
Abstract
Currently, cancer prevails as a prime health matter worldwide. Selecting the appropriate biomarkers for early cancer detection might improve patient care and have often driven revolutions in medicine. Statistics and machine learning techniques have been broadly investigated for biomarker identification, especially feature selection where researchers try to identify the most distinguishing genes that can achieve better predictive performance of cancer subtypes. The robustness of the selected signature remains a crucial goal in personalized medicine. Ensemble and parallel feature selection are promising techniques to overcome this problem in which they have seen an increasing use in biomarker discovery. We focus in this chapter on the principal aspects of using ensemble feature selection in biomarker discovery. Furthermore, we propose a massively parallel meta-ensemble of filters (MPME-FS) to select a robust and parsimonious subset of genes. Two types of filters (ReliefF and Information Gain) are investigated in this study. The performances of the proposed approach in terms of robustness, classification power and the biological meaning of the selected signatures on five publicly available cancer datasets are explored. The results attest that the MPME-FS approach can effectively identify a small subset of biomarkers and improve both robustness and classification accuracy.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- Bioinformatics
- Biomarker discovery
- Ensemble feature selection
- Meta learning
- ReliefF
- Information gain
- Cancer classification
1 Introduction
Over the last two decades, the general rate of deaths to new cancer cases persists as high as 49 % overall. Thus, current bioinformatics efforts are focusing on biomarker discovery which is the key element of personalized medicine, where the genetic constitution is used to guide therapeutic approaches [1]. Therefore, the discovery of more effective cancer biomarkers is urgently needed, since the development and the effective use of biomarkers in clinical practice will certainly lead to tailor treatments for the disease in an individual [2].
The application of omics high-throughput technologies for cancer biomarker discovery is being rapidly expanded in current biomedical research, including DNA microarrays, Next Generation Sequencing (NGS) and MicroRNAs which are able to capture a substantial fraction of a cell state [3]. These technologies allow monitoring the expression levels of thousands of genes simultaneously in healthy and diseased cells, as well as are essential to biomarker discovery.
Gene expression data can effectively help to differentiate between cancer subtypes and then serve as an effective tool for diagnostic purposes in clinical practice. However, the identification of the smallest possible set of genes that could be used as biomarkers is a crucial problem in bioinformatics and personalized medicine. These genes must be the most informative for cancer prediction through supervised classification models [4]. The identification is generally referred to as a feature selection problem which is desirable to provide the features that contribute most to both classification and prediction [5]. It is a vital preprocessing step in data mining tasks, to reduce the effect of noise and improve the quality of data processing as well as considered to be one of current challenges in statistical machine learning for high-dimensional data.
A major challenge in the analysis of gene expression data is due to their sizes: a very small number of samples, of the order of tens, versus thousands of genes associated to all samples. This is commonly known as the “curse-of-dimensionality” which is also characterized by a large number of irrelevant, redundant and noisy genes that mislead or impede diagnosis efficiency [6]. Thus, only a fraction of genes contains useful biological interpretations and further gives a high accuracy for cancer diagnosis. Another challenge concerns the biological variations in real clinical tests which require the development of more stable feature selection methods [7]. In other words, selection of informative genes and an appropriate assessment of robustness, classification accuracy and biological meaning of the results are the most important matters in this field.
Ensemble-based learning is a robust and popular technique, due to the immense success of many ensemble methods in bioinformatics applications. It has the broad advantage of overcoming the curse-of-dimensionality in gene expression data, thereby offer higher accuracy and stability than conventional feature selection algorithm can achieve. Therefore, the use of ensemble methods to feature selection problem has been one of the recent growing trends. It consists of performing multiple diverse selectors with different subsamples, and then aggregates their results using a consensus function to obtain a final best subset of biomarkers [8]. Another benefit of applying ensemble feature selection, that it is naturally susceptible to parallelism, as well as we can easily undertake their parameters in parallel. The parallel implementation of ensemble methods can certainly speedup the computational time of the selection and allow solving large-scale problems by involving multiprocessors to execute the different parts of the ensemble in parallel [9].
This chapter will focus on the different aspects of the application of ensemble feature selection methods to biomarker discovery from gene expression data. Furthermore, we propose a massively parallel meta-ensemble based feature selection method that can select robust and accurate biomarkers from DNA-microarrays datasets and can be generalized to several genomics studies. Two types of filter-based feature selection algorithms are investigated in this study: ReliefF and Information Gain. We also discuss the results in terms of robustness, classification power and the biological meaning of the selected signatures.
2 Application of Ensemble Feature Selection to Biomarker Discovery
In analogy with ensemble methods in supervised machine learning which combine multiple learned models to achieve high classification accuracy such as bagging and boosting [10]. Ensemble feature selection has received much attention recently. We mainly present here the different aspects to be considered in ensemble-based feature selection for biomarker discovery in which can help researchers to classify any method of them. The main critical problems in this category of methods are both the construction of diverse local selectors and the consensus function used to combine the different subsets of features [7, 11]. Therefore, the first aspect to be examined is the diversity design within the ensemble. This criterion divides ensemble feature selection methods into three classes:
-
Ensemble based on data diversity: where we run the same selector with different subsamples generated from the original dataset [12, 13].
-
Ensemble based on functional diversity: where different selectors are performed on the whole set of data (without sampling) [14].
-
Ensemble based on data and functional diversity: here both data and functional diversity are combined in which multiple feature selection algorithms are performed on different subsamples.
Another aspect to be considered in ensemble feature selection methods is the representation used by the different selectors, since the notation of the results is not the same in all feature selection algorithms. This has a great impact on the consensus function to be used to aggregate the results [12]. Typically, we can observe three types of representations:
-
Feature subset representation: subset containing only selected features (generally with different size)
-
Feature ranking representation: subset of ranked features (a threshold is necessary for the selection)
-
Feature weighting representation: a subset of pairs feature/weight which can easily converted to feature ranking representation.
Recent studies have focused on ensemble methods using wrapper-based selectors [15, 16]. It prompts us to consider the dependence of the selectors to any classifier as an important aspect in ensemble feature selection methods. This criterion influences the quality of solutions within the ensemble and the overall computational cost of the selection, as well as it divides ensemble feature selection methods into:
-
Filter/ranking-based ensembles: they are simple, fast and independent of any classifier [14].
-
Wrapper-based ensembles: they are very computationally intensive and have the risk of over-fitting due to high dimensionality of data as well as include the interaction between feature subset search and the mining algorithm. Moreover, they have the ability to take into account feature dependencies [17].
-
Embedded-based ensembles: they use internal information of the classifier to perform selection and show a better computational complexity than wrapper methods [13].
-
Hybrid-based ensembles: they are a combination of filter and wrapper methods which use the ranking information obtained using filters to guide the search in the optimization algorithms used by wrapper methods [16].
3 Massively Parallel Meta-Ensemble Feature Selection
Feature selection is an important preprocessing step in many machine learning applications including bioinformatics and computational biology, where it is generally used to find the smallest subset of features that extremely increases the performance of the classification model. In this section, we focus on ensemble of ensembles learning techniques which work by aggregating the outcomes of different ensembles into a final agreed decision through one or more consensus functions. The main objective is to attempt high performance of computer-aided diagnosis (CAD), by selecting a few genes with high predictive power and high sensibility to variations in real clinical tests. The selected biomarkers will be directly used by the CAD system for cancer diagnosis or others predictive goals.
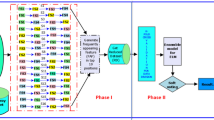
For this purposes, a new parallel framework of feature selection is explored (see Fig. 1). In analogy with meta-ensemble models for supervised learning [10], the proposed approach is designed as an ensemble of ensembles of different selectors which perform selection in parallel through various ensembles and two consensus functions. In the following, we introduce our parallel framework for biomarker discovery in detail. We first, formulate both problem and representation of our solution under the proposed framework and then the general framework is explored including the parallel construction of the ranked lists as well as the related consensus functions.

Parallel model of the MPME-FS
Accordingly, biomarker discovery from gene expression is the problem of selecting subset of representative biomarkers from a large dataset. Given a set \( X \) of K features with K very large, the problem consists in finding out the minimal subset \( X_{s}^{*} \subset X \) that contains the more relevant yet non redundant features. Ensemble feature selection is a promising technique for addressing these complex structures of data and alleviates the problems of small sample size and high dimensionality [18].
The use of an ensemble of ensembles of filters leads to several subsets. Let us denote by \( X_{sj}^{i} \) the subset j of selected features using filter i. Therefore, two matters need to be addressed. The first one is related to the importance of each feature and the second one is related to the way subsets are aggregated to lead to the final subset of features. In order to properly deal with these two issues, we propose a two stage approach that uses ranking and consensus functions. At a first step various subsets of ranked lists of features are constructed using several filters then aggregation of these subsets is performed at two levels to form ensembles and then the meta-ensemble. More formally, the output of the first step can be represented as:
\( f_{ij}^{k} \) represents the rank of the feature k in the ensemble j using filter i. Its relevance is given by the weight \( w_{ij}^{k} \). The global weight of feature k within the ensemble j is denoted as \( w_{j}^{k} \). Three subsets of pairs of features and their weights are needed.
-
The first is \( Lbest_{j} \) that represents the local best features in each ensemble.
$$ Lbest_{j} = \left\{ {\left( {k,w_{j}^{k} } \right)\;{\text{where}}\;j = 1 \ldots M\;{\text{and}}\;k = 1 \ldots K} \right\} $$(2) -
This subset is the result of an aggregation process over \( {\text{X}}_{\text{sj}}^{i = 1 \ldots N} \) subsets.
-
The second is Gbest. It represents the global best features over the meta-ensemble. It is the result of an aggregation process over \( Lbest_{j} \).
-
The third is \( Fbest = X_{s}^{*} \subset X \). It represents the final selected features given by MPME-FS method.
3.1 General Framework of MPME-FS for Biomarker Discovery
The general framework of MPME-FS consists of multiple ensembles of filters performed in parallel each of which employs a robust consensus function to select the best subset within each ensemble. The next step is to aggregate the outcome of all ensembles using a second consensus function and finally select features with higher scores given by all filters from all ensembles as shown in Fig. 1. The selection processes starts by the construction of M sub-samples Si = 1…M from the whole dataset. Then, the parallel selection is initiated in all the M ensembles. At this stage, each ensemble j constructs N ranked lists \( X_{sj}^{i} \) by using filter i. To achieve the goal of both functional and data diversity when constructing the \( X_{sj}^{i} \) lists, we have used data partitioning with overlap allowing creating a reduced dataset to each filter i .
Data perturbation involves generating subsamples by removing instances from the original datasets randomly. Knowing that, the overlap represents the percentage of samples belonging to the original dataset [11]. Subsequently, a consensus function within each ensemble is applied in order to aggregate these ranked lists \( {{X}}_{{sj}}^{{i}} \) and finally obtain the local best features in the ensemble j (Lbest j ) (see Fig. 2). Note that the construction of the ranked lists within each ensemble can be performed in parallel and through different filters (Information Gain, Gain ratio, Fisher Ratio, Symmetric uncertainty, ReliefF).

Ensemble feature selection
The following step consists of the aggregation of all local best features over the meta-ensemble, to construct the global best subset (Gbest) of features using the consensus function 2 alongside with the accumulation of scores associated to these features. Finally, we select the best ranked features which constitute the subset Fbest, from the subset Gbest ranked based on features’ global weights
3.2 Consensus Functions
Recent work in biomarker identification has seen an increasing use of ensemble based feature selection due to their power to give higher accuracy and stability than a single algorithm can achieve. It can also dealing with small sample size and complex data structures. The key idea in ensemble methods is how to combine subsets of different selectors to lead to the final subset of features. This problem has received considerable attention in recent years [19]. Aggregation methods depend on the representation of the outcome of selectors which can be divided in three types: feature subset, feature ranking and feature weighting-score [11]. Based on these representations there exist many consensus functions, some of them include weighted voting, mean aggregation and threshold based aggregation for both rank and weighting-score representations, and counting the most frequently selected features for feature subset representation [12, 20].
Certainly, choosing the appropriate consensus function is a difficult task in ensemble methods. In our work, we use two consensus functions; the first one in the ensembles level and the second function is in the meta-ensemble level. Both are based on features ranking and their global weights which lead to a more robust and parsimonious final selection.
The first consensus function aggregates the ranked lists created by filters in the same ensemble. This function is inspired from both counting the most frequently selected features and weighted voting aggregation functions, but with hard selection by using the intersection over the entire ranked lists. For the intersection purpose, we use a threshold denoted by TS1 in order to select only features belonging to TS1 first ranked ones. Afterward, the weights of all selected features over the entire ensemble j denoted by \( w_{i,j }^{k} \) are accumulated in order to obtain the global weights of selected features \( w_{j}^{k} \) over all ensembles [18]. More formally, the consensus function 1 can be described as follows:
By this way, we obtain the set Lbestj containing pairs of the best selected features with their global weights in the ensemble \( {j}\;\{ \left( {k,\;w_{\text{j }}^{\text{k}} } \right)\} \). This latter will be the input of the second consensus function in the meta-ensemble level, to construct the subset Gbest which contribute to the final selection.
The second consensus function consists primarily of aggregation the M subsets (Lbest j ) generated in the parallel previous step. The Gbest subset represents pairs of features and their accumulated weights belonging to the union of the M local best subsets of features, which can be calculated as follows:
Finally, we select the best TS2 ranked features from Gbest that represent the final selected features to be validated in the validation step. The pseudo-code of the whole process can be summarized as follows:

4 Experiments and Discussions
In the following sections, the analysis of classification performances, robustness and biological interpretation of the MPME-FS method on large feature and small sample size microarrays are presented. First, the data sets and the experimental settings used in this analysis are briefly described. Second, we analyze the classification performances in terms of accuracy, sensitivity and specificity using different classifiers. After that, we study the robustness of the selected signatures. Finally, we perform a biological interpretation of the selected genes.
4.1 Datasets and Experiment Setting
All experiments were conducted using MATLAB®’s Parallel Computing Toolbox (PCT). The proposed MPME-FS was evaluated by means of five publicly available DNA microarray datasets which can be divided into binary and multiclass types. The binary datasets are the most prominent and can separate healthy patients from cancer patients, while multiclass datasets are used to differentiate the various types of cancers based on gene expressions. Therefore, the datasets were collected from both Kent Ridge bio-medical data repositoryFootnote 1 and Gene Expression Model Selector, from Vanderbilt UniversityFootnote 2. The main datasets characteristics are shown in Table 1.
To assess the performances of our parallel meta-ensemble feature selection method, we use in the experiments two well-known and successful filters: information gain and ReliefF. Based on an empirical evaluation using different settings of the proposed method, the best parameters setting of MPME-FS which is adopted in this study is depicted in Table 2.
4.2 Classification Accuracy Analysis
The first experiment is devoted to assess the performance of the MPME-FS in terms of accuracy, sensitivity, specificity and the number of selected biomarkers using 10-fold cross validation technique. The latter is a common choice in the specialized literature [21], which splits the whole set of data into many subsets to evaluate the goodness of the selected signature.
To achieve high level evaluation of the classification ability of the selected genes, we first use different classifiers separately (SVM, KNN, ANN). Then we have employed an ensemble of different classifiers (SVM, KNN and ANN) with majority voting as consensus function. Accordingly, the results indicated in Fig. 3 represent the average accuracies of MPME-FS given by SVM, KNN, ANN and the ensemble of classifiers described above. For comparison reasons, we have used in this experiment both Information Gain and ReliefF filters to perform selection by the proposed approach. Form this figure we observe that the MPME-FS performs better using information Gain filter than ReliefF filter in the five datasets.

Average 10 cross-validation classification accuracy using: SVM, KNN, ANN and an ensemble of different classifiers over the five datasets
A second observation which can be made is that the ensemble of classifier gives higher accuracy than single classifiers in almost cases that are not surprising since it combines the efforts of the three classifiers. Furthermore, Fig. 4 shows boxplots of MPME-FS using SVM classifier over thirty runs for both Information Gain and ReliefF (Fig. 4a, b successively). As desired, the performance variance between runs reaches an almost completely stable result through the two filters (among 0.001 and 0.04).

Boxplots of MPME-FS method on colon, leukemia, DLBCL, SRBCT and ovarian datasets across 30 runs. a Informain gain filter and b ReliefF filter
We also provide in Table 3 the average performance of MPME-FS using the different classifiers of both information Gain and ReliefF filters. In the last column the number of selected biomarkers on the five datasets is shown. We observe that both sensitivity and specificity of our selection are convergent among the different classifiers. The previous experiments were performed on 30 independent runs to have statistically meaningful conclusions as our approach is stochastic.
4.3 Robustness Analysis
We explore and discuss in the present study the robustness of the selected signature by the MPME-FS approach. Therefore, we assess the similarity between the outputs of different independent executions of our method. The global stability is defined as the average over all pairwise similarity comparisons between the different feature selectors as follows [12]:
where \( {\text{f}}_{i} \) represents the outcome of the feature selection method applied to subsamplei (1 ≤ i ≤ 20), and \( {\text{S}}({\text{f}}_{{\begin{array}{*{20}c} i \\ \\ \\ \end{array} }} , {\text{f}}_{\text{j}} ) \) represents a similarity measure between \( {\text{f}}_{i} \) and \( {\text{f}}_{\text{j}} \). Mainly, for feature subsets selection (as in our case), we use the Jaccard index (JI) which can be calculated as follows:
A set of experiment assess the overall stability of the selected signature on the five datasets using both Information Gain and ReliefF filters which is shown in the Fig. 5a. Results in term of Jaccard index show that the MPME-FS performed using information gain is generally more robust over the most datasets. To provide a better robustness analysis, we assess the effect of data perturbation rate when creating subsamples on the stability of the signature. In this experiment we use Information Gain as filter over the five datasets of which the results can be seen in Fig. 5b, which indicates that the robustness decreases as the perturbation rate is decreased.

Average robustness results of the MPME-FS in term of Jaccard index over 20 independent runs on the five datasets. a Information Gain filter versus ReliefF filter b Jaccard index versus perturbation rate of subsampling between the independent runs (80, 90 and 100 %)
4.4 Biological Interpretation of the Results
In this section, we address biological analysis of the selected biomarkers. We focus in this experiment on the analysis of the selected biomarkers from Colon and Leukemia datasets which are widely studied in the literature. Accordingly, Tables 4 and 5 list and describe the top thirty ranked genes over 30 independent runs which have a complete frequency level (freq = 30) from Colon and Leukemia datasets successively. Furthermore, the selected genes are considered informative in most well-known methods in the literature. Specially, in Leukemia dataset which has been widely studied in this area.
As a result, genes selected from Leukemia dataset listed in boldface in Table 5, were also selected among the top 25 most relevant genes by Wu et al. [4] and they are considered useful to discriminate between the two class label AML and ALL. Moreover, eight out of these thirty top genes selected by our method, i.e., M23197_at, M27891_at, U05259_rna1_at, U46499_at, X95735_at, L09209_s_at, M31523_at and M89957_at were deemed as relevant by Zhu et al. [21]. The selected genes can now be validated by biologists through clinical trials. We expect these discoveries may offer useful information for biologists and medical experts.
5 Conclusion
In summary, we considered in this chapter the application of ensemble feature selection methods to biomarker identification. Indeed, the most reviewed ensemble feature selection methods attest that this technique is a promising direction for more stable and accurate selection in cancer gene identification. We have also proposed a massively parallel approach based on meta-ensemble of filters for biomarker discovery from high dimensional data. The MPME-FS is different from other ensemble feature selection methods since it performs a parallel selection in two steps: the first one within each ensemble by the aggregation of results of different selectors, the second step is the aggregation of the outcomes of all ensembles using a second consensus function. The final selected biomarkers are employed to construct a classification model that will be used as an effective tool to handle patients and diagnose cancer subclasses.
In addition, the proposed MPME-FS is very fast and is computationally efficient as it is massively parallel and no learning algorithm is used in the selection process. Instead, we have employed filter model which is usually exploited when the number of features becomes very large especially for high dimensional data. Clearly, the MPME-FS can be performed using any ranking based feature selection algorithm and applied to any feature selection problem.
The experiments over five DNA microarrays datasets revealed that good results can be achieved through MPME-FS in terms of classification performance and robustness. Biological analysis of the results shows that MPME-FS provides the selection of highly informative genes which have biological meanings and are also selected by the other approaches.
References
Zhang, X., et al.: Integrative omics technologies in cancer biomarker discovery. Omics Technol. Cancer Biomark. Discov. 129 (2011)
Nair, M., Sandhu, S.S., Sharma, A.K.: Prognostic and predictive biomarkers in cancer. Curr. Cancer Drug Targets (2014)
Mäbert, K., Cojoc, M., Peitzsch, C., Kurth, I., Souchelnytskyi, S., Dubrovska, A.: Cancer biomarker discovery: current status and future perspectives. Int. J. Radiat. Biol. (0), 1–48 (2014)
Wu, M.Y., Dai, D.Q., Shi, Y., Yan, H., Zhang, X.F.: Biomarker identification and cancer classification based on microarray data using laplace naive bayes model with mean shrinkage. IEEE/ACM Trans. Comput. Biol. Bioinf. (TCBB) 9(6), 1649–1662 (2012)
Bolón-Canedo, V., Sánchez-Maroño, N., Alonso-Betanzos, A.: A review of feature selection methods on synthetic data. Knowl. Inf. Syst. 34(3), 483–519 (2013)
Bolón-Canedo, V., Sánchez-Maroño, N., et al.: A review of microarray datasets and applied feature selection methods. Inf. Sci. 282, 111–135 (2014)
He, Z., Yu, W.: Stable feature selection for biomarker discovery. Comput. Biol. Chem. 34(4), 215–225 (2010)
Guan, D., Yuan, W., Lee, Y.K., Najeebullah, K., Rasel, M.K.: A review of ensemble learning based feature selection. IETE Tech. Rev. 31(3), 190–198 (2014)
Upadhyaya, S.R.: Parallel approaches to machine learning—a comprehensive survey. J. Parallel Distrib. Comput. 73(3), 284–292 (2013)
Yang, P., Hwa Yang, Y., B Zhou, B., Y Zomaya, A.: A review of ensemble methods in bioinformatics. Curr. Bioinf. 5(4), 296–308 (2010)
Awada, W., Khoshgoftaar, T.M., et al.: A review of the stability of feature selection techniques for bioinformatics data. In: Information Reuse and Integration (IRI), 13th International Conference, 356–363 (2012)
Saeys, Y., Abeel, T., Van de Peer, Y.: Robust feature selection using ensemble feature selection techniques. In: Machine Learning and Knowledge Discovery in Databases, pp. 313–325. Springer, Berlin (2008)
Abeel, T., Helleputte, T., et al.: Robust biomarker identification for cancer diagnosis with ensemble feature selection methods. Bioinformatics 26(3), 392–398 (2010)
Bolón-Canedo, V., Sánchez-Maroño, N., Alonso-Betanzos, A.: Data classification using an ensemble of filters. Neurocomputing 135, 13–20 (2014)
Yang, P., Liu, W., Zhou, B. B., Chawla, S., Zomaya, A.Y.: Ensemble-based wrapper methods for feature selection and class imbalance learning. In: Advances in Knowledge Discovery and Data Mining, pp. 544–555. Springer, Berlin (2013)
Xu, J., Sun, L., Gao, Y., Xu, T.: An ensemble feature selection technique for cancer recognition. Bio-Med. Mater. Eng. 24(1), 1001–1008 (2014)
Ghorai, S., et al.: Cancer classification from gene expression data by NPPC ensemble. IEEE/ACM Trans. Comput. Biol. Bioinf. 8(3), 659–671 (2011)
Boucheham, A., Batouche, M.: Robust biomarker discovery for cancer diagnosis based on meta-ensemble feature selection. In: The Proceedings of Science and Information Conference, IEEE, pp. 452–460 (2014). ISBN: 978-0-9893193-1-7
Boulesteix, A.L., Slawski, M.: Stability and aggregation of ranked gene lists. Briefings Bioinf. 10(5), 556–568 (2009)
Haury, A.C., Gestraud, P., Vert, J.P.: The influence of feature selection methods on accuracy, stability and interpretability of molecular signatures. PLoS ONE 6(12), e28210 (2011)
Zhu, Z., Ong, Y.S., et al.: Identification of full and partial class relevant genes. Comput. Biol. Bioinf. IEEE/ACM Trans. 7(2), 263–277 (2010)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this paper
Cite this paper
Boucheham, A., Batouche, M. (2015). Massively Parallel Feature Selection Based on Ensemble of Filters and Multiple Robust Consensus Functions for Cancer Gene Identification. In: Arai, K., Kapoor, S., Bhatia, R. (eds) Intelligent Systems in Science and Information 2014. SAI 2014. Studies in Computational Intelligence, vol 591. Springer, Cham. https://doi.org/10.1007/978-3-319-14654-6_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-14654-6_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-14653-9
Online ISBN: 978-3-319-14654-6
eBook Packages: EngineeringEngineering (R0)