
Abstract
Geochemical processes involve reactions which mediate and control the dissolution/release and (re)formation/sorption of mineral elements at geological as well as soil levels. The role of trace elements, copper among others, in soil chemical changes is still a matter of detailed studies. Primary copper-bearing minerals and their secondary weathering stage compounds should potentially release Cu via CO2 injection into deep geological structures or mining activities. The latter ones initiate slow or intensive spread of Cu compounds into the environment.
Multiple-phase Cu evaluation was additionally detailed in a case study of natural (uncontaminated) and anthropogenic (contaminated) soil Cu concentrations. Langmuir-based sorption density (SD) models and surface charge density (SCD) were also used in the geochemical Cu processes assessment. The simplified Elovich model showed a good data fit, which is suggestive of bulk and intraparticle diffusion processes. Copper in contaminated and uncontaminated soils was characterised by a negative hysteresis indicative of relatively high copper desorbability. This process should be strengthened under acidic conditions and high copper contamination. Energies of activation for the adsorption (E aa) process were generally lower for uncontaminated as compared to contaminated soils. Copper required low energies to desorb, and the activation energy for desorption (E ad) values did not exceed 15 kJ mol−1. Such low desorption energies indicate that copper is disposed to enhance mobility. Geochemical aspects were substantially outlined.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Cation Exchange Capacity
- Surface Charge Density
- Flame Atomic Absorption Spectrometry
- Copper Smelter
- Uncontaminated Soil
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Geological Aspects
1.1 Geogenesis
Considerations given to the Earth’s geochemical structure might be referred to one of the most widely recognised hypotheses on its creation, i.e., initially the Earth was a melted mass having been maintained in its semifluid state by bombing planetoids. The chemistry of the Earth at the early forming stage was similar to an average compound of meteorites with the predominance of magnesium and iron silicates, nickel-bearing iron in the metallic state and numerous other elements. Referring to the commonly agreed threshold of 0.01 %, elements such as O, Si, Al, Fe, Ca, Na, Mg, K, C are considered as major since their concentrations are higher than this value. In the case of Ni, Zn, Cu, Co, Pb, Sn, As and Mo, their level is below 0.01% mm−1 and they are designated as rare earth metals or trace elements. Copper levels in the rocks of the Earth’s crust are estimated to range from 5 to 100 mg kg−1, with the average of 55 mg kg−1 (Konstantynowicz 1979; Kabata-Pendias and Pendias 1999).
Copper as free metal is rarely encountered in the environment. Due to its chemical affinity for sulphur, copper is classified as the so-called chalcophylic elements and is mostly found in the form of sulphide minerals. In terms of usefulness, the greatest significance is attributed to chalcopyrite CuFeS2, chalcocite Cu2S and bornite Cu5FeS4. These minerals originate from magma processes whose quantitative predominance in nature is recognised, although the richest concentrations of copper are related to its secondary concentration at the weathering stage (Polański and Smulikowski 1969). Copper, being a weak basic oxide, forms also minerals of hydroxycarbonate nature with high utility significance such as azurite Cu3(CO3)2(OH)2 and malachite Cu2CO3(OH)2. In this case they are mainly found in sedimentary rocks as products of hydrothermal transformations.
1.2 Geological CO2 Sequestration Mediated Mineral Solubilisation
Processes of rock and minerals’ weathering in conditions of underground injection of CO2 to deep geological structures are considered to be new and of local character. In fact, these practices have occurred in different parts of our globe for over 40 years (Drobek et al. 2008). Carbon dioxide injection to deep geological structures including uncultivated saline aquifers initiates chemical processes in the pattern: CO2-rock, CO2-brine and brine (saturated CO2)-rock, resulting in chemical changes of both underground waters and surrounding rocks. The effect of the diphasic system of CO2-brine on geological milieu is based on bicarbonate balance according to the following reactions:
A targeted example of such process is the surface aquifer of copper deposits in the Legnicko-Glogowski Okręg Miedziowy (LGOM, Poland) with average amounts of chalcophylic elements Cu 0.08, Zn 0.50 and Pb 0.045 mg dm−3 (Wilk and Bocheńska 2003). For metals of similar concentrations in the places of carbon dioxide injection in underground waters, copper and other metals will precipitate in the form of virtually dissoluble or hardly soluble hydroxycarbonate salts which are subject to the process of secondary dissolving, due to hydrogeochemical balance (Drobek et al. 2008).
The effect of two-phase brine (saturated CO2)-rock system on geological environment is a subsequent mechanism of chemical processes occurring during CO2 deposition under the ground. Carbonate minerals will gradually dissolve causing an increase in permeability of the surrounding rocks; thus, in the environment of brine supersaturated with carbon dioxide, precipitation of metals including dissoluble and hardly soluble copper hydroxycarbonates will take place. In this case the bicarbonate balance in brine environment will be again a potential source of co-precipitation of the hydroxycarbonate form of copper, loamy minerals and silicate (Huijgen et al. 2006; Ibsen and Jacobsen 1996).
The reported cases of chemical processes accompanying CO2 injection to deep geological structures point out that such anthropogenic interference into the environment may generate changes in the forms of elements’ mineralogy, copper among others. Taking into account great dispersions of copper in the Earth’s crust, these processes are rather of local character and their magnitude should not generate significant changes in the environment at present.
2 Solid-Soil Solution Interface: Processes and Evaluation
Soil in its whole entity is one of the more complex naturally formed medium. The heterogeneity of its constituents, their mutual interactions and last but not the least the direct or indirect influence of external factors make this complexity quite unbelievable to define. In all natural heterogeneous soil environments, the following generalised mechanisms and phenomena occur simultaneously: (a) dissolution, (b) adsorption/desorption, (c) complexation, (d) migration, (e) precipitation, (f) occlusion, (g) diffusion, (h) absorption by microbiota and (i) volatilisation. The developments in soil chemistry started with the pioneering studies of Thomas Way on Base Exchange published in 1850 as On the Power of Soils to Absorb Manure (Forrester and Giles 1971). Detailed informations on geochemical reactions are reported by Gapon (1933), Gaines and Thomas (1953), Sokołowska (1989), Sposito (1984), Sparks (1989), Mulder and Cresser (1994) and Barrow (1999).
Soil chemistry has traditionally focused on the chemical reactions in soils that affect plant growth and plant nutrition. However, as concerns increased about inorganic and organic contaminants in water and soil, the emphasis of soil chemistry is now on environmental soil chemistry. This holds to the definition suggested by McBride (1994) and Sparks (1995) as the study of chemical reactions between soils and environmentally important plant nutrients, radionuclides, heavy metals and organic chemicals.
Environmental concern in the 1970s led to the adaptation of simulative/predictive models for metals solid-phase/aqueous-phase interactions and their probable behaviour in natural environments. Transport of heavy metals and copper among others through soil layers has long presented great interest to both environmental and soils scientists (Murali and Aylmore 1983a, b; Ogwada and Sparks 1986; Diatta et al. 2000). The complexity of the soil matrix makes it difficult to selectively choose interactions, which mostly contribute to the adsorption of a specific metal (Diatta et al. 2012). This problem creates more difficulty in the process of formulating meaningful soil models for the prediction of metal transport.
Most of soil chemical reactions are time dependent and may differ notably in rates. The frequently concerned mechanisms of ion movement through soil layer are diffusion-based processes, the rates of which are closely related to the homogeneity and heterogeneity of the systems (Lai and Mortland 1968; Sparks 2000). The following illustration (Weber 1984) shows rate-determining steps of soil chemical processes (Fig. 3.1):

Rate-determining steps in soil chemical reactions (Weber 1984 with permission from ASCE)
There are three basic ways to determine rate constants (Skopp 1986; Sparks 1989, 1995). The first is the use of initial rates by plotting the concentration of a reactant or product over a short reaction time period during which the concentrations of the reactants change so little that the instantaneous rate is hardly affected. The second way is by directly applying integrated equations and graphing the data, and the last one is by using the nonlinear least square analysis.
In recent years, research on metal (copper among others) adsorption/desorption processes has been based mainly on the study of equilibrium conditions. The kinetic approach of these processes in soil systems is also reported (Bunzl et al. 1976; Elkhatib et al. 1992; Violante et al. 2008). The kinetics of metal adsorption (copper among others) at its first phase is found to be basically influenced by sorbent amounts, types and pH as well (Basta and Tabatabai 1992). The impact of organic matter on the kinetics of copper sorption by peat humic substances was extensively reported by Sapek (1976). The author reported that the time course of these processes is partly attributed to the occurrence of specific high- and low-energy adsorbing sites at the surface of soil organic colloids. It is of common view that the pool of high-energy sites is more developed than that expected for the low energy one. This acts as a limiting factor during adsorption process. At a longer time, more and more other low-energy sites become successively saturated (Tiller et al. 1984). On the basis of kinetic studies, it was formulated that kinetic adsorption is characterised by a two-phase process, a rapid exchange followed by a slower reaction of copper ions with organic sorbents (Sapek and Zebrowski 1976; Boehringer 1980; Aringhieri et al. 1985).
The reaction rate theory assumes that colliding molecules (e.g. reactions between solution ions and ions adsorbed in the exchange complex) must be in a high-energy state before a reaction can occur. This energy of activation is of van der Waals repulsive forces that take place as two ions approach each other (Sparks 1989). Then the energy of activation may be considered as a measure of the magnitude of the forces to be overcome during the process of adsorption/desorption. Thermodynamic approach can predict only the final state of a soil system from an initial nonequilibrium state and has been used to characterise exchange equilibria on clay and soil particle surfaces (El-Sayed et al. 1970). An alternative that should be examined is the use of kinetics to follow the course of adsorption/desorption reactions. This technique has the potential of providing both the mechanistic and thermodynamic informations. However, application of kinetic theory to determine thermodynamic parameters (e.g. for phosphorus and potassium) has received more attention (Evans and Jurinak 1976; Pavlatou and Polyzopoulos 1988; Ogwada and Sparks 1986; Liu and Huang 2000; Zhang et al. 2000) than for heavy metals (El-Sayed et al. 1970; Elkhatib et al. 1993).
A number of models have been elaborated for describing adsorption/desorption reactions of various elements in soils (Gapon 1933; Stawiński 1977; Mattigod and Sposito 1979; Kinniburgh 1986; Amacher et al. 1986; Sokołowska 1989; Dube et al. 2001; Diatta et al. 2003). The most well-known sorption models may be classified into two groups: (1) models which take account of electrostatic forces and bonding and (2) models which do not include these parameters. Models of the first group have some bug: they are complicated—it means that they require many parameters at the beginning of modelling—or they are simply physical equalisations of transport inside the soil profile without consideration of interaction between contaminants and soil matrix. To the second group belong the classical models, i.e. Freundlich and Langmuir isotherms with their suggestive extensions (Veith and Sposito 1977; Sposito 1982, 1984; Sparks 1995; Sarbak 2000). These isotherms are generally based on the amount of adsorbed element (quantity) plotted against its equilibrium concentration (intensity) and the resulting slopes and intercepts to be indicative of various soil sorption/desorption parameters (Fig. 3.2).

Schematic representation of various sorption and buffering parameters that could be characterised during equilibration studies. S = amount retained (i.e. adsorbed) or released (desorbed). Q = quantity, Q o = intercept on the Q axis of the TBCe tangent, C e = equilibrium concentration, I = intensity, I o = no retention (i.e. adsorption) and no release (desorption), MBC = maximum buffering capacity (C e → 0), TBCe = tangential buffering capacity, ABC = average buffering capacity
3 Copper Status in Contaminated and Uncontaminated Soils
Dynamic development of civilisation during the past century led to excessive accumulation of heavy metals, among others copper, in the environment, creating serious ecological problems. Gradual increase of soil contamination with copper and its relative mobility in the environment threatens with the risk of this metal introduction into trophic cycle (Kabata-Pendias 1978; Markert and Friese 2000; Markert et al. 2003; Nriagu et al. 2012). Several phosphate fertilisers and pests’ products contain copper either as impurity or as active constituent (Alloway 1995; Robert 1996), but one of the major copper pollution sources is attributed to mining activity and dust emissions from metallurgical industries (Geiger et al 1993; Kabata-Pendias A. 1993; Grzebisz and Chudziński 1996; Diatta et al. 2000).
Heavy metals (copper among others) are ubiquitous and fundamental components of life on Earth and part of all food chain. Because their levels in soils naturally range between 0.1 and 100.0 mg kg−1 (Cox 1989; Baker 1990; Kabata-Pendias and Pendias 1992), they are also called “trace elements” (Adriano 1986; Duffus 2002). Some metals such as copper and zinc are essential to life, but in high concentrations, however, the same metals can exhibit toxicity effects. The natural concentration of copper in soil solution is found to be very low, being in the range of 1 × 10−8 to 60 × 10−8 moles dm−3. Hodgson et al. (1966) reported that more than 98 % of soil solution copper was complexed with organic matter and more strongly bound to organic matter than are other micronutrient cations like Zn2+ and Mn2+. The affinity to organic matter regulates Cu mobility and availability in the soil. In a fractionation study of Cu in soils, McLaren and Crawford (1973a, b) have found that a high proportion of total copper was in occluded or lattice form. Irrespective of the forms in which copper may occur, its behaviour and shift from natural copper nontoxicity to anthropogenic copper toxicity are intrinsically related to metal specificity and man activities (Adriano 1986; Kabata-Pendias 1993; Chudziński 1995; Prasad 2008).
The behaviour of heavy metals and copper among others is governed mainly by two groups of adversely acting phenomena. The first group involves processes such as: sorption of copper by soil mineral and organic sorptive complex, precipitation of insoluble compounds and biogenic accumulation, which aim at reducing copper solubility and mobility (Adriano 1986; Brümmer et al. 1986; Logan 1990). The second group consists of processes that tend to increase copper mobility, i.e. desorption, solubility and mineralisation of organic compounds (Ram and Verloo 1985). Copper solubility and desorption in soil systems are generally strengthened by a decrease of pH (Lindsay 1979) which increases its activity manifold than for soil with neutral or weakly acid soils (Xian and Shokohifard 1989; Msaky and Calvet 1990; McBride 2001).
Copper retention by the organo-mineral soil complex has been a topic of several reports (McLaren and Crawford 1973a, b; Cavallaro and McBride 1978; Sapek and Sapek 1980; Logan et al. 1997; Diatta et al. 2000). Several works dealt with copper organic complexes of soil systems in which the organic part is quite dominating. Clay minerals are also potential binding agents for copper as they exhibit large specific surface area and enhanced cation exchange capacity. In general clay minerals are found to possess both a pH-dependent and permanent negative surface charge, which is among others a resultant of isomorphous substitution. The permanent charge is for the most part compensated by cations intercalated in the interstitial space (McBride 1994; Sparks 1995). Therefore, the metal cations bound in such a way can readily be exchanged by other cations, e.g. Ca2+, Mg2+ and NH4 + (Van Bladel et al. 1993).
The mechanisms of copper adsorption are pH dependent and may additionally depend on the concentrations of copper in the soil solution. At relatively high pH and low copper concentrations, specific adsorption is often the predominant binding mechanism (McBride 1989; Diatta et al. 2004, 2012). Surface complex is thought to occur via interaction of a surface functional group with an ion or molecule present in the soil solution, which creates a stable molecular entity. If water molecule is present between the surface functional group and the bound ion or molecule, the surface complex is termed outer sphere (Sposito 1984). If there is not a water molecule present between the ion or molecule and the surface functional group to which it is bound, this is an inner sphere complex. It is important to note that inner sphere complexes can be monodentate (metal is bound to only one oxygen) and bidentate (metal is bound to two oxygens) as illustrated below (Fig. 3.3).

Illustration of surface complexes’ formation between inorganic ions and hydroxyl groups of an oxide surface (with permission from Elsevier for Sparks 1995: Environmental Soil Chemistry, p. 99–139)
The prediction of the mobility of metals in soils may also be assessed by simulative natural diffusion studies (Helios-Rybicka and Jędrzejczyk 1995) or desorption. It is often observed that desorption is a more difficult process than adsorption and that the adsorbate is not entirely desorbed. So the reactions (adsorption process) appear to be irreversible. Ion exchange is in essence a reversible reaction in which cations are stoichiometrically exchanged between the electrolyte solution and the surface phase of the exchanger material. Exchange irreversibility is sometimes observed and has been attributed to colloidal aggregation and the formation of quasi-crystals (Van Bladel and Laudelout 1967), which could make exchange sites inaccessible. Such apparent irreversibility is commonly referred to as hysteresis that means adsorption and desorption isotherms corresponding to the forward and backward reaction would not coincide (Verburg and Baveye 1994). Hysteresis is often reported for soil-pesticide interactions (Barriuso et al. 1994; Zhu and Selim 2000) and also for metal reactions with soils or soil components (Ainstworth et al. 1994).
Informations about hysteretic behaviour of copper in soils are scarce. Schultz et al. (1987) reported about a substantial hysteresis in the desorption of Cu, Ni, Pb and Zn and Cr sorbed to hydrous ferric oxide. They found that the magnitude of the hysteresis was dependent on pH at which the adsorption process took place and the length of time of contact of sorbent-solute prior to initiation of the desorption process. However, Maqueda et al. (1998) have found that copper desorption isotherms on a montmorillonite showed a very high hysteresis, and in the same order a substantial sorption/desorption hysteresis was also observed by Wu et al. (1999), who reported that Cu has formed high-energy bonds with organic matter and layer silicate surfaces.
4 Site-Specific Copper Geochemistry: A Case Study
4.1 Site Characterisation and Soil Sampling Procedure
The study area is located in the Głogów proglacial stream valley (N 51°41′03″ and E 15°57′12″) situated between Sudetes (North) and the Małopolska Plateau (South) (Kondracki 1978). Soil samplings were performed within the years 2000–2001. The investigated area is represented by two major soil groups: Fluvisols and Luvisols (WRB 1998), as reported in Table 3.1. The Głogów copper smelter (GCS) was established in the 1970s, and the primary cause of high metal accumulation was the emission of dusts and gases by smelters. In the 1970s the Głogów copper smelter annually discharged into the atmosphere over 15,000 tons of dusts containing heavy metals, sulfuric acid and sulphur dioxide, and from early 1990s a rapid reduction of dust emissions occurred. Copper content of dust discharges amounted to 2,968, 204, 23, 9.7 tons for the years 1980, 1990, 2000 and 2012, respectively (www.kghm.pl/index. Access 20.07.2014).
These tremendous amounts of copper accumulated progressively in soils for many years, and their negative environmental impact is still lasting. The zone mostly contaminated by heavy metals (copper mainly) was taken out of agricultural utilisation and afforested (poplars and maples), establishing since 1987 the “sanitary belt”. Earlier investigations carried out in this area showed a copper distribution pattern consistent with the wind rose, where southwest (SW) and west (W) winds—referring to Głogów copper smelter position—were dominating (Kabata-Pendias 1978; Grzebisz et al. 1997; Geochemical Atlas 1999) (Fig. 3.4).

Spatial location of sampling sites (numbers in circles) within the investigated area
It should be mentioned in the past all soils of the area under study were arable, some have currently changed management purposes. Since most of sampling sites were located in arable soils, the sampling depth of 0–20 cm was applied due to agricultural practices. The same sampling depth was considered also for sampling sites other than those under agricultural practices.
4.2 Physical and Chemical Soil Tests
Prior to basic analyses, soil samples were air-dried and crushed to pass through a 1 mm mesh sieve. Total copper (CuTot) content was determined in soil samples (International Standard 1995). On the basis of total copper content (CuTot), it was decided to elaborate a “consensual” share of soils into two operational groups: Group I represents the moderate and highly contaminated soils (from the sanitary belt) with total copper content varying from 76.1 to 334.0 mg kg−1, except one soil sample with CuTot of 1,048 mg kg−1. To the group I belong soil Nos. 1–8 considered as contaminated. The group II consists of soils collected beyond the “Sanitary Belt”, where CuTot was below 30.0 mg kg−1. The mentioned group involved soil Nos. 9–16 and is designated as uncontaminated (Table 3.2). Copper from soils was additionally extracted by using DTPA solution (diethylenetriaminepentaacetic acid) (Lindsay and Norvell 1978; Kociałkowski et al. 1999). Copper in filtrates was determined by the FAAS method (Flame Atomic Absorption Spectrometry, Varian SpectrAA 250 plus).
Particle-size distribution was determined according to the method of Bouyoucos-Casagrande (Ryan et al. 2001). The texture classes were established according to USDA classification (Soil Taxonomy 1975) on the basis of the following separates: sand (1.0–0.05 mm), silt (0.05–0.002 mm) and clay (<0.002 mm). Organic carbon was determined by the dichromate wet oxidative method (Nelson and Sommers 1996) and soil pH potentiometrically, according to Polish Standard (1994). Soil samples were additionally analysed for carbonate content (Loeppert and Suarez 1996), whereas the cation exchange capacity (CEC) was assessed by the barium chloride method as suggested by Hendershot and Duquette (1986). The cation exchange capacity of the soils was obtained by summation of exchangeable alkaline cations and acidity according to Thomas (1982). The specific surface area (SSA) of the soils was determined according to the ethylene glycol monoethyl ether (EGME) procedure, whereas the external surface area was measured by the BET method (Gregg and Sing 1967; Carter et al. 1986; de Jong 1999).
4.3 Dynamic Phase Evaluation of Copper in Soils
4.3.1 Batch Tests
Copper concentrations of 0.2, 0.4, 0.6, 0.8, 1.2, 1.6, 2.0, 2.4, 2.8 and 3.2 mmolc dm−3 designated as initial solutions (C i ) were prepared by dissolving appropriate amounts of Cu(NO3)2·3H2O p.a. in 0.010 mole Ca(NO3)2·4H2O (p.a.) as a background electrolyte. The use of 0.010 mole Ca(NO3)2·4H2O was an attempt to approximate the electrolyte environment of soils in the field (Jopony and Young 1994). Nitrate was chosen because of its poor ability to complex metallic cations (Sillén and Martell 1971). Solutions were added to homogenised soils at soil/solution ratios (w/v) of 1:25 into a series of polyethylene centrifuge tubes and shaken in a rotative shaker for 2 h. Suspensions were allowed to equilibrate for 22 h after which filtration followed. The concentrations of Cu of the filtrates at equilibrium concentration (C e ) were determined by FAAS method (Flame Atomic Absorption Spectrometry, Varian SpectrAA 250 plus). The solid-phase Cu was calculated as the difference between the initial Cu concentration (C i ) and that remaining in the solution after equilibration (i.e. C e ) accordingly:
where
-
S—amount of Cu adsorbed (mmolc kg−1)
-
C i —initial Cu concentration in solution before reaction (mmolc dm−3)
-
C e —equilibrium Cu concentration in solution after reaction (mmolc dm−3)
-
V—volume of the solution (dm3)
-
W—weight of soil sample (kg)
Langmuir and Freundlich adsorption constants were computed by using Sorption Softwares (IZOTERMY© 1993), especially elaborated for sorption studies.
4.3.2 Kinetics of Copper Adsorption and Desorption
Studies on the kinetics of adsorption were carried out using soil samples described earlier. Prior to these studies, soil samples were separated into two sets on the basis of copper equilibrium concentrations (C e ) at the highest initial copper concentration (C i ) as indicated in Table 3.3.
Appropriate triplicate 1.00 g of soil samples were placed in 100 cm3 polyethylene centrifuge tubes and equilibrated with 50 cm3 of 0.8, 1.6, 2.4 and 3.2 mmolc dm−3 as solution of Cu(NO3)2·3H2O in 0.010 mole Ca(NO3)2·4H2O used as a background electrolyte to reduce variability and simulate natural soil conditions (Table 3.4). The suspensions were vigorously shaken for 30 s at the beginning of the kinetic experiment to ensure a uniform mixture and allowed to settle for 96 h (whole kinetic adsorption time) at temperature conditions of 21.0 °C (294 K). Two (2) cm3 aliquots for Cu determination (FAAS method) were collected at the following time intervals: 0.5, 2, 8, 24, 48, 72 and 96 h. Amounts of Cu adsorbed during the adsorption kinetic phases were calculated by using Eq. (3.3).
The kinetics of desorption studies started immediately at the end of the kinetics of adsorption and were conducted on all samples used above. The supernatants at 96 h (end of kinetic adsorption study) were centrifuged at 4,000 rpm for 5 min, 5 cm3 aliquots were collected, and the remaining decant was discarded. The polyethylene centrifuge tubes with soil samples were weighted and 50 cm3 of 0.010 mole Ca(NO3)2·4H2O Cu-free solution was placed after which the suspensions were shaken for 30 min. After this time the suspensions were centrifuged and 5 cm3 aliquots were collected for Cu analysis. This step was repeated six more times using 0.010 mole Ca(NO3)2·4H2O Cu-free solution. Amounts of copper desorbed at each desorption stage were calculated on the basis of the amounts of copper initially adsorbed (at t = 96 h).
4.3.3 Langmuir and Freundlich Models (Isotherms)
Solute adsorption mechanisms at solid surfaces are observed to bear several shapes basically divided into four main classes, namely, S, L, H and C, as widely outlined by Giles et al. (1960). With an S-type isotherm, the slope initially increases with adsorbate concentration, but eventually decreases and becomes zero as vacant adsorbent sites are occupied. The L-type (Langmuir) isotherm is characterised by a decreasing slope as concentration increases since vacant adsorption sites decrease as the adsorbent surface becomes covered. The H-type (high affinity) isotherm is indicative of strong adsorbate-adsorbent interactions such as inner sphere complexes. The C-type isotherms are indicative of a partitioning mechanism whereby adsorbate ions or molecules are linearly distributed or partitioned between the solid phase (adsorbent) and the solution phase without any specific bonding between the adsorbent and adsorbate.
Of these classes, the L-type isotherm is described mathematically by the Langmuir equation (Sposito 1984), as follows:
where
-
S—amount of Cu adsorbed (mmolc kg−1)
-
C e —equilibrium Cu concentration in solution after reaction (mmolc dm−3)
-
a max—adsorption maximum (mmolc kg−1)
-
b—bonding term relative to interaction energies (dm3 mmolc −1)
In the linear form, Eq. (3.4) may be rewritten as follows:
where
a max · b expresses the maximal buffering capacity (MBC, dm3 kg−1) exhibited by an adsorbent (soil) for a given adsorbate (Cu) when the equilibrium concentration is very low (tends to 0). The slope is equal to \( \frac{1}{a_{\max }} \) and the intercept (when C e tends closely to 0) is expressed by \( \frac{1}{a_{\max}\cdot b}. \) The value of the Langmuir term related to the interaction energies (b) may be calculated from the ratio of the slope and the intercept and is equal to \( \frac{\mathrm{slope}}{\mathrm{intercept}} \) Diatta (2002). It is noteworthy that the calculation of the maximum sorption capacity a max can involve errors of 50 % and more (Harter 1984) if the isotherm does not have correct Langmuir shape and only low concentration data (Schulthess and Dey 1996) is used for the calculation.
The Freundlich isotherm as an empirical equation is given by
where
-
K F —Freundlich partition constant (dm3 kg−1)
-
C e —equilibrium Cu concentration in solution after reaction (mmolc dm−3)
-
m—sorption constant (shape intensity factor, dimensionless) expressing the slope of the adsorption isotherm. This positive-valued empirical constant is reported to lie between 0 and 1 (Elzinger et al. 1999; Diatta et al. 2003). By taking the log of Eq. (3.6), the linear form of the Freundlich equation is obtained as
4.3.4 Sorption and Surface Charge Densities
Adsorption and desorption processes predominantly occur at surfaces of organic and inorganic colloids. Therefore, the expression of these processes in terms of “concentration” of substances (e.g. ions) per unit area may give more practical approach. Copper sorption densities at any point were calculated as reported by Schulte and Beese (1994) and Zehetner and Wenzel (2000):
where
-
SDSSA—sorption density based on the specific surface area (ions m−2)
-
S—amount of Cu adsorbed (mmolc kg−1)
-
N A —Avogadro’s number (6.023 × 1023 ions mol−1)
-
SSA—specific surface area (m2 g−1)
Sorption processes, especially non-specific adsorption through ion exchange, are influenced by the surface charge, of which CEC is a measure. The surface charge density (SCD) as pointed out by Poonia and Talibudeen (1977) and Laird et al. (1992) is expressed as follows:
where
-
SCD—surface charge density (mmolc m−2)
-
CEC—cation exchange capacity (cmolc kg−1)
By dividing SDSSA (Eq. 3.8) by the surface charge density (SCD), we obtain the charge-based sorption density (SDCEC):
where
SDCEC—charge-based sorption density (ions molc −1)
4.3.5 Development of the Simplified Elovich Equation
The simplest form of Elovich equation (Allen and Scaife 1966) is generally expressed as
where S is the amount of Cu adsorbed at time t and α and β are constants during one experiment. The constant α can be regarded as initial rate since \( \frac{dS}{dt}\to \alpha \) as \( S\to 0 \), i.e. a rapid adsorption not governed by the exponential law (Low 1960). The integrated form of the Eq. (3.11) gives
in which t o is the integration constant for which sometimes the symbol k is given (to avoid confusion with the time variable). Assuming t o ≈ 0 indicates that no other processes besides Elovichian ones are occurring, that is, the boundary condition S = 0 at t = 0 applies (Allen and Scaife 1966; Sparks 1989). In this case the assumption t o ≈ 0 leads directly to the simplified equation:
Chien and Clayton (1980) reached the simplified Eq. (3.13) by obtaining the form:
from Eq. (3.12), by first assuming that the boundary condition S = 0 at t = 0 applies and then by making the assumption that \( \alpha\;\beta\;t\gg 1 \).
However, as pointed out by Allen and Scaife (1966), t o can never be exactly zero without loss of the physical significance of Eq. (3.11). If this approximation is used, important information may be lost. Equation (3.13) was used to test the applicability of the simplified Elovich equation for the kinetics of Cu adsorption and desorption of the soils. Thus, a plot of S versus ln t should give a linear relationship with the slope of \( \frac{1}{\beta } \) and intercept of \( \frac{1}{\beta}\; \ln \left(\alpha \beta \right). \)
4.3.6 Kinetics of Copper Adsorption and Desorption and Hysteresis Approach
It was assumed that copper adsorption during the kinetic course should be monitored at some selected time intervals by sampling given equilibrium solution volumes for Cu determination. Therefore, it should be necessary to correct for this volume loss in the calculation of the amount of metal adsorbed at each subsequent sampling time (Amacher et al. 1986) as shown below:
and
where
-
ΔS kin—change in amount of Cu adsorbed by the soil from one sampling time to the next (mmolc kg−1)
-
S kin—amount of Cu adsorbed by the soil at each sampling time (mmolc kg−1)
-
C e prev—concentration of Cu in solution at previous sampling time (mmolc dm−3)
-
C e pres—concentration of Cu in solution at present sampling time (mmolc dm−3)
-
V—initial total solution volume (dm3)
-
v—volume of solution removed at each sampling time for Cu determination (dm3)
-
n—number of times sampled
-
S prev—amount of Cu adsorbed by the soil at previous sampling time (mmolc kg−1)
Copper partition parameters during the kinetics of the adsorption phase were calculated at each sampling time on the basis of Freundlich equation:
where
-
K kin-ads—Freundlich partition parameter for copper kinetics of adsorption (dm3 kg−1)
-
N kin-ads—empirical kinetics of adsorption parameter (dimensionless)
It was assumed that the starting point of the desorption isotherm is a point on the adsorption isotherm and that Freundlich adsorption and desorption coefficients are not independent as outlined by O’Connor et al. (1980) and Laird et al. (1992). Then:
where
-
S kin-max—amount of Cu adsorbed at the end of kinetic adsorption process (i.e. t = 96 h) (mmolc kg−1)
-
C e-max—equilibrium copper concentration at the end of kinetic adsorption process (i.e. t = 96 h) (mmolc dm−3)
By solving Eq. (3.18) for K kin-des, it comes
where
-
K kin-des—Freundlich partition constant for copper kinetics of desorption (dm3 kg−1)
-
N kin-des—empirical kinetics of desorption constant (dimensionless)
By taking into account the amount of copper adsorbed, Eq. (3.19) bears the form
The ratio of Freundlich exponents (N kin-des/N kin-ads) gives an indication of adsorption/desorption hysteresis (O’Connor et al. 1980).
4.3.7 Activation Energies: Processes Evaluation
All adsorption and desorption kinetic studies were run in triplicate under isothermal conditions at 294 K. Activation energies were calculated based on the simplified Elovich equation using the following relationship as reported by Pavlatou and Polyzopoulos (1988) and Zhang et al. (2000):
where
-
E aa/ad—activation energy for kinetics of Cu adsorption/desorption processes (J mol−1)
-
S kin—amount of Cu adsorbed or desorbed during the kinetic study (mmolc kg−1)
-
R—gas constant (8.314 J mol−1 K−1)
-
T—absolute temperature (273 K)
-
E slope—slope of the simplified Elovich linear part of S kin versus ln t plots for Cu adsorption and desorption processes (mmolc kg−1)
5 Evaluation and Quantification
The basic physical and chemical properties of investigated soils are reported in Tables 3.1 and 3.2. Contaminated soils (Nos. 1–8) were predominantly neutral to alkaline with pH ranging from 6.4 to 7.3 (except for soil Nos. 8 with pH = 5.0). Organic carbon (C org) content fluctuated within the range 5.6–18.3 g kg−1, whereas the specific surface area (SSAEGME), external (SSABET) and internal (SSAINT), varied from 9.4 to 72.0, 1.3 to 17.1 and 7.1 to 54.9 m2 g−1, respectively. Soil particle distribution showed a general prevalence of the sand fraction varying from 420 to 860 g kg−1 over the silt and clay ones. Clay fractions were relatively low and were in the range 20–160 g kg−1 in which most of soils were characterised by clay content below 100 g kg−1. The particle distribution and the relatively low organic carbon content promoted a cation exchange capacity (CEC), which varied from 9.8 to 21.8 cmolc kg−1 with quite all soils characterised by a CEC higher than 10.0 cmolc kg−1 (except for soils No. 8).
Most of uncontaminated soils (Nos. 9–16) were slightly alkaline but their pH values fluctuated from pH 5.1 to 7.6. The organic carbon content ranged within the interval 5.8–12.2 g kg−1, where 75 % of soil samples did not exceed 10.0 g kg−1. The clay fraction content of soils was below 100 g kg−1 (from 20 to 90 g kg−1) with a relatively high content of the sand fraction varying from 750 to 900 g kg−1. About 75 % of soils were characterised by CEC values higher than 10.0 cmolc kg−1 and relatively high specific surface area (SSAEGME) with 75 % of soils possessing more than 15.0 m2 g−1. The occurrence of carbonates in some soils could be attributed to frequent liming in order to reduce acidity.
The average copper content in uncontaminated soils around the world is estimated to vary from 20 to 30 mg kg−1, whereas in Poland the mean level is about 12 mg kg−1 (Kabata-Pendias and Pendias 1992). On the other hand, there is great concern of copper contamination from metal processing plants, where a site pollution of 1,400–3,700 mg Cu kg−1 in Ontario was reported by Freeman and Hutchinson (1980). Local copper pollution in Poland was also pointed out by Roszyk and Szerszeń (1988) at the surroundings of Legnica copper smelter, with a copper content ranging from 25 to 9,800 mg kg−1, and in the case of Głogów copper smelters, authors have reported a relatively lower level of 30–3,280 mg kg−1. Copper chemistry in soils is ruled by several factors which may be classified into two main groups: (1) soil properties, in which pH plays one of the key roles (Cavallaro and McBride 1980; Harter and Lehmann 1983; Msaky and Calvet 1990), organic matter and clay content (Eriksson 1988) and redox potential (McBride 1989; Diatta 2008), and (2) copper content and its forms such as Cu2+, CuOH+, Cu(OH)2, CuO, CuCO3 and Cu(OH)2CO3 (Adriano 1986; McBride 1989).
The distribution of sorptive sites on soil colloids is generally not uniform (Sparks 1995; Wu et al. 1999; Diatta et al. 2000). For adsorption, cations are held either through electrostatic attraction, this giving rise to ion exchange with surrounding ions or by specific adsorption through surface complexation on organic and mineral substrates (Benjamin and Leckie 1981). The distribution of charges per square metre of the specific surface area (SSA) is reported in Table 3.5 for contaminated and uncontaminated soils.
As it could be observed, the higher the reactive surface, the apparently lower the number of free active sites, as pointed out especially for soils No. 4 and No. 5 (contaminated soils) with surface charge density (SCDSSA) amounting to 3.5 and 3.0 × 10−3 mmolc m−2, characterised relatively by SSA of 37.4 and 72.0 m2 g−1, respectively. A reverse case occurred for soils No. 3, No. 6 and No. 8, which developed apparently higher SCDSSA, accordingly 8.7, 8.6 and 10.4 × 10−3 mmolc m−2. If one considers the SCDSSA = 5.0 × 10−3 mmolc m−2 as targeted reference, it comes out that only two contaminated soils should be tentatively classified as “highly” charged and the remaining as “moderately” charged. Uncontaminated soils behave similarly as the contaminated ones, that is, the lowest the specific surface area, the apparently highest the surface charge density as shown for soils No. 11 and No. 15 with proper SCDSSA values of 10.2 and 12.0 × 10−3 mmolc m−2. Referring to the targeted SCDSSA = 5.0 × 10−3 mmolc m−2 as earlier reported, only two of eight soils, basically, i.e. No. 12 and No. 16 (and soil No. 10 inclusive), should be considered as “highly” charged in opposite to the “moderately” ones.
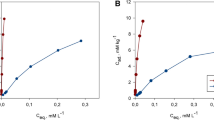
Plots of these sorption densities (SD) versus copper equilibrium concentration (C e ) resulted in at least one linear regression for most of the soils (Fig. 3.5a, b). Such plots are indicative of the occurrence of at least one pool of adsorption sites, the first highly energetic and the consecutive with lower adsorption energy.


Relationships between sorption density (SDSSA) and Cu equilibrium concentrations (Ce) for selected (a) contaminated soils (Nos. 1, 5 and 8) and (b) uncontaminated soils (Nos. 12, 14 and 15)
The use of surface-based sorption density isotherms for heavy metal ecotoxicology assessment should be based on the relationship of metal ion densities at the soil colloid surface versus equilibrium concentration (C e ), as suggested by Schulte and Beese (1994) for Cd and Zehetner and Wenzel (2000) for Cu and Ni. In the current work, plots of SDSSA versus C e for the studied soils gave the best visualisation of copper adsorption process. A general analysis of plots revealed that the lower the SDSSA and C e , the highest the potential reactive sites for copper, and inversely. A simulative computation based on regressions of soils No. 1, No. 5 and No. 14, for instance, has shown that these soils attained a C e = 2.0 μmolc dm−3 at an average SDSSA of 1.2 × 1017 ions m−2 (soil No. 1), whereas for soils No. 5 and No. 14, it was 0.4 × 1017 and 2.3 × 1017 ions m−2, respectively.
Practically a marked increase in the solution concentration following copper additions (for soils No. 1 and No. 5) is not to be expected as long as a targeted ion density (e.g. 2.3 × 1017 ions m−2 for soil No. 14) limit has not been reached. These approaches emphasise on the possibility of copper equilibrium monitoring based on sorption densities’ application.
Copper adsorption and its equilibrium concentrations, both equilibria are pH dependent, pointed out that soils exhibited in fact more charges than evaluated on the basis of SCDSSA (Table 3.5). Therefore the complex reaction of copper ions with deprotonated surface OH and COOH groups as bidentate and monodentate ligands could be expectable as pointed out by Schindler et al. (1976) and Davis and Leckie (1978). The adsorption of copper at energetically high sorptive sites can be formulated:

The saturation of the energetically high sorptive sites runs with the emergence of energetically low sorptive sites, and the latter ones may be presumably attributed to isomorphous substitution charge (Forbes et al. 1976; Bowden et al. 1977) as indicated below:

-
[XO4]—exchange site due to isomorphous substitution charge, X to represent Si, Al
Such approach is in agreement with the reports of Msaky and Calvet (1990), who stated that copper adsorption systematically decreased as the surface coverage increased. So it may be deduced that copper ions were primarily attracted by high energetically charged sites, what coincided to the first intensive adsorption process leading to a marked removal of copper ions from the equilibrium solution. These sorption mechanisms showed some limitation of the formerly applied Langmuir and Freundlich isotherms for soil systems (Diatta et al. 2004, 2012).
Langmuir constants, i.e. a max (adsorption maximum) and b (constant related to bonding energies) spread widely, mostly in the case of b values (Table 3.6). For contaminated soils, it was noted that the lowest b value was obtained for the soil No. 8 and accounted for 6.7 dm3 mmolc −1, whereas the highest one, amounting for 40.2 dm3 mmolc −1, was attributed to soil No. 7. It is also important to note that both a max and b constants directly influence the whole soils’ maximal buffering capacities (MBC) towards Cu. Soil No. 5 with the highest a max (111.7 mmolc kg−1) was characterised by an MBC of 2,412 dm3 kg−1 with a respective b = 21.6 dm3 mmolc −1, whereas soil No. 7 at a max = 81.5 mmolc kg−1 has developed the highest maximal buffering capacity of 3,276 dm3 kg−1 (for b = 40.2 dm3 mmolc −1). Soil No. 8 deserves special attention, since all sorption constants are strikingly low as a result of combined low pH, organic carbon content (C org) and specific surface area on one hand and high copper content (1,048.0 mg kg−1), on the other hand. Uncontaminated soils were also characterised by a great variation of both Langmuir constants, which for adsorption maximum—a max—ranged from 14.0 to 115.1 mmolc kg−1 and for the bonding energy term, b, from 11.4 to 26.0 dm3 mmolc −1 and exceptionally to 236.6 dm3 mmolc −1, obtained for soil No. 15 with a resulting high buffering capacity (MBC = 2,454 dm3 kg−1). Assuming that small amounts of copper should be strongly fixed by soil colloids, therefore, the notably low a max simultaneously with high b explains the case encountered for this soil. The involved mechanisms resulted basically from the heterogeneous character of soil sorptive surfaces (Sokołowska 1989; Sparks 1995; Diatta et al. 2003).
The Freundlich constant m expresses the intensity of isotherms slope (Table 3.6). Most of m values were ≪1, that means isotherm shapes were less steepy (occurrence of curvature), typically for soils in which most of high energetic sorptive sites have been just occupied by copper ions, the remaining ones being characterised by low adsorption energies with increasing surface coverage. Such cases were frequently reported also for Langmuir isotherms (Wu et al. 1999). Since slope intensity clearly indicated differences in the capacities of soils for copper adsorption, it appeared necessary to find out any relationships which may be helpful in determining Cu adsorption maximum as equated below:
where a max is the Langmuir adsorption maximum constant and m is the Freundlich slope intensity.
6 Kinetic Reaction (Adsorption/Desorption) Rate Constants
Kinetics is the study of the time-dependent processes, which in heterogeneous systems such as soil components occur simultaneously and consecutively over a wide time scale (Amacher 1991). The time-interval set for kinetics of copper adsorption in this study ranged from 0.5 up to 96 h. The 8 h time used for the estimation showed that amounts of adsorbed copper varied from 84 to 93 % for contaminated soils (with 57 % for soil No. 8) and from 78 to 90 % for uncontaminated soils (with soil No. 15 about 52 %). Experimental data fitted the simplified Elovich equation that proves its utility for empirical prediction. According to Sparks (1989), the chemical significance of these constants has not been clearly resolved β constants, but they can be used for comparing adsorption rates in different soils (Chien and Clayton 1980; Elkhatib et al. 1992; Dang et al. 1994; Taylor et al. 1995).
The constants β calculated from the slopes \( \frac{1}{\beta } \) of the Elovich equation were also a function of both soil textural group and initial copper concentration. The values of the slopes \( \frac{1}{\beta } \) increased gradually when increasing copper concentrations in the solution with a simultaneous decrease of β values. When comparing contaminated and uncontaminated soils, it was found that the decrease in β between C i = 0.8 and 1.6 mmolc dm−3 averaged 69 % in comparison with the relatively small decrease (31 %) observed between C i = 2.4 and 3.2 mmolc dm−3. The “sensitivity” of the slope and hence constants β relied on the degree of surface coverage by copper ions. The relatively high coefficients of determination (R 2) resulting from multiple variable interactions (Table 3.7) on the constant β pointed out the fact that copper chemistry of contaminated as well as uncontaminated soils should be similarly described by simplified Elovich equation, which is generally considered to characterise a number of different processes including bulk diffusion.
7 Hysteresis of Copper in Investigated Soils
Ion exchange reactions in soils are by nature reversible, but this reversibility may appear to be apparently irreversible (Van Bladel and Laudelout 1967), or quite irreversible, that means occurrence of hysteresis (Fripiat et al. 1965; Verburg and Baveye 1994; Zhu and Selim 2000). The total amount of copper desorbed at the end of the desorption process was relatively high and averaged 22 and 26 % at C i = 2.4 and 3.2 mmolc dm−3, respectively, for contaminated soils, except for soil No. 1, where copper was less desorbed (i.e. 6.0 and 10 % at the respective C i levels). Interestingly higher desorption rates, i.e. 28 and 35 %, were obtained even for uncontaminated soils at C i = 2.4 and 3.2 mmolc dm−3, respectively, whereas soil No. 12 desorbed accordingly low copper in the order 4.0 and 8 %. On the other hand Wu et al. (1999) have found that 35 % of formerly adsorbed copper was desorbed from a fine clay fraction, while 27 and 25 % of copper were desorbed from medium and coarse clay, respectively. The relatively high levels of copper desorbed in the current study did not confirm the occurrence of any irreversibility (hysteresis) as earlier reported by Ainstworth et al. (1994). The presence of carbonates in the case of soils No. 1 and No. 12 resulted in a limited copper desorbability (i.e. limited irreversibility).
According to O’Connor et al. (1980) and Barriuso et al. (1994), if there is no hysteresis the ratio N kin-des/N kin-ads = 1 and K kin-ads > K kin-des and in case of negative hysteresis, i.e. enhanced desorbability, the ratio N kin-des/N kin-ads > 1 and K kin-ads > K kin-des, as also observed in this study by the fact that the increase of N kin-des/N kin-ads induced an increase of K kin-ads parameter (Table 3.8). Such conditions could explain the relatively high copper desorbability (exception for soils No. 1 and No. 12). As surface charge density (SCDSSA) increases, the N kin-des/N kin-ads ratio decreases, implying that copper was more tightly adsorbed by high charge density soil colloids than by low charge density ones (Wu et al. 1999; Diatta et al. 2002). This is evidenced by the negative slopes (SCDSSA versus N kin-des/N kin-ads) as follows:
For N kin-des/N kin-ads parameters, refer to Eqs. (3.17, 3.19 and 3.20).
Therefore, if we assume the pool of high-density charges to be pH dependent, since 88 % of contaminated as well as uncontaminated soils exhibited pH > 6.0, the high-density charges could probably be limited in number.
8 Activation Energies for Copper Geochemical Processes
The reaction rate theory assumes that colliding molecules (e.g. reactions between solution ions and ions held in the soil sorptive complex) must be in a high energy before a reaction occurs (El-Sayed et al. 1970; Diatta et al. 2000). This energy of activation measures the magnitude of the forces that must be overcome during the process of adsorption and desorption reactions (Elkhatib et al. 1993). Energies of activation for copper adsorption (E aa) and desorption (E ad) are grouped in three operational ranges, respectively, for C i levels (0.8, 1.6, 3.2 mmolc dm−3), both for contaminated and uncontaminated soils: E aa < 50, 51 < E aa < 150, E aa > 151 kJ mol−1, namely, range I (RI), range II (RII) and range III (RIII), respectively, as follows:
C i = 0.8 mmolc dm−3 | |
Contaminated | 12 % of soil belong to RI, 63 % to RII and 25 % to RIII |
Uncontaminated | 25 % of soils belong to RI, 63 % to RII and 12 % to RIII |
C i = 1.6 mmolc dm−3 | |
Contaminated | 25 % of soils belong to RI, 63 % to RII and 12 % to RIII |
Uncontaminated | 63 % of soils belong to RI, 25 % to RII and 12 % to RIII |
C i = 3.2 mmolc dm−3 | |
Contaminated | 75 % of soils belong to RI, 25 % to RII |
Uncontaminated | 100 % of soils belong to RI |
According to Barrow and Whelan (1989) and Barrow (1998), the E aa for investigated reactions varied from 80 to 100 kJ mol−1. The activation energy of a diffusion-controlled process in a solution is about 25 kJ mol−1 (Sparks 1986). Activation energies for copper adsorption (E aa) ranged from 11.81 to 453.81 kJ mol−1 and for copper desorption (E ad), between 3.79 and 13.59 kJ mol−1 (Fig. 3.6a, b).

(a) Energy flow for Cu adsorption/desorption at C i = 2.4 mmolc dm−3, AC = activated complex, E aa and E ad = adsorption and desorption activation energy, respectively. E T = energy transfer (energy needed for Cu to move from the solution to the solid phase). (a) Contaminated soils (Nos. 1, 3, 5 and 8) and (b) Uncontaminated soils (Nos. 12 and 15)
The energy transfer (E T ) expresses a measure of the amount of energy needed to transfer copper ions from the solution to activated complex, i.e. soil solid state (Ogwada and Sparks 1986). The E T value outcomes for soils No. 1 and No. 12 amounted for 97.78 and 93.65 kJ mol−1, respectively, and were notably higher than those obtained for soils No. 3, No. 5, No. 8 and No. 15, amounting for 18.96, 39.71, 5.85 and 4.99 kJ mol−1, respectively. A suggestive endothermic reaction could have been occurring in these soils. The endothermic nature of the reaction and the trend with surface coverage (a decrease of E aa and E ad with increasing initial copper concentration) may be attributed to the potential energy available for adsorption and the fact that most energetic sites were occupied first (Msaky and Calvet 1990). Since the latter become limited as surface coverage increases, adsorbed copper ions at lower interaction energy will be readily desorbable due to weak attraction. So relatively low energy is necessary for copper ions to transfer back to the ambient soil solution.
Investigated soils both contaminated and uncontaminated were mostly characterised by relatively weak buffering properties towards copper ions and should be managed with care: a reduction of copper inputs may increase its E ad and as a result slow down copper transfer to soil solution. Mass transfer and diffusion reaction control seem to be of prime order. The multiple-phase evaluation pointed out in the current study showed the complexity of mechanisms involved in copper dynamics in soil systems.
9 Summary
Geochemical processes involve reactions which mediate and control the dissolution/release and (re)formation/sorption of mineral elements at geological as well as soil levels. The role of trace elements, copper among others, in soil chemical changes is still a matter of detailed studies. Copper-bearing minerals (chalcopyrite, CuFeS2; chalcocite, Cu2S, and bornite, Cu5FeS4) predominate in nature, although the richest Cu concentrations are related to the secondary weathering stage compounds [azurite, Cu3(CO3)2(OH)2, and malachite, Cu2CO3(OH)2, for instance]. The release of Cu from these minerals may operate via CO2 injection into deep geological structures or mining activities. The latter ones initiate slow or intensive spread of Cu compounds into the environment.
For decades, geochemists have been facing challenges related to the quantification of both natural (uncontaminated) and anthropogenic (contaminated) soil Cu concentrations. The current chapter was enriched with a case study which outlines in detail multiple steps involved in the evaluation of copper geochemical processes. The approach suggested herein takes into account the medium concentration, time dependency, model application and data fitting for Cu phase characterisation. Equilibria, exchange mechanisms, retention versus release, hysteresis as well as Cu complex formation (energy transfer) and the resulting (geo)environmental concerns were treated with substantial care.
References
Adriano DC (1986) Trace elements in the environment. Springer-Verlag, New York, p 553
Ainstworth CC, Pilon LJ, Gassman PL, Van Der Sluys W (1994) Cobalt, cadmium and lead sorption to hydrous iron oxide: residence time effect. Soil Sci Soc Am J 58:1615–1623
Allen JA, Scaife PH (1966) The Elovich equation and chemisorption kinetics. Aust J Chem 19:2015–2023
Alloway BJ (1995) Heavy metals in soils, 2nd edn. Blackie Academic & Professional, London, p 360
Amacher MC (1991) Methods of obtaining and analysing kinetic data. In: Sparks DL, Suarez DL (eds) Rates of soil chemical processes. SSSA, Madison WI, pp 19–59, Spec. Publ. Nr 27. Soil Sci. Soc. Am
Amacher MC, Kotuby-Amacher J, Selim HM, Iskandar IK (1986) Retention and release of metals by soils. Evaluation of several models. Geoderma 38:131–154
Aringhieri R, Carrai P, Petruzzelli G (1985) Kinetics of Cu2+ and Cd2+ adsorption by an Italian soil. Soil Sci 139(3):197–204
Baker DE (1990) Copper. In: Alloway BJ (ed) Heavy metals in soils. Halsted, Blackie, NY, USA, pp 151–176
Barriuso E, Laird DA, Koskinen CW, Dowdy RH (1994) Atrazine desorption from smectites. Soil Sci Soc Am J 58:1632–1638
Barrow NJ (1998) The effect of time and temperature on the sorption of cadmium, zinc, cobalt and nickel by a soil. Aust J Soil Res 36:941–950
Barrow NJ (1999) The four laws of soil chemistry: the Leeper lecture 1998. Aust J Soil Res 37:787–829
Barrow NJ, Whelan BW (1989) Testing a mechanistic model. VIII. The effect of time and temperature of incubation on the sorption and subsequent desorption of selenite and selenate by a soil. J Soil Sci 40:29–37
Basta NT, Tabatabai MA (1992) Effect of cropping systems on adsorption of metals by soils: II. Effect of pH. Soil Sci 153:195–204
Benjamin MM, Leckie JO (1981) Conceptual model for metal-ligand-surface interactions during adsorption. Environ Sci Tech 15:1050–1056
Boehringer IM (1980) Echanges cationique avec des métaux lourds sur la tourbe acide. Thèse de Doctorat Ecole Polytechnique, Fédérale, Zurich; No. 6700. p 131
Bowden JW, Posner AM, Quirk JP (1977) Ionic adsorption on variable charge mineral surfaces. Theoretical-charge development and titration curves. Aust J Soil Res 15:121–136
Brümmer GW, Gerth J, Herms U (1986) Heavy metal species, mobility and availability in soils. Z Pflanzenernehr Bodenk 149:382–398
Bunzl K, Schmidt W, Sansoni B (1976) Kinetics of ion exchange in soil organic matter. IV. Adsorption and desorption of Pb2+, Cu2+, Cd2+, Zn2+ and Ca2+ by peat. J Soil Sci 27:32–41
Carter DL, Mortland MM, Kemper WD (1986) Specific surface. In: Klute A (ed) Methods of soils analysis, part I—physical and mineralogical methods, 2nd edn. Am Soc Agron, Madison, WI, pp 413–423
Cavallaro N, McBride MB (1978) Copper and cadmium adsorption characteristics of selected acid and calcareous soils. Soil Sci Soc Am J 42:550–556
Cavallaro N, McBride MB (1980) Activities of Cu2+ and Cd2+ in soil solutions as affected by pH. Soil Sci Soc Am J 44:729–732
Chien SH, Clayton WR (1980) Application of the Elovich equation to the kinetics of phosphate release and sorption in soils. Soil Sci Soc Am J 44:265–268
Chudziński B (1995) Assessment of heavy metals phytotoxicity on the basis of their availability, yields and quality of cereals. PhD Thesis, IOR Poznan (Poland). 166 p. (in Polish).
Cox PA (1989) The elements. Their origin, abundance and distribution. Oxford University Press, Oxford, p. 207
Dang YP, Dalal RC, Edwards DG, Tiller KG (1994) Kinetics of Zinc desorption from vertisols. Soil Sci Soc Am J 58:1392–1399
Davis JA, Leckie JO (1978) Surface ionization and complexation at the oxide-water interface. II. Surface properties of amorphous iron oxyhydroxide and adsorption of metal ions. J Colloid Interface Sci 67:90–107
de Jong E (1999) Comparison of three methods of measuring surface area of soils. Can J Soil Sci 79:345–351
Diatta JB (2002) Evaluation of adsorption parameters and charge densities of some selected soils: application to lead. EJPAU Environ Dev 5(Issue 2). http://www.ejpau.media.pl/series/volume5/issue2/environment/art-03.html
Diatta JB (2008) Mutual Cu, Fe and Mn solubility control under differentiated soil moisture status. J Elementol 13(4):473–489
Diatta JB, Kociałkowski WZ, Grzebisz W (2000) Copper distribution and quantity-intensity parameters of highly contaminated soils in the vicinity of a copper plant. Polish J Environ Stud 9(5):355–361
Diatta JB, Kociałkowski WZ, Grzebisz W (2003) Lead and zinc partition coefficients of selected soils evaluated by Langmuir, Freundlich and linear isotherms. Commun Soil Sci Plant Anal 34(17 & 18):2419–2439
Diatta JB, Grzebisz W, Wiatrowska K (2004) Competitivity, selectivity, and heavy metals-induced alkaline cation displacement in soils. Soil Sci Plant Nutr 50(6):899–908
Diatta JB, Komisarek J, Wiatrowska K (2012) Evaluation of heavy metals competitive sorption and potential mobility on the basis of Cu/Cd and Zn/Pb binary systems. Fresenius Environ Bullet 21(5):1105–1109
Drobek L, Bukowska M, Borecki T (2008) Chemical aspects of CO2 sequestration in deep geological structures. Gospod Surowcami Min 24(3/1):425–438
Dube A, Zbytniewski R, Kowalkowski T, Cukrowska E, Buszewski B (2001) Adsorption and migration of heavy metals in soil. Polish J Environ Stud 10(1):1–10
Duffus JH (2002) Heavy metals—a meaningless term. IUPAC technical report. Pure Appl Chem 74(5):793–807
Elkhatib EA, Elshebiny GM, Balba MA (1992) Comparison of four equations to describe the kinetics of lead desorption from soils. Z Pflanzenernahr Bodenk 155:285–291
Elkhatib EA, Elshebiny GM, Elsubruiti GM, Balba MA (1993) Thermodynamics of lead sorption and desorption in soils. Z Pflanzenernahr Bodenk 156:461–465
El-Sayed MH, Burau RG, Babcock KL (1970) Thermodynamics of copper (II)—calcium exchange on bentonite clay. Soil Sci Soc Am Proc 34:397–400
Elzinger EJ, van Grinsven MJ, Swartjes FA (1999) General of Freundlich isotherms for cadmium, copper and zinc in soils. Eur J Soil Sci 50:139–149
Eriksson JE (1988) The effects of clay, organic matter and time on adsorption and plant uptake of cadmium added to the soil. Water Air Soil Pollut 40:359–373
Evans RL, Jurinak JJ (1976) Kinetics of phosphate release from a desert soil. Soil Sci 121:205–211
Forbes EA, Posner AM, Quirk JP (1976) The specific adsorption of divalent Cd, Co, Cu, Pb and Zn on goethite. J Soil Sci 27:154–166
Forrester SD, Giles CH (1971) From manure heaps to monolayers: the earliest development of solute-solid adsorption studies. Chem Ind, 1314–1321
Freedman B, Hutchison TC (1980) Pollutant inputs from the atmosphere and accumulations in soils and vegetation near a nickel-copper smelter at Sudbury, Ontario. Canada Can J Bot 58:108–132
Fripiat JJ, Cloos P, Poncelet A (1965) Comparison entre les propriétés d’échange de la montmorillonite et d’une résine vis-à-vis des cations alcalins et alcalino-terreux. I. Réversibilité des processus. Bull Soc Chim Fr, 208–215
Gaines GL, Thomas HC (1953) Adsorption studies on clay minerals. II. A formulation of a thermodynamics of exchange-adsorption. J Chem Phys 21:714–718
Gapon EN (1933) Theory of exchange adsorption in soils. J Gen Chem (USSR) 3(2):144–152
Geiger G, Federer P, Sticher H (1993) Reclamation of heavy metal contaminated soils—field studies and germination experiments. J Environ Qual 22:201–207
Geochemical Atlas of Legnica-Głogów Copper District (1999) State Geological Institute. Scale 1:250 000. Warszawa (in Polish)
Giles CH, MacEwan TH, Nakhwa SN, Smith D (1960) Studies in adsorption. Part XI. A system of classification of solution adsorption isotherms, and its use in diagnosis of adsorption mechanisms and in measurements of specific surface areas of solids. J Chem Soc 786:3973–3993
Gregg SJ, Sing KSW (1967) Adsorption, surface area and porosity. Academic, London, UK, pp 44–50
Grzebisz W, Chudziński B (1996) Crop aided biological remediation of soils contaminated by a copper smelter. In: Gărban Z, Drăgan P (eds) Proceedings of second international symposium on metal elements in environment, medicine and biology, Publishing House “Eurobit” Timişoara—Romania, 1st edition. 1996. ISBN 973-9336-15-9, pp 63–72
Grzebisz W, Kociałkowski WZ, Chudzinski B (1997) Copper geochemistry and availability in cultivated soils contaminated by a copper smelter. J Geochem Explor 58:301–307
Harter RD (1984) Curve-fit errors in Langmuir adsorption maxima. Soil Sci Soc Am J 48:749–752
Harter R, Lehmann R (1983) Use of kinetics for the study of exchange reactions in soils. Soil Sci Soc Am J 47:666–669
Helios-Rybicka E, Jędrzejczyk B (1995) Preliminary studies on mobilisation of copper and lead from contaminated soils and readsorption on competing sorbents. Appl Clay Sci 10(3):259–268
Hendershot WH, Duquette M (1986) A simple barium chloride method for determining cation exchange capacity and exchangeable cations. Soil Sci Soc Am J 50:605–608
Hodgson JF, Lindsay WL, Trierweiler JF (1966) Micronutrient cation complexing in soil solution. II. Complexing of zinc and copper in displacing solution from calcareous soils. Soil Sci Am Proc 30:723–726
Huijgen WJJ, Comans RNJ, Witkamp GJ (2006) Mechanisms of aqueous wollastonite carbonation as a possible CO2 sequestration process. Chem Eng Sci 61:4242–4251
Ibsen KH, Jacobsen FL (1996) The Linde Structure, Denmark an example of a CO2-depository with a secondary chalk cap rock. Energ Convers Manag 37(6–8):1161–1166
International Standard (1995) Soil quality—extraction of trace elements soluble in aqua regia, Geneva ISO11466
Izotermy© (1993) Wersje 1.0; 1.1; 1.2. Biuro Projektów Informatyki, A. Ratajczak, Poznań
Jopony M, Young SD (1994) The solid-solution equilibria of lead and cadmium in polluted soils. Eur J Soil Sci 45:59–70
Kabata-Pendias A (1978) The impact of copper mining and industrial activity of Lower Silesia on the chemical composition of plants. Final report—IUNG, p 173
Kabata-Pendias A (1993) Behavioural properties of trace metals in soils. Appl Geochem Suppl Issue 2, 3–9
Kabata-Pendias A, Pendias H (1992) Trace elements in soils and plants, 2nd edn. CRC Press, Boca Raton, p 365
Kabata-Pendias A, Pendias H (1999) Biogeochemia pierwiastków śladowych. Wydawnictwo Naukowe PWN, Warszawa
Kabata-Pendias A, Motowicka-Terelak T, Piotrowska M, Terelak H, Witek T (1993) Ocena stopnia zanieczyszczenia gleb i roślin metalami ciężkimi i siarką. IUNG-Puławy, str. 20
Kinniburgh DG (1986) General purpose adsorption isotherms. Environ Sci Technol 20:895–904
Kondracki J (1978) Geografia fizyczna Polski. PWN, Wyd. II, Warszawa, p 463
Kociałkowski WZ, Diatta JB, Grzebisz W (1999) Assessment of lead sorption by acid agroforest soils. Pol J Environ Stud 8(6):403–408
Konstantynowicz E (1979) Geologia surowców mineralnych. Tom 2. Złoża rud metali, Uniwersytet Śląski, Katowice
Lai TM, Mortland MM (1968) Cationic diffusion in clay minerals: I. Homogeneous and heterogeneous systems. Soil Sci Soc Am Proc 32:56–61
Laird DA, Barriuso E, Dowdy RH, Koskinen WC (1992) Adsorption of atrazine on smectites. Soil Sci Soc Am J 56:62–67
Lindsay WL, Norvell WA (1978) Development of a DTPA soil test for zinc, iron, manganese and copper. Soil Sci Soc Am J 42:421–428
Lindsay WL (1979) Chemical equilibria in soils. Wiley, New York, p. 449
Liu C, Huang PM (2000) Kinetics of phosphate adsorption on iron oxide formed under the influence of citrate. Can J Soil Sci 80:445–454
Loeppert RH, Suarez DL (1996) Carbonate and gypsum. In: Bartels JM et al (eds) Methods of soil analysis: part 3. Chemical methods, vol 5, 3rd edn. ASA and SSSA, Madison, WI, pp 437–474
Logan TJ (1990) Chemical degradation of soil. Adv Soil Sci 11:187–219
Logan EM, Pulford ID, Cook GT, McKenzie AB (1997) Complexation of Cu2+ and Pb2+ by peat and humic acid. Eur J Soil Sci 48:685–696
Low WJD (1960) Kinetics of chemisorption of gases on solids. Chem Rev 60:267–312
Maqueda C, Undabeytia T, Morrillo E (1998) Retention and release of copper on montmorillonite as affected by the presence of a pesticide. J Agric Food Chem 46(3):1200–1204
Markert B, Friese K (eds) (2000) Trace elements. Their distribution and effects in the environment. Elsevier, Amsterdam
Markert B, Breure A, Zechmeister H (eds) (2003) Bioindicators & biomonitors. Principles, concepts and applications. Elsevier, Amsterdam
Mattigod SV, Sposito G (1979) Chemical modeling of trace metal equilibria in contaminated soil solutions using the computer program GEOCHEM. Chemical modeling in aqueous systems. Henne E. A. Editions, Washington D.C. Am Chem Soc 93:837–856
McBride MB (1989) Reactions controlling heavy metal solubility in soils. Adv Soil Sci 10:1–56
McBride MB (1994) Environmental chemistry of soils. Oxford University Press, New York, p 406
McBride MB (2001) Cupric ion activity in peat soil as a toxicity indicator for maize. J Environ Qual 30:78–84
McLaren RG, Crawford DV (1973a) Studies on soil copper. I. The fractionation of copper in soils. J Soil Sci 24(2):172–181
McLaren RG, Crawford DV (1973b) Studies on soil copper. II. The specific adsorption of copper by soils. J Soil Sci 24(4):443–452
Msaky JJ, Calvet R (1990) Adsorption behaviour of copper and zinc in soils: influence of pH on adsorption characteristics. Soil Sci 150(2):513–522
Mulder J, Cresser MS (1994) Soil and soil solution chemistry. In: Moldan B, Cerny J (eds) Biogeochemistry of small catchments: a tool for environmental research. Wiley, Chichester, pp 107–131
Murali V, Aylmore LAG (1983a) Competitive adsorption during solute transport in soils. 1: mathematical models. Soil Sci 135(3):143–150
Murali V, Aylmore LAG (1983b) Competitive adsorption during solute transport in soils. 2: simulation of competitive adsorption. Soil Sci 135(4):203–213
Nelson DW, Sommers LE (1996) Total carbon, organic carbon and organic matter. In: Sparks DL (ed) Methods of soil analysis. Part 3. Chemical methods. SSSA, Madison, WI, pp 961–1010, SSA Book Ser. 5
Nriagu J, Pacyna P, Szefer P, Markert B, Wuenschmann S, Namiesnik J (eds) (2012) Heavy metals in the environment. Maralte Books, The Netherlands
O’Connor GA, Wierenga PJ, Cheng HH, Doxtader KG (1980) Movement of 2,4,5-T through large soil columns. Soil Sci 130:157–162
Ogwada RA, Sparks DL (1986) A critical evaluation on the use of kinetics for determining thermodynamics of ion exchange. Soil Sci Soc Am J 50:300–305
Pavlatou A, Polyzopoulos NA (1988) The role of diffusion in kinetics of phosphate desorption: the relevance of the Elovich equation. J Soil Sci 39:425–436
Polański A, Smulikowski K (1969) Geochemia. Wydawnictwa Geologiczne, Warszawa
Polish Standard (1994) Polish Standardisation Committee, ref. PrPN-ISO 10390 (E): soil quality and pH determination, 1st edn (in Polish)
Poonia SR, Talibudeen O (1977) Sodium-calcium exchange equilibria in salt-affected and normal soils. J Soil Sci 28:276–288
Prasa MNV (2008) Trace elements as contaminants and nutrients. Consequences in ecosystems and human health. Wiley, Hoboken, p. 777
Ram N, Verloo M (1985) Effect of various organic materials on the mobility of heavy metals in soils. Environ Pollut 10:241–248
Robert M (1996) Le sol: Interface dans l’Environnement, Ressource pour le développement. Masson, Paris, p 244
Roszyk E, Szerszeń L (1988) Accumulation of heavy metals in arable soil layers of the copper smelters sanitary zones, part II: “Głogów”. Rocz Gleboz 39(4):147–158 (in Polish)
Ryan J, Estefan G, Rashid A (2001) Soil and plant analysis laboratory manual, 2nd edn. International Center for Agricultural Research in the Dry Areas (ICARDA), the National Agricultural Research Center (NARC), Aleppo, Syria, p 172
Sapek B (1976) Study on the copper sorption kinetics by peat-muck soils. In: Proceedings of the 5th international peat congress—Poznań: new recognition of peatlands and peats, vol 2. pp 236–245
Sapek B, Sapek A (1980) The application of ion-selective electrodes to investigations of binding of copper and cadmium by humic substances from peat-muck soils. Pol J Soil Sci 13(2):125–134
Sapek B, Zebrowski W (1976) Comparison of copper binding rate by peat-muck soils at various transformation stages. Pol J Soil Sci 9(2):93–99
Sarbak Z (2000) Adsorption and adsorbents. Theory and application. Wydawnictwo Nauko we UAM, Seria Chemia Nr 71., 167 str. (in Polish)
Schindler PW, Fürst B, Dick R, Wolf PU (1976) Ligand properties of surface silanol groups I. Surface complex formation with Fe3+, Cu2+, Cd2+ and Pb2+. J Colloid Interface Sci 55:469–475
Schulte A, Beese F (1994) Isotherms of cadmium sorption densities. J Environ Qual 23:712–718
Schulthess CP, Dey DK (1996) Estimation of Langmuir constants using linear and nonlinear least squares regression analyses. Soil Sci Soc Am J 60:433–442
Schultz MF, Benjamin MM, Ferguson JF (1987) Adsorption and desorption of metals on ferrihydrite: reversibility of the reaction and sorption properties of the generated solid. Environ Sci Technol 21:863–869
Sillén LG, Martell AE (1971) Stability constants of metal-ion complexes. Suppl. 1, pt. I and II. The Chemical Society, Metcalfe and Cooper, Ltd, London, p 865
Skopp J (1986) Analysis of time dependent chemical processes in soils. J Environ Qual 15:205–213
Soil Taxonomy (1975) A basic system of soil classification for making and interpreting soil surveys: soil survey staff. U.S. Dept Agric. Handb. 436. U.S. Govt Print. Off. Washington, DC
Sokołowska Z (1989) The role of surface uniformity in adsorption processes occurring in soils. Problemy Agrofizyki 58:1–170, (in Polish) kinetics
Sparks DL (1986) Kinetics of reactions in pure and mixed systems. In: Sparks DL (ed) Soil physical chemistry. CRC Press, Boca Raton, FL, pp 83–178
Sparks DL (1989) Kinetics of soil chemical processes. Academic, San Diego, CA, p 210
Sparks DL (1995) Environmental soil chemistry. Academic, San Diego, CA, p 267
Sparks DL (2000) New frontiers elucidating the kinetics and mechanisms of metals and oxyanion sorption at the soil mineral/water interface. J Plant Nutr Soil Sci 163:563–570
Sposito G (1982) On the use of the Langmuir equation in the interpretation of adsorption phenomena II. The “Two-surface” Langmuir equation. Soil Sci Soc Am J 46:1147–1152
Sposito G (1984) The surface chemistry of soils. Oxford University Press, New York, p 234
Stawiński J (1977) Interrelationships between the specific surface area and some physico-chemical properties of soils. Zesz Probl Post Nauk Roln 197:229–240
Taylor RW, Kamaleldin H, Mehadi AA, Shoford WJ (1995) Kinetics of zinc sorption by soils. Commun Soil Sci Plant Anal 26(11 & 12):1761–1771
Thomas GW (1982) Exchangeable cations. In: Page AL, Miller RH, Keeney DR (eds) Methods of soil analysis, part 2. Chemical and microbial properties (No 9), 2nd edn. ASA-SSSA, Madison, WI, pp 159–165
Tiller KG, Gerth J, Brummer G (1984) The sorption of Cd, Zn and Ni by soil clay fractions: procedures for partition of bound forms and their interpretation. Geoderma 34:1–16
Van Bladel R, Laudelout H (1967) Apparent reversibility of ion-exchange reactions in clay suspension. Soil Sci 104:134–137
Van Bladel R, Halen H, Cloos P (1993) Calcium-zinc and calcium–cadmium exchange in suspensions of various types of clays. Clay Miner 28:33–38
Veith JA, Sposito G (1977) On the use of the Langmuir equation in the interpretation of “Adsorption” phenomena. Soil Sci Soc Am J 41:697–702
Verburg K, Baveye P (1994) Hysteresis in the binary exchange of cations on 2:1 clay minerals. A critical review. Clay Miner 42:207–220
Violante A, Krishnamurti GSR, Pigna M (2008) Factors affecting the sorption/desorption of trace elements in soil environments. In: Violante A, Ming Huang P, Gadd GM (eds) Biophysico-chemical processes of heavy metals and metalloids in soil environments. John Wiley & Sons, Inc., New York, pp 169–213
Weber WJ Jr (1984) Evolution of a technology. J Environ Eng Div 110:899–917
Wilk Z, Bocheńska T (2003) Hydrogeology of Polish mineral deposits and hydrological mining problems. Ed. AGH University of Science and Technology Press, Cracow, (Poland), 478 p. ISBN: 83-89388-21-9 (in Polish)
WRB (1998) World reference base for soil resources. 84 World soil resources reports. FAO-ISSS-ISRIC
Wu J, Laird DA, Thompson ML (1999) Sorption and desorption of copper on soil clay components. J Environ Qual 29(1):334–338
www.kghm.pl. Accessed 2 February 2015
Xian X, Shokohifard G (1989) Effect of pH on chemical forms and plant availability of cadmium, zinc and lead in polluted soils. Water Air Soil Pollut 45:265–273
Zehetner F, Wenzel WW (2000) Nickel and copper sorption in acid soils. Soil Sci 165(6):463–472
Zhang M, Nyborg M, Malhi SS, McKenzie RH, Solberg E (2000) Phosphorus release from coated monoammonium phosphate: effect of coating thickness, temperature, elution medium, soil moisture and placement method. Can J Soil Sci 80:127–134
Zhu H, Selim HM (2000) Hysteretic behavior of metolachlor adsorption-desorption in soils. Soil Sci 165(8):632–645
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Diatta, J.B., Chudzińska, E., Drobek, L., Wojcicka-Półtorak, A., Markert, B., Wünschmann, S. (2015). Multiple-Phase Evaluation of Copper Geochemistry. In: Sherameti, I., Varma, A. (eds) Heavy Metal Contamination of Soils. Soil Biology, vol 44. Springer, Cham. https://doi.org/10.1007/978-3-319-14526-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-14526-6_3
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-14525-9
Online ISBN: 978-3-319-14526-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)