
Abstract
Antibody–drug conjugates (ADCs) seek to exploit the desirable features of their components, i.e., specificity in targeting tumor cells and long half-life in plasma conferred by the antibody and the cytotoxic potency of the small effector molecule. A linker tethers the antibody to a cytotoxic agent, and allows stable circulation of the cytotoxin in the form of a prodrug. Many linkers have been designed over the years, which showcase features that are more than a mere physical linkage between the two constituent parts. Linkers can confer several advantages to ADCs, such as modulating the rate of cytotoxin release in tumor cells, overcoming multidrug resistance, enhancing tumor penetration, and a better safety profile in humans. This chapter highlights the progress made in linker technology, and how it impacts the design of optimal ADCs for clinical evaluation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Antibody–drug conjugate
- Noncleavable linker
- Cleavable linker
- Reducible linker
- Peptide linker
- Acid-labile linker
- Bystander activity
- Lysosomal processing
- MDR
- Liver detoxification
1 Introduction
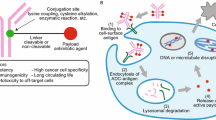
Antibody–drug conjugates (ADCs) are composed of a cytotoxic agent conjugated to a monoclonal antibody (mAb) raised against an antigen that is preferentially expressed on cancer cells relative to normal tissues (Fig. 4.1; Lambert 2013). A linker provides a covalent bridge between the cytotoxic agent and the antibody, which serves to deliver the cytotoxic agent specifically to cancer cells. The function of a linker is to keep the cytotoxic molecule stably attached to the antibody during formulation and storage of the drug product, and during circulation in plasma following administration. However, a linker must also allow rapid and efficient release of the cytotoxic agent upon internalization of the ADC within cancer cells. This fine balance of extracellular stability and intracellular release is a necessary starting point for the design of a linker for ADCs.

Antibody–drug conjugate
Contemporary linker technologies have taken the linker beyond its basic role as a physical bridge between the antibody and a cytotoxic moiety. Linkers have been modified to improve the activation of ADCs, thereby allowing ADCs to release the cytotoxic agents inside the cells at a faster rate with higher efficiency utilizing different mechanisms for cytotoxin release. Modulation of the linker to effect a change in the polarity or charge of the final metabolite has also allowed improved activity toward multidrug resistant (MDR ) cells owing to better retention of the cytotoxic agent inside the cells. Some linker designs have facilitated the generation of catabolites that are capable of diffusing into proximal tumor cells, inducing bystander killing that results in greater in vivo antitumor activity. This chapter reviews various linkers that have been designed for ADCs and the impact of linkers on the activity and safety of ADCs as cancer therapeutics.
2 Sites of Attachment and Reactive Chemical Moieties on Linkers
Sites of attachment on the antibody and the nature of the chemical groups on linkers to effect such attachment are important considerations in ADC design. They not only affect the conjugation efficiency and the ease of production of ADCs, but also the integrity and stability of the conjugate during production/storage as well as in patients. In addition, the linker often remains as an integral part of the metabolites and plays a key role in the activity and safety of ADCs in patients.
Lysine and cysteine residues have been utilized as sites of attachment on antibodies. Lysines are abundantly present on the antibody (80–100 lysines per antibody), and their primary amine readily reacts with N-hydroxysuccinimide esters, a chemical group often incorporated into linkers , to form stable amide bonds. Cysteines are present in antibodies in the form of disulfide bonds, which mediate intrastrand and interstrand bridges connecting light and heavy chains. For cysteines to be used as sites for conjugation, typically the interstrand disulfide bonds need to be reduced to generate reactive thiols, which can undergo the Michael reaction with a maleimide group commonly utilized in linkers to form thioether bonds. Both conjugation sites/methods allow efficient reactions and generate conjugates as a heterogeneous mixture with varying number of cytotoxic molecules per antibody. The distribution of the cytotoxic molecules per antibody can be highly reproducible from batch to batch, and typically follows a binomial function (Fig. 4.2; Singh and Erickson 2009; Wang et al. 2005).

Mass spectrometry profile of an antibody–drug conjugates (ADCs). The profile depicts a maytansinoid–ADC made with an average maytansinoid-to-antibody ratio of 3.5. The n of Dn refers to the number of cytotoxins attached to the antibody
Recent efforts to make homogeneous conjugates have explored diverse sites of attachment. An engineered cysteine on heavy and light chains of the antibody has been utilized as a site of attachment via maleimide linker chemistry (Junutula et al. 2008; Kung Sutherland et al. 2013). The reactive thiol of an engineered cysteine is not readily available for conjugation, because during antibody production, the sulfhydryl group of the engineered cysteine forms a mixed disulfide through exchange with cystine in the media. Before conjugation can proceed, a free thiol of the engineered cysteine must be generated by reduction of the antibody. The reduced antibody is then subject to partial oxidation to restore the intrachain and interchain disulfides that are important for antibody integrity, while maintaining the engineered cysteine in its thiol form. The thiol can then be utilized to react with a maleimide group on linkers , a conjugation strategy used to produce ADCs from the Thiomab (Junutula et al. 2008) or a similarly engineered antibody (Kung Sutherland et al. 2013). Alternatively, the free thiol of cysteine within a specific recognition sequence can be modified by a formylglycine-generating enzyme posttranslationally to produce an aldehyde-bearing amino acid, which can be conjugated to a cytotoxic agent via a hydrazine linker in a process called “aldehyde tagging” (Rabuka et al. 2012).
Carbohydrate moieties that are naturally present on antibodies have been used as sites of conjugation, especially for antibodies that tend to lose binding to antigen upon conjugation via lysine residues (Walus et al. 1996). Conjugation via glycans involves oxidation of the carbohydrate using sodium periodate, followed by reaction of the resulting aldehyde with a linker bearing a hydrazide group. Although efficient, this reaction suffered from undesired oxidation of the protein. A new method for glycan modification has emerged in recent years. Mannose or galactose containing an azido substituent is introduced into the endogenous glycosylation at Asp297 of the antibody either by expressing the antibody in the presence of azido-sugar (Ac4ManNAz or Ac4GalNAz) or by chemical remodeling using beta(1,4)-galactosyltransferase (Boeggeman et al. 2009). Use of the azido group ushered in the development of linkers with strained alkynes (e.g., cycloalkynes) that can react with the azide in copper-free click chemistry. The use of click chemistry gained popularity in conjugation to engineered sites, as the chemistry provides specificity without affecting the reactivity of the endogenous amino acids on antibodies.
Recently, nonnatural amino acids have been introduced into specific sites in antibodies either by the use of bacterial strains with orthogonal transfer RNAs (tRNAs) that can be charged with the nonnatural amino acid (Hutchins et al. 2011) or by cell-free extracts containing such tRNA and appropriate aminoacyl tRNA synthetases (Zimmerman et al. 2014). Click chemistry, along with oxime ligation, has been used for conjugation to these engineered nonnatural amino acids. A nonnatural amino acid containing an azido group can react with a linker containing a cycloalkyne, or a nonnatural amino acid containing a hydroxylamine can react with linkers containing an aldehyde. The latter oxime ligation has been used to generate a homogeneous anti-Her2 conjugate (Axup et al. 2012).
Glutamine has been used as an acceptor for conjugation when a bacterial enzyme, transglutaminase, was employed (Dennler et al. 2014; Strop et al. 2013). Transglutaminase catalyzes the formation of a covalent bond between the acyl side chain of glutamine (e.g., endogenous Q295 of human immunoglobulin G (IgG)) and a free amino group (e.g., of lysine) on a linker. An antibody can be engineered by introducing a glutamine tag to scan for sites that favor transglutaminase reaction anywhere on antibodies (Strop et al. 2013), or the native glutamine residue (Q295) can be exposed for conjugation by deglycosylation at N297 that typically hinders the accessibility of the glutamine (Dennler et al. 2014).
Conjugation chemistry has become sophisticated in recent years, and different sites on antibody and various functional groups on linkers have been explored. As a result, many claims have been made regarding the effect of these approaches (sites of conjugation and accompanying conjugation chemistry, as well as the consequence of homogeneous vs. heterogeneous conjugates) on the biological activity and potential safety of these ADCs (Junutula et al. 2008; Zimmerman et al. 2014). Thus far, there are no clinical data to support the various claims, and hence there is no clear understanding of the effect of these strategies on the activity and safety of ADCs.
3 Types of Linkers
Linkers are generally categorized into noncleavable or cleavable linker s. Noncleavable linkers represent linkers which remain intact during intracellular metabolism. ADCs with such linkers require lysosomal degradation of the antibody to release the cytotoxic agent. The metabolite retains the amino acid residue that served as the site of attachment to the antibody. Cleavable linkers are linkers that cleave during intracellular metabolism, generating metabolites that contain the cytotoxic agent with or without a portion of the linker. Cleavable linkers may be cleaved by hydrolysis, enzymatic reaction, or reduction, and include acid-labile hydrazone linkers, peptide-based linkers, and disulfide linkers, respectively.
3.1 Noncleavable Linkers
Thioethers are a widely used format for noncleavable linker s because of the facile reaction of maleimides with thiols under mild, neutral aqueous conditions. Thioethers can also be generated by reacting thiols with haloacetamido groups, although this reaction needs harsher conditions (e.g., higher pH and excess haloacetamido reagents; Alley et al. 2008). Thioether linkage has been used for microtubule-targeting agents, e.g., thiol derivatives of maytansine (Fig. 4.3; Singh and Erickson 2009) and auristatin (monomethyl auristatin F or MMAF; Alley et al. 2008).

Derivatives of maytansinoid
Ado-trastuzumab emtansine (T-DM1 or Kadcyla ® ) is a conjugate approved by the Food and Drug Administration (FDA) for the treatment of human epidermal growth factor receptor 2 (HER2)-positive, metastatic breast cancer in patients previously treated with trastuzumab and a taxane. T-DM1 is composed of trastuzumab and DM1 linked by a heterobifunctional linker, SMCC (N-succinimidyl-4-(maleimidomethyl) cyclohexane-1-carboxylate; Fig. 4.4). The SMCC–DM1 conjugate is prepared by reacting lysine residues on the antibody with the N-hydroxysuccinimide ester moiety of SMCC, and linking the thiol of DM1 with the maleimide group on SMCC (Fig. 4.4). The antibody–SMCC–DM1 format is currently used in other ADCs. Epidermal growth factor receptor (EGFR)-targeting IMGN289, CD37-targeting IMGN529, and CD70-targeting AMG172 are being evaluated in the clinic. The sole metabolite of SMCC–DM1 conjugates is Lys–SMCC–DM1 (Fig. 4.5; Erickson et al. 2006).

Conjugation scheme for antibody–SMCC–DM1. SMCC N-succinimidyl-4-(maleimidomethyl) cyclohexane-1-carboxylate

Antibody–SMCC–DM1 generates a single metabolite Lys–SMCC–DM1. SMCC N-succinimidyl-4-(maleimidomethyl) cyclohexane-1-carboxylate
An analogous thioether linker in which a hydrophilic tetraethylene glycol (PEG4) replaced the hydrophobic cyclohexyl group of SMCC was developed (Fig. 4.6). When conjugated to an anti-epithelial cell adhesion molecule (EpCAM) antibody , the ADC with the hydrophilic linker showed higher potency in cells expressing P-glycoprotein, which is responsible for the MDR phenotype (Kovtun et al. 2010). This PEG4Mal–DM1 conjugate generates a single metabolite, Lys–PEG4Mal–DM1 (Kovtun et al. 2010). A similar PEG linker was also used to conjugate DM1 to two engineered cysteines in the heavy chain of trastuzumab (Junutula et al. 2010). The homo-bifunctional BMPEO (bis-maleimido-trioxyethylene glycol) linker used contained a triethylene glycol (PEG3) spacer with two maleimido groups, used for conjugating to the thiol of DM1 and the thiol of a reduced free cysteine, to generate a homogeneous site-specific conjugate.

Conjugation scheme for antibody–PEG4–Mal–DM1. PEG4 hydrophilic tetraethylene glycol
A thioether linkage has also been used for conjugation of antibodies to the tubulin inhibitor, auristatin. Anti-CD70–maleimidocaproyl (mc)–MMAF was generated by reaction of the thiol groups of native cysteines on the antibody with a maleimide-containing linker (Fig. 4.7). The anti-CD70–mc–MMAF conjugate was evaluated in the clinic for the treatment of lymphomas and renal cell carcinomas, and contained an average of four MMAF molecules per antibody. The anti-CD70 antibody was evaluated preclinically with another noncleavable linker . When a bromoacetamidecaproyl (bac) group replaced the mc to generate anti-CD70–bac–MMAF, it showed an improved plasma stability and 25 % higher intratumoral drug exposure compared with a similar conjugate bearing mc–MMAF. However, despite this improvement, there was no statistically significant difference in the efficacy of these two conjugates in a subcutaneous 786-O renal cell carcinoma xenograft model (Alley et al. 2008).

Antibody–mc–MMAF. MMAF Monomethyl auristatin, mc maleimidocaproyl
3.2 Cleavable Linkers
3.2.1 Acid-labile Linkers
Hydrazone linkers are designed to be stable in a neutral pH environment, such as in plasma, but hydrolyzed to release the cytotoxic agent in an acidic pH environment, such as in late endosomes and lysosomes. Potent DNA-targeting cytotoxic agents, e.g., calicheamicin or doxorubicin (Fig. 4.8), have been conjugated to antibodies via hydrazone linkers.

DNA-targeting calicheamicin and doxorubicin
N-acetyl-γ1 I-calicheamicin is a type of enediyne antibiotics that associates with the minor groove of DNA and causes double-strand breaks in a sequence-specific manner (Greenberg 2014). Calicheamicin has been evaluated in the context of antibodies to CD33, CD22, and Muc1, all of which were conjugated via hydrazone linkers (Ricart 2011; Hamann et al. 2005a).
The first FDA-approved ADC , gemtuzumab ozogamicin (Mylotarg® ), was used for the treatment of patients with acute myelogenous leukemia (AML). Gemtuzumab ozogamicin is composed of an anti-CD33 IgG4 conjugated to calicheamicin via a bifunctional linker 4-(4’acetylphenoxy)-butanoic acid. The N-hydroxysuccinimide ester of N-acetyl-γ-calicheamicin dimethyl hydrazide 4-(4acetylphenoxy)-butanoic acid reacts with lysine residues on the antibody , leading to an average loading of 4–6 calicheamicins per antibody, but with 50 % of the antibody remaining unconjugated (Hinman et al. 1993). In an acidic environment, calicheamicin is released from the ADC upon hydrolysis of the hydrazone linker, and undergoes thiol-mediated reduction, followed by Masume–Bergman cyclization. This reaction generates ρ-benzyne biradicals that abstract hydrogen atoms from the phosphodiester backbone of DNA and leads to single- and double-strand lesions (Fig. 4.9). Mylotarg was voluntarily withdrawn from the market when a confirmatory phase III clinical trial failed to demonstrate clinical benefit. A new regimen of fractionated doses for better efficacy and safety is being evaluated in the clinic (Pilorge et al. 2014). Anti-CD22 antibody was also conjugated to calicheamicin via the same acid-labile 4-(4’acetylphenoxy)-butanoic acid linker, generating inotuzumab ozogamicin (CMC-544). CMC-544 was evaluated in the clinic for treatment of B-lymphoid malignancies (DiJoseph et al. 2004a, 2004b; Jain et al. 2014); however, its evaluation for the treatment of CD22-positive aggressive non-Hodgkin lymphoma was halted due to lack of benefit in overall survival. Another example for the hydrazone linker is highlighted in the anti-Muc1 antibody, CTM01, which was conjugated to calicheamicin via a hydrazone link to periodate oxidized carbohydrates on the antibody (Hamann et al. 2005b).

Mechanism of release and action for antibody conjugated to calicheamicin via a hydrazone linker
Similarly, doxorubicin, a DNA intercalator that elicits cytotoxicity by inhibiting topoisomerase II, has been conjugated to antibodies via a hydrazone linker. Thiols from the reduced interchain disulfides of the anti-CD74 antibody , IMMU-110, are conjugated to doxorubicin via a 4-[N-maleimidomethyl] cyclohexane-1 carboxylhydrazide, yielding an average of eight doxorubicin per antibody. This conjugate, milatuzumab–dox, was evaluated for the treatment of patients with multiple myeloma (Sapra et al. 2005), but was recently withdrawn for lack of efficacy.
3.2.2 Reducible Linkers
Reducible disulfide linkers take advantage of the difference in reduction potential in plasma versus the intracellular compartment. They are designed to keep conjugates intact during systemic circulation in patients, while efficiently allow reduction/cleavage of the disulfide bond inside cells. Reducible disulfide linkers generate neutral metabolites that can freely diffuse into neighboring cells and elicit bystander killing aiding the tumor penetration of the cytotoxic anticancer agent (see Sect. 4.4.3).
The most abundant low molecular thiol in blood is cysteine, the concentration of which is low at ~ 5 µM (Mills and Lang 1996). Serum albumin is another source of reducing potential in the blood, and its concentration is higher at ~ 0.6 mM (Turell et al. 2009). However, despite its relative abundance, the thiols in albumin are buried and inaccessible to thiol–disulfide exchange, thus it is not a major source of disulfide cleavage (for more discussion, see the linker stability section). In contrast to blood, the reduction potential inside cancer cells is much higher. Reduced glutathione is present at 1–10 mM (Wu et al. 2004), and cells also contain enzymes of the protein disulfide isomerase family, which may contribute to reduction of the disulfide bond in cellular compartments. This differential allows stable circulation in plasma, but efficient release inside the cells upon internalization of the ADC .
An important consideration for designing disulfide linkers is the steric hindrance of the disulfide bond and its impact on the stability of the linker. The HuC242 antibody has been used as a model system to evaluate the effect of hindrance on the reactivity of disulfide linkers to thiol–disulfide exchange in vitro and in plasma (Kellogg et al. 2011). The humanized C242 antibody recognizes CanAg, which is abundantly expressed in colorectal carcinoma. Various numbers of methyl groups were introduced on the carbon atoms bearing the disulfide bond (Fig. 4.10), and the rate of reduction of these disulfide bonds in ADCs by dithiothreitol (DTT) was measured. The study led to the conclusion that the stability of the disulfide linker, in the presence of reducing agent, increased with the level of steric hindrance. The introduction of one methyl group on each side (total two methyl groups) of the disulfide bond was shown to confer greater stability than two methyl groups on one side of the disulfide. The difference in stability despite the same total number of methyl groups/hindrance highlights the importance of the position of hindrance (Kellogg et al. 2011).

Increasing methyl hindrance and disulfide bond stability
Where are disulfide linkers reduced? The pKa of the thiol of reduced glutathione is 9.65. Hence, it is expected that the reduction will be inefficient in a low pH environment such as late endosomes and lysosomes (pH ~ 5). By contrast, the reduction is expected to be more efficient in a higher pH environment, as in cytoplasm (pH 7.4; Fig. 4.11). Consistent with this, no significant amount of reduction was reported to occur in lysosomes when probed with a conjugate linked to a pH-sensitive fluorophore via a disulfide linker (Austin et al. 2005; Yang et al. 2006). However, it has been demonstrated that in certain cell lines, there is some degree of reduction of the disulfide bond in endosomes, allowing an alternative pathway of metabolism before lysosomes and consequential advantage in cytotoxic effect (Maloney et al. 2009). Accordingly, significant reduction takes place in the endosomes for folate conjugated to fluorescence resonance energy transfer (FRET) fluorophores via a linker with a disulfide bond (Yang et al. 2006).

Metabolism of antibody–SPDB–DM4. SPDB N-succinimidyl-4-(2-pyridyldithio) butanoate
3.2.3 Peptide Linkers
Peptide linkers are attractive for several reasons. First, peptide linker s may allow easier release of the cytotoxic molecule, as cleavage of one bond within the peptide linker is sufficient to free the metabolite within cells, as opposed to noncleavable linker s that require a cleavage of two bonds, both the N- and C-termini of the conjugated amino acid residue. Second, unlike hydrophobic synthetic linkers, peptide linkers, with the appropriate choice of amino acids, can offer hydrophilicity allowing higher cytotoxin loading per antibody , and/or better solubility in combination with unusually hydrophobic cytotoxins.
Microtubule-targeting MMAE is often conjugated to an antibody via a linker containing a valine–citrulline (Val–Cit) dipeptide and a self-immolative ρ-aminobenzylcarbamate (PABC) spacer (Fig. 4.12). Upon hydrolysis of the Val–Cit peptide by lysosomal proteases such as cathepsin B, PABC–MMAE is released and subsequently undergoes self-immolation at the PABC site to generate MMAE, which can diffuse into neighboring cells and elicit bystander cytotoxicity (Fig. 4.13). When compared with antibodies conjugated to MMAE via the hydrazone of 5-benzoylvaleric acid-AE ester (AEVB), the ADC with the Val–Cit dipeptide linker showed better stability and greater specificity of ADC activity in vitro and in vivo (Doronina et al. 2003). Val–Cit–PABC is the linker used in brentuximab vedotin (SGN-35, Adcetris™), a CD30-targeting ADC (Fig. 4.14; Senter and Sievers 2012). Val–Cit with PABC spacer has also been conjugated to the DNA-interacting cytotoxin, doxorubicin, in the context of mAbs c1F6 (anti-CD70) and cAC10 (anti-CD30) for targeting renal cell carcinoma and anaplastic large cell lymphoma, respectively (Jeffrey et al. 2006).

mc–Val–Cit–PABC–MMAE. mc maleimidocaproyl, Val valine, Cit citrulline, MMAE monomethyl auristatin E

Metabolism and self-immolation of mc–Val–Cit–PABC–MMAE. mc maleimidocaproyl, Val valine, Cit citrulline, MMAE monomethyl auristatin E

Brentuximab vedotin
In addition to Val–Cit, variations of the dipeptide have been evaluated. Antibodies recognizing Lewis Y and CD30 on carcinomas were conjugated to MMAE via a linker containing phenylalanine–lysine with a PABC spacer. In a head-to-head comparison using the same antibodies, the ADC with the Phe–Lys linker conferred lower stability in mouse and human plasma compared to that with a Val–Cit linker as measured ex vivo, with projected half-lives of 12.5 days versus 30 days in mouse plasma, and 80 days versus 230 days in human plasma, respectively (Doronina et al. 2003).
Peptide linkers can be modified to confer hydrophilicity for conjugation of hydrophobic drugs such as DNA minor-groove-binding drugs (MGBs). The valine–lysine–tetraethyleneglycol and valine–lysine–para-aminobenzyl ether self-immolative spacer allowed the conjugation of MGBs to antibodies without forming aggregates (Jeffrey et al. 2005).
3.2.4 β-Glucuronide Linker
A β-glucuronic acid-based linker was used to conjugate MMAE, MMAF, and doxorubicin propyloxazoline individually to antibodies c1F6 (anti-CD70) and cAC10 (anti-CD30; Jeffrey et al. 2007). β-Glucuronides are hydrolyzed by β-glucuronidase, an enzyme that is abundantly present in lysosomes and is overexpressed in some tumors (Fig. 4.15; Albin et al. 1993), and is reported to be the main drug metabolizing enzyme systems in human breast tumors and peritumoral tissues. The hydrophilicity of this linker allows the generation of monomeric ADCs with as many as eight cytotoxins per antibody . In addition, the hydrophilicity of the glucuronide linker afforded conjugation of hydrophobic drugs such as DNA minor-groove binders (Jeffrey et al. 2005, 2007).

Metabolism of antibody-β glucuronide-cytotoxin
4 Effect of Linkers on the Activity and Safety of ADC
4.1 Linker and Intracellular Processing/Activation
Ideally, ADCs behave as prodrugs: They are designed to be inactive during systemic circulation in plasma, but become activated upon internalization inside cells (Fig. 4.16). In the first step, ADCs bind to antigens on the cell surface, and undergo endocytosis. Upon clathrin-mediated vesicle formation, ADCs are transported to endosomes (pH ~ 5–6). Subsequently, endosomes fuse with lysosomes (pH ~ 4), the compartment that is rich in enzymes responsible for degradation of proteins, e.g., proteases and esterases. Inside lysosomes, ADCs are degraded to generate metabolites that consist of the cytotoxic agent covalently linked to the amino acid site of conjugation (Erickson et al. 2006). For example, T-DM1 generates Lys–SMCC–DM1 , as the conjugation occurred between the primary amine of lysine and N-hydroxysuccinimide ester of SMCC (Fig. 4.5; Erickson et al. 2012).

Intracellular processing of antibody–drug conjugates
Linkers can be modified to take advantage of different modes of activation/metabolism. For example, conjugates containing peptide linker s may allow a faster rate of activation, as cleavage of one bond in the peptide linker is sufficient to release the cytotoxic agent, as opposed to ADCs with a noncleavable linker , which necessitates cleavage of two bonds at both N- and C-termini of the amino acid of attachment. Peptide linkers may also be designed so that the conjugates are metabolized in both endosomes and lysosomes, by altering the peptide sequence to match the substrate specificity of enzymes that are present in both compartments. For example, conjugates containing a peptide linker that can be cleaved by cathepsin B may be metabolized in both endosomes and lysosomes since cathepsin B is present in both compartments (Diederich et al. 2012). This may or may not be advantageous for the activity of ADC for the following reasons. Metabolism in both endosomes and lysosomes may allow faster activation that may afford an advantage in activity. However, if the linker is cleaved in the endosomes, it may also be susceptible to cleavage in the endosomal compartment during Fc recycling. Antibodies are recycled via neonatal Fc receptor (FcRN) binding in endosomes and are transported back to the cell surface thereby avoiding degradation in lysosomes. Such recycling of antibodies via FcRN is responsible for their long half-life in plasma (Lencer and Blumberg 2005). If the linker is cleaved in endosomes during recycling, the number of molecules of cytotoxic agent linked per antibody will decrease, which leads to (i) delivery of a lower amount of cytotoxic agent per antibody to cancer cells and (ii) toxicity caused by early release of free cytotoxin in normal tissues, with the degree of toxicity correlating to membrane permeability of the free cytotoxic agent. A similar activity and toxicity concern may apply to the acid-labile hydrazone linker, with an added concern for the lack of specificity; rather than relying on the enzyme specificity and its localized compartments (e.g., endosome), hydrazone linkers can be cleaved in any acidic environment.
4.2 Linkers to Overcome MDR
Treatment of cancer patients with chemotherapeutic reagents often leads ultimately to an MDR phenotype. The mechanism of the MDR phenotype varies, but overexpression of multidrug transporter MDR1 (also called P-glycoprotein) is the most commonly observed phenotype in the clinic. MDR1 is a membrane-associated transporter that confers drug resistance by mediating efflux of cytotoxic compounds. Many compounds used for ADCs, including calicheamicin (Matsui et al. 2002; Walter et al. 2003), doxorubicin, taxanes (Szakacs et al. 2006), maytansinoids (Tang et al. 2009), and analogs of dolastatin (Toppmeyer et al. 1994), are substrates of multidrug transporter MDR1, which poses a barrier to effective treatment of cancer patients with an ADC .
Linkers can be designed to evade the MDR1-mediated drug resistance. In a study to understand the effect of linkers on MDR1-dependent drug resistance, cell lines ranging from those naturally expressing a high level of MDR1, e.g., the colon adenocarcinoma HCT-15 and the renal adenocarcinoma UO-31, to an engineered cell line that mimics the high expression of MDR1, COLO 205MDR (MDR positive; parental COLO 205 is MDR negative), were used (Kovtun et al. 2010). The presence of MDR1 led to a 6–18-fold reduction in sensitivity to tubulin inhibitors including maytansine, paclitaxel, and vinblastine. Similarly, cells became resistant to anti-EpCAM–SMCC–DM1 compared to those co-treated with cyclosporin A that inhibits MDR1, indicating that MDR1 is also effective against the metabolite of the ADC , Lys–SMCC–DM1. The replacement of SMCC with PEG4Mal (Fig. 4.6) in an anti-EpCAM ADC led to enhanced cytotoxic activity in vitro and in HCT-15 and COLO 205MDR xenograft models in vivo. The sole metabolite generated by anti-EpCAM–PEG4Mal–DM1 was Lys–PEG4Mal–DM1, suggesting that the evasion of MDR phenotype is due to the hydrophilicity of the linker. The potency of the metabolites is expected to be the same for Lys–SMCC–DM1 and Lys–PEG4Mal–DM1, as anti-EpCAM–SMCC–DM1 and anti-EpCAM–PEG4Mal–DM1 display similar cytotoxic potency in non-MDR cell lines (Kovtun et al. 2010).
Evasion of MDR1-mediated drug resistance is not limited to noncleavable linker s. Recently, a hydrophilic disulfide linker was generated by modifying N-succinimidyl-4-(2-pyridyldithio) butanoate, SPDB, with a sulfonate group positioned distal to the disulfide bond (Fig. 4.17). When evaluated as an anti-EpCAM–sulfo-SPDB–DM4 conjugate, it showed better activity in the COLO 205MDR cell line and xenograft model than anti-EpCAM–SPDB–DM4, suggesting that the sulfo-SPDB linker is effective in overcoming MDR1-mediated drug resistance. A similar enhanced activity in MDR1-positive cell lines was also observed for huC242–sulfo-SPDB–DM4 targeting CanAg, a novel glycoform of Muc1 (Zhao et al. 2011).

Sulfo-SPDB linker that evades MDR resistance. SPDB N-succinimidyl-4-(2-pyridyldithio) butanoate, MDR multidrug resistant
In addition to MDR1-resistance, the hydrophilicity of PEG4Mal and sulfo-SPDB affords conjugation of a higher number of maytansinoid molecules per antibody (8–9 drugs per antibody; (Zhao et al. 2011)). Thus, these linkers may be particularly beneficial in conjugation of hydrophobic cytotoxins that may not have been previously feasible using other linkers. The ADC targeting the folate receptor (IMGN853) using DM4 linked to the antibody via a sulfo-SPDB linker is currently undergoing clinical evaluation in patients with ovarian carcinoma and nonsmall cell lung cancer.
4.3 Linkers to Improve Activity in Solid Tumors
Solid tumors are architecturally complex, often heterogeneous, and composed of different tissue types. These inherent properties of solid tumors can lead to heterogeneous expression of the target antigen, which limits the population of cancer cells that can be targeted by ADC . Even for tumors expressing the target antigen homogeneously, it has been documented that tumor penetration is not efficient due in part to the large molecular size of antibody -based therapies and uneven vasculature within tumors. When anti-Her2 antibody penetration was monitored in a MDA-435/LCC6HER2 xenograft model, despite homogeneous expression and immunohistochemical (IHC) staining of Her2, the fraction of Her2 bound by anti-Her2 antibody was patchy and localized (Baker et al. 2008). Thus, even solid tumors with homogeneous expression of the target antigen suffer from incomplete tumor penetration, which could limit the effectiveness of ADCs.
Linkers can be designed to help compensate for this. Thus far, two types of linkers have been designed to release cell-permeable free cytotoxic agents that can diffuse into neighboring cells and cause “bystander killing,” irrespective of whether these neighboring cells express target antigen. ADCs with bystander killing have often shown better activity in vivo compared with those without a bystander effect. This advantage in vivo has not always been apparent in vitro, where the bystander killing cannot be sufficiently recapitulated due to a difference in the architecture of two-dimensional tissue culture versus three-dimensional tumors.
One type of linker that can elicit bystander killing are disulfide linkers (Kellogg et al. 2011). The disulfide bonds in ADCs of the maytansinoid DM4 can be reduced by intracellular thiols to generate DM4 that can freely diffuse into neighboring cells, and if dividing, kill them. Furthermore, it has been demonstrated that DM4 undergoes methylation to form S-methyl DM4 in vitro and in vivo (Fig. 4.11; Erickson et al. 2006, 2010). Capping of DM4 through methylation may lead to an improved bystander effect: (i) methylation leads to formation of a noncharged compound that is readily membrane permeable and (ii) capping the free thiol of DM4 by methylation prevents possible disulfide exchange with endogenous disulfides such as cystine, which creates a hydrophilic charged compound, cysteinyl-DM4, that is not as membrane permeable. The ADC , huC242–N-succinimidyl 4-(2-pyridyldithio)pentanoate (SPP)–DM1 , wherein DM1 is linked via a disulfide bond using the SPP linker, displayed bystander killing as a result of the reduction of the disulfide to release DM1. This ADC showed better efficacy than the conjugate with a noncleavable linker , huC242–SMCC–DM1 in COLO 205 and HT-29 xenograft models expressing the CanAg antigen, suggesting that the bystander effect plays an important role in the antitumor activity in vivo (Kellogg et al. 2011).
Peptide linkers have been utilized to generate free cytotoxins that can elicit bystander killing. Conjugates containing Val–Cit–PABC–MMAE are cleaved at Val–Cit dipeptide and release p-aminobenzyloxycarbonyl (PABC)–MMAE, which can undergoes self-immolation to generate a noncharged MMAE molecule that can penetrate neighboring cells (Fig. 4.13; Sievers and Senter 2013).
4.4 Linker Stability in Plasma
Noncleavable linker s often employ a thioether bond formed by a Michael reaction between free sulfhydryl and maleimide groups, the examples of which are evident in maytansinoid and auristatin conjugates. Noncleavable linkers are considered relatively stable during circulation in plasma. However, recent data suggest that the stability of the thioether bond may vary for different linkages.
1F6-C4v2-mc–MMAF, an ADC targeting the CD70 antigen on lymphomas and renal cell carcinoma, is generated by reacting a maleimido group of mc–MMAF with the reduced thiol of cysteines that normally form interchain disulfides in IgG. When compared with IF6-C4v2–bac–MMAF, an ADC that uses a haloacetamido group instead of a maleimide, the IF6–C4v2–mc–MMAF conjugate showed a reduced serum concentration and drug exposure, and it was found that a portion of mc–MMAF becomes conjugated to cysteine 34 of serum albumin during incubation in plasma. It is speculated that IF6–C4v2–mc–MMAF undergoes a retro-Michael reaction, which releases the maleimide drug that subsequently reacts with cysteine of serum albumin (Alley et al. 2008).
A similar retro-Michael reaction has been implicated for the instability of site-specific conjugate with a Thiomab that utilizes engineered cysteine as a reaction site for maleimide. Interestingly, a conjugation site-dependent instability of the thiol–maleimide bond was observed. When conjugated to a solvent-exposed cysteine residue S396C of Fc, mc–vc–MMAE or mc-Alexa488 was released from the antibody at a higher rate than the same chemical moiety conjugated to V205C on a light chain that is located in a positively charged environment. This site-dependent loss of conjugated moiety from the antibody was accompanied by conjugation of the released “payload” to serum albumin, as modeled utilizing mc–Alexa488 conjugates, suggesting that a maleimide exchange has occurred between antibody and albumin. It was speculated that the thiol–maleimide bond in a solvent-exposed environment readily undergoes maleimide exchange in plasma, whereas maleimide in a positively charged environment undergoes succinimidyl ring opening, which prevents maleimide exchange (Shen et al. 2012b).
Maytansinoid conjugates have proven to be an exception. Although the thiol-containing maytansinoid, DM1 , utilizes the same thiol–maleimide chemistry, it is much more resistant to the retro-Michael reaction, likely owing to the higher pKa of the thiol donor, compared to cysteine residues on the antibody . A study to further understand the stability of the SMCC–DM1 thioether linkage led to the observation that free DM1 is released only by β-elimination following oxidation of the thioether bond to sulfoxide, which likely occurs in ex vivo conditions (Fishkin et al. 2011). It is suggested that the thioether oxidation is a potential ex vivo artifact that is less likely to occur in vivo where the redox potential of plasma is more tightly regulated.
The stability of the disulfide linkers depends on hindrance of the disulfide. As discussed previously (see Sect. 4.3.2.2.), the greater the degree of hindrance around the disulfide bond, the lower the propensity to reduction in vitro by DTT, and this stability correlates well with pharmacokinetics (PK) of the ADCs in vivo. HuC242–SPDB–DM4 (two methyl groups at the carbon next to the disulfide bond) shows a longer half-life in mouse plasma than huC242–SPDB–DM3 or huC242–SPP–DM1 (both contain one methyl group at the carbon adjoining the disulfide bond; Kellogg et al. 2011).
4.5 Linker Stability and Activity of ADCs
Do stable linkers provide better activity for ADCs? Preclinical findings suggest that although stable linkers may increase exposure of tumors to ADC , it is the careful balance between the resistance to extracellular cleavage (e.g., in plasma) and facility of intracellular cleavage (upon cellular internalization ) of linkers that provides the maximal activity.
The study of reducible linker s demonstrates this point elegantly. First, an ADC with SPP–DM4 with three methyl groups around the disulfide bond shows better stability against the thiol–disulfide exchange in vitro and longer half-life in plasma of 218 h compared with a SPP–DM1 conjugate, which has only one methyl group on the carbon atom adjacent to the disulfide bond (half-life at 47 h). Accordingly, the exposure for SPP–DM4 conjugate is greater than that of SPP–DM1, with AUC being 22,712 and 5186 h µg/mL, respectively. Yet, the huC242–SPP–DM1 shows better efficacy compared with huC242 antibody conjugated to SPP–DM4 in subcutaneous COLO 205 and HT-29 xenograft models. It is hypothesized that SPP–DM1 releases catabolites more readily than SPP–DM4 inside cells, such that the higher exposure of SPP–DM4 cannot compensate for the faster intracellular activation of SPP–DM1 (Kellogg et al. 2011).
Similar results were observed for αv integrin-targeting conjugates. CNTO365, using SPDB–DM4 with two methyl groups hindering thiol–disulfide exchange, showed better efficacy than CNTO366, using SPP–DM4 with three methyl groups hindering thiol–disulfide exchange, in HT-29 colon cancer and A-549 human lung cancer xenograft models (Chen et al. 2007).
Recent studies with T-DM1 illustrates that the linker stability alone does not predict efficacy. When compared against trastuzumab–SPP–DM1 (or T–SPP–DM1), T-DM1 (T–SMCC–DM1) showed better stability in plasma and longer half-life (Fig. 4.18). However, despite faster clearance and less total conjugate localization to tumors, T–SPP–DM1 showed a similar amount of metabolites generated at tumors (Fig. 4.18). As such, T-SPP-DM1 demonstrated similar efficacy as T–SMCC–DM1 in the BT474-EEI xenograft model (Erickson et al. 2012). It is clear that rate of activation inside the tumors and the total amount of metabolites are important predictive factors in antitumor activity. Moreover, different mechanisms of action for cell killing, i.e., bystander activity for T–SPP–DM1, but not for T-DM1, should also be considered in interpreting the efficacy data.

Pharmacokinetics of trastuzumab-SPP-DM1 (T-SPP-DM1) and T-DM1 (T-SMCC-DM1) and accumulation of the metabolites in the BT474 EEI-derived tumor xenograft model. SMCC N-succinimidyl-4-(maleimidomethyl) cyclohexane-1-carboxylate
In conclusion, efficacy conferred by the linker cannot be predicted based on one aspect of ADC behavior, such as PK, as a number of different factors can contribute to the activity of ADC. Thus, each ADC with different linkers must be tested empirically to determine the combined effect of PK, exposure, rate of intracellular activation, and mode of killing (e.g., bystander, etc.) in the context of each target.
4.6 Linker Stability and Safety
4.6.1 Effect of Linker on Liver Detoxification of ADC and Biodistribution
The liver is the primary site of antibody metabolism, and indeed, a significant amount of metabolites from ADCs are found in the liver. The anti-CD56 antibody, huN901, conjugated to maytansinoid via various linkers was used to study the effect of a linker on detoxification of ADC (Sun et al. 2011). A radioactive tracer, tritium, was incorporated at the C-20 methoxy group of maytansinoid to allow for the detection of metabolites. HuN901-SMCC-[3H]DM1 with a noncleavable linker was metabolized in liver to Lys–SMCC–[3H]DM1, which is more than 50-fold less cytotoxic than the parental compound due to poor cell penetration. Both huN901–SPP–[3H]DM1 and huN901–SPDB–[3H]DM4 containing disulfide linkers also generate initially the analogous lysine-linked maytansinoid. However, subsequent reduction, S-methylation, and nicotinamide adenine dinucleotide phosphate (NADPH)-dependent oxidation in the liver leads to the formation of S-methyl sulfoxide and S-methyl sulfone derivatives of maytansinoid (Fig. 4.19). When tested in vitro, these oxidized maytansinoids were found to be 5- to 50-fold less cytotoxic than parental maytansine in many human cancer cell lines, illustrating efficient detoxification of ADCs in liver (Sun et al. 2011).

Liver detoxification of antibody–maytansinoid conjugates
Biodistribution of ADC and the effect of linkers on the tissue distribution was assessed using the huC242 antibody targeting CanAg antigen. The unconjugated antibody, antibody–SPP–DM1 , and antibody–SPDB–DM4 were labeled with 125I on the antibody backbone to track the localization of ADC to various tissues. Following a single bolus injection of 4.16 mg/kg, it was found that the biodistribution profile is similar among all conjugates and the unconjugated antibody, demonstrating that ADC distribution is dictated by the antibody component (Xie et al. 2004; Xie and Blattler 2006). A closer look at huC242–SPDB–DM4 with a tritium label at the C-20 methoxy group of maytansinoid to follow the drug portion of ADC led to the finding that 30–50 % of injected dose/gram was recovered in the gall bladder from 2 h to 2 days post dosing, which is consistent with hepatobiliary elimination of maytansinoid (Erickson and Lambert 2012). Similar observations were made when rats were administered with a single bolus injection of T-[3H]DM1; up to 80 % of the radioactivity was recovered in the feces over 7 days, consistent with hepatobiliary elimination of maytansinoid (Shen et al. 2012a). The biodistribution profile of huC242–SMCC–[3H]DM1 resembled the profile of huC242–SPDB–[3H]DM4 (Erickson and Lambert 2012), suggesting a lack of significant contribution of linkers on biodistribution for the linkers tested. Thus, different degrees of toxicity conferred by SPP, SPDB, and SMCC linkers in mice may be due to the cell permeability and subsequent potency of the metabolites, rather than linker-dependent distribution of the ADCs.
4.6.2 Effect of Linkers on Safety in Clinic: Inverse Correlation of Stability and Safety
There are many ADCs with different linkers in the clinic today. The PK of the ADC in humans has been a reflection of both stability of the linker and antigen-mediated clearance. For maytansinoid conjugates containing disulfide linkers, the PK in humans has been consistent with the susceptibility of the linker to reduction by DTT and the PK observed in preclinical animals. Cantuzumab or anti-huC242 targeting CanAg antigen provides an ideal example, in which the same antibody has been conjugated with two linkers and the resulting conjugates have been evaluated in phase I clinical trials . Cantuzumab mertansine (huC242–SPP–DM1 ), which contains mildly hindered disulfide, has a half-life of 2 days in human plasma (Rodon et al. 2008). Cantuzumab ravtansine (huC242–SPDB–DM4 ), which contains a highly hindered disulfide, shows a half-life of 4.6 days (Qin et al. 2008). The PK of these conjugates in humans reflects the difference in the linker stability of SPP–DM1 and SPDB–DM4 in the context of CanAg. SAR3419 (huB4–SPDB–DM4) targeting CD19 shows 7.9 days of half-life in human plasma (Younes et al. 2009; Ribrag et al. 2014), indicating that the faster clearance of huC242–SPDB–DM4 (half-life of 4.6 days) is likely due to some contribution of antigen-mediated clearance rather than the inherent instability of the SPDB–DM4 linker–drug combination. Ado-trastuzumab emtansine has a half-life of 4.4 days in human plasma (Krop et al. 2010), reflecting largely the antigen-mediated clearance.
The maximum tolerated dose (MTD) can be affected by target-dependent toxicity, i.e., target expression on normal tissues could contribute to the final tolerable level of dose. However, different ADCs with the same linker-cytotoxic agent pairing directed against unrelated targets with diverse expression in tissues demonstrate that pairings can create an upper limit for the highest administered dose. For example, the MTD for auristatin conjugates containing dipeptide Val–Cit linker is typically close to 2 mg/kg (Younes et al. 2010) due to neutropenia and/or peripheral neuropathy. Maytansinoid conjugates also show a strong linker impact on tolerability. T-DM1 and AMG595, which use the SMCC–DM1 pairing, both have dose-limiting toxicity (DLT) of reversible thrombocytopenia; the MTD for T-DM1 is 3.6 mg/kg (Q3W; Krop et al. 2010) and AMG595 has been dosed to 3.0 mg/kg (Q3W). SAR3419 or huB4–SPDB–DM4 with a cleavable disulfide linker shows an MTD of 4.3 mg/kg (Q3W) with the DLT of reversible ocular toxicity (Younes et al. 2009). Cantuzumab mertansine or anti-huC242–SPP–DM1 with the most readily cleavable disulfide linker had an MTD of 6.4 mg/kg (Q3W) with the DLT of reversible elevation of liver transaminases (Rodon et al. 2008). Interestingly, there is an inverse correlation of the tolerability and the stability of linkers (Fig. 4.20). The chemical stability of the linkers in plasma (in vivo) can be ranked as SMCC > SPDB > SPP, with SMCC being the most stable linker and SPP being the least stable linker. In contrast to the chemical stability in plasma, the tolerability as demonstrated by the MTDs in the clinic for the maytansinoid conjugates listed above can be ranked as SPP > SPDB > SMCC. More clinical data are needed to determine whether (i) this stands true for all maytansinoid conjugates that may yield different small molecular weight metabolites during their eventual elimination and (ii) whether a similar trend can be found for other cytotoxin payloads. These findings suggest that it should not be hastily concluded that the most stable linker is the best linker for clinical development , and as has discussed above, stability, efficacy, and safety must be all factored in for consideration of the optimal linker for ADCs.

Inverse correlation of the linker stability and MTD in human. Number of compounds refers to those that have been evaluated in the clinic. MTD maximum tolerated dose
References
Albin N, Massaad L, Toussaint C, Mathieu MC, Morizet J, Parise O, Gouyette A, Chabot GG (1993) Main drug-metabolizing enzyme systems in human breast tumors and peritumoral tissues. Cancer Res 53(15):3541–3546
Alley SC, Benjamin DR, Jeffrey SC, Okeley NM, Meyer DL, Sanderson RJ, Senter PD (2008) Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug Chem 19(3):759–765. doi:10.1021/bc7004329
Austin CD, Wen X, Gazzard L, Nelson C, Scheller RH, Scales SJ (2005) Oxidizing potential of endosomes and lysosomes limits intracellular cleavage of disulfide-based antibody-drug conjugates. Proc Natl Acad Sci USA 102(50):17987–17992. doi:10.1073/pnas.0509035102
Axup JY, Bajjuri KM, Ritland M, Hutchins BM, Kim CH, Kazane SA, Halder R, Forsyth JS, Santidrian AF, Stafin K, Lu Y, Tran H, Seller AJ, Biroc SL, Szydlik A, Pinkstaff JK, Tian F, Sinha SC, Felding-Habermann B, Smider VV, Schultz PG (2012) Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc Natl Acad Sci USA 109(40):16101–16106. doi:10.1073/pnas.1211023109
Baker JH, Lindquist KE, Huxham LA, Kyle AH, Sy JT, Minchinton AI (2008) Direct visualization of heterogeneous extravascular distribution of trastuzumab in human epidermal growth factor receptor type 2 overexpressing xenografts. Clinical Cancer Res 14(7):2171–2179. doi:10.1158/1078-0432.CCR-07-4465
Boeggeman E, Ramakrishnan B, Pasek M, Manzoni M, Puri A, Loomis KH, Waybright TJ, Qasba PK (2009) Site specific conjugation of fluoroprobes to the remodeled Fc N-glycans of monoclonal antibodies using mutant glycosyltransferases: application for cell surface antigen detection. Bioconjug Chem 20(6):1228–1236. doi:10.1021/bc900103p
Chen Q, Millar HJ, McCabe FL, Manning CD, Steeves R, Lai K, Kellogg B, Lutz RJ, Trikha M, Nakada MT, Anderson GM (2007) Alphav integrin-targeted immunoconjugates regress established human tumors in xenograft models. Clinical Cancer Res 13(12):3689–3695. doi:10.1158/1078-0432.CCR-07-0026
Dennler P, Chiotellis A, Fischer E, Bregeon D, Belmant C, Gauthier L, Lhospice F, Romagne F, Schibli R (2014) Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody-drug conjugates. Bioconjug Chem 25(3):569–578. doi:10.1021/bc400574z
Diederich S, Sauerhering L, Weis M, Altmeppen H, Schaschke N, Reinheckel T, Erbar S, Maisner A (2012) Activation of the Nipah virus fusion protein in MDCK cells is mediated by cathepsin B within the endosome-recycling compartment. J Virol 86(7):3736–3745. doi:10.1128/JVI.06628-11
DiJoseph JF, Armellino DC, Boghaert ER, Khandke K, Dougher MM, Sridharan L, Kunz A, Hamann PR, Gorovits B, Udata C, Moran JK, Popplewell AG, Stephens S, Frost P, Damle NK (2004a) Antibody-targeted chemotherapy with CMC-544: a CD22-targeted immunoconjugate of calicheamicin for the treatment of B-lymphoid malignancies. Blood 103(5):1807–1814. doi:10.1182/blood-2003-07-2466
DiJoseph JF, Goad ME, Dougher MM, Boghaert ER, Kunz A, Hamann PR, Damle NK (2004b) Potent and specific antitumor efficacy of CMC-544, a CD22-targeted immunoconjugate of calicheamicin, against systemically disseminated B-cell lymphoma. Clinical Cancer Res 10(24):8620–8629. doi:10.1158/1078-0432.CCR-04-1134
Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, DeBlanc RL, Gearing RP, Bovee TD, Siegall CB, Francisco JA, Wahl AF, Meyer DL, Senter PD (2003) Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol 21(7):778–784. doi:10.1038/nbt832[pii]
Erickson HK, Lambert JM (2012) ADME of antibody-maytansinoid conjugates. Aaps J 14(4):799–805. doi:10.1208/s12248-012-9386-x
Erickson HK, Park PU, Widdison WC, Kovtun YV, Garrett LM, Hoffman K, Lutz RJ, Goldmacher VS, Blattler WA (2006) Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res 66(8):4426–4433. doi:10.1158/0008-5472.CAN-05-4489
Erickson HK, Widdison WC, Mayo MF, Whiteman K, Audette C, Wilhelm SD, Singh R (2010) Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody-maytansinoid conjugates. Bioconjug Chem 21(1):84–92. doi:10.1021/bc900315y
Erickson HK, Lewis Phillips GD, Leipold DD, Provenzano CA, Mai E, Johnson HA, Gunter B, Audette CA, Gupta M, Pinkas J, Tibbitts J (2012) The effect of different linkers on target cell catabolism and pharmacokinetics/pharmacodynamics of trastuzumab maytansinoid conjugates. Mol Cancer Ther 11(5):1133–1142. doi:10.1158/1535-7163.MCT-11-0727
Fishkin N, Maloney EK, Chari RV, Singh R (2011) A novel pathway for maytansinoid release from thioether linked antibody-drug conjugates (ADCs) under oxidative conditions. Chem Commun (Camb) 47(38):10752–10754. doi:10.1039/c1cc14164c
Greenberg MM (2014) Abasic and oxidized abasic site reactivity in DNA: enzyme inhibition, cross-linking, and nucleosome catalyzed reactions. Acc Chem Res 47(2):646–655. doi:10.1021/ar400229d
Hamann PR, Hinman LM, Beyer CF, Greenberger LM, Lin C, Lindh D, Menendez AT, Wallace R, Durr FE, Upeslacis J (2005a) An anti-MUC1 antibody-calicheamicin conjugate for treatment of solid tumors. Choice of linker and overcoming drug resistance. Bioconjug Chem 16(2):346–353. doi:10.1021/bc049795f
Hamann PR, Hinman LM, Beyer CF, Lindh D, Upeslacis J, Shochat D, Mountain A (2005b) A calicheamicin conjugate with a fully humanized anti-MUC1 antibody shows potent antitumor effects in breast and ovarian tumor xenografts. Bioconjug Chem 16(2):354–360. doi:10.1021/bc049794n
Hinman LM, Hamann PR, Wallace R, Menendez AT, Durr FE, Upeslacis J (1993) Preparation and characterization of monoclonal antibody conjugates of the calicheamicins: a novel and potent family of antitumor antibiotics. Cancer Res 53(14):3336–3342
Hutchins BM, Kazane SA, Staflin K, Forsyth JS, Felding-Habermann B, Schultz PG, Smider VV (2011) Site-specific coupling and sterically controlled formation of multimeric antibody fab fragments with unnatural amino acids. J Mol Biol 406(4):595–603. doi:10.1016/j.jmb.2011.01.011
Jain N, O'Brien S, Thomas D, Kantarjian H (2014) Inotuzumab ozogamicin in the treatment of acute lymphoblastic leukemia. Front Biosci (Elite Ed) 6:40–45
Jeffrey SC, Torgov MY, Andreyka JB, Boddington L, Cerveny CG, Denny WA, Gordon KA, Gustin D, Haugen J, Kline T, Nguyen MT, Senter PD (2005) Design, synthesis, and in vitro evaluation of dipeptide-based antibody minor groove binder conjugates. J Med Chem 48(5):1344–1358. doi:10.1021/jm040137q
Jeffrey SC, Nguyen MT, Andreyka JB, Meyer DL, Doronina SO, Senter PD (2006) Dipeptide-based highly potent doxorubicin antibody conjugates. Bioorg Med Chem Lett 16(2):358–362. doi:10.1016/j.bmcl.2005.09.081
Jeffrey SC, Nguyen MT, Moser RF, Meyer DL, Miyamoto JB, Senter PD (2007) Minor groove binder antibody conjugates employing a water soluble beta-glucuronide linker. Bioorg Med Chem Lett 17(8):2278–2280. doi:10.1016/j.bmcl.2007.01.071
Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, Lu Y, Meng YG, Ng C, Yang J, Lee CC, Duenas E, Gorrell J, Katta V, Kim A, McDorman K, Flagella K, Venook R, Ross S, Spencer SD, Lee Wong W, Lowman HB, Vandlen R, Sliwkowski MX, Scheller RH, Polakis P, Mallet W (2008) Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol 26(8):925–932. doi:10.1038/nbt.1480
Junutula JR, Flagella KM, Graham RA, Parsons KL, Ha E, Raab H, Bhakta S, Nguyen T, Dugger DL, Li G, Mai E, Lewis Phillips GD, Hiraragi H, Fuji RN, Tibbitts J, Vandlen R, Spencer SD, Scheller RH, Polakis P, Sliwkowski MX (2010) Engineered thio-trastuzumab-DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2-positive breast cancer. Clinical Cancer Res 16(19):4769–4778. doi:10.1158/1078-0432.CCR-10-0987
Kellogg BA, Garrett L, Kovtun Y, Lai KC, Leece B, Miller M, Payne G, Steeves R, Whiteman KR, Widdison W, Xie H, Singh R, Chari RV, Lambert JM, Lutz RJ (2011) Disulfide-linked antibody-maytansinoid conjugates: optimization of in vivo activity by varying the steric hindrance at carbon atoms adjacent to the disulfide linkage. Bioconjug Chem 22(4):717–727. doi:10.1021/bc100480a
Kovtun YV, Audette CA, Mayo MF, Jones GE, Doherty H, Maloney EK, Erickson HK, Sun X, Wilhelm S, Ab O, Lai KC, Widdison WC, Kellogg B, Johnson H, Pinkas J, Lutz RJ, Singh R, Goldmacher VS, Chari RV (2010) Antibody-maytansinoid conjugates designed to bypass multidrug resistance. Cancer Res 70(6):2528–2537. doi:10.1158/0008-5472.CAN-09-3546
Krop IE, Beeram M, Modi S, Jones SF, Holden SN, Yu W, Girish S, Tibbitts J, Yi JH, Sliwkowski MX, Jacobson F, Lutzker SG, Burris HA (2010) Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol 28(16):2698–2704. doi:10.1200/JCO.2009.26.2071
Kung Sutherland MS, Walter RB, Jeffrey SC, Burke PJ, Yu C, Kostner H, Stone I, Ryan MC, Sussman D, Lyon RP, Zeng W, Harrington KH, Klussman K, Westendorf L, Meyer D, Bernstein ID, Senter PD, Benjamin DR, Drachman JG, McEarchern JA (2013) SGN-CD33A: a novel CD33-targeting antibody-drug conjugate utilizing a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood. doi:10.1182/blood-2013-03-491506
Lambert JM (2013) Drug-conjugated antibodies for the treatment of cancer. Br J Clin Pharmacol 76(2):248–262. doi:10.1111/bcp.12044
Lencer WI, Blumberg RS (2005) A passionate kiss, then run: exocytosis and recycling of IgG by FcRn. Trends Cell Biol 15(1):5–9. doi:10.1016/j.tcb.2004.11.004
Maloney E, Fishkin N, Chari R, Singh R (2009) Abstract B120: designing potent antibody-drug conjugates: the impact of lysosomal processing efficiency and conjugate linker selection on anticancer activity. Mol Cancer Ther 8(Suppl 1):B120. doi:10.1158/1535-7163.targ-09-b120
Matsui H, Takeshita A, Naito K, Shinjo K, Shigeno K, Maekawa M, Yamakawa Y, Tanimoto M, Kobayashi M, Ohnishi K, Ohno R (2002) Reduced effect of gemtuzumab ozogamicin (CMA-676) on P-glycoprotein and/or CD34-positive leukemia cells and its restoration by multidrug resistance modifiers. Leukemia 16(5):813–819. doi:10.1038/sj.leu.2402459
Mills BJ, Lang CA (1996) Differential distribution of free and bound glutathione and cyst(e)ine in human blood. Biochem Pharmacol 52(3):401–406. doi:0006-2952(96)00241-9[pii]
Pilorge S, Rigaudeau S, Rabian F, Sarkozy C, Taksin AL, Farhat H, Merabet F, Ghez S, Raggueneau V, Terre C, Garcia I, Renneville A, Preudhomme C, Castaigne S, Rousselot P (2014) Fractionated gemtuzumab ozogamicin and standard dose cytarabine produced prolonged second remissions in patients over the age of 55 years with acute myeloid leukemia in late first relapse. Am J Hematol 89(4):399–403. doi:10.1002/ajh.23653
Qin A, Watermill J, Mastico RA, Lutz RJ, O’Keeffe J, Zildjian S, Mita AC, Phan AT, Tolcher AW (2008) The pharmacokinetics and pharmacodynamics of IMGN242 (huC242-DM4) in patients with CanAg-expressing solid tumors. Paper presented at the ASCO Meet Abstr
Rabuka D, Rush JS, deHart GW, Wu P, Bertozzi CR (2012) Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat Protoc 7(6):1052–1067. doi:10.1038/nprot.2012.045
Ribrag V, Dupuis J, Tilly H, Morschhauser F, Laine F, Houot R, Haioun C, Copie C, Varga A, Lambert J, Hatteville L, Ziti-Ljajic S, Caron A, Payrard S, Coiffier B (2014) A dose-escalation study of SAR3419, an anti-CD19 antibody maytansinoid conjugate, administered by intravenous infusion once weekly in patients with relapsed/refractory B-cell non-Hodgkin lymphoma. Clinical Cancer Res 20(1):213–220. doi:10.1158/1078-0432.CCR-13-0580
Ricart AD (2011) Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clinical Cancer Res 17(20):6417–6427. doi:10.1158/1078-0432.CCR-11-0486
Rodon J, Garrison M, Hammond LA, de Bono J, Smith L, Forero L, Hao D, Takimoto C, Lambert JM, Pandite L, Howard M, Xie H, Tolcher AW (2008) Cantuzumab mertansine in a three-times a week schedule: a phase I and pharmacokinetic study. Cancer Chemother Pharmacol 62(5):911–919. doi:10.1007/s00280-007-0672-8
Sapra P, Stein R, Pickett J, Qu Z, Govindan SV, Cardillo TM, Hansen HJ, Horak ID, Griffiths GL, Goldenberg DM (2005) Anti-CD74 antibody-doxorubicin conjugate, IMMU-110, in a human multiple myeloma xenograft and in monkeys. Clinical Cancer Res 11(14):5257–5264. doi:10.1158/1078-0432.CCR-05-0204
Senter PD, Sievers EL (2012) The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat Biotechnol 30(7):631–637. doi:10.1038/nbt.2289
Shen BQ, Bumbaca D, Saad O, Yue Q, Pastuskovas CV, Khojasteh SC, Tibbitts J, Kaur S, Wang B, Chu YW, LoRusso PM, Girish S (2012a) Catabolic fate and pharmacokinetic characterization of trastuzumab emtansine (T-DM1): an emphasis on preclinical and clinical catabolism. Curr Drug Metab 13(7):901–910
Shen BQ, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu SF, Mai E, Li D, Tibbitts J, Baudys J, Saad OM, Scales SJ, McDonald PJ, Hass PE, Eigenbrot C, Nguyen T, Solis WA, Fuji RN, Flagella KM, Patel D, Spencer SD, Khawli LA, Ebens A, Wong WL, Vandlen R, Kaur S, Sliwkowski MX, Scheller RH, Polakis P, Junutula JR (2012b) Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol 30(2):184–189. doi:10.1038/nbt.2108
Sievers EL, Senter PD (2013) Antibody-drug conjugates in cancer therapy. Annu Rev Med 64:15–29. doi:10.1146/annurev-med-050311-201823
Singh R, Erickson HK (2009) Antibody-cytotoxic agent conjugates: preparation and characterization. Methods Mol Biol 525:445–467, xiv. doi:10.1007/978-1-59745-554-1_23
Strop P, Liu SH, Dorywalska M, Delaria K, Dushin RG, Tran TT, Ho WH, Farias S, Casas MG, Abdiche Y, Zhou D, Chandrasekaran R, Samain C, Loo C, Rossi A, Rickert M, Krimm S, Wong T, Chin SM, Yu J, Dilley J, Chaparro-Riggers J, Filzen GF, O'Donnell CJ, Wang F, Myers JS, Pons J, Shelton DL, Rajpal A (2013) Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol 20(2):161–167. doi:10.1016/j.chembiol.2013.01.010
Sun X, Widdison W, Mayo M, Wilhelm S, Leece B, Chari R, Singh R, Erickson H (2011) Design of antibody-maytansinoid conjugates allows for efficient detoxification via liver metabolism. Bioconjug Chem 22(4):728–735. doi:10.1021/bc100498q
Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM (2006) Targeting multidrug resistance in cancer. Nat Rev Drug Discov 5(3):219–234. doi:10.1038/nrd1984
Tang R, Cohen S, Perrot JY, Faussat AM, Zuany-Amorim C, Marjanovic Z, Morjani H, Fava F, Corre E, Legrand O, Marie JP (2009) P-gp activity is a critical resistance factor against AVE9633 and DM4 cytotoxicity in leukaemia cell lines, but not a major mechanism of chemoresistance in cells from acute myeloid leukaemia patients. BMC Cancer 9:199. doi:10.1186/1471-2407-9-199
Toppmeyer DL, Slapak CA, Croop J, Kufe DW (1994) Role of P-glycoprotein in dolastatin 10 resistance. Biochem Pharmacol 48(3):609–612
Turell L, Carballal S, Botti H, Radi R, Alvarez B (2009) Oxidation of the albumin thiol to sulfenic acid and its implications in the intravascular compartment. Braz J Med Biol Res 42(4):305–311
Walter RB, Raden BW, Hong TC, Flowers DA, Bernstein ID, Linenberger ML (2003) Multidrug resistance protein attenuates gemtuzumab ozogamicin-induced cytotoxicity in acute myeloid leukemia cells. Blood 102(4):1466–1473. doi:10.1182/blood-2003-02-0396
Walus LR, Pardridge WM, Starzyk RM, Friden PM (1996) Enhanced uptake of rsCD4 across the rodent and primate blood-brain barrier after conjugation to anti-transferrin receptor antibodies. J Pharmacol Exp Ther 277(2):1067–1075
Wang L, Amphlett G, Blattler WA, Lambert JM, Zhang W (2005) Structural characterization of the maytansinoid-monoclonal antibody immunoconjugate, huN901-DM1, by mass spectrometry. Protein Sci 14(9):2436–2446. doi:10.1110/ps.051478705
Wu G, Fang YZ, Yang S, Lupton JR, Turner ND (2004) Glutathione metabolism and its implications for health. J Nutr 134(3):489–492
Xie H, Blattler WA (2006) In vivo behaviour of antibody-drug conjugates for the targeted treatment of cancer. Expert Opin Biol Ther 6(3):281–291. doi:10.1517/14712598.6.3.281
Xie H, Audette C, Hoffee M, Lambert JM, Blattler WA (2004) Pharmacokinetics and biodistribution of the antitumor immunoconjugate, cantuzumab mertansine (huC242-DM1), and its two components in mice. J Pharmacol Exp Ther 308(3):1073–1082. doi:10.1124/jpet.103.060533
Yang J, Chen H, Vlahov IR, Cheng JX, Low PS (2006) Evaluation of disulfide reduction during receptor-mediated endocytosis by using FRET imaging. Proc Natl Acad Sci U S A 103(37):13872–13877. doi:10.1073/pnas.0601455103
Younes A, Gordon L, Kim S, Romaguera J, Copeland AR, de Castro Farial S, Kwak L, Fayad L, Hagemeister F, Fanale M, Lambert J (2009) Phase I multi-dose escalation study of the anti-CD19 maytansinoid immunoconjugate SAR3419 administered by intravenous (IV) infusion every 3 weeks to patients with relapsed/refractory B-Cell non-Hodgkin's lymphoma (NHL). Paper presented at the ASH Annu Meet Abstr
Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL, Forero-Torres A (2010) Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med 363(19):1812–1821. doi:10.1056/NEJMoa1002965
Zhao RY, Wilhelm SD, Audette C, Jones G, Leece BA, Lazar AC, Goldmacher VS, Singh R, Kovtun Y, Widdison WC, Lambert JM, Chari RV (2011) Synthesis and evaluation of hydrophilic linkers for antibody-maytansinoid conjugates. J Med Chem 54(10):3606–3623. doi:10.1021/jm2002958
Zimmerman ES, Heibeck TH, Gill A, Li X, Murray CJ, Madlansacay MR, Tran C, Uter NT, Yin G, Rivers PJ, Yam AY, Wang WD, Steiner AR, Bajad SU, Penta K, Yang W, Hallam TJ, Thanos CD, Sato AK (2014) Production of site-specific antibody-drug conjugates using optimized non-natural amino acids in a cell-free expression system. Bioconjug Chem 25(2):351–361. doi:10.1021/bc400490z
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 American Association of Pharmaceutical Scientists
About this chapter
Cite this chapter
Hong, E., Chari, R. (2015). Linker Design for Antibody–Drug Conjugates. In: Wang, J., Shen, WC., Zaro, J. (eds) Antibody-Drug Conjugates. AAPS Advances in the Pharmaceutical Sciences Series, vol 17. Springer, Cham. https://doi.org/10.1007/978-3-319-13081-1_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-13081-1_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-13080-4
Online ISBN: 978-3-319-13081-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)