
Abstract
Cancer immunotherapy aims to harness the innate ability of the immune system to recognize and destroy malignant cells. Immunotherapy for malignant gliomas is an emerging field that promises the possibility of highly specific and less toxic treatment compared to conventional chemotherapy. In addition, immunotherapy has the added benefit of sustained efficacy once immunologic memory is induced. Although there are numerous therapeutic agents that boost general immune function and facilitate improved antitumor immunity, to date, immunotherapy for gliomas has focused primarily on active vaccination against tumor-specific antigens. The results of numerous early phase clinical trials demonstrate promising results for vaccine therapy, but no therapy has yet proven to improve survival in a randomized, controlled trial. The major barrier to immunotherapy in malignant gliomas is tumor-induced immunosuppression. The mechanisms of immunosuppression are only now being elucidated, but clearly involve a combination of factors including regulatory T cells, tumor-associated PD-L1 expression, and CTLA-4 signaling. Immunomodulatory agents have been developed to combat these immunosuppressive factors and have demonstrated efficacy in other cancers. The future of glioma immunotherapy likely lies in a combination of active vaccination and immune checkpoint inhibition.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Cancer Immunotherapy
Under normal conditions, the immune system scavenges the body not only for infection, but also for mutations and malignant degeneration of its own cells. Immune surveillance and immunoediting keeps mutations in check, preventing true malignancy before it begins [1]. Most systemic cancers arise only when neoplastic cells escape immune control [2]. It is well recognized that patients with defects in normal immune function are prone to developing multiple malignancies [3].
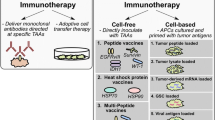
Cancer immunotherapy is broadly defined as any therapy that utilizes the immune system to destroy malignant cells, thereby reestablishing immune control of tumor growth [4]. Immunotherapy can be passive or active. Passive immunotherapy employs administration of antibodies or activated immune cells that target a specific antigen expressed by the tumor. For example, monoclonal antibodies such as trastuzumab target the HER2/neu receptor in breast cancer leading to signaling blockade and arrest of tumor proliferation [5]. Adoptive transfer of modified chimeric antigen receptor (CAR) T cells utilizes immune cells that are generated and expanded ex vivo to attack and destroy malignancies [6]. These passive immunotherapy strategies rely on introduction of exogenous immune factors not replicated by the patient’s own immune system. Their limitation, like any chemotherapeutic agent, is that efficacy only lasts as long as the agent is given. In addition, they target a single tumor marker, selecting for growth of tumor cells that do not express this marker. In contrast, active immunotherapy employs the immune system’s innate response to the tumor. Active immunotherapy may consist of immunomodulatory agents that boost already established but insufficient immune responses, or may involve vaccination against tumor antigens to educate the immune system to recognize new tumor targets [7]. Active immunotherapy may have a single or multiple antigenic targets, and can have sustained efficacy long after the therapy is given.
The primary effector of the innate immune response to malignancy is the cytolytic CD8+ T cell (CTL) [8]. Clonal populations of CTLs with T cell receptors that specifically recognize tumor antigens can bind to class I major histocompatibility complexes (MHC) expressing the antigen on the tumor surface. This results in tumor cell destruction through a variety of mechanisms [9]. Education of tumor-specific CTLs begins in the peripheral circulation, where antigen presenting cells (APC) displaying tumor-specific peptides encounter naïve T cells. When this binding occurs in the presence of the appropriate costimulatory factors, clonal expansion and activation of tumor-specific T cells results.
Some immunomodulatory agents nonspecifically enhance the natural process of T cell education and activation/expansion to boost the innate immune response. Agents such as interleukin-2 (IL-2) facilitate clonal expansion of T cells once appropriate binding to the APC occurs [10]. IL-2 immunotherapy has shown significant promise for the treatment of renal cell carcinoma and melanoma [11, 12]. Other agents such as interferons and toll-like receptor (TLR) agonists, nonspecifically activate proinflammatory pathways in multiple immune cells, resulting in increased T cell activity [13]. The latest trend in immunotherapy has been modulation of immune checkpoints intended to prevent overactivation of the immune response and autoimmunity [14]. Agents such as ipilumumab that inhibit CTLA-4 signaling appear to boost T cell-mediated immunity and have shown significant responses in patients with melanoma [15]. An alternative approach to nonspecifically boosting immunity, is to direct an immune response against a specific tumor antigen by vaccination. The first cancer vaccine, which utilized dendritic cells (DCs) pulsed with antigenic peptides targeting prostate, was approved by the FDA in 2010 [16]. Tumor vaccines can be generated to target tumor-specific markers, and have the added benefit of generating immunologic memory for sustained efficacy even after the vaccination period is over.
1.1 Immunotherapy for Gliomas
Active immunotherapy relies on systemic exposure to tumor-specific antigens and sufficient costimulus for activation of CTLs. For most systemic cancers, recognition of tumor antigens occurs naturally as part of routine immunoediting [17]. In contrast, the brain is a relatively immune-privileged space, protected behind the blood—brain barrier (BBB). Rather than being scavenged by DCs, foreign antigen recognition in the brain is usually left up to resident microglia and astrocytes that express TLRs [18, 19]. Only once an inflammatory response is initiated and cytokines are produced by resident cells, does the BBB become permeable to peripheral immune cells [20]. High-grade gliomas, associated with significant necrosis and inflammation, are known to be infiltrated by peripheral immune effector cells [21]. The majority of immune cells in high-grade tumors are macrophages, with T cells representing only 5–10 % of infiltrating cells [22]. Therefore, unlike most systemic tumors that are recognized by the immune system prior to escaping immune surveillance, glial tumor antigens may not be recognized by the peripheral immune system until the tumors have substantially progressed and become highly inflammatory. Even once high-grade tumors are infiltrated by immune effectors, it is unclear how well tumor antigens are presented by APCs in circulation. Therefore, the primary modality of immunotherapy for gliomas to date has been active tumor vaccination rather than the use of immunomodulatory agents. Vaccination ensures that the immune system is educated against tumor-specific peptides rather than nonspecifically activated.
2 Glioma Vaccines
Numerous vaccines targeting glioblastoma (GBM) have been tested in clinical trials (Table 1). The various vaccines differ in the antigenic peptides that they target and the method by which the antigen is delivered to the immune system. Ultimately, a successful vaccine must deliver a tumor-specific peptide(s) to the patient’s APCs such that the peptide is displayed on class I MHC and presented to naïve T cells in circulation for education and clonal expansion. This can be accomplished by delivering tumor cell fragments or naked peptides systemically along with an immune adjuvant that stimulates uptake by circulating APCs and T cell proliferation. Alternatively, DCs can be extracted by plasmapheresis and pulsed with tumor antigens ex vivo. The antigen presenting DCs can then be reintroduced into circulation to activate T cells. Finally, antigens can be delivered to DCs in circulation using specialized antigen carriers such as heat shock proteins (HSP). Each of these approaches has been used as a vaccine modality for GBM in a clinical trial.
2.1 Peptide Vaccines
Systemic delivery of tumor-specific peptides or cell fragments can be used to educate and activate CTLs through uptake and presentation by dermal APCs. The process of uptake and expression of the antigenic peptides is enhanced by conjugating them to immune stimulants, such as keyhole limpet hemocyanin (KLH), and/or simultaneously administering leukocyte growth factors, such as GM-CSF or IL-2. The key to this approach is delivering peptides that are significantly expressed and highly specific for the tumor. Vaccines can be created by selecting antigenic target(s) and generating synthetic peptides, by extracting specific peptides from tumor lysates, or by nonspecifically delivering tumor cell lysates with a variety of antigenic peptides.
The most investigated target of a specific peptide vaccine in GBM is the tumor variant epidermal growth factor receptor (EGFRvIII). Amplification and overactivation of EGFR is a common mutation seen in GBM [23]. Approximately 30–40 % of GBM patients express an aberrant receptor, EGFRvIII, which remains constitutively active regardless of ligand binding, driving cell activity [24]. EGFRvIII is only expressed by GBM cells, making it an ideal target for vaccine therapy. A peptide vaccine containing a 14 amino acid antigenic sequence for EGFRvIII conjugated to KHL (rindopepimut) has been studied in phase II and III trials for newly diagnosed and recurrent GBM [25]. In the ACTIVATE trial, 18 patients received the vaccine and concurrent GM-CSF for newly diagnosed GBM following surgical resection and standard concurrent temozolomide chemotherapy and conformal radiotherapy (NCT00643097). The median progression-free survival (PFS) in the study was 12.3 months. In the ACT II trial, 22 patients received the vaccine after standard therapy and went on to adjuvant temozolomide therapy after progression (NCT00643097). Median PFS was 15.3 months and overall survival (OS) was 20.5 months [26]. The primary outcome measure in these trials was PFS, which faired favorably against historical control data from disease-matched patients with a median PFS of 6.4 months. These results led to the ACT III study, a phase II trial of the vaccine following surgery and chemoradiotherapy for newly diagnosed GBM, given concurrently with maintenance temozolomide (NCT00458601). The primary outcome for this trial was also PFS, with a median PFS of 12.3 months and a median OS of 21.8 months. A phase III trial of the EGFRvIII vaccine with adjuvant temozolomide versus placebo with temozolomide is currently ongoing (ACT IV; NCT01480479). Additionally, a phase II trial of the vaccine with bevacizumab versus bevacizumab alone for recurrent GBM in adults is ongoing (ReACT; NCT01498328).
In addition to EGFR, a number of other target proteins have been used to develop peptide vaccines for GBM. A Japanese phase II trial of a peptide vaccine targeting the Wilms Tumor (WT1) protein in recurrent gliomas demonstrated a median PFS of 5 months [27]. A phase I trial of a WT vaccine for advanced solid malignancies including GBM is now underway in the United States. Other phase I trials targeting a survivin peptide, CMV antigens, and telomerase are ongoing (see Table 1). The challenge for peptide vaccines with a single target is that their use is limited to patients who express the target. In the case of EGFRvIII, only 30–40 % of patients are eligible to receive the vaccine based on target protein expression. Furthermore, GBM is known to have significant heterogeneity in gene expression from cell to cell within the tumor [28]. Targeting a single protein may lead to eradication of all cells expressing that target, but other cells may survive, resulting in recurrence with selection for tumor that is not recognized by the immune response. This was demonstrated with the EGFRvIII peptide vaccine in the phase II trial [26]. All patients in the trial were histologically proven to have EGFRvIII expression at enrollment; however, of the 11 patients in the trial who underwent biopsy/re-resection at recurrence, 9 of 11 (82 %) had no evidence of EGFRvIII expression in the recurrent tumor [26]. These results suggest that the vaccine was effective in eradicating its target, but facilitated selection of a resistant tumor at recurrence.
To address the concern of limited efficacy with a single antigenic target, a new generation of multivalent peptide vaccines is now in clinical trials for GBM. The peptide vaccine SL-701 is a proprietary multivalent vaccine that has been tested for a mixed group of pediatric high-grade gliomas in a phase I trial with evidence of a positive immunologic response in 81 % of patients [29]. A phase I/II study is now enrolling adult patients with recurrent GBM (NCT02078648). The IMA950 platform contains a proprietary group of 11 synthetic HLA-A2 restricted tumor-associated peptides (TUMAPs) identified by screening a large number of GBM samples [30]. This multivalent peptide vaccine is being studied in patients with newly diagnosed GBM when given in combination with GM-CSF (NCT01222221). A phase I/II study of IMA950 in combination with Poly-ICLC is now recruiting patients as well (NCT01920191). Enrollment in this trial is still limited to HLA-A2 positive patients and it is unclear how many of the target peptides the average individual patient actually expresses.
2.2 Dendritic Cell Vaccines
In contrast to peptide vaccines that rely on endogenous APCs to uptake and display the peptide for T cell stimulation, DC vaccines control this crucial step by ex vivo manipulation. DC vaccines are generated after harvesting a patient’s autologous DCs by plasmapheresis. Cells are stimulated ex vivo and antigenic peptides are introduced by pulsing them with DCs in culture. Activated DCs expressing the antigenic peptides on class I MHC are then reintroduced systemically, facilitating education of naïve T cells. This approach was used for the first cancer vaccine approved by the FDA, developed for the treatment of prostate cancer [16]. In comparison to peptide vaccines that can be given “off the shelf”, this personalized vaccine approach is much more labor intensive and expensive. However, while peptide vaccines require predetermination of the antigenic target to synthesize the immune-stimulatory peptide, DCs can be pulsed with selected peptides or whole tumor lysate, allowing development of a unique, multivalent vaccine targeting the most highly expressed antigens in a particular patient’s tumor.
A number of early phase clinical trials utilizing DC vaccine for GBM have been completed and reported (Table 1). The ICT-107 vaccine utilizes a panel of six HLA-A1/2 restricted peptides known to be highly expressed in glioma stem cells that are pulsed into autologous DCs for generation of the vaccine. In a small phase I study, the vaccine demonstrated highly hopeful results in 17 newly diagnosed GBM patients with a median PFS of 16.9 months and median OS of 38.4 months [31]. This led to a randomized, placebo-controlled phase IIb trial for newly diagnosed GBM (NCT01280552). Although not yet published, the results have been reported, demonstrating that the median OS in 81 vaccine treated patients was only 18.3 months with no significant differences in the treatment and placebo arms. Other DC vaccines with selected glioma-associated antigens have shown similar median OS of approximately 18 months in early phase trials with mixed high-grade gliomas [32].
Capitalizing on the advantages of the DC approach, a number of studies have utilized whole tumor lysate pulsed into DCs to develop a patient-specific multivalent vaccine. Single arm phase I/II trials utilizing this approach in mixed populations of newly diagnosed and recurrent GBM have demonstrated median PFS of 8–18 months with median OS of 18–34 months [33, 34]. It is difficult to assess efficacy in these small, single arm trials with mixed populations of newly diagnosed and recurrent GBM patients, as well as some anaplastic astrocytoma patients. These trials do, however, clearly demonstrate a robust immune response in the majority of patients in response to vaccination [33]. A number of other early phase trials of DC vaccine with more homogenous populations are currently ongoing (Table 1). In addition, a phase III, randomized, placebo-controlled trial of the DCVax-L vaccine, an autologous tumor lysate pulsed DC vaccine, is currently underway for newly diagnosed GBM patients (NCT00045968). The results of this trial are highly awaited and will be an important determinant of the viability of DC vaccines for the treatment of GBM.
2.3 Heat Shock Protein Vaccines
An alternative method to deliver antigenic peptides to APCs for presentation to naïve T cells is the use of heat shock proteins. HSPs are intracellular chaperones involved in trafficking peptides throughout an active cell. They are ubiquitously expressed in all cells, but particularly in cells under stress, such as neoplasms. Members of the HSP family, such as HSP 70 and 96, have specialized mechanisms to deliver antigenic peptides to APCs for presentation [35]. Peptides bound to HSP-96 in the extracellular environment can be internalized into endogenous DCs through the CD91 receptor, resulting in cleavage of the peptide and expression of the antigen on class I and II MHC [36]. By extracting HSP-96 with its associated peptides from whole tumor lysate, a personalized polyvalent vaccine can be generated. HSP vaccines are easier and more cost-effective to produce than DC vaccine, while maintaining the personalized polyvalent antigen expression not available with peptide vaccines.
A heat shock protein peptide complex-96 (HSPPC-96) vaccine has been developed and tested in newly diagnosed and recurrent GBM in phase I/II trials. In a phase I study, 11 of 12 vaccinated patients were found to have a significant peripheral immune response to intradermal vaccination [37]. In a phase II study for recurrent GBM in 41 patients, median PFS was 5 months and median OS was 10.5 months [38]. The phase II study for newly diagnosed GBM has not been published, but early results in 46 patients demonstrate a median PFS of 16 months and median OS of 23.3 months [39]. These single arm results are promising, but randomized, controlled phase II/III trials are necessary to assess the true clinical benefit. A phase IIb trial of the HSPPC-96 vaccine with bevacizumab versus bevacizumab alone for recurrent GBM is currently ongoing (NCT01814813).
3 Immune Modulators
While the results of early phase clinical trials for glioma vaccines demonstrate promising results, the survival benefit among highly selected patient populations is on the order of months, rather than years or decades. In most of these trials, a positive immune-stimulatory response to vaccination has been measured. So, why are not the benefits of immunotherapy greater? In general, cancer is known to be highly immunosuppressive. This is particularly true of gliomas. Multiple studies have demonstrated that patients with gliomas have reduced leukocyte counts and impaired leukocyte function, a phenomenon that worsens with the grade of the tumor [40–42]. The mechanisms of local immunoresistance and systemic immunosuppression are multifactorial, but a few key factors have been identified in GBM and other cancers.
3.1 Regulatory T Cells
Regulatory T cells (Tregs) are a subclass of CD4+ T cells that exert an immunosuppressive effect on APCs and effector T cells through the production of immunosuppressive cytokines such as IL-10 and TGF-b [43]. Tregs (defined as CD4+, CD25+, FoxP3+) are enriched in the blood and tumor of patients with GBM, establishing an immunosuppressive environment [44–46]. Soluble factors secreted from the tumor have been shown to recruit and expand Tregs, and therefore the degree of immunosuppression is proportional to the tumor burden [47]. Experimental depletion of Tregs in animal models of gliomas has been shown to improve survival [48]. Humanized monoclonal antibodies targeting the alpha subunit of the IL-2 receptor (CD25) are now available for clinical use. Although developed to modulate autoimmune diseases, these agents have been used in small pilot studies for GBM in combination with vaccine immunotherapy to deplete Tregs. When daclizumab was given to patients with newly diagnosed GBM in combination with an EGFRvIII peptide vaccine, a significant reduction in Tregs was demonstrated relative to saline-injected control patients [49]. Additionally, patients with decreased Tregs mounted a greater humoral response to vaccination, as had been previously shown in other studies [50]. A phase I study of basiliximab in combination with a DC vaccine is currently ongoing (NCT00626483).
3.2 CTLA-4
Normal activation of effector T cells involves binding of the specific T cell receptor to its antigenic target displayed on the MHC of an APC. Activation also requires binding of a cofactor (B7.1/B7.2) on the APC with its receptor (CD28) on the T cell. However, B7 can also bind to the CTLA-4 receptor on T cells resulting in the opposite effect, T cell inactivation [51]. The balance of binding to CD28 versus CTLA-4 determines the relative activity of systemic T cells. Inhibiting CTLA-4 can increase overall T cell reactivity and boost systemic immunity [4]. Inhibitors of CTLA-4, such as ipilimumab, have been shown to modulate the immune response to cancer in melanoma patients and can successfully improve survival when used as monotherapy or in combination with vaccines [52]. Although there is limited data on CTLA-4 expression in circulating T cells in GBM, the use of ipilumumab in GBM has been suggested and is part of a 3-arm trial of immunomodulators versus bevacizumab for recurrent GBM currently in a phase II trial (NCT02017717). Since Tregs are also known to express high levels of CTLA-4, CTLA-4 inhibitors, may function in part by modulating Treg activity as well [15].
3.3 PD-L1
T cell activity is also modulated by the immune checkpoint regulator programmed death ligand 1 (PD-L1), also known as B7 homologue 1 (B7-H1). PD-L1 is normally expressed on a variety of immune cells and can bind to its receptor, programmed death 1 (PD-1), on T cells, inducing T cell apoptosis or anergy. It is now well recognized that expression of PD-L1 on the surface of cancer cells results in immunoresistance in the tumor microenvironment [53]. Inhibitors of PD-L1 and the PD-1 receptor have been tested in early phase clinical trials for a variety of advanced solid organ tumors, demonstrating significant tumor regression in a small subset of patients [54, 55]. In GBM, expression of PD-L1 on the surface of tumor cells has been linked to loss of PTEN and overactivation of the PI3(k)-Akt pathway [56]. Tumor expression of PD-L1 contributes to local immunoresistence in GBM in a subset of patients with elevated expression [57]. However, most GBM patients are known to be systemically immunosuppressed with T cell dysfunction in circulation [41]. Recently, it has been identified that GBM patients also have increased PD-L1 expression on circulating monocytes and tumor-infiltrating macrophages, leading to a tumor-independent mechanism of immunosuppression [58]. Expression of PD-L1 on circulating monocytes has been shown to correlate with significantly worsened survival in patients who received the HSPPC-96 vaccine for newly diagnosed GBM [39]. A phase II trial of nivolumab, a PD-1 inhibitor, is currently ongoing for patients with recurrent GBM (NCT02017717).
4 Discussion
Immunotherapy for the treatment of cancer offers the possibility of a highly specific, minimally toxic alternative to chemotherapy, with the benefit of sustained efficacy provided that immunologic memory is induced. While immunotherapy comes in many forms, immune recognition of tumor-specific antigens by endogenous exposure or exogenous introduction of antigenic peptides is necessary for efficacy. Due to the lack of systemic metastasis by gliomas and their immune-privileged space behind the BBB, it is often believed that active vaccination is necessary to mount an immune response to gliomas. Nearly all active immunotherapy trials of GBM to date have been vaccine trials with or without an immune stimulating adjuvant. As presented, the results of a number of phase I/II trials for glioma vaccines demonstrate moderate improvement in survival as compared to historical controls. Dramatic effects of glioma vaccines are limited by the challenge of immunosuppression and local tumor immunoresistance. The mechanisms underlying the immunosuppression, including expansion of Tregs, CTLA-4 expression, and PD-L1/PD-1 interaction, have just recently been fully elucidated. Targeting these factors with immune modulating therapy has been successful in other cancers, but those other tumors are known to metastasize hematogenously and to be highly immunoreactive at baseline. Although a trial of PD-1 inhibition with and without CTLA-4 inhibition is currently ongoing for recurrent GBM, it is not clear that immune checkpoint modulation alone is sufficient to mount a robust immune response in GBM. More likely, a combined approach of active vaccination with immunomodulation to boost the response will be the most effective therapy for gliomas. Such combined therapy is not currently part of any active clinical trial, but is being planned for a number of vaccine approaches. Immunotherapy for cancer, and particularly for gliomas, is still in its early stages, but as our understanding of the critical factors in cancer immunology matures, immunotherapy will likely play a larger part in the treatment of malignant gliomas.
References
Dunn GP, Old LJ, Schreiber RD (2004) The immunobiology of cancer immunosurveillance and immunoediting. Immunity 21:137–148. doi:10.1016/j.immuni.2004.07.017, pii:S1074761304002092
Schreiber RD, Old LJ, Smyth MJ (2011) Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331:1565–1570. doi:10.1126/science.1203486, pii:331/6024/1565
Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM (2007) Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet 370: 59–67. doi:10.1016/S0140-6736(07)61050-2, pii:S0140-6736(07)61050-2
Raval RR, Sharabi AB, Walker AJ, Drake CG, Sharma P (2014) Tumor immunology and cancer immunotherapy: summary of the 2013 SITC primer. J Immunother Cancer 2:14. doi:10.1186/2051-1426-2-14, pii:2051-1426-2-14
Slamon DJ, Leyland-Jones B, Shak S et al (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344:783–792. doi:10.1056/NEJM200103153441101
Peinert S, Kershaw MH, Prince HM (2009) Chimeric T cells for adoptive immunotherapy of cancer: using what have we learned to plan for the future. Immunotherapy 1:905–912. doi:10.2217/imt.09.69
Mellman I, Coukos G, Dranoff G (2011) Cancer immunotherapy comes of age. Nature 480:480–489. doi:10.1038/nature10673, pii:nature10673
Mukherji B, Chakraborty NG, Sivanandham M (1990) T-cell clones that react against autologous human tumors. Immunol Rev 116:33–62
Berke G (1995) The CTL’s kiss of death. Cell 81(1):9–12. doi:10.1016/0092-8674(95)90365-8,pii:0092-8674(95)90365-8
Grimm EA, Owen-Schaub L (1991) The IL-2 mediated amplification of cellular cytotoxicity. J Cell Biochem 45:335–339. doi:10.1002/jcb.240450405
McDermott DF, Regan MM, Clark JI et al (2005) Randomized phase III trial of high-dose interleukin-2 versus subcutaneous interleukin-2 and interferon in patients with metastatic renal cell carcinoma. J Clin Oncol 23:133–141. doi:10.1200/JCO.2005.03.206, pii:23/1/133
Sparano JA, Fisher RI, Sunderland M et al (1993) Randomized phase III trial of treatment with high-dose interleukin-2 either alone or in combination with interferon alfa-2a in patients with advanced melanoma. J Clin Oncol 11:1969–1977
Lu H (2014) TLR agonists for cancer immunotherapy: tipping the balance between the immune stimulatory and inhibitory effects. Front Immunol 5:83. doi:10.3389/fimmu.2014.00083
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12:252–264. doi:10.1038/nrc3239, pii:nrc3239
Curran MA, Montalvo W, Yagita H, Allison JP (2010) PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA 107:4275–4280. doi:10.1073/pnas.0915174107, pii:0915174107
Kantoff PW, Higano CS, Shore ND et al (2010) Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 363:411–422. doi:10.1056/NEJMoa1001294
Dunn GP, Old LJ, Schreiber RD (2004) The three Es of cancer immunoediting. Annu Rev Immunol 22:329–360. doi:10.1146/annurev.immunol.22.012703.104803
Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD (2005) Differential activation of astrocytes by innate and adaptive immune stimuli. Glia 49:360–374. doi:10.1002/glia.20117
Dong Y, Benveniste EN (2001) Immune function of astrocytes. Glia 36(2):180–190. doi:10.1002/glia.1107,pii:glia.1107
Pachter JS, de Vries HE, Fabry Z (2003) The blood-brain barrier and its role in immune privilege in the central nervous system. J Neuropathol Exp Neurol 62:593–604
Giometto B, Bozza F, Faresin F, Alessio L, Mingrino S, Tavolato B (1996) Immune infiltrates and cytokines in gliomas. Acta Neurochir (Wien) 138:50–56
Parney IF, Waldron JS, Parsa AT (2009) Flow cytometry and in vitro analysis of human glioma-associated macrophages. Laboratory investigation. J Neurosurg 110:572–582. doi:10.3171/2008.7.JNS08475, pii:10.3171/2008.7.JNS08475
Hatanpaa KJ, Burma S, Zhao D, Habib AA (2010) Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 12:675–684
Gan HK, Kaye AH, Luwor RB (2009) The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci 16:748–754. doi:10.1016/j.jocn.2008.12.005, pii:S0967-5868(09)00046-0
Heimberger AB, Sampson JH (2009) The PEPvIII-KLH (CDX-110) vaccine in glioblastoma multiforme patients. Expert Opin Biol Ther 9:1087–1098. doi:10.1517/14712590903124346
Sampson JH, Heimberger AB, Archer GE et al (2010) Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol 28:4722–4729. doi:10.1200/JCO.2010.28.6963, pii:JCO.2010.28.6963
Izumoto S, Tsuboi A, Oka Y et al (2008) Phase II clinical trial of Wilms tumor 1 peptide vaccination for patients with recurrent glioblastoma multiforme. J Neurosurg 108:963–971. doi:10.3171/JNS/2008/108/5/0963
Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavare S (2013) Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA 110:4009–4014. doi:10.1073/pnas.1219747110, pii:1219747110
Pollack IF, Jakacki RI, Butterfield LH et al (2014) Antigen-specific immune responses and clinical outcome after vaccination with glioma-associated antigen peptides and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in children with newly diagnosed malignant brainstem and nonbrainstem gliomas. J Clin Oncol. doi:10.1200/JCO.2013.54.0526, pii:JCO.2013.54.0526
Dutoit V, Herold-Mende C, Hilf N et al (2012) Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain 135:1042–1054. doi:10.1093/brain/aws042, pii:aws042
Phuphanich S, Wheeler CJ, Rudnick JD et al (2013) Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother 62:125–135. doi:10.1007/s00262-012-1319-0
Prins RM, Wang X, Soto H et al (2013) Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in malignant glioma patients. J Immunother 36:152–157. doi:10.1097/CJI.0b013e3182811ae4
Liau LM, Prins RM, Kiertscher SM et al (2005) Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res 11:5515–5525. doi:10.1158/1078-0432.CCR-05-0464, pii:11/15/5515
Wheeler CJ, Black KL, Liu G et al. (2008) Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res 68:5955–5964. doi:10.1158/0008-5472.CAN-07-5973, pii:68/14/5955
Srivastava PK, Callahan MK, Mauri MM (2009) Treating human cancers with heat shock protein-peptide complexes: the road ahead. Expert Opin Biol Ther 9:179–186. doi:10.1517/14712590802633918
Binder RJ, Srivastava PK (2004) Essential role of CD91 in re-presentation of gp96-chaperoned peptides. Proc Natl Acad Sci USA 101:6128–6133. doi:10.1073/pnas.0308180101, pii:0308180101
Crane CA, Han SJ, Ahn B et al (2013) Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin Cancer Res 19:205–214. doi:10.1158/1078-0432.CCR-11-3358, pii:1078-0432.CCR-11-3358
Bloch O, Crane CA, Fuks Y et al (2014) Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro Oncol 16:274–279. doi:10.1093/neuonc/not203, pii:not203
Bloch O, Kaur R, Aghi MK, McDermott MW, Berger MS, Parsa AT (2013) Glioma-induced immunosuppression shortens progression-free survival in a trial of immunotherapy for glioblastoma. In: American association of neurological surgeons annual meeting, New Orleans, LA
Brooks WH, Netsky MG, Normansell DE, Horwitz DA (1972) Depressed cell-mediated immunity in patients with primary intracranial tumors. Characterization of a humoral immunosuppressive factor. J Exp Med 136:1631–1647
Elliott LH, Brooks WH, Roszman TL (1984) Cytokinetic basis for the impaired activation of lymphocytes from patients with primary intracranial tumors. J Immunol 132:1208–1215
Gousias K, Markou M, Arzoglou V, Voulgaris S, Vartholomatos G, Kostoula A, Voulgari P, Polyzoidis K, Kyritsis AP (2010) Frequent abnormalities of the immune system in gliomas and correlation with the WHO grading system of malignancy. J Neuroimmunol 226: 36–42. doi:10.1016/j.jneuroim.2010.05.027, pii:S0165-5728(10)00211-0
Facciabene A, Motz GT, Coukos G (2012) T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res 72:2162–2171. doi:10.1158/0008-5472.CAN-11-3687, pii:72/9/2162
Fecci PE, Mitchell DA, Whitesides JF et al (2006) Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res 66:3294–3302. doi:10.1158/0008-5472.CAN-05-3773, pii:66/6/3294
Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, Yang DS, Sun W, Qiao W, Hiraoka N, Fuller GN (2008) Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin Cancer Res 14:5166–5172. doi:10.1158/1078-0432.CCR-08-0320, pii:14/16/5166
Learn CA, Fecci PE, Schmittling RJ et al. (2006) Profiling of CD4+, CD8+, and CD4+ CD25+ CD45RO+ FoxP3+ T cells in patients with malignant glioma reveals differential expression of the immunologic transcriptome compared with T cells from healthy volunteers. Clin Cancer Res 12:7306–7315. doi:10.1158/1078-0432.CCR-06-1727, pii:12/24/7306
Crane CA, Ahn BJ, Han SJ, Parsa AT (2012) Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: implications for immunotherapy. Neuro Oncol 14:584–595. doi:10.1093/neuonc/nos014, pii:nos014
El Andaloussi A, Han Y, Lesniak MS (2006) Prolongation of survival following depletion of CD4+ CD25+ regulatory T cells in mice with experimental brain tumors. J Neurosurg 105:430–437. doi:10.3171/jns.2006.105.3.430
Sampson JH, Schmittling RJ, Archer GE et al (2012) A pilot study of IL-2Ralpha blockade during lymphopenia depletes regulatory T-cells and correlates with enhanced immunity in patients with glioblastoma. PLoS One 7:e31046. doi:10.1371/journal.pone.0031046, pii:PONE-D-11-20574
Fong B, Jin R, Wang X, Safaee M, Lisiero DN, Yang I, Li G, Liau LM, Prins RM (2012) Monitoring of regulatory T cell frequencies and expression of CTLA-4 on T cells, before and after DC vaccination, can predict survival in GBM patients. PLoS One 7:e32614. doi:10.1371/journal.pone.0032614, pii:PONE-D-11-22564
Ceeraz S, Nowak EC, Noelle RJ (2013) B7 family checkpoint regulators in immune regulation and disease. Trends Immunol 34:556–563. doi:10.1016/j.it.2013.07.003, pii:S1471-4906(13)00110-5
Hodi FS, O’Day SJ, McDermott DF et al (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363:711–723. doi:10.1056/NEJMoa1003466, pii:NEJMoa1003466
Dong H, Strome SE, Salomao DR et al (2002) Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 8:793–800. doi:10.1038/nm730, pii:nm730
Brahmer JR, Tykodi SS, Chow LQ et al (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366:2455–2465. doi:10.1056/NEJMoa1200694
Topalian SL, Hodi FS, Brahmer JR et al (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366:2443–2454. doi:10.1056/NEJMoa1200690
Parsa AT, Waldron JS, Panner A et al (2007) Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 13:84–88. doi:10.1038/nm1517, pii:nm1517
Wintterle S, Schreiner B, Mitsdoerffer M, Schneider D, Chen L, Meyermann R, Weller M, Wiendl H (2003) Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res 63:7462–7467
Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT (2013) Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res 19:3165–3175. doi:10.1158/1078-0432.CCR-12-3314, pii:1078-0432.CCR-12-3314
Vik-Mo EO, Nyakas M, Mikkelsen BV, Moe MC, Due-Tønnesen P, Suso EM, Sæbøe-Larssen S, Sandberg C, Brinchmann JE, Helseth E, Rasmussen AM, Lote K, Aamdal S, Gaudernack G, Kvalheim G, Langmoen IA (2013) Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer ImmunolImmunother 62(9):1499–1509. doi:10.1007/s00262-013-1453-3
Fadul CE, Fisher JL, Hampton TH, Lallana EC, Li Z, Gui J, Szczepiorkowski ZM, Tosteson TD, Rhodes CH, Wishart HA, Lewis LD, Ernstoff MS (2011) Immune response in patients with newly diagnosed glioblastomamultiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J Immunother 34(4):382–389. doi:10.1097/CJI.0b013e318215e300
Wen PY, Reardon DA, Phuphanich S, Aiken R, Landolfi JC, Curry WT, Zhu JJ, Glantz MJ, Peereboom DM, Markert J, LaRocca RV, O’Rourke D, Fink KL, Kim LJ, Gruber ML, Lesser GJ, Pan E, Kesari S, Hawkins ES, Yu J (2014) A randomized, double-blind, placebo-controlled phase 2 trial of dendritic cell (DC) vaccination with ICT-107 in newly diagnosed glioblastoma (GBM) patients. J Clin Oncol 32:5s (suppl; abstract number 2005)
Lai R, Recht LD, Reardon DA, Paleologos N, Groves MD, Rosenfeld MR, Meech S, Davis TA, Pavlov D, Sampson JH (2010) Interim data for ACT III: phase II trial of PF-04948568 (CDX-110) in combination with temozolomide (TMZ) in patients with glioblastoma (GBM). J Clin Oncol 28:15s (suppl; abstract number 2014)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Bloch, O. (2015). Immunotherapy for Malignant Gliomas. In: Raizer, J., Parsa, A. (eds) Current Understanding and Treatment of Gliomas. Cancer Treatment and Research, vol 163. Springer, Cham. https://doi.org/10.1007/978-3-319-12048-5_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-12048-5_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-12047-8
Online ISBN: 978-3-319-12048-5
eBook Packages: MedicineMedicine (R0)