are internalized and could be degraded by lysosomal enzymes. 6 CSC’s adopt quiescence by shutting down replication and remaining in a dormant state with no metabolic activity. 7 The cancer microenvironment protects CSC’s from therapeutics or other stress. The CSC niche includes stromal fibroblasts, immune cells, mesenchymal stem cells, extra cellular matrix, growth factors and cytokines released by these cells and also physiologic factors like hypoxia and pH
2.4.1 Drug Efflux
Cells that efflux drugs termed as side population (SP) are identified in resistant cancer cells. In cancer stem cells these MDR proteins are up-regulated and, thus, show resistance towards many chemotherapeutic drugs. The ABCG2 transporter has been reported to be involved in resistance to various drugs like methotrexate, doxorubicin, imatinib, daunorubicin, topotecan, mitoxantrone etc [39]. The ABC family of transporters was also associated with resistance in targeted therapies such as the tyrosine kinase inhibitors Sorafenib, Imatinib, Nilotinib, Gefitinib and Erlotinib [38]. Cancer stem cells use drug efflux mechanisms actively to prevent the drug from acting and this property is used to even sort CSC populations based on their ability to efflux dyes like Hoechst 33342.
2.4.2 Detoxification and Cellular Repair
Resistance to radiation therapy has been associated with increased involvement of ROS scavenging mechanisms which enhance cell survival by eliminating ROS generated during therapy. The level of glutathione, one of the ROS scavengers has been correlated in gastrointestinal cancer cells and HNSCC. Other genes involved in ROS scavenging like superoxide dismutase, glutathione peroxidase and catalase were reportedly upregulated in CD44 + CD24−breast CSCs [38]. CSC’s were consistently found to display high ALDH activity which is involved in detoxification and are associated with drug resistance. Levels of ALDH1 have been frequently used as a marker for identification and sorting of CSC’s. Administration of ALDH1 inhibitors could reduce tumor growth and resistance [40] .
Damage-associated molecular patterns (DAMP’s) are molecules released by damaged cells that initiate repair and survival mechanisms in cells. These molecules are recognized by pattern recognition receptors (PRR). Hombach-Klonisch et al. [41] , suggest that DAMP signaling via several PRR may be one of the major tumor survival response associated with cell proliferation, inflammatory and autophagy responses in cancer stem cells.
2.4.3 DNA Repair and Modification
Cancer stem cells often display highly efficient DNA repair systems. Higher expression of DNA repair genes have been reported to be involved in chemo-resistance. Cells with higher expression of High Mobility Group (HMG) proteins show enhanced DNA repair mechanisms and thus evade killing by drugs. Cojoc et al. [38] has reviewed several studies which showed that DNA repair mechanisms are activated in cancer stem cells in glioma, nasopharyngeal carcinoma, lung, breast and mouse mammary tumors. Also, the checkpoint mechanisms have been induced in cancer stem cells via the kinase signaling pathways ATM-Chk2 and ATR-Chk1.
In another study genes involved in chromatin modification, such as KDM5A/Jarid1A, a histone H3K demethylase and histone deacetylases (HDACs) were over expressed in drug resistant cells in Non-small cell lung cancer cell lines [42]. Thus, chromatin modifications and epigenetic changes can be partly responsible for resistance [43]. The unlimited replicative potential is one of the hallmarks of cancer and it requires activation of telomere maintenance mechanisms (TMMs). Two TMMs are currently known in human cancer, namely, telomerase activity and the alternative lengthening of telomere (ALT) mechanisms. Although both TMMs appear to be equivalent in their ability to support immortalization, their contribution to tumor growth and survival and consequently patientsʼ prognosis may differ [44].
Telomeres are specialized DNA-protein structures located at the end of eukaryotic chromosomes. They are essential for continued cell proliferation. Indeed, telomere attrition, which occurs within each cell division, represents a molecular clock that counts the number of times a cell can divide and determines its entry into senescence [45]. Other than acting as a mitotic clock, telomeres play an important role in the maintenance of genomic integrity. As suggested by Feijoo et al. [46] , telomere erosion in a context of impaired cell cycle checkpoint may constitute an important mechanism during tumoriogenesis.
2.4.4 Survival Pathways
Different studies have proven the involvement of developmental pathways, wingless-type MMTV integration site family (WNT), Notch signaling and Hedgehog pathways in cancer stem cells and resistance. These pathways are involved in self-renewal of normal stem cells. Activators of Wnt, Notch and Hedgehog signaling pathways could induce proliferation of HSC’s. Inhibition of β-catenin of the Wnt pathway by axin was found to reduce the self-renewal capacity [47]. Cojoc et al. [38] in his review compiled the studies that have shown over expression of genes involved in these pathways in cancer stem cells. Inhibitors of these pathways like gamma-secretase inhibitors (GSIs) and cyclopamine rendered the cells susceptible to treatment .
2.4.5 Autophagy and EMT
Autophagy is a lysosomal degradation pathway which is involved in the degradation of intracellular materials and the removal of damaged organelles, protein aggregates or microbes. It plays a major role in cell survival in metabolic stress and preventing apoptosis in cancer cells. The epithelial Mesenchymal transition (EMT) which has also been associated with resistance to cancer therapy is known to be related to autophagy and stemness. Cancer stem cells utilize alternative mechanisms of survival to manage environmental stress, autophagy being one of them. Cojoc et al. [38], have shown increased resistance in prostate and pancreatic cancer cell lines by the induction of autophagy by Neurophilin-2.
2.4.6 Quiescence
One of the important features of CSC’s is their dormancy or quiescence. Dormant CSC’s are extremely slow cycling with an arrest in the GO phase and, hence, have a minimum energy requirement. They have been reported to show highest capacity for self-renewal. Quiescence was considered as a major factor responsible for the ability of CSC’s to survive harsh conditions and anti-cancer therapy. Drug resistance was suggested to be due to the fact that most drugs target DNA replication and proliferation of cells or metabolic pathways which are greatly reduced in these cells. Their dormancy could be broken by the addition of cytokines involved in activating dormant CSC’s during injury like G-CSF and IFNα, which induced proliferation. This has been used as a strategy to eliminate CSC’s [48].
2.4.7 Microenvironment
CSC’s are often hidden in the hypoxic core of cancer tissue in a unique niche that contributes to its survival. This niche called the microenvironment is vital for their existence as they are surrounded by stromal fibroblasts and an extra cellular matrix (ECM) that release cytokines and signaling factors. Tumor and stromal-derived factors have been shown to play a key role in CSC maintenance and therapy resistance . CXCL12/CXCR4 signaling and TGF-β/SMAD signaling are major pathways induced by these growth factors. Growth factors released in the tumor niche like PDGF, IL1β, TNF, TGF-β, chemokine CXCL12 and MMPs were involved in the development and regulation of CSC’s [38].
CSC’s are protected from the environmental stress and attack by therapeutic agents in the microenvironment. One of the major reasons for radio resistance was found to be hypoxia in the CSC niche since oxygen is required for radiation-induced killing. Also, the hypoxia-inducible factor (HIF) signaling is activated in hypoxic condition which in turn activates survival pathways.
2.5 Strategies Used to Overcome Resistance
2.5.1 Inhibitors of Anti-apoptotic Proteins
Blocking anti-apoptotic proteins Bcl-2, Bcl-xl, and Bcl-w is an important strategy used by many investigators to combat resistance. Oblimersen, was used to target Bcl-2, showed improvement in GO treatment in 25 % of AML patients in a phase II clinical trial [49]. IL-2/granzyme A fusion protein improved doxorubicin sensitivity of the MDR + lm1-mdr cell line by inducing caspase-independent apoptosis [50].
In a detailed study by Traini et al. [26] , ABT-737 and PE immunotoxin could inhibit apoptosis in combination. ABT-737 could bind to the hydrophobic core of Bcl-2 proteins Bcl-2, Bcl-xl, and Bcl-w while the immunotoxin degraded Mcl-1 protein, thus releasing the inhibition of the apoptotic pathway. ABT-263 as well as ABT-737 have been reported to show synergistic killing with PE immunotoxin targeting transferring receptor in Small Cell Lung Cancer (SCLC) cell lines that were otherwise resistant to the immunotoxin . Killing was observed in 6 h with loss of Mcl-1. The same effect was observed in vivo also when the immunotoxin was administered in combination with ABT-737 in nude mice with H69AR tumor [51].
TNF-related apoptosis-inducing ligand (TRAIL) reportedly induces apoptosis independent of major pathways controlling chemotherapy resistance [52]. The MDR + subline MDR-U2OS was shown to be TRAIL sensitive due to reduced AKT activation [53]. Apoptosis could be induced in resistant cells with low Bak using mesothelin conjugated with anti-TRAIL receptor 2 [54]. Anti-sense oligonucleotides have been employed in clinical studies to sensitize cancer cells to apoptotic triggers [55, 56]. However, there is emerging evidence to support the novel mechanism of death inhibition by Bcl-2 involving its ability to modulate cellular redox status and mitochondrial metabolism.
2.5.2 Blocking Membrane Drug Transporters
Hamada and Tsuruo [57] developed two monoclonal antibodies (MRK16 and MRK17) against the membrane transporter P-glycoprotein . Fitzgerald et al. [58] , reported the use of MRK16 coupled with PE toxin in killing multi-drug resistant KB cell lines. The anti-P-glycoprotein monoclonal antibody MRK16 could overcome bone marrow resistance against daunomycin, doxorubicin, vincristine, vinblastine, etoposide, and taxol in multi-drug resistant transgenic mice. The MRK16-PE conjugate was also successfully shown to kill bone marrow cells in a dose-dependant manner [59]. MRK16 was used along with Saponin immunotoxin that could eliminate 99 % of MDR cells [60]. A recombinant single-chain Fv fragment against P-gp was developed by Niv et al. [61] .
It was proven that the combination of antibody conjugates with chemosensitisers (cyclosporin A, D, G) that block P-gp transporters restored the sensitivity of MDR cell lines [62]. Two inhibitors of ABCG2 and ABCB1 transporters, GF120918 and tariquidar, have been approved for clinical studies. Although CD33 is expressed in 90 % of AML patients, more than 50 % of patients show remission due to GO resistance mediated via the membrane transporters [63]. Addition of U0126, a MEK1/2 inhibitor, was reported to prevent GO resistance induced in HL-60/GO resistant cells. Combination of the MDR modifiers PSC833 or MS209 with Gemtuzumab ozogamicin (CMA-676) was observed to reverse resistance in CD33 + AML with P-gp-related MDR [64] by inhibiting the efflux of therapeutic agents.
2.5.3 Delivery and Intracellular Trafficking
HPMA hydrogels were used successfully to prolong the delivery of antibody-drug conjugates with different targeting moieties (anti-CD71, anti-thymocyte globulin, anti-CD4, transferrin) tested on human multidrug resistance (MDR) cell lines [62]. The trafficking route via specific organelles was found to play a major role in the case of LMB2, an IT comprising PE38 and an Fv against IL2 receptor [65].
ABT-737 showed 20-fold enhanced killing of resistant cell lines by PE IT’s by increasing the delivery of IT from the ER to the cytosol by a mechanism poorly understood. However, it was hypothesized that ABT-737 induces ER stress and facilitates its transport to the cytosol [26]. Recently, IT named RG7787, a PE-based toxin targeting mesothelin was reported to be efficient due to resistance to lysosomal degradation in breast and gastric cancers [66] .
2.5.4 Inhibition of DNA Repair and Telomerase Activity
Inhibition of DNA repair and DNA damage checkpoint mechanisms like the kinase pathways ATM-Chk2 and ATR-Chk1 were utilized by some workers. The chk1 inhibitor AZD7762, debromohymenial-disine (DBH) that inhibits both Chk1 and Chk2 kinases and the ATM inhibitor KU55933 were effective against resistant populations along with chemo or radiotherapy [67]. Santambrogio et al. [68] used microRNAs to impair telomerase activity or to affect telomere functions in cancer cells. Crees et al. [69] , Romaniuk et al. [70] and Uziel and Lahav [71] described the approaches developed during the last decades to inhibit telomerase, aimed to interfere with the enzyme's catalytic activity. Overall, accumulating evidence from preclinical studies on the effects of telomerase inhibition in human cancer has provided persuasive arguments to indicate that the enzyme is a well-validated cancer target and an ideal tumor-associated antigen [45].
2.5.5 Combination Therapy
Two treatment strategies were devised based on cancer cell genetic findings. It involves the serial use of two anti-cancer drugs, the first to weaken or "prime" the cancer cells, and the second to deliver an added impact. To prime the cancer cells, researchers used Palbociclib (which selectively inhibits two cell-cycle promoting proteins, CDK4 and CDK6) to slow down the cancerʼs growth and sensitize cells being targeted by the second drug. Previous clinical studies have shown that palbociclib itself can significantly inhibit the growth of mantle cell lymphoma. In the cells with a mutated BTK, palbociclib was administered first, and then the second drug idelalisib. In lymphoma cells lacking the BTK mutation, the investigators also started with palbociclib, followed by ibrutinib, since both drugs are well tolerated by the patients.
7-hydroxystaurosporine (UCN-01) is a novel protein kinase inhibitor that increases chemotherapy-induced apoptosis in vitro and is in early phases of clinical development [72]. In vitro, UCN-01 is synergistic with multiple cytotoxic agents and increases fludarabine-induced apoptosis in a human breast cell line. These results suggest that UCN-01 sensitized the lymphoma to the cytotoxic effects of EPOCH, possibly by modulating the “threshold” for apoptosis, and may illustrate a new paradigm for reversal of drug resistance.
Immunotoxins were also used in combination with other drugs. Anti-CD138 IT B-B4-SO6 with doxorubicin were used as a combination therapy for the drug-resistant multiple myeloma (MM)-derived cell line RPMI8226. The authors conclude that combination of IT and chemotherapy could prevent drug resistance that arises due to exposure to chemotherapy alone [73]. In another early study, the combination of ricin conjugated IT targeting CD19 (anti-B4 blocked ricin) combined with drugs like cisplatin, cyclophosphamide and etoposide showed long term cure in vivo in SCID mice with disseminated tumors of the multidrug-resistant human B-cell lymphoma Namalwal/mdr-1 [74].
IT containing anti-melanoma antibody ZME-018 recognizing a 240-kDa surface glycoprotein (gp 240) and the plant toxin gelonin was tested in resistant human melanoma cells (A375-M). Combination with cisplatin, IFN-γ, IFN-α, and etoposide were observed to enhance the cytotoxic effects of ZME-gelonin against resistant cells [75]. Other combinations with GO include G-CSF that induced AML cells to enter G2/M and hypodiploid phase and Valproic acid, a histone deacetylase inhibitor [63]. As mentioned earlier, the combination of ABT-737 with immunotoxin could enhance killing by 20-fold in resistant cell lines by neutralizing anti-apoptotic proteins and by increasing the delivery of the immunotoxin from the ER to the cytosol [26].
2.5.6 Nanotechnology
Nanotechnology holds great promise in establishing efficacious, innovative strategies to overcome chemoresistance and may facilitate complementary treatment methods and cancer diagnostics. Various nanomedical devices are being introduced and evaluated, demonstrating encouraging results. While stealth liposomes serve as a benchmark, astonishing progress is witnessed in polymeric nanovehicles. It can be also combined with low molecular weight surfactants, inhibiting drug resistance in addition to solubilizing drugs. A nanocrystalline silver particle (8 nm) modified with TAT (AgNP-TAT) was developed for MDR cancer cell treatment. The antitumor activity was reported in both MDR cells and non- resistant cells [76]. AgNP-TAT showed significant enhancement in tumor cell killing, up to 24-fold higher cytotoxic effect compared to its counter-part lacking the TAT conjugation. AgNP-TAT NPs were able to effectively inhibit tumor growth in mice bearing malignant melanoma at a dose of 1 nmol/kg (compared with 4.3 μmol/kg of DOX), and showed significantly reduced adverse toxicity in vivo [77]. Various nanoparticle-based approaches have been investigated to overcome efflux-mediated resistance . These include the use of formulation excipients that inhibit transporter activity and co-delivery of the anticancer drug with a specific inhibitor of transporter function or expression [78].
2.5.7 Other Novel Strategies
Oncolytic viruses (OV) are promising anti-cancer agents, capable of selectively targeting replication in tumor cells. Genetically modified oncolytic viruses (OVs) kill tumor cells via completely unique mechanisms compared to small molecule chemotherapeutics typically used in lung cancer treatment and can also be used to deliver specific toxic, therapeutic or immunomodulatory genes to tumor cells. Recent pre-clinical and clinical studies with oncolytic vaccine approaches have revealed promising combination strategies that enhance oncolysis of tumor cells and circumvent tumor resistance mechanisms [79]. Synergistic effects of therapy based on combining OV and various cytostatics are in preclinical studies and have shown promising results.
Over-expression of recombinant GlcCer synthase (GCS) confers resistance to adriamycin and to ceramide in GlcCer synthase-transfected human breast cancer cells , suggesting that drug resistance is related to stimulation of glycosylation of ceramide and the resultant inhibition of drug induced apoptotic signalling. Blocking glycosylation of ceramide has been shown to increase cancer cell sensitivity to cytotoxic drugs. Drug combinations that enhance ceramide generation and limit glycosylation have been shown to enhance effectiveness of chemotherapy by inducing apoptosis in cancer cell models
Targeting intracellular compartments is another challenging approach. A particularly interesting direction which shows promise for targeted anticancer nanomedicine is the use of viral components against drug resistant cancer cells. Hence, newly discovered anticancer- and antimetastatic drugs may be combined with a broad spectrum of molecules which includes small-molecule inhibitors, interfering RNA molecules, microRNA, oncolytic viruses, and also naturally occurring substances. This combination with anti-inflammatory and adjuvant therapies seems to be a very promising treatment approach [80].
2.6 Targeting Cancer Stem Cells
It is clear from various studies that cancer stem cells play a major role in resistance against all kinds of therapy. Almost all the factors listed as responsible for resistance in IT therapy are found to overlap in CSC’s. It is also quite evident that CSC’s are not only responsible for resistance against chemo and radiotherapy but also against IT therapy since they can use any of the following ways to handle IT’s conveniently:
-
i)
They can be protected from exposure to IT in their microenvironment
-
ii)
Efflux IT’s using membrane transporters,
-
iii)
Utilize autophagy to degrade and get rid of recombinant IT’s,
-
iv)
Use detoxification and repair pathways to circumvent the damage,
-
v)
Recruit anti-apoptotic proteins to prevent apoptosis,
-
vi)
Use alternative survival pathways to escape cell death and
-
vii)
Remain quiescent with inactive cellular machinery.
Although the strategies mentioned earlier have been successful to some extent in avoiding resistance, it is highly unlikely that these strategies alone would be completely effective in dealing resistance since the major contributor to resistance remains hidden and active. Hence, currently several groups are studying the possibility of targeting CSC’s to destroy cancer permanently. The targets include proteins involved in signaling pathways in CSC’s like WNT, NOTCH and Hedgehog pathways. drug transporters, CSC specific surface markers, ALDH, quiescence factors, anti-apoptotic proteins and factors involved in the CSC niche [81, 82].
2.6.1 Targeting Signaling Pathways in CSC’s
Various modes of therapies are being investigated to kill CSC’s which that have been summarized in the review by Han et al. [81] . Inhibition of the Hedgehog pathway with drugs like GDC-0449, LDE225 and GSIs like RO4929097 and MK-0752 have been used along with chemotherapy with paclitaxel, carboplatin, capecitabine, cinblastine, gemcitabine and temozolo-mide [38]. The steroid-like compound, cyclopamine, was used to target SMO of hedgehog signaling that could eliminate prostate cancer cells in mice xenograft tumors in vivo and was shown to be effective in killing CSC’s in glioma sphere cells. Arsenic trioxide (As2O3) inhibits the glioma-associated oncogene homolog (Gli) and has been used in combination with the SMO inhibitors cyclopamine and GDC-0449. (-)-epigallocatechin-3-gallate (EGCG) with quercetin could inhibit self- renewal capacity in CSC’s by inhibiting the sonic hedgehog (SHh) pathway.
Inhibition of the Notch signaling using the γ-secretase inhibitor GSI-18 could eliminate CD133 + medulloblastoma cells while MRK-003 was effective in killing CSC’s in breast cancer. The Wnt signaling pathway has also been widely targeted like cAMP response-element binding protein (CBP)/b-catenin antagonist ICG-001, used to target leukemic stem cells [81]. Targeting mTOR involved in PI3/AKT using rapamycin could deplete leukemic stem cells [82]. The Notch signaling pathway has been inhibited by several other groups using GSI, siRNA or antibody against the Notch ligand, delta-like 4 ligand (DLL4), which either reduced the CSC population or rendered the CSC’s susceptible to drug therapy [38]. Recent study have reported that c-Met silencing could inhibit CSC’s in head and neck squamous carcinoma by down regulation of the Wnt/β-catenin signaling [83].
NF-κB is activated during lymphoid development and is used as a target in few studies. Inhibition of NF-κB activation using the proteasome inhibitors, bortezomib or MG-132, and inhibition of IκB kinase (IKK) by Parthenolide were used to target AML stem cells. However, CML stem cells were resistant to ABL kinase inhibitors imatinib and its derivative nilotinib [47].
2.6.2 Targeting Apoptosis and Cellular Repair Mechanisms in CSC’s
Inducing apoptosis by MSC’s expressing TNF-related apoptosis-inducing factor (TRAIL) along with mitoxantrone was effective in putative CSC’s. In our laboratory , Madhumathi et al. , (unpublished data) have successfully used TRAIL-based immunotoxins to induce apoptosis in CSC’s isolated from leukemic cell lines by culturing cells in the presence of methotrexate. The Methotrexate resistant side population was found to be enriched in the CSC population. Inducing apoptosis selectively in CSC’s using IT’s conjugated with TRAIL, targeting different surface markers of CSC’s has been a promising strategy used in our laboratory for all cancers.
Inhibition of ALDH activity using all-trans retinoic acid (ATRA), synthetic retinoids, disulfiram, 4- diethylaminobenzaldehyde (DEAB) or ALDH1A1 shRNA were used in combination with chemotherapeutic drugs in various studies. Targeting ROS scavengers by buthionine sulfoximine (BSO) reduced radioresistance in CSC’s by inhibiting glutamate-cysteine ligase [38]. Sorafenib and sulforaphane could be used to inhibit ALDH1 activity and thus was postulated as potential drugs for CSC’s [40].
2.6.3 Targeting Autophagy and Microenvironment in CSC’s
Autophagy has been inhibited in another strategy of killing CSC’s using lysosomotropic anti-malaria drug chloroquine/hydroxychloroquine. Targeting tumor microenvironment or hypoxic niche by improving tumor oxygenation has also been tested along with radio and chemotherapy . Inhibition of cytokine and chemokine receptors like IL-8 receptor CXCR1 by antibody or by repertaxin was successful in reducing breast CSC’s. Inhibition of TGF-β/SMAD pathway also showed reduction in CSC’s. Mab against VEGF, bevacizumab in mice glioma cell xenografts could decrease CD133+ cancer stem cells by anti-angiogenesis while treatment with IFN-α alone could kill side population in ovarian cancer [82]. Quiescence of CSC’s has been inhibited using Arsenic trioxide, G-CSF or IFN α as an alternative strategy [48].
2.6.4 Targeting Membrane Transporters and CSC Surface Markers
ABC transporters have been inhibited by drugs like phosphodiesterase-5 inhibitors and fumitremorgin-type indolyl diketopiperazine, dofequidar fumarate, Ko143, ABCG2 siRNA, or ABCG2 inhibitor YHO-13351, in different types of cancers which could be used to target CSC’s since they over-express these transporters [38]. The monoclonal antibody (Mab) H90 targeting CD44 could bind and kill leukemic stem cells in AML in vivo. Since GO targets the CD33 receptors which are highly expressed in CSC’s, it was presumed that the activity of GO could be due to killing of CD33 + AML stem cells. Micro-RNAs have also been shown to be involved in inhibiting CSC’s [84, 85]. Lentiviral-mediated shRNA was used to target the neuronal cell surface adhesion molecule LiCAM in CD133 + glioma stem cells [86].
Antibodies against other cell surface molecules like VLA-4 and CLL-1 (C-type lectin-like molecule-1) are being evaluated as potential targets [47]. Immunotoxins targeting CSC’s have been recently developed using ligands or antibodies that specifically bind CSC’s. IL3 conjugated with diphtheria toxin (DT) , targeting CD123 receptor that is over-expressed in leukemic stem cells, has been used for AML [87].
2.7 Conclusion
It is evident from the factors involved in resistance, that CSC’s are the major contributors of therapy resistance for all kinds of treatments—either conventional or targeted therapies. Many mechanisms observed in immunotoxin resistance were also identified as a major feature of CSC’s like drug efflux, anti-apoptotic pathways, lysosomal degradation, etc. Thus, it could be concluded that CSC’s are responsible for resistance against all treatment modalities since they have innumerable ways to handle all kinds of stress. They have mechanisms to evade any attack in order to survive in adverse conditions. Targeting two or three of these key survival strategies together by means of combination therapies would be ideal in managing cancer in the future, instead of targeting only one factor. Targeted therapy using immunotoxins should be combined with blocking other alternative survival pathways for maximum efficacy in treatment.
Disclosure of Potential Conflicts of Interest
No conflicts of interest to disclose.
Abbreviations
- ABC:
-
ATP-Binding Cassette
- AML:
-
Acute myeloid leukemia
- BCRP:
-
Breast cancer resistance protein
- BTK:
-
Bruton tyrosine kinase
- CSC:
-
Cancer stem cells
- DAMP:
-
Damage-associated molecular patterns
- DT:
-
Diphtheria toxin
- EMT:
-
Epithelial Mesenchymal transition
- GCS:
-
GlcCer synthase
- GO:
-
Gemtuzumab ozogamicin
- GSIs:
-
Gamma-secretase inhibitors
- HIF:
-
Hypoxia inducible factor
- IT:
-
Immunotoxin
- MCL:
-
Mantle cell lymphoma
- MDR:
-
Multi Drug Resistance
- MRP1:
-
Multidrug resistance protein 1
- OV:
-
Oncolytic viruses
- P-gp:
-
P-glycoprotein
- SCLC:
-
Small Cell Lung Cancer
- SCNP:
-
Single cell network profiling
- T-ALL:
-
T cell acute lymphoblastic leukemia
- TMM:
-
Telomere maintenance mechanisms
- Tnfaip3:
-
Tumor necrosis factor alpha induced protein 3
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- VEGF:
-
Vascular endothelial growth factor
References
Harris AL, Hochhauser D Mechanisms of multidrug resistance in cancer treatment. Acta Oncol. 1992;31(2):205–13.
Jamroziak K, Robak T. Pharmacogenomics of MDR1/ABCB1 gene: the influence on risk and clinical outcome of haematological malignancies. Hematology. 2004;9(2):91–105.
Leighton JC Jr, Goldstein LJ. P-glycoprotein in adult solid tumors. Expression and prognostic significance. Hematol Oncol Clin North Am. 1995;9(2):251–73.
Hanahan D, Weinberg RA. Hallmarks of Cancer: the next generation. Cell. 2011;144(5):646–74.
Giaccia AJ, Schipani E. Role of carcinoma-associated fibroblasts and hypoxia in tumor progression. Curr Top Microbiol Immunol. 2010;345:31–45.
da Silva CG, Minussi DC, Ferran C, Bredel M. A20 expressing tumors and anticancer drug resistance. Adv Exp Med Biol. 2014;809:65–81.
Pujari R, Hunte R, Khan WN, Shembade N. A20-mediated negative regulation of canonical NF-κB signaling pathway. Immunol Res. 2013;57(1–3):166–71.
Perez-Galan P, Dreyling M, Wiestner A. Mantle cell lymphoma: biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood. 2011;117:26–38.
Liang DC, Shih LY, Hung IJ, Yang CP, Chen SH, Jaing TH, Liu HC, Wang LY, Chang WH. FLT3-TKD mutation in childhood acute myeloid leukemia. Leukemia. 2003;17(5):883–6.
Piovan E, Yu J, Tosello V, Herranz D, Ambesi-Impiombato A, Silva AC D, Sanchez-Martin M, Perez-Garcia A, Rigo I, Castillo M, Indraccolo S, Cross JR, de Stanchina E, Paietta E, Racevskis J, Rowe JM, Tallman MS, Basso G, Meijerink JP, Cordon-Cardo C, Califano A, Ferrando AA. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell. 2013;24(6):766–76.
Small S. Ibrutinib approved for the treatment of mantle cell lymphoma. Clin Adv Hematol Oncol. 2013;11(12):808.
Wilson WH, Sorbara L, Figg WD, Mont EK, Sausville E, Warren KE, Balis FM, Bauer K, Raffeld M, Senderowicz AM, Monks A. Modulation of clinical drug resistance in a B cell lymphoma patient by the protein kinase inhibitor 7-hydroxystaurosporine: presentation of a novel therapeutic paradigm. Clin Cancer Res. 2000;6(2):415–21.
Chiron D, Liberto M D, Martin P, Huang X, Sharman J, Blecua P, Mathew S, Vijay P, Eng K, Ali S, Johnson A, Chang B, Ely S, Elemento O, Mason CE, Leonard JP, Chen-Kiang S. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 2014;4(9):1022–35.
Williams AB, Nguyen B, Li L, Brown P, Levis M, Leahy D, Small D. Mutations of FLT3/ITD confer resistance to multiple tyrosine kinase inhibitors. Leukemia. 2013;27(1):48–55.
Chen CJ, Chin JE, Ueda K, Clark DP, Pastan I, Gottesman MM, Roninson IB. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell. 1986;47(3):381–9.
Coley HM. Overcoming multidrug resistance in cancer: clinical studies of p-glycoprotein inhibitors. Methods Mol Biol. 2010;596:341–58.
Pietro A D, Dayan G, Conseil G, Steinfels E, Krell T, Trompier D, Baubichon-Cortay H, Jault J. P-glycoprotein-mediated resistance to chemotherapy in cancer cells: using recombinant cytosolic domains to establish structure-function relationships. Braz J Med Biol Res. 1999;32(8):925–39.
Sarkadi B, Ozvegy-Laczka C, Nemet K, Varadi A. ABCG2- a transporter for all seasons. FEBS Lett. 2004;567:116–20.
Yasuda H. Solid tumor physiology and hypoxia-induced chemo/radio-resistance: novel strategy for cancer therapy: nitric oxide donor as a therapeutic enhancer. Nitric Oxide. 2008;19(2):205–16.
Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 2007;26(2):225–39.
Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. 1994;269(38):23757–63.
Walter RB, Raden BW, Kamikura DM, Cooper JA, Bernstein ID. Influence of CD33 expression levels and ITIM-dependent internalization on gemtuzumab ozogamicin-induced cytotoxicity. Blood. 2005;105:1295–302.
Barok M, Joensuu H, Isola J. Trastuzumab emtansine: mechanisms of action and drug resistance. Breast Cancer Res. 2014;16(2):209.
Haag P, Viktorsson K, Lindberg ML, Kanter L, Lewensohn R, Stenke L. Deficient activation of Bak and Bax confers resistance to gemtuzumab ozogamicin-induced apoptotic cell death in AML. Exp Hematol. 2009;37(6):755–66.
Linenberger ML. CD33-directed therapy with gemtuzumab ozogamicin in acute myeloid leukemia: progress in understanding cytotoxicity and potential mechanisms of drug resistance. Leukemia. 2005;19(2):176–82.
Traini R, Ben-Josef G, Pastrana DV, Moskatel E, Sharma AK, Antignani A, Fitzgerald DJ. ABT-737 overcomes resistance to immunotoxin-mediated exotoxin-based proteins to the cell cytosol apoptosis and enhances the delivery of pseudomonas. Mol Cancer Ther. 2010;9(7):2007–15.
Cianfriglia M, Mallano A, Ascione A, Dupuis ML. Multidrug transporter proteins and cellular factors involved in free and mAb linked calicheamicin-Á1 (gentuzumab ozogamicin, GO) resistance and in the selection of GO resistant variants of the HL60 AML cell line. Int J Oncol. 2010;36(6):1513–20.
Walter RB, Raden BW, Hong TC, Flowers DA, Bernstein ID, Linenberger ML. Multidrug resistance protein attenuates gemtuzumab ozogamicin-induced cytotoxicity in acute myeloid leukemia cells. Blood. 2003;102(4):1466–73.
McGrath MS, Rosenblum MG, Philips MR, Scheinberg DA. Immunotoxin resistance in multidrug resistant cells. Cancer Res. 2003;63(1):72–9.
Weldon JE, Xiang L, Chertov O, Margulies I, Kreitman RJ, FitzGerald DJ, Pastan I. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood. 2009;113(16):3792–800.
Hazes B, Read RJ. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. BioChemistry. 1997;36(37):11051–4.
Engert A, Brown A, Thorpe P. Resistance of myeloid leukaemia cell lines to ricin A-chain immunotoxins. Leuk Res. 1991;15(11):1079–86.
Hu X, Wei H, Xiang L, Chertov O, Wayne AS, Bera TK, Pastan I. Methylation of the DPH1 promoter causes immunotoxin resistance in acute lymphoblastic leukemia cell line KOPN-8. Leuk Res. 2013;37(11):1551–6.
Wei H, Xiang L, Wayne AS, Chertov O, FitzGerald DJ, Bera TK, Pastan I. Immunotoxin resistance via reversible methylation of the DPH4 promoter is a unique survival strategy. Proc Natl Acad Sci U S A. 2012;109(18):6898–903.
Wei H, Bera TK, Wayne AS, Xiang L, Colantonio S, Chertov O, Pastan IA. Modified form of diphthamide causes immunotoxin resistance in a lymphoma cell line with a deletion of the WDR85 gene. J Biol Chem. 2013;288(17):12305–12.
Rosen DB, Harrington KH, Cordeiro JA, Leung LY, Putta S, Lacayo N, Laszlo GS, Gudgeon CJ, Hogge DE, Hawtin RE, Cesano A, Walter RB. AKT signalling as a novel factor associated with in vitro resistance of human AML to gemtuzumab ozogamicin. PLoS ONE. 2013;8(1):e53518.
Ding K, Banerjee A, Tan S, Zhao J, Zhuang Q, Li R, Qian P, Liu S, Wu ZS, Lobie PE, Zhu T. Artemin, a member of the glial cell line-derived neurotrophic factor family of ligands, is HER2-regulated and mediates acquired trastuzumab resistance by promoting cancer stem cell-like behavior in mammary carcinoma cells. J Biol Chem. 2014;289(23):16057–71.
Cojoc M, Mäbert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Semin Cancer Biol. 2014;31:16–27.
Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–84.
Vinogradov S, Wei X Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine (Lond). 2012;7(4):597–615.
Hombach-Klonisch S, Natarajan S, Thanasupawat T, Medapati M, Pathak A, Ghavami S, Klonisch T. Mechanisms of therapeutic resistance in cancer (stem) cells with emphasis on thyroid cancer cells. Front Endocrinol (Lausanne). 2014;5:37.
Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, Wong KK, Brandstetter K, Wittner B, Ramaswamy S, Classon M, Settleman J. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141(1):69–80.
Borst P. Cancer drug pan-resistance: pumps, cancer stem cells, quiescence, epithelial to mesenchymal transition, blocked cell death pathways, persisters or what? Open Biol. 2012;2(5):120066.
Folini M Editorial: targeting telomere maintenance mechanisms in cancer therapy. Curr Pharm Des. 2014;20(41):6359–60.
Reddel RR. Telomere maintenance mechanisms in human cancer: clinical implications. Curr Pharm Des. 2014;20(41):6361–74.
Feijoo P, Dominguez D, Tusell L, Genesca A. Telomere-dependent genomic integrity: evolution of the fusion-bridge-breakage cycle concept. Curr Pharm Des. 2014;20(41):6375–85.
Krause DS, Van Etten RA. Right on target: eradicating leukemic stem cells. Trends Mol Med. 2007;13(11):470–81.
Essers MAG, Trumpp A. Targeting leukemic stem cells by breaking their dormancy. Mol Oncol. 2010;4:443 -450.
Moore J, Seiter K, Kolitz J, Stock W, Giles F, Kalaycio M, Zenk D, Marcucci G. A phase II study of Bcl-2 antisense (oblimersen sodium) combined with gemtuzumab ozogamicin in older patients with acute myeloid leukemia in first relapse. Leuk Res. 2006;30:777–83.
Grodzovski I, Lichtenstein M, Galski H, Lorberboum-Galski H. IL-2-granzyme A chimeric protein overcomes multidrug resistance (MDR) through a caspase 3-independent apoptotic pathway. Int J Cancer. 2011;128(8):1966–80.
Mattoo AR, FitzGerald DJ. Combination treatments with ABT-263 and an immunotoxin produce synergistic killing of ABT-263-resistant small cell lung cancer cell lines. Int J Cancer. 2013;132(4):978–87.
Secchiero P, Vaccarezza M, Gonelli A, Zauli G. TNF-related apoptosis-inducing ligand (TRAIL): a potential candidate for combined treatment of hematological malignancies. Curr Pharm Des. 2004;10(29):3673–81.
Cenni V, Maraldi NM, Ruggeri A, Secchiero P, Del Coco R, De Pol A, Cocco L, Marmiroli S. Sensitization of multidrug resistant human ostesarcoma cells to Apo2 Ligand/TRAIL-induced apoptosis by inhibition of the Akt/PKB kinase. Int J Oncol. 2004;25(6):1599–608.
Du X, Xiang L, Mackall C, Pastan I. Killing of resistant cancer cells with low Bak by a combination of an antimesothelin immunotoxin and a TRAIL receptor 2 agonist antibody. Clin Cancer Res. 2011;17:5926–34.
Yamanaka K, Rocchi P, Miyake H, Fazli LA, So A, Zangemeister-Wittke U, Gleave ME. Induction of apoptosis and enhancement of chemosensitivity in human prostate cancer LNCaP cells using bispecific antisense oligonucleotide targeting Bcl-2 and Bcl-xL genes. BJU Int. 2006;97:1300–8.
Indran IR, Tufo G, Pervaiz S, Brenner C. Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim Biophys Acta. 2011;1807:735–45.
Hamada H, Tsuruo T Functional role for the 170- to 180-kDa glycoprotein specific to drug-resistant tumor cells as revealed by monoclonal antibodies. Proc Natl Acad Sci U S A. 1986;83(20):7785–9.
FitzGerald DJ, Willingham MC, Cardarelli CO, Hamada H, Tsuruo T, Gottesman MM, Pastan I. A monoclonal antibody-Pseudomonas toxin conjugate that specifically kills multidrug-resistant cells. Proc Natl Acad Sci U S A. 1987;84:4288–92.
Mickisch GH, Pai LH, Gottesman MM, Pastan I. Monoclonal antibody MRK16 reverses the multidrug resistance of multi-drug resistant transgenic mice. Cancer Res. 1992;52(16):4427–32.
Dinota A, Tazzari PL, Michieli M, Visani G, Gobbi, M, Bontadini A, Tassi C, Fanin R, Damiani D, Grandi M, Pileri S, Bolognesi A, Stirpe F, Baccarani M, Tsuruo T, Tura S. In vitro bone marrow purging of multidrug-resistant cells with a mouse monoclonal antibody directed against Mr 170,000 glycoprotein and Saporin-conjugated anti-mouse antibody. Cancer Res. 1990;50:4291–4.
Niv R, Assaraf YG, Segal D, Pirak E, Reiter Y. Targeting multidrug resistant tumor cells with a recombinant single-chain FV fragment directed to P-glycoprotein. Int J Cancer. 2001;94(6):864–72.
St’astný M, Strohalm J, Plocová D, Ulbrich K, Ríhová B. A possibility to overcome P-glycoprotein (PGP)-mediated multidrug resistance by antibody-targeted drugs conjugated to N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer carrier. Eur J Cancer. 1999;35(3):459–66.
Takeshita A. Efficacy and resistance of gemtuzumabozogamicin for acute myeloid leukemia. Int J Hematol. 2013;97(6):703–16.
Matsui H, Takeshita A, Naito K, Shinjo K, Shigeno K, Maekawa M, Yamakawa Y, Tanimoto M, Kobayashi M, Ohnishi K, Ohno R. Reduced effect of gemtuzumabozogamicin (CMA-676) on P-glycoprotein and/or CD34-positive leukemia cells and its restoration by multidrug resistance modifiers. Leukemia. 2002;16(5):813–9.
Tortorella LL, Pipalia NH, Mukherjee S, Pastan I, Fitzgerald D, Maxfield FR. Efficiency of immunotoxin cytotoxicity is modulated by the intracellular itinerary. PLoS ONE. 2012;7:e47320.
Alewine C, Xiang L, Yamori T, Niederfellner G, Bosslet K, Pastan I. Efficacy of RG7787, a next generation mesothelin-targeted immunotoxin, against triple-negative breast and gastric cancers. Mol Cancer Ther. 2014;13(11):2653–61.
Folini M Editorial: targeting telomere maintenance mechanisms in cancer therapy. Curr Pharm Des. 2014;20(41):6359–60.
Santambrogio F, Gandellini P, Cimino-Reale G, Zaffaroni N, Folini M. MicroRNA-dependent regulation of telomere maintenance mechanisms: a field as much unexplored as potentially promising. Curr Pharm Des. 2014;20(41):6404–21.
Crees Z, Girard J, Rios Z, Botting GM, Harrington K, Shearrow C, Wojdyla L, Stone AL, Uppada SB, Devito JT, Puri N. Oligonucleotides and G-quadruplex stabilizers: targeting telomeres and telomerase in cancer therapy. Curr Pharm Des. 2014;20(41):6422–37.
Romaniuk A, Kopczy_ski P, Ksi_ek K, Rubi B. Telomerase as a target for anticancer therapies. Curr Pharm Des. 2014;20(41):6438–51.
Uziel O, Lahav M. Conventional anticancer therapeutics and telomere maintenance mechanisms. Curr Pharm Des. 2014;20(41):6452–65.
Jabbour E, Ottmann OG, Deininger M, Hochhaus A. Targeting the phosphoinositide 3-kinase pathway in hematologic malignancies. Haematologica. 2014;99(1):7–18.
Post J, Vooijs WC, Bast BJ, De Gast GC. Efficacy of an anti-CD138 immunotoxin and doxorubicin on drug-resistant and drug-sensitive myeloma cells. Int J Cancer. 1999;83(4):571–6.
Liu C, Lambert JM, Teicher BA, Blättler WA, O’Connor R. Cure of multidrug-resistant human B-Cell lymphoma xenografts by combinations of anti-B4-blocked ricin and chemotherapeutic drugs. Blood. 1996;87(9):3892–8.
Rosenblum MG, Cheung L, Kim SK, Mujoo K, Donato NJ, Murray JL. Cellular resistance to the antimelanoma immunotoxin ZME-gelonin and strategies to target resistant cells. Cancer Immunol Immunother. 1996;42(2):115–21.
Wagstaff KM, Jans DA. Protein transduction: cell penetrating peptides and their therapeutic applications. Curr Med Chem. 2006;13(12):1371–87.
Liu J, Zhao Y, Guo Q, Wang Z, Wang H, Yang Y, Huang Y. TAT-modified nanosilver for combating multidrug-resistant cancer. Biomaterials. 2012;33(26):6155–61.
Kirtane AR, Kalscheuer SM, Panyam J. Exploiting nanotechnology to overcome tumor drug resistance: challenges and opportunities. Adv Drug Deliv Rev. 2013;65(13–14):1731–47.
Beljanski V, Hiscott J. The use of oncolytic viruses to overcome lung cancer drug resistance. Curr Opin Virol. 2012;2(5):629–35.
Pavelic J. Editorial: combined cancer therapy. Curr Pharm Des. 2014;20(42):6511–2.
Han L, Shi S, Gong T, Zhang Z, Sun X. Cancer stem cells: therapeutic implications and perspectives in cancer therapy. Acta Pharmaceutica Sinica B. 2013;3(2):65–75.
Ischenko I, Seeliger H, Schaffer M, Jauch KW, Bruns CJ. Cancer stem cells: how can we target them? Curr Med Chem. 2008;15:3171–84.
Sun S, Liu S, Duan SZ, Zhang L, Zhou H, Hu Y, Zhou X, Shi C, Zhou R, Zhang Z. Targeting the c-Met/FZD8 signaling axis eliminates patient-derived cancer stem-like cells in head and neck squamous carcinomas. Cancer Res. 2014;74(24):7546–59
Huang H, Hu M, Li P, Lu C, Li M. Mir-152 inhibits cell proliferation and colony formation of CD133 + liver cancer stem cells by targeting KIT. Tumour Biol. 2014;36(2):921–8.
Bao B, Azmi AS, Ali S, Zaiem F, Sarkar FH. Metformin may function as anti-cancer agent via targeting cancer stem cells: the potential biological significance of tumor associated miRNAs in breast and pancreatic cancers. Ann Transl Med. 2014;2(6):59.
Bao S, Wu Q, Li Z, Sathornsumetee S, Wang H, McLendon RE, Hjelmeland AB, Rich JN. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008;68(15):6043–8.
Hogge DE, Feuring-Buske M, Gerhard B, Frankel AE. The efficacy of diphtheria-growth factor fusion proteins is enhanced by co-administration of cytosine arabinoside in an immunodeficient mouse model of human acute myeloid leukemia. Leuk Res. 2004;28(11):1221–6.
Acknowledgement
This research was supported in part by Department of Science and Technology and Department of Biotechnology, Government of India.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Devilakshmi, S., Madhumathi, J., Verma, R. (2015). Immunotoxins, Resistance and Cancer Stem Cells: Future Perspective. In: Verma, R., Bonavida, B. (eds) Resistance to Immunotoxins in Cancer Therapy. Resistance to Targeted Anti-Cancer Therapeutics, vol 6. Springer, Cham. https://doi.org/10.1007/978-3-319-17275-0_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-17275-0_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-17274-3
Online ISBN: 978-3-319-17275-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)