
Abstract
High serum leptin levels characterize obesity, an established risk factor for many cancers. There is compelling and growing peer-reviewed scientific evidence that leptin plays a direct role in the development and progression of specific cancers. This chapter discusses some of this evidence. The mediating molecular mechanisms are often complex and in specific cases involve dysregulation of the cell cycle. Our current understanding of the role of leptin in carcinogenesis has already revealed aspects for potential clinical applications.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Leptin
- Cell cycle
- Cancer
- Esophageal adenocarcinoma
- Colon cancer
- Adiponectin
- Signal transduction
- Acid
- Obesity
- Adipocytes
- Cyclooxygenase-2
Introduction
Leptin is one of the best known adipocytokines. Successful cloning of the leptin gene was first reported in 1994 [1]. Since then it has become an important target of study by researchers interested in understanding the biological basis for the strong association between obesity and many cancers. Some of the cancers associated with obesity include colon cancer [2–5], breast cancer [6], endometrial cancer [7, 8], esophageal adenocarcinoma [9–12], kidney cancer [13–17], leukemia [18], melanoma [19–23], multiple myeloma [24], non-Hodgkin’s lymphoma [25–28], pancreatic cancer [29–32], prostate cancer [33, 34] rectal cancer [35, 36] and thyroid cancer [37–40]. The most strongly associated cancers with obesity in men are esophageal adenocarcinoma, thyroid cancer, colon cancer, and kidney cancer [41]. In women, the cancers most strongly associated with obesity are endometrial cancer, esophageal adenocarcinoma, and kidney cancer [41]. There is increasing evidence indicating that high circulating leptin levels characteristic of obesity may be promoting the development and progression of many of these obesity-associated cancers. Some of the observational and laboratory-based mechanistic data will be discussed in later sections of this chapter. The discussion will be particularly focused on the role of leptin in the development and progression of esophageal adenocarcinoma and colon cancer, although Table 13.1 summarizes the main cancer types where increased leptin and leptin receptor expression have been reported. The current state of scientific knowledge regarding how leptin regulates signal transduction and the cell cycle to promote carcinogenesis will be described. The main molecular mechanisms underlying the role of leptin in carcinogenesis are listed in Table 13.2. For completion, it must be mentioned that there are other adipocytokines that may regulate carcinogenesis. These include adiponectin, resistin, chemerin, collagenous repeat-containing sequence of 26 kDa protein (CORS-26), omentin, and visfatin [42–44].
Leptin and Esophageal Adenocarcinoma
Esophageal adenocarcinoma is one of two types of esophageal cancer. The more common type of esophageal cancer is esophageal squamous cell carcinoma. However, the incidence of esophageal squamous cell carcinoma has been decreasing whereas the incidence of esophageal adenocarcinoma (EAC) has been rapidly increasing worldwide [45]. This has especially been the case in the western world [45]. In fact, some researchers feel that the incidence of EAC worldwide is rising faster than that of any other solid cancer.
As mentioned above, there is a very strong association between obesity and esophageal adenocarcinoma. One of the earlier well-performed studies in this area was done in Sweden. The study showed that the odds ratio between obese persons (those with body mass index (BMI) greater than 30 kg/m2) as compared to the leanest persons (individuals with BMI less than 22 kg/m2) was 16.2 [46]. Around the same time, a group of researchers in the USA conducted a population-based case–control study of 589 patients of esophageal and gastric adenocarcinoma and 695 healthy control volunteers. After adjusting for age, sex, race, and cigarette smoking, they found that the odds ratio for esophageal adenocarcinoma for men in the highest quartile of BMI as compared to those in the lowest quartile was 3.0 (95 % confidence interval was 1.7–5.0) while the odds ratio for women in the highest quartile as compared to those in the lowest quartile was 2.6 (95 % confidence interval was 0.8–8.5) [47]. Many other studies performed since then have consistently showed that obesity is strongly associated with EAC [9, 10, 12, 45, 48]. Some have suggested that the reason for this association is the increased incidence of gastro-esophageal reflux disease (GERD) among obese people and that increased GERD leads to increased Barrett’s esophagus (BE) and consequently increased EAC. It should be noted, however, that the association between obesity and EAC is stronger than that between GERD and EAC [46, 49]. Obesity is, therefore, a direct risk factor for EAC independent of GERD [50]. Consequently, it is very likely that there is a more direct link between obesity and EAC.
Adipocytokines are dysregulated in obesity and are known to exert effects on a wide variety of peripheral tissues [51–53]. Therefore, any or a combination of adipocytokines may provide a direct biological link between obesity and EAC. Leptin is a very strong candidate for this role because serum leptin concentrations correlate with percentage body mass and BMI [51, 54]. Consequently, many researchers have specifically investigated the direct role of leptin in the development and progression of many cancers. Although only few studies have specifically investigated EAC, their results have been clear and consistent.
In 2003, Somasundar et al. reported that leptin stimulated the proliferation of two different EAC cell lines but they were not able to detect an anti-apoptotic effect using their experimental approach [55]. However, 3 years later and working with a different EAC cell line, Ogunwobi et al. showed very clearly that leptin stimulated proliferation of an EAC cell line in a dose-dependent manner [56]. They were also able to detect an anti-apoptotic effect for leptin on the EAC cells. More importantly, they elucidated a cascade of intracellular signaling mechanisms via which leptin stimulates proliferation and inhibits apoptosis of EAC cells. Their studies showed that leptin stimulation resulted in the activation of cyclooxygenase-2 (COX-2) and prostaglandin E2 (PGE2) production in a manner dependent upon activation of extracellular signal-regulated kinase (ERK), p38 mitogen-activated protein kinase (MAPK), phosphatidylinositol 3′-kinase (PI3K)/Akt, and janus kinase (JAK) 2. They also showed that the PGE2 produced was subsequently involved in the transactivation of the epidermal growth factor receptor and c-Jun amino terminal kinase (JNK) [56]. It is noteworthy that all of the signaling pathways activated by leptin in the Ogunwobi et al. study have been shown by multiple studies and research groups to be important in the development and progression of EAC [57–59]. Furthermore, the leptin receptor is expressed in the OE33 EAC cell line used in the Ogunwobi et al. study. Moreover, leptin receptor expression has been demonstrated throughout the mouse gastrointestinal epithelium including the esophagus [60]. More importantly, leptin receptor expression has been demonstrated in the epithelium of the human lower esophagus [61] and it has been shown to increase as the lower esophagus changes from normal to Barrett’s esophagus and finally to development of EAC [62].
Mention should also be made of data that suggest that hyperleptinemia in obesity likely cooperates with GERD in contributing to the development and progression of EAC. Beales et al. investigated the combined effects of acid and leptin exposure on EAC cells [63]. They found that there was a positive synergistic effect on proliferation and inhibition of apoptosis [63]. Additionally, these effects were via synergistic activation of the epidermal growth factor receptor (EGFR) and ERK [63].
Taken together, these studies provide a strong basis for concluding that increased circulating leptin in obese people may play a direct role in the increased incidence and progression of EAC in obesity.
Leptin and Colon Cancer
The role of leptin as a direct promoter of colon cancer is also clear. Expression of the leptin receptor in the epithelium of the human colon was described by Hardwick et al. in their 2001 publication [64]. In that paper, they also demonstrated that treatment of mice with leptin resulted in significant increase in colonic epithelial cell proliferation [64]. More recently, Koda and colleagues reported that leptin is overexpressed in human colon cancer [65]. Multiple research groups have independently demonstrated that leptin is able to promote proliferation, motility, invasiveness, and inhibit apoptosis of colonic epithelial cells by activation of a complex cascade of intracellular signaling mechanisms that have all been implicated in the development of human colon cancer [64, 66–69]. It is noteworthy that many of the intracellular signaling mechanisms activated by leptin in esophageal epithelial cells are also activated by leptin in colonic epithelial cells. However, one striking difference is that whereas leptin-induced proliferation is dependent on activation of the COX-2 pathway in esophageal epithelial cells [56], in colonic epithelial cells leptin is able to potently induce proliferation and inhibit apoptosis independent of activation of COX-2 [67].
The epidemiologic data supporting leptin’s role in the promotion of colon cancer is also compelling. In Stattin et al’s 2003 Sweden-based case–control study, they measured the prediagnostic plasma leptin levels of 75 men, 93 women, and 327 control volunteers. They performed statistical analysis using logistic regression and found that plasma leptin was associated with increased risk of colon cancer in men. The odds ratio (OR) between the highest and lowest quartiles was 1.96 (95 % confidence interval (CI) was 0.72–5.29) [70]. Subsequently, in 2005 Tamakoshi and colleagues performed a separate case–control study in Japan where they found an association between plasma leptin and colon cancer risk in women. Their study assessed 58 colon cancer patients and 145 colon cancer-free volunteers matched for study area and age. They also used logistic regression for their statistical analysis. In addition, however, they also accounted for BMI, life-style factors, reproductive factors, and insulin-like growth factor and its binding protein. Their analysis revealed that the ORs of female colorectal cancer risk for the second and third quintiles combined and the fourth and fifth quintiles combined relative to the first quintile were 1.40 (95 % CI was 0.41–4.78) and 4.84 (95 % CI was 1.29–18.1) respectively [71]. Around the same time, Stattin and colleagues reported a case–control study they performed in Norway wherein they analyzed serum levels of leptin in cryopreserved prediagnostic sera of 235 men with colon cancer and 378 control volunteers matched for age and date of blood collection. This time they performed logistic regression analysis and their data showed about a threefold increase in the risk of colon cancer associated with increasing concentrations of leptin. The odds ratio between the lowest and highest quartiles was up to 2.72 (95 % CI was 1.44–5.12); p (trend) was 0.008 [72].
Consequently, there is compelling evidence that circulating leptin concentrations correlate with colon cancer risk and that leptin directly promotes colon cancer.
Leptin and Cell Cycle in Cancer
Dysregulation of the cell cycle is undoubtedly a fundamental factor in the development and progression of cancer. This is especially the case with the uncontrolled proliferation and the genetic alterations that are characteristic of cancer cells (reviewed in [73]). It is, therefore, not surprising that the role of leptin in promoting many cancers often involves dysregulation of the cell cycle.
Consider a few examples. In an ovarian cancer study that used leptin receptor-expressing OVCAR-3 ovarian cancer cells, leptin was found to stimulate proliferation by upregulating cell cycle progression genes and suppressing cell cycle inhibitor genes. The authors also reported that leptin promoted cell cycle progression, as demonstrated by an increased cell population in both the S and G2/M phases [74]. In a separate study that used a mouse model of spontaneous thyroid cancer, consumption of a high-fat diet resulted in the development of obesity and elevated serum leptin levels. This in turn resulted in increased speed of tumor growth and reduced survival. Interestingly, thyroid tumor cell proliferation was increased via cyclin D1-dependent cell cycle progression [38]. Also, in an endometrial cancer model, leptin exposure reduced the number of cells in the G0/G1 phase while increasing the number of cells in the S-phase of the cell cycle. These regulatory effects on the cell cycle were also associated with differential regulation of cyclin D1 and the cyclin-dependent kinase inhibitor, p21: there was upregulation of cyclin D1 and downregulation of p21 [75]. Furthermore, in a separate study using the MCF-7 human breast cancer cell line, leptin treatment led to upregulation of the cell cycle genes cyclin D, cyclin G, cyclin-dependent kinase 2, p27, p21, and p16 as well as downregulation of the cell cycle suppressor, transforming growth factor beta [76]. The authors of the study further demonstrated that leptin down-regulates apoptosis-inducing genes and inhibits apoptosis. And they concluded that these tumorigenic effects of leptin in the MCF-7 human breast cancer model are mediated in part via regulation of the cell cycle [76].
The above examples clearly show that leptin is able to dysregulate the cell cycle to promote the development and progression of cancer.
Leptin and Adiponectin Cross-Talk to Regulate Carcinogenesis
As mentioned above, obesity is an established risk factor for many cancers. And it is characterized by hyperleptinemia and hypoadiponectinemia. It is, therefore, possible that either elevated serum leptin levels or reduced serum adiponectin levels may independently contribute to the development of obesity-associated cancers. In the previous sections of this chapter, compelling evidence indicating that leptin directly promotes development and progression of obesity-related cancers has been presented. It should be noted that there is also independent evidence that reduced circulating levels of adiponectin may contribute to the development of obesity-associated cancers [77–80]. Nevertheless, it is most likely that a variety of molecular factors inter-play to regulate the development and progression of cancers. Consequently, it is not surprising that there is increasing experimental evidence that leptin and adiponectin cross-talk to regulate the development and progression of obesity-associated cancers.
For example, using multiple cellular models of esophageal adenocarcinoma (EAC), Ogunwobi and Beales demonstrated that the globular form of adiponectin inhibited leptin-induced proliferation of EAC cells in a manner dependent on the adipoR1 receptor isoform and 5′-AMP-activated protein kinase [81]. In a more recent study of the OE33 esophageal adenocarcinoma model, Beales et al. further reported that the globular form of adiponectin inhibited leptin-induced proliferation, migration, and anti-apoptosis via protein tyrosine phosphatase 1B-mediated downregulation of signal transducer and activator of transcription 3 (STAT3) transcriptional activity [82]. Similar findings have been reported in other cancer types. A recent case–control study of 516 Chinese women specifically evaluated potential correlation between serum leptin and adiponectin levels with the presence of endometrial carcinoma. The study found that patients with endometrial carcinoma had higher serum leptin and lower serum adiponectin concentrations than normal healthy volunteers. After adjusting for cholesterol, triglycerides, fasting insulin, serum glucose, BMI, and age, logistic regression analysis of their data revealed that high serum leptin concentration and low serum adiponectin concentration are both significantly associated with endometrial carcinoma [83]. Data from the study by Jardé and colleagues published in 2009 are also very interesting [84]. They found that leptin mRNA expression in the MCF-7 breast cancer model was 34.7 times higher than adiponectin mRNA expression [84]. Furthermore, they found that adiponectin treatment resulted in loss of expression of leptin and its receptors whereas leptin treatment resulted in loss of expression of the adiponectin receptor, adipoR1 [84]. They also found that adiponectin treatment inhibited the proliferation of the MCF-7 breast cancer cells whereas leptin treatment stimulated their proliferation [84]. In addition, adiponectin inhibited leptin-induced proliferation of the MCF-7 breast cancer cells [84].
Consequently, there is epidemiological as well as experimental evidence suggesting that leptin likely is not the only adipocytokine regulating tumor development and progression. The role of leptin in cancer development and progression is probably dependent in ways not yet fully understood on adiponectin activity.
Potential Clinical Impact of Understanding the Role of Leptin in Carcinogenesis
Understanding the pathophysiological role of leptin in cancer development and progression is undoubtedly important. It is especially important that we understand the molecular signaling mechanisms mediating leptin activity as it relates to cancer. This may reveal novel insights into cancer development and progression in general. More importantly, it is possible that some of these mechanisms may be exploitable for screening, diagnostic, therapeutic, or monitoring purposes.
The good news is that there is already indication that knowledge gained so far from researching the role of leptin in cancer may be useful clinically. For example, a retrospective analysis of leptin and leptin receptor expression in normal lung tissue in comparison to non-small cell lung cancer tissue revealed that leptin expression significantly affected survival time and that leptin expression is an independent prognostic factor in non-small cell lung cancer [85]. Also, a study of 71 consecutive colon cancer patients that examined serum leptin and leptin receptor levels found that serum leptin and leptin receptor levels correlate with advanced tumor stage [86], thus indicating that information regarding serum leptin and tissue leptin receptor expression may be clinically useful in this setting. Data from breast cancer studies also suggest potential clinical usefulness in that setting. Several research groups have reported that leptin and leptin receptor expression may be useful as prognostic factors in breast cancer [87, 88]. Interestingly, there are even data suggesting that leptin levels may predict response or resistance to anti-estrogen therapy during breast cancer treatment [89]. Also, in gastric cancer, there is evidence that leptin expression may be useful clinically as a prognostic factor [90].
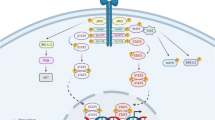
Therefore, there is real potential for the use of leptin or its receptor as clinical biomarkers. And our understanding of its pathophysiologic signaling mechanisms in cancer may also lead to clinical applications. Figure 13.1 summarizes the key signaling pathways that may be important in the role of leptin in carcinogenesis. These are also potential molecular targets that can be exploited for clinical applications.

Summary of key signaling pathways that may be important in the role of leptin in carcinogenesis
Contrarian Reports
There is, of course, some data (though fewer) that may suggest that leptin does not play a role or that it plays an anti-tumorigenic role in certain cancers. For example, a 2013 report of a case–control study and meta-analysis of the association between leptin and colorectal adenoma or colon cancer mentioned that there was a positive association of serum leptin with colorectal adenoma, but not colorectal cancer [91]. It should be noted, though, that even the authors of the report explicitly mentioned that there was significant heterogeneity between the studies they analyzed and that there may have been multiple effect modifiers [91]. Another 2013 report partly focused on the association between leptin and prostate cancer [92]. The authors used nested case–control methodology and utilized both conditional and unconditional logistic regression for their analyses [92]. And they reported that there was no compelling evidence of an association between leptin and prostate cancer stage [92]. An earlier study that examined breast, esophageal, prostate, and pancreatic cancer cell lines found that the effects of leptin may be cell line specific. They found that whereas leptin had a proliferative effect on the breast, esophageal, and prostate cancer cell lines, it had an anti-proliferative effect on the pancreatic cancer cell lines [93]. More recently, it was reported that leptin exerted significant anti-tumor effects in hepatocellular carcinoma [94]. In athymic nude mice implanted with Hep3B human hepatocellular carcinoma cells, leptin significantly reduced tumor size and improved animal survival [94]. Furthermore, it was reported that leptin exerted anti-proliferative effects on the MDA-MB-231 breast cancer cell line via downregulation of protein kinase A [95].
These reports, although not exhaustive, certainly indicate that in some organs or cell types leptin may not have a pro-carcinogenic role.
Conclusion
There is much more that can be said about the role of leptin in the cell cycle and cancer development and progression. This chapter has, somewhat narrowly, focused on only some of the peer-reviewed scientific evidence that leptin does indeed play a role in cancer development and progression. As discussed in the previous section of this chapter, there is some data that may suggest that leptin does not play a role in certain cancers. Nevertheless, the evidence that leptin is an important player in carcinogenesis is compelling and is very likely to continue to grow.
References
Zhang YY, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homolog. Nature. 1994;372(6505):425–32.
Ma YL, Yang YZ, Wang F, Zhang P, Shi CZ, Zou Y, et al. Obesity and risk of colorectal cancer: a systematic review of prospective studies. PLoS One. 2013;8(1):16.
Campbell PT, Jacobs ET, Ulrich CM, Figueiredo JC, Poynter JN, McLaughlin JR, et al. Case-control study of overweight, obesity, and colorectal cancer risk, overall and by tumor microsatellite instability status. J Natl Cancer Inst. 2010;102(6):391–400.
Moghaddam AA, Woodward M, Huxley R. Obesity and risk of colorectal cancer: a meta-analysis of 31 studies with 70,000 events. Cancer Epidemiol Biomarkers Prev. 2007;16(12):2533–47.
LeMarchand L, Wilkens LR, Kolonel LN, Hankin JH, Lyu LC. Associations of sedentary lifestyle, obesity, smoking, alcohol use, and diabetes with the risk of colorectal cancer. Cancer Res. 1997;57(21):4787–94.
Nichols HB, Trentham-Dietz A, Egan KM, Titus-Ernstoff L, Holmes MD, Bersch AJ, et al. Body mass index before and after breast cancer diagnosis: associations with all-cause, breast cancer, and cardiovascular disease mortality. Cancer Epidemiol Biomarkers Prev. 2009;18(5):1403–9.
Reeves KW, Carter GC, Rodabough RJ, Lane D, McNeeley SG, Stefanick ML, et al. Obesity in relation to endometrial cancer risk and disease characteristics in the Women’s Health Initiative. Gynecol Oncol. 2011;121(2):376–82.
Meredith I, Sarfati D, Ikeda T, Atkinson J, Blakely T. High rates of endometrial cancer among Pacific women in New Zealand: the role of diabetes, physical inactivity, and obesity. Cancer Causes Control. 2012;23(6):875–85.
Duggan C, Onstad L, Hardikar S, Blount PL, Reid BJ, Vaughan TL. Association between markers of obesity and progression from Barrett’s esophagus to esophageal adenocarcinoma. Clin Gastroenterol Hepatol. 2013;11(8):934–43.
Ryan AM, Duong M, Healy L, Ryan SA, Parekh N, Reynolds JV, et al. Obesity, metabolic syndrome and esophageal adenocarcinoma: epidemiology, etiology and new targets. Cancer Epidemiol. 2011;35(4):309–19.
Lofdahl HE, Lane A, Lu YX, Lagergren P, Harvey RF, Blazeby JM, et al. Increased population prevalence of reflux and obesity in the United Kingdom compared with Sweden: a potential explanation for the difference in incidence of esophageal adenocarcinoma. Eur J Gastroenterol Hepatol. 2011;23(2):128–32.
Veugelers PJ, Porter GA, Guernsey DL, Casson AG. Obesity and lifestyle risk factors for gastroesophageal reflux disease, Barrett esophagus and esophageal adenocarcinoma. Dis Esophagus. 2006;19(5):321–8.
Ildaphonse G, George PS, Mathew A. Obesity and kidney cancer risk in men - a meta-analysis (1992-2008). Asian Pac J Cancer Prev. 2009;10(2):279–86.
Mathew A, George PS, Ildaphonse G. Obesity and kidney cancer risk in women - a meta-analysis (1992-2008). Asian Pac J Cancer Prev. 2009;10(3):471–8.
Pan SY, DesMeules M, Morrison H, Wen SW, Canadian Canc Registries E. Obesity, high energy intake, lack of physical activity, and the risk of kidney cancer. Cancer Epidemiol Biomarkers Prev. 2006;15(12):2453–60.
Chow WH, Gridley G, Fraumeni JF, Jarvholm B. Obesity, hypertension, and the risk of kidney cancer in men. N Engl J Med. 2000;343(18):1305–11.
McCredie M, Stewart JH. Risk-factors for kidney cancer in New South Wales, Australia. 2. Urologic disease, hypertension, obesity, and hormonal factors. Cancer Causes Control. 1992;3(4):323–31.
Castillo JJ, Reagan JL, Ingham RR, Furman M, Dalia S, Merhi B, et al. Obesity but not overweight increases the incidence and mortality of leukemia in adults: a meta-analysis of prospective cohort studies. Leuk Res. 2012;36(7):868–75.
Brandon EL, Gu J-W, Cantwell L, He Z, Wallace G, Hall JE. Obesity promotes melanoma tumor growth Role of leptin. Cancer Biol Ther. 2009;8(19):1871–9.
Dennis LK, Lowe JB, Lynch CF, Alavanja MCR. Cutaneous melanoma and obesity in the Agricultural Health Study. Ann Epidemiol. 2008;18(3):214–21.
Jung J, Her S, Park J. High-fat diet-induced obesity increases solid tumor growth and metastasis of melanoma cells in C57BL/6 mice. Mol Biol Cell. 2011;22.
Pandey V, Vijayakumar MV, Ajay AK, Malvi P, Bhat MK. Diet-induced obesity increases melanoma progression: involvement of Cav-1 and FASN. Int J Cancer. 2012;130(3):497–508.
Sergentanis TN, Antoniadis AG, Gogas HJ, Antonopoulos CN, Adami H-O, Ekbom A, et al. Obesity and risk of malignant melanoma: a meta-analysis of cohort and case-control studies. Eur J Cancer. 2013;49(3):642–57.
Friedman GD, Herrinton LJ. Obesity and multiple myeloma. Cancer Causes Control. 1994;5(5):479–83.
Chu D-M, Wahlqvist ML, Lee M-S, Chang H-Y. Central obesity predicts non-Hodgkin’s lymphoma mortality and overall obesity predicts leukemia mortality in adult Taiwanese. J Am Coll Nutr. 2011;30(5):310–9.
Larsson SC, Wolk A. Obesity and risk of non-Hodgkin’s lymphoma: a meta-analysis. Int J Cancer. 2007;121(7):1564–70.
Maskarinec G, Erber E, Gill J, Cozen W, Kolonel LN. Overweight and obesity at different times in life as risk factors for non-Hodgkin’s lymphoma: the multiethnic cohort. Cancer Epidemiol Biomarkers Prev. 2008;17(1):196–203.
Pan SY, Mao Y, Ugnat AM, Canadian Canc R. Physical activity, obesity, energy intake, and the risk of non-Hodgkin’s lymphoma: a population-based case-control study. Am J Epidemiol. 2005;162(12):1162–73.
Bracci PM. Obesity and pancreatic cancer: overview of epidemiologic evidence and biologic mechanisms. Mol Carcinog. 2012;51(1):53–63.
Gong Z, Holly EA, Bracci PM. Obesity and survival in population-based patients with pancreatic cancer in the San Francisco Bay Area. Cancer Causes Control. 2012;23(12):1929–37.
Gukovsky I, Li N, Todoric J, Gukovskaya A, Karin M. Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144(6):1199.
White PB, Ziegler KM, Swartz-Basile DA, Wang SS, Lillemoe KD, Pitt HA, et al. Obesity, but not high-fat diet, promotes murine pancreatic cancer growth. J Gastrointest Surg. 2012;16(9):1680–5.
Golabek T, Bukowczan J, Chlosta P, Powroznik J, Dobruch J, Borowka A. Obesity and prostate cancer incidence and mortality: a systematic review of prospective cohort studies. Urol Int. 2014;92(1):7–14.
De Nunzio C, Albisinni S, Freedland SJ, Miano L, Cindolo L, Agro EF, et al. Abdominal obesity as risk factor for prostate cancer diagnosis and high grade disease: a prospective multicenter Italian cohort study. Urol Oncol Semi Orig Investig. 2013;31(7):997–1002.
Mao Y, Pan SY, Wen SW, Johnson KC. Candian Canc Epidemiology Res G. Physical inactivity, energy intake, obesity and the risk of rectal cancer in Canada. Int J Cancer. 2003;105(6):831–7.
Larsson SC, Wolk A. Obesity and colon and rectal cancer risk: a meta-analysis of prospective studies. Am J Clin Nutr. 2007;86(3):556–65.
Han JM, Kim TY, Jeon MJ, Yim JH, Kim WG, Song DE, et al. Obesity is a risk factor for thyroid cancer in a large, ultrasonographically screened population. Eur J Endocrinol. 2013;168(6):879–86.
Kim WG, Park JW, Willingham MC, Cheng S-y. Diet-induced obesity increases tumor growth and promotes anaplastic change in thyroid cancer in a mouse model. Endocrinology. 2013;154(8):2936–47.
Marcello MA, Sampaio AC, Geloneze B, Vasques ACJ, Assumpcao LVM, Ward LS. Obesity and excess protein and carbohydrate consumption are risk factors for thyroid cancer. Nutr Cancer. 2012;64(8):1190–5.
Zhao ZG, Guo XG, Ba CX, Wang W, Yang YY, Wang J, et al. Overweight, obesity and thyroid cancer risk: a meta-analysis of cohort studies. J Int Med Res. 2012;40(6):2041–50.
Khandekar MJ, Cohen P, Spiegelman BM. Molecular mechanisms of cancer development in obesity. Nat Rev Cancer. 2011;11(12):886–95.
Garcia JM, Splenser AE, Kramer J, Alsarraj A, Fitzgerald S, Ramsey D, et al. Circulating inflammatory cytokines and adipokines are associated with increased risk of Barrett’s esophagus: a case-control study. Clin Gastroenterol Hepatol. 2014;12(2):229.
Ntikoudi E, Kiagia M, Boura P, Syrigos KN. Hormones of adipose tissue and their biologic role in lung cancer. Cancer Treat Rev. 2014;40(1):22–30.
Wurm S, Neumeier M, Weigert J, Schaeffler A, Buechler C. Plasma levels of leptin, omentin, collagenous repeat-containing sequence of 26-kDa protein (CORS-26) and adiponectin before and after oral glucose uptake in slim adults. Cardiovasc Diabetol. 2007;6:7.
Dubecz A, Solymosi N, Stadlhuber RJ, Schweigert M, Stein HJ, Peters JH. Does the incidence of adenocarcinoma of the esophagus and gastric cardia continue to rise in the twenty-first century?-a SEER database analysis. J Gastrointest Surg. 2014;18(1):124–9.
Lagergren J, Bergstrom R, Nyren O. Association between body mass and adenocarcinoma of the esophagus and gastric cardia. Ann Intern Med. 1999;130(11):883.
Chow WH, Blot WJ, Vaughn TL, Risch HA, Gammon MD, Stanford JL, et al. Body mass index and risk of adenocarcinomas of the esophagus and gastric cardia. J Natl Cancer Inst. 1998;90(2):150–5.
Merry AHH, Schouten LJ, Goldbohm RA, van den Brandt PA. Body mass index, height and risk of adenocarcinoma of the oesophagus and gastric cardia: a prospective cohort study. Gut. 2007;56(11):1503–11.
Lagergren J, Bergstrom R, Lindgren A, Nyren O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med. 1999;340(11):825–31.
Lagergren J. Adenocarcinoma of oesophagus: what exactly is the size of the problem and who is at risk? Gut. 2005;54:I1–5.
Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum immunoreactive leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334(5):292–5.
Heinonen MV, Purhonen AK, Miettinen P, Paakkonen M, Pirinen E, Alhava E, et al. Apelin, orexin-A and leptin plasma levels in morbid obesity and effect of gastric banding. Regul Pept. 2005;130(1–2):7–13.
Baratta M. Leptin–from a signal of adiposity to a hormonal mediator in peripheral tissues. Med Sci Monit. 2002;8(12):RA282–92.
Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, et al. Leptin levels in human and rodent - measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1(11):1155–61.
Somasundar P, Riggs D, Jackson B, Vona-Davis L, McFadden DW. Leptin stimulates esophageal adenocarcinoma growth by nonapoptotic mechanisms. Am J Surg. 2003;186(5):575–8.
Ogunwobi O, Mutungi G, Beales I. Leptin stimulates proliferation and inhibits apoptosis in Barrett’s esophageal adenocarcinoma cells by cyclooxygenase-2-dependent, prostaglandin-E2-mediated transactivation of the epidermal growth factor receptor and c-Jun NH2-terminal kinase activation. Endocrinology. 2006;147(9):4505–16.
Abdalla SI, Sanderson IR, Fitzgerald RC. Effect of inflammation on cyclooxygenase (COX)-2 expression in benign and malignant oesophageal cells. Carcinogenesis. 2005;26(9):1627–33.
Moons LMG, Kuipers EJ, Rygiel AM, Groothuismink AZM, Geldof H, Bode WA, et al. COX-2 CA-haplotype is a risk factor for the development of esophageal adenocarcinoma. Am J Gastroenterol. 2007;102(11):2373–9.
Tuynman JB, Buskens CJ, Kemper K, ten Kate FJW, Offerhaus GJA, Richel DJ, et al. Neoadjuvant selective COX-2 inhibition down-regulates important oncogenic pathways in patients with esophageal adenocarcinoma. Ann Surg. 2005;242(6):840–50.
Dal Farra C, Zsurger N, Vincent JP, Cupo A. Binding of a pure I-125-monoiodoleptin analog to mouse tissues: a developmental study. Peptides. 2000;21(4):577–87.
Francois F, Roper J, Goodman AJ, Pei Z, Ghumman M, Mourad M, et al. The association of gastric leptin with oesophageal inflammation and metaplasia. Gut. 2008;57(1):16–24.
Ahmed S, Khan AL, Pazmany L, Bogder K. Epithelial cells over-express the leptin receptor during malignant progression in Barrett’s oesophagus. Gastroenterology. 2006;130(4):A564.
Beales ILP, Ogunwobi OO. Lept in synergistically enhances the anti-apoptotic and growth-promoting effects of acid in OE33 oesophageal adenocarcinoma cells in culture. Mol Cell Endocrinol. 2007;274(1–2):60–8.
Hardwick JCH, Van den Brink GR, Offerhaus GJ, Van Deventer SJH, Peppelenbosch MP. Leptin is a growth factor for colonic epithelial cells. Gastroenterology. 2001;121(1):79–90.
Koda M, Sulkowska M, Kanczuga-Koda L, Surmacz E, Sulkowski S. Overexpression of the obesity hormone leptin in human colorectal cancer. J Clin Pathol. 2007;60(8):902–6.
Jaffe T, Schwartz B. Leptin promotes motility and invasiveness in human colon cancer cells by activating multiple signal-transduction pathways. Int J Cancer. 2008;123(11):2543–56.
Ogunwobi O, Beales I. Cyclo-oxygenase-independent inhibition of apoptosis and stimulation of proliferation by leptin in human colon cancer cells. Dig Dis Sci. 2007;52(8):1934–45.
Ogunwobi OO, Beales ILP. The anti-apoptotic and growth stimulatory actions of leptin in human colon cancer cells involves activation of JNK mitogen activated protein kinase, JAK2 and PI3 kinase/Akt. Int J Colorectal Dis. 2007;22(4):401–9.
Rouet- Benzineb P, Aparicio T, Guilmeau S, Pouzet C, Descatoire V, Buyse M, et al. Leptin counteracts sodium butyrate-induced apoptosis in human colon cancer HT-29 cells via NF-kappaB signaling. J Biol Chem. 2004;279(16):16495–502.
Stattin P, Palmqvist R, Soderberg S, Biessy C, Ardnor B, Hallmans G, et al. Plasma leptin and colorectal cancer risk: a prospective study in Northern Sweden. Oncol Rep. 2003;10(6):2015–21.
Tamakoshi K, Toyoshima H, Wakai K, Kojima M, Suzuki K, Watanabe Y, et al. Leptin is associated with an increased female colorectal cancer risk: a nested case-control study in Japan. Oncology. 2005;68(4–6):454–61.
Stattin P, Lukanova A, Biessy C, Soderberg S, Palmqvist R, Kaaks R, et al. Obesity and colon cancer: does leptin provide a link? Int J Cancer. 2004;109(1):149–52.
Williams GH, Stoeber K. The cell cycle and cancer. J Pathol. 2012;226(2):352–64.
Ptak A, Kolaczkowska E, Gregoraszczuk EL. Leptin stimulation of cell cycle and inhibition of apoptosis gene and protein expression in OVCAR-3 ovarian cancer cells. Endocrine. 2013;43(2):394–403.
Catalano S, Giordano C, Rizza P, Gu G, Barone I, Bonofiglio D, et al. Evidence that leptin through STAT and CREB signaling enhances cyclin D1 expression and promotes human endometrial cancer proliferation. J Cell Physiol. 2009;218(3):490–500.
Perera CN, Chin HG, Duru N, Camarillo IG. Leptin-regulated gene expression in MCF-7 breast cancer cells: mechanistic insights into leptin-regulated mammary tumor growth and progression. J Endocrinol. 2008;199(2):221–33.
Aleksandrova K, Boeing H, Jenab M, Bueno-de-Mesquita HB, Jansen E, van Duijnhoven FJB, et al. Total and high-molecular weight adiponectin and risk of colorectal cancer: the European Prospective Investigation into Cancer and Nutrition Study. Carcinogenesis. 2012;33(6):1211–8.
Pinthus J, Hopmans S, Paschos A, Austin R, Duivenvoorden W. ERP46 mediates prostate cancer tumorigenesis in vitro by inhibiting adiponectin-induced tumor-suppressive effects - linking obesity to prostate cancer. J Urol. 2013;189(4):E128.
Chung SJ, Saxena N, Sharma D. Adiponectin induces autophagic cell death in breast cancer cells through SIRT1 mediated deacetylation of LKB1 leading to ATG1 activation. Cancer Res. 2013;73(8, Suppl 1):1673.
Erdogan S, Sezer S, Baser E, Eryilmaz OG, Gungor T, Uysal S, et al. Low serum adiponectin and vaspin levels as novel risk factors for postmenopausal endometrial cancer. Int J Gynecol Cancer. 2013;23(8).
Ogunwobi OO, Beales ILP. Globular adiponectin, acting via adiponectin receptor-1, inhibits leptin-stimulated oesophageal adenocarcinoma cell proliferation. Mol Cell Endocrinol. 2008;285(1–2):43–50.
Beales ILP, Garcia-Morales C, Ogunwobi OO, Mutungi G. Adiponectin inhibits leptin-induced oncogenic signalling in oesophageal cancer cells by activation of PTP1B. Mol Cell Endocrinol. 2014;382(1):150–8.
Ma Y, Liu Z, Zhang Y, Lu B. Serum leptin, adiponectin and endometrial cancer risk in Chinese women. J Gynecol Oncol. 2013;24(4):336–41.
Jarde T, Caldefie-Chezet F, Goncalves-Mendes N, Mishellany F, Buechler C, Penault-Llorca F, et al. Involvement of adiponectin and leptin in breast cancer: clinical and in vitro studies. Endocr Relat Cancer. 2009;16(4):1197–210.
Xu Y-J, Shao Y-F, Zhao X, Geng Y-T, Wang K, Yin Y-M. Expression and clinical significance of leptin, the functional receptor of leptin (OB-Rb) and HER-2 in non-small-cell lung cancer: a retrospective analysis. J Cancer Res Clin Oncol. 2011;137(12):1841–8.
Tutino V, Notarnicola M, Guerra V, Lorusso D, Caruso MG. Increased soluble leptin receptor levels are associated with advanced tumor stage in colorectal cancer patients. Anticancer Res. 2011;31(10):3381–3.
Miyoshi Y, Funahashi T, Tanaka S, Taguchi T, Tamaki Y, Shimomura I, et al. High expression of leptin receptor mRNA in breast cancer tissue predicts poor prognosis for patients with high, but not low, serum leptin levels. Int J Cancer. 2006;118(6):1414–9.
Ishikawa M, Kitayama J, Nagawa H. Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin Cancer Res. 2004;10(13):4325–31.
Maccio A, Madeddu C, Gramignano G, Mulas C, Floris C, Massa D, et al. Correlation of body mass index and leptin with tumor size and stage of disease in hormone-dependent postmenopausal breast cancer: preliminary results and therapeutic implications. J Mol Med. 2010;88(7):677–86.
Zhao X, Huang K, Zhu Z, Chen S, Hu R. Correlation between expression of leptin and clinicopathological features and prognosis in patients with gastric cancer. J Gastroenterol Hepatol. 2007;22(8):1317–21.
Gialamas SP, Sergentanis TN, Antonopoulos CN, Dessypris N, Chrousos GP, Petridou ET. Circulating leptin levels and risk of colorectal cancer and adenoma: a case-control study and meta-analysis. Cancer Causes Control. 2013;24(12):2129–41.
Burton A, Martin RM, Holly J, Lane JA, Donovan JL, Hamdy FC, et al. Associations of adiponectin and leptin with stage and grade of PSA-detected prostate cancer: the ProtecT study. Cancer Causes Control. 2013;24(2):323–34.
Somasundar P, Yu AK, Vona-Davis L, McFadden DW. Differential effects of leptin on cancer in vitro. J Surg Res. 2003;113(1):50–5.
Elinav E, Abd-Elnabi A, Pappo O, Bernstein I, Klein A, Engelhardt D, Rabbani E, Ilan Y. Suppression of hepatocellular carcinoma growth in mice via leptin, is associated with inhibition of tumor cell growth and natural killer cell activation. J Hepatol. 2006;44(3):529–36.
Naviglio S, Di Gesto D, Illiano F, Chiosi E, Giordano A, Illiano G, Spina A. Leptin potentiates antiproliferative action of cAMP elevation via protein kinase A down-regulation in breast cancer cells. J Cell Physiol. 2010;225(3):801–9.
Acknowledgements
Thanks to Pascal DuBois for helping with literature search, preparation of Fig. 13.1, and proofreading of the manuscript. Thanks too to Dr. Ian Beales for his outstanding supervision of Dr. Ogunwobi’s initial work on the role of leptin in cancer. Work in Dr. Ogunwobi’s laboratory at Hunter College of The City University of New York is currently supported by a NIH RCMI grant to Hunter College, and Pilot Project and Innovation Seed Funding awards from the NIMHD-funded Center for Translational and Basic Research at Hunter College.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Ogunwobi, O.O. (2015). Leptin, Cell Cycle, and Cancer. In: Dagogo-Jack, MD, S. (eds) Leptin. Springer, Cham. https://doi.org/10.1007/978-3-319-09915-6_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-09915-6_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-09914-9
Online ISBN: 978-3-319-09915-6
eBook Packages: MedicineMedicine (R0)