
Abstract
With the advent of next-generation sequencing (NGS), we have the promise of a complete genetic description of patient tumors to optimally direct therapy. Of hundreds to thousands of somatic mutations that exist in each cancer genome, a large number are unique and nonrecurrent variants. Prioritizing and annotating genetic variants identified via NGS technologies remains a major challenge. Some variants occur in tumor genes that have well-established biological and clinical relevance and are putative targets of therapy. However, most variants have limited evidence as predictive markers or are still of unknown significance. Furthermore, how to prioritize therapy when multiple potentially targetable aberrations and/or coexisting resistance mechanisms are identified in a patient’s tumor still remains largely a heuristic task. In this context, there is a growing need for the biomedical research community to have access to curated and up-to-date cancer pharmacogenomic associations. In addition, the community needs to remain cognizant of the potential consequences of misuse or overinterpretation of genomic data. Herein, I describe a systematic framework for variant annotation and prioritization and propose a structured molecular pathology report using standardized terminology in order to best inform oncology clinical practice.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Clinical laboratories increasingly view large cancer gene panels and NGS as a cost-effective—and tissue-saving—alternative to running a series of multiple single-gene companion tests. Large amounts of genomic data are being generated as these assays enter the clinical realm, challenging molecular pathologists and cancer genomicists in charge of interpreting and reporting the results. Manually annotating each single variant in terms of clinical significance in every possible tumor type is a daunting task. In addition, the strain on the turnaround time drives the need for prioritization strategies for the identification and reporting of clinically significant genetic variants.
Routine testing of full gene sequences as opposed to hot spots frequently identifies mutations of low frequency and unknown functional consequences, most of which are likely to be neutral or passenger alterations. On the other hand, some variants occur in cancer genes that have well-established clinical utility, driving tumorigenesis, and tumor progression. The available scientific knowledge on these mutations should be presented in the report, so that physicians and patients can make evidence-based decisions in a responsible fashion. Genetic results may provide a strong rationale for treatment with matched targeted agents in clinical trials, with the potential of directly benefitting the patient and accelerating the drug development process [1]. Consolidating so much information into a very discrete report that emphasizes the clinical significance while preserving observations that can be further looked into by the clinician is not an easy undertaking. As physicians trained in fields other than genetics are playing a more central role in the ordering and reviewing of genetic test results, the importance of translating genomic data into informative reports is further increased.
Performing NGS in the clinical laboratory is a multistep process that typically involves sample acquisition and quality control, DNA extraction, library preparation, sequencing, and genomic data generation. The process continues with three dynamic pipelines for data analysis: (1) bioinformatics tools for variant identification, (2) variant annotation and prioritization, and (3) interpretation of clinical significance and reporting to clinicians [2, 3]. In this chapter, I propose a framework for clinical interpretation of somatic cancer variants and describe how genomic data can be translated into structured evidence-based reports after a detailed variant annotation and prioritization process.
2 Prioritizing Cancer Genomic Variants
Following variant identification using bioinformatics pipelines, a computational engine is needed in order to parse the variants and suppress those that are irrelevant, highlight the ones which need manual curation, and identify pertinent “wild types” in each tumor sample. In the first step of variant prioritization, as summarized in Fig. 1, molecular pathologists have to define what is considered a “reportable” variant. Several annotation and prioritization parameters are taken into consideration so as to provide a stronger estimation of the functional significance of unknown and novel mutations. Useful tools include sequencing metric variables, external germ line single nucleotide polymorphisms (SNPs), and cancer databases for comparison of variants across populations, as well as prediction models for defining damaging/deleterious or potentially driver mutations, as discussed below.

Variant analysis flowchart of NGS tests performed in clinical laboratories. The bioinformatics pipeline identifies real and tumor-specific variants. During the variant annotation and prioritization pipeline, curated databases, predefined thresholds, and functional prediction models serve as filters, with reportable variants as final output. The clinical interpretation pipeline involves careful literature review and reporting of actionable variants
2.1 Upstream Filtering Tools
In the case of exome or whole genome sequencing, pairwise comparison with germ line DNA plays a pivotal role. Subtracting the genetic variation of a noncancerous “normal” genome from its cancerous counterpart allows the identification of the somatic mutations. In parallel, eliminating known harmless variants that are present in public or in-house polymorphism databases is a very helpful strategy for reducing the candidate list of deleterious mutations. The next step involves prioritizing missense, nonsense, or splice-site mutations over synonymous and intronic variants. Different bioinformatic adjustments can be used in order to improve variant detection and deal with library preparation or sequencing artifacts along with sample characteristics, including tumor purity and heterogeneity. In order to consider the variant as real and reportable, it is also advised to establish a minimum threshold of mutant allele fraction (MAF), the number of alternate reads at the genomic position divided by the total number of reads—coverage—at the same site. This threshold should take into consideration tumor cellularity and also clinical context, as rare resistant subclones in the treatment-refractory setting might be of relevance. Therefore, known gene variants previously clinically annotated are generally prioritized irrespective of MAF.
The most useful annotation tool for somatic variant interpretation involves the assessment of published cancer databases. The software used for variant prioritization should directly link genetic alterations to the Cancer Gene Census (http://cancer.sanger.ac.uk/cancergenome/projects/census/) or similar catalogues of genes for which mutations have been causally implicated in cancer [4], as well as the Catalogue of Somatic Mutations in Cancer (COSMIC) (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/), International Cancer Genome Consortium (ICGC) (https://dcc.icgc.org/), and The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/; http://www.cbioportal.org/), large cancer databases that present prevalence of gene variants in different tumor types. Assessing whether a newly discovered alteration may be functionally relevant rests heavily on how many times it has been reported in these international cancer genomics studies, supporting further clinical interpretation.
2.2 Downstream Filtering Tools
Prediction of the putative functional effect of a mutation is a common problem already addressed in the context of germ line SNP association studies, and several tools have been used for this purpose. These models annotate variants specifically with respect to evolutionary conservation, biochemical deleteriousness, and functional importance scores, thereby facilitating the differentiation between functional and nonfunctional variants [5–7]. At present, for alleles without prior functional analysis in genes that have been related to human cancer, such as non-hot spot/novel variants in known oncogenes and tumor suppressor genes, prediction algorithms based on evolutionary conservation patterns are often used. Sorting Intolerant from Tolerant (SIFT) [8] and MutationAssessor [9] exploit the fact that sequences observed among living organisms are those that have not been removed by natural selection and sites with fewer observed substitutions are inferred to be under tighter constraints, having more deleterious effects when mutated. On the other hand, mutations in non-conserved residues are likely neutral. Other resources for predicting the effects of protein-coding sequence changes typically exploit the physicochemical properties of amino acids and information about the role of amino acid side chains in protein structure. These in silico protein sequence-based algorithms, such as PolyPhen2 [10], are capable of leveraging both evolutionary and biochemical information. Despite having high sensitivity for the detection of damaging variants, prediction tools that rely on conservation and structure should be used with caution. In addition to the low specificity, these methods generally have limited value in annotating gain-of-function or switch-of-function mutations [11]. Furthermore, most of these algorithms have been designed for research purposes with germ line variants, and very few databases present clinically oriented molecular annotation. As an alternative, machine learning scoring methods attempt to increase the predictive precision of somatic mutations in cancer. One example is the cancer-specific high-throughput annotation of somatic mutation (CHASM) tool, specifically designed to distinguish driver from passenger somatic missense variants [12]. It is trained on a positive class of drivers curated from the COSMIC database and a negative class of passenger variants generated in silico based on background base substitution in specific tumor types. Limitations include reduced coverage as compared to traditional algorithms—restriction to missense mutations—and the understanding that driver and passenger mutations are tumor type and context dependent, possibly changing roles during cancer evolution and therapy [7]. Whether cancer-trained methods outperform more general predictors still needs further investigation. Recent studies suggest that no method or combination of methods exceeds ~80 % accuracy [13, 14], indicating that there is still significant room for improvement in functional prediction, possibly with the development of specific algorithms for different classes of mutations.
To summarize, complex criteria involving multiple annotation sources should be used in order to select or filter out variants. Part of this process can be automated, although most of the work still needs to be done manually. As the most valuable tool consists in leveraging the cancer literature, either generated in-house or derived from publicly available databases, the genomic prioritization engine needs to be dynamic in nature, recognizing driver cancer mutations that have been previously annotated and reported. Additional tumor-specific variants with very low MAFs and those considered silent mutations are typically excluded from further clinical interpretation. Novel variants in genes that have been causally implicated in cancer are prioritized when functional models predict damaging/deleterious scores, the alteration is in the phosphorylation loop of an oncogenic kinase, or it alters the reading frame of a tumor suppressor gene.
3 Interpreting Results with Clinical Perspective
After narrowing down the list of candidate variants, the biggest challenge is to interpret the remaining genomic alterations within a biological context. Potentially “reportable” variants can be grouped in three categories: (1) those that may have a direct impact on patient care and are considered “actionable,” (2) those that may have “biological relevance” but are not clearly actionable, and (3) those that are of “unknown significance.” Different groups have varying definitions for clinically “actionable.” This category can be restricted to variants matched to drugs that have been approved by regulatory agencies for the tumor that is being studied, but may also include those directing to off-label use of approved drugs, as well as variants that are matched to drugs being investigated in clinical trials. Academic laboratories should adopt the most inclusive definition of an actionable mutation—which accounts for variants that support treatment recommendation and enrollment in a particular clinical trial or have prognostic or diagnostic implications—even knowing that it may increase challenges in clinical decision-making, as the results sometimes lead to regulatory issues regarding the use of targeted drugs in unapproved indications.
Importantly, variants should not be reported in an uncategorized format, which can be confusing to clinicians and detrimental to patients. For actionable mutations to be fully curated, a team of experts with strong background in cancer biology and access to up-to-date knowledge resources is mandatory. Clinical interpretation of most variants identified in NGS-based cancer diagnostic tests involves the burdensome procedure of manually reviewing the published literature on four different layers: (1) gene, (2) specific variant, (3) drug or class-of-agent sensitivity/resistance patterns, and (4) tumor-type context. To facilitate this process, several groups have implemented “Sequencing Tumor Boards” or “Molecular Rounds” with up to 15 faculty members that share expertise in cancer genomics, bioinformatics, pathology, clinical genetics, bioethics, and clinical oncology as well as experimental therapeutics. Rigorous analysis of comprehensive genomic data is a time-consuming and labor-intensive task, considering that not many mutations have been validated with a high enough level of evidence to predict for response to targeted treatment. Experts should prioritize the knowledge on mutations in tumor-specific contexts, but curation of data derived from other tumor types and preclinical experiments—when clinical validation is under way—usually gives valuable information to clinicians. Unfortunately, most resources currently available cover information at limited levels: some focus on gene-tumor associations, others only on gene-drug or drug-target relationships. Moreover, databases originally developed to enable preclinical research or annotate germ line variants are of limited applicability for clinical oncology curation. Alternatively, associations on predictive, prognostic, or diagnostic variants in cancer can be retrieved in clinically oriented databases, such as My Cancer Genome (http://www.mycancergenome.org/), Targeted Cancer Care (http://www.targetedcancercare.org/), and Personalized Cancer Therapy (https://pct.mdanderson.org/). These websites are the result of large institutional efforts to provide information on cancer types, aberrant genes, and variants that are targeted by approved or experimental therapies. However, information available in these databases does not cover all genes, variants, and tumor types. In addition, it is not accessible for download, mainly because it is presented in a descriptive format, without standardized terminology.
In order to deal with these limitations, some groups have developed internal knowledge databases with more comprehensive annotations on consensus and emerging clinical/preclinical predictive genomic markers linked to targeted therapies. When integrated to the variant prioritization computational engine and report generation system, the curated information on somatic variants that have been classified for clinical reporting is stored for future use. Maintenance of these databases involves a regular and systematic review of drug regulatory and approval status, consensus guidelines, peer-reviewed publications, and clinical trial databases. One example of detailed cancer genomics knowledge database is available for download through Synapse (https://www.synapse.org/#!Synapse:syn2370773), the collaborative cloud-based repository developed at Sage Bionetworks. As many academic groups are independently working on similar projects, an international consortium on curated cancer genomic data matching genomic aberrations to targeted therapies could have a huge clinical impact. Ideally, the information should be released as an interactive web-based tool, subjected to editing, validation, and critique from the medical community.
4 Generating NGS Reports
Previous studies evaluating single-gene reports have suggested that patient care may be compromised as a consequence of poor communication between laboratories and clinicians [15]. Developing a framework to content-rich NGS reports is complicated. The traditional “narrative” style reporting is too cumbersome for the amount of data generated by large cancer gene panels. In addition, medical oncologists prefer structured reports with results displayed in a more straightforward manner rather than detailed descriptions of each genomic alteration. Consequently, web-enabled technologies are a good alternative to text reports as they enable dynamic and interactive display of the NGS results, which could be accessed by providers and patients in different formats. Embedding links to internal and external databases allows members of the team to further explore the results and the evidence used to guide the interpretation, including more detailed information on the gene, the variant, the drug, or the clinical trial matched to a particular genomic alteration and tumor type, as well as records of PubMed identification numbers for relevant clinical literature. Unfortunately, most laboratory information systems and electronic medical records (EMR) to date do not support data formatting and metadata (data associated with the result). Therefore, reports may need to be oversimplified to a static format for inclusion in the EMR.
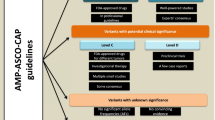
Wagle et al. reported the first framework to segregate genetic alterations derived from NGS tests on the basis of their predicted clinical utility [16]. The actionable category includes variants that predict tumor sensitivity or resistance to approved (tier 1) or experimental therapies (tier 2). As shown in Fig. 2a, the mutational categories are organized based on the strength of evidence supporting its predictive value. An alternative classification is presented in Fig. 2b, which represents a simplified gene-oriented approach developed to facilitate clinical decision-making [17]. Reports based on this framework provide the information in a hierarchical/categorical format, and results can be structured in tabular view. The content is formatted in such a way as to draw the clinician’s attention to associations with the highest level of evidence. As exemplified in Fig. 3, all actionable—predictive, prognostic, and diagnostic—markers are displayed first, followed by biologically relevant gene variants that warrant detailed annotation and pertinent negatives in the tumor being tested. Details are discussed in the following sections.

Examples of somatic variant classification system for NGS reports. (a) Wagle et al. The actionable category includes variants that predict tumor sensitivity or resistance to approved (tier 1) or experimental therapies (tier 2) and those that have prognostic/diagnostic implications. (b) Dienstmann et al. Reportable variants can be grouped in three categories: (1) actionable, which support treatment recommendation (therapeutic consensus) and enrollment in clinical trials (therapeutic emerging) and/or have prognostic or diagnostic implications; (2) biologically relevant but not clearly actionable; and (3) unknown significance

Illustrative example of sequencing results describing somatic cancer variants with structured evidence-based classification. Using the framework described in Fig. 2b, results are presented in a hierarchical and tabular format, drawing clinician’s attention to associations with different levels of actionability
4.1 Predictive Associations
Consensus predictive associations include those (1) linked to drugs approved or rejected by regulatory agencies in the context of a specific gene variant and tumor type or (2) described in national guidelines as predicting response or resistance to specific therapies. Emerging predictive associations were classified in a hierarchical way based on the strength of evidence: (1) late trials, including evidence derived from trials that prospectively recruited patients based on genomic profiling as well as large trials with robust data suggesting sensitivity/resistance to targeted therapies based on retrospective analysis of biomarkers; (2) early trials, referring to phase 1 or 2 studies with genomically selected patients that show preliminary signs of efficacy (or lack of efficacy); (3) case reports of dramatic responses to targeted therapies in a specific genomic context; and (4) strong preclinical data that is being explored in clinical trials. The magnitude of the biomarker-drug effects for clinical associations is classified as “responsive,” “resistant,” or “not responsive” (when an expected responsive effect is not observed). In preclinical models, biomarker-drug associations are graded as “sensitive,” “reduced sensitivity,” or “resistant.”
Some of the questions that scientists involved in clinical interpretation of genomic data have to deal with include:
-
Is this an activating or inactivating mutation?
-
Does this mutation engender sensitivity to targeted therapeutics—and what is the agent with highest potency?
-
How to select therapy in case of multiple genomic alterations and/or coexisting resistance mechanisms?
-
Is the association tumor type or context specific (treatment-naïve versus refractory setting) after exposure to which targeted agents?
Ideally, reports of NGS tests in oncology should include a list of clinical trials recruiting patients that harbor the specific genomic aberrations identified in the individual tumor sample. These are matched targeted therapies available either on-site or as part of multi-institutional collaborations. A current limitation for matching a patient’s tumor genotype to clinical trials is the lack of molecular annotations in notices of national registries, such as the US National Cancer Institute clinical trial locator (www.clinicalTrials.gov). As an example, the search term “PIK3R1” does not identify any matched trial, even though many PI3K pathway inhibitors in clinical development have a clear rationale for testing in tumors that harbor PIK3R1 inactivating mutations.
4.2 Prognostic and Diagnostic Associations
Medical oncologists are usually concerned about reporting detailed information on prognostic associations of genomic markers in cancer. First, the literature is full of inconsistent and even opposing results based on retrospective studies. Second, as patients have access to the report, bad prognostic associations could lead to misinterpretation and anxiety, emphasizing the idea that this information should be discussed in person taking into consideration additional clinical parameters. Therefore, only prognostic markers with well-established associations in the same tumor type should be reported, preferably without description of the related outcome information. Common diagnostic associations should also be described, mainly those favoring a specific tumor subtype.
4.3 Variants with Biological Relevance
Many variants in well-known cancer genes do not fall into the prior categories but still might be causally associated with the malignant phenotype. Their relevance is justified by known biological implications (pathway activation/inactivation) or by “theoretical” actionability, when agents potentially targeting novel activating mutations in oncogenes or the downstream effects of loss-of-function mutations in tumor suppressor genes are available for clinical testing. Therefore, the expected effect of the variant on protein function (gain- or loss-of-function) is also presented in the report, as it might give insights to the ordering physician with regard to therapeutic interventions in the investigational setting. Nevertheless, until functionality is validated in preclinical studies, it is appropriate to report these novel variants as non-actionable.
4.4 Pertinent Negative Variants
Genes that have clear predictive, prognostic, or diagnostic associations in a specific tumor type and are found to be “wild type” in the NGS test should be described in the report.
4.5 Variants of Unclear Significance
The accelerated pace of advances in our understanding of cancer genomics justifies the description of all “reportable” variants in the final NGS report, even those not classified as actionable or biologically relevant when the assay is performed. These variants may become biomarkers in the near future or may be of particular interest in research settings. The most practical approach to handle variants of unknown biological/clinical significance is to present them according to the main pathway affected by the alteration. Key gene-pathway associations are increasingly being highlighted in the cancer genomics literature [18, 19]. As an example, in renal cell carcinomas, mutations in genes involved in histone modification/chromatin remodeling might dominate a report, warning the medical oncologist-translational researcher about the importance of aberrations in this pathway during cancer progression.
4.6 Germ Line Variants
The American College of Medical Genetics and Genomics (ACMG) recently published a minimum list of genes that should be reported to the patient when an incidental germ line mutation associated with heritable risk of cancer or other diseases is identified and confirmed [20]. The group prioritized disorders where preventive measures and/or treatments were available and those in which individuals with pathogenic mutations might be asymptomatic for long periods of time. Only pathogenic mutations should be reported, considering the challenges of interpreting variants of unknown significance as incidental findings. Notably, the group acknowledged the fact that insufficient data on penetrance and clinical utility support these recommendations. Considerable personnel resources, including genetic counselors with specialized training, may be needed to ensure that patients understand the potential benefits and risks of receiving somatic and germ line data and to support physicians in conveying such information.
4.7 Performance Characteristics of the Test
Specific regions interrogated by the assay and the coverage metrics by sample and target—including median depth, uniformity, and percentage of target covered at the minimum level—should be described in every NGS assay, regardless of application or platform. Minimum depth of coverage should be established during the test validation process and will depend upon the required sensitivity of the assay as well as the targeting/sequencing method. Regions of sequence not meeting the required read depth, especially genes with highest priority (see “pertinent negatives” above), should be clearly reported as indeterminate. Importantly, medical oncologists still need to be educated for the proper interpretation of MAF counts. This information is very useful in the research setting, reflecting clonal evolution and selection when NGS tests are performed in different samples and time points over the course of a disease and therapy. Of note, continued medical education is an important aspect in the process of implementing NGS reports in a clinical lab, so that physicians are trained to understand molecular profile results.
5 Conclusion
NGS tests were initially developed for research or investigational purposes but will eventually become part of cancer care. During the process of clinical implementation of these assays, many technical, legal, and ethical challenges have to be overcome. Clinical Laboratory Improvement Amendment (CLIA) or Good Clinical Laboratory Practice (GCLP) certification is required for clinical centers and consulting biotechnology companies offering NGS-based cancer diagnostic tests. Several professional societies have generated guidelines for the implementation of NGS tests, with a focus on analytical validity or patient privacy rules. Nonetheless, recommendations for the use of computational tools and bioinformatics pipelines and reporting of somatic cancer variants are still missing. A major challenge is how to convey the amount of data obtained from NGS tests and all the information reviewed for interpretation within a reasonable time frame, so that it can be translated into a useful clinical tool. Effective communication of results with interactive reports can promote appropriate clinical decision-making and minimize the potential for patient harm. Unfortunately, at the present time, validated evidence on specific gene variants linked to predictive, prognostic, or diagnostic associations in cancer is limited. In addition, genomics knowledge is currently ahead of our ability to therapeutically target tumors, given that many mutations identified by sequencing either are linked to unapproved drugs or are not targetable by currently available molecular therapy.
Importantly, while sequencing can identify druggable targets, clinicians are often left with the task of further interpretation, treatment prioritization, and decision-making in the context of additional clinical information. When the best option is to offer the patient genomic-driven clinical trials, additional logistical challenges need to be overcome, including too strict eligibility criteria in phase 1 trials or slots not available at the time of referral and geographical limitations to access drug development units. These difficulties explain why only a small number of patients are ultimately enrolled in a specific trial based on the results of NGS assays, even when actionable genomic alterations are identified in the majority of the tumor samples tested [21]. Multi-institutional trial networks assessing novel agents that target specific mutations are needed in order to deal with these issues. Alternatively, when physicians and patients agree on off-label use of targeted therapies, another aspects that go beyond reimbursement concerns need to be taken into consideration. There is an inherent bias to publish positive results—case reports showing that sequencing results are associated with responses to off-label use of a targeted agent—and mechanisms to annotate lack of response in this setting are missing. One option is to create national formularies of targeted agents against common aberrations, so that every patient receiving a matched therapy in the off-label setting can be tracked and become a “cancer information donor.” These pharmacy exchange programs could generate ever-growing data banks integrating the genomic information with therapeutic response and outcome [22]. The information derived from these registries should be added to knowledge databases such as My Cancer Genome or Personalized Cancer Therapy and become readily available to oncologists worldwide, providing annotated predictive genomic markers in cancer and potentially changing the paradigm of drug approval process.
In conclusion, structured reporting of clinically relevant variants may help addressing the current limitations of NGS to directly guide patient care. With standardized terminology and an expanding knowledge database, variant annotation, prioritization, and clinical interpretation become a fluid process with the potential to open new therapeutic options.
References
Dienstmann R, Rodon J, Tabernero J. Biomarker-driven patient selection for early clinical trials. Curr Opin Oncol. 2013;25:305–12.
Watt S, Jiao W, Brown AM, et al. Clinical genomics information management software linking cancer genome sequence and clinical decisions. Genomics. 2013;102:140–7.
Van Allen EM, Wagle N, Levy MA. Clinical analysis and interpretation of cancer genome data. J Clin Oncol. 2013;31:1825–33.
Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501.
Cooper GM, Shendure J. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat Rev Genet. 2011;12:628–40.
Frousios K, Iliopoulos CS, Schlitt T, et al. Predicting the functional consequences of non-synonymous DNA sequence variants – evaluation of bioinformatics tools and development of a consensus strategy. Genomics. 2013;102:223–8.
Zhang J, Liu J, Sun J, et al. Identifying driver mutations from sequencing data of heterogeneous tumors in the era of personalized genome sequencing. Brief Bioinform. 2014;15:244–55.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Flanagan SE, Patch AM, Ellard S. Using SIFT and PolyPhen to predict loss-of-function and gain-of-function mutations. Genet Test Mol Biomarkers. 2010;14:533–7.
Wong WC, Kim D, Carter H, et al. CHASM and SNVBox: toolkit for detecting biologically important single nucleotide mutations in cancer. Bioinformatics. 2011;27:2147–8.
Gnad F, Baucom A, Mukhyala K, et al. Assessment of computational methods for predicting the effects of missense mutations in human cancers. BMC Genomics. 2013;14:S7.
Gonzalez-Perez A, Lopez-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88:440–9.
Lubin IM, Caggana M, Constantin C, et al. Ordering molecular genetic tests and reporting results: practices in laboratory and clinical settings. J Mol Diagn. 2008;10:459–68.
Wagle N, Berger MF, Davis MJ, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012;2:82–93.
Dienstmann R, Dong F, Borger D, et al. Standardized decision support in next generation sequencing reports of somatic cancer variants. Mol Oncol. 2014;8:859–73.
Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37.
Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science. 2013;339:1546–58.
Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–74.
Tran B, Brown AM, Bedard PL, et al. Feasibility of real time next generation sequencing of cancer genes linked to drug response: results from a clinical trial. Int J Cancer. 2013;132:1547–55.
Schilsky RL. Implementing personalized cancer care. Nat Rev Clin Oncol. 2014;11:432–8.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Dienstmann, R. (2015). Standardized Decision Support in NGS Reports of Somatic Cancer Variants. In: Wu, W., Choudhry, H. (eds) Next Generation Sequencing in Cancer Research, Volume 2. Springer, Cham. https://doi.org/10.1007/978-3-319-15811-2_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-15811-2_5
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-15810-5
Online ISBN: 978-3-319-15811-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)