
Summary
Pathogenic Salmonella species reside in the phagosomes of infected cells and inject various virulence factors into the phagosome and cytoplasm of the host cell to evade the defense mechanisms. During infection of mice with Salmonella enterica serovar Typhimurium (S. typhimurium), which mimics the typhoid disease in humans, type I IFN signaling leads to rapid host fatality, and abrogation of this signaling leads to prolonged host survival. The detrimental role of type I IFN during Salmonella infection appears to be related to the modulation of inflammatory response.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
TLR-Dependent Induction of Type I IFNs
During infectious disease a host can recognize pathogens by various receptors unified under the term pathogen recognition receptors (PRR). These receptors initiate signaling cascades to alert the host immune system of the imminent danger associated with the invading pathogen, which commits the immune system to first restraining and ultimately, clearing off the pathogen [1]. Such signaling results in activation of the innate immune response, which in turn leads to amplification of the adaptive branch of the immune system [2]. Among the best described PRRs is the family of the Toll-like receptors (TLRs). These receptors are expressed by most if not all host cells, are localized on the host cell surface and at the endosomal compartment or both cell surface and endosomes, depending on the cell type. For recognition and activation of TLR4 by LPS, a set of adaptor proteins, MD2 and CD14, are necessary. These adaptor proteins are located extracellularly. The MD2–TLR4 complex is able to distinguish smooth or rough forms of LPS [3], where CD14 relays the signal accordingly [4]. For the rough form, the signaling through TLR4 is MyD88-dependent, and when smooth LPS serves as ligand, the TRIF-mediated signaling cascade is the dominant form of downstream gene activation [4]. The distinction between smooth and rough form is based on the oligosaccharide component of the LPS [5, 6]. LPS on the surface of Salmonella is of the smooth form, which suggests that the TRIF pathway is the predominant mechanism of type I IFN expression. The lipid A component of LPS is also highly inflammatory, which activates the MyD88 pathway, and synthetic structures with modification of lipid A have been shown to selectively induce the TRIF pathway [7]. Furthermore, the modifications of lipid A also determine how potent if any the signaling cascades are activated. Namely, LPS comprised of hexa- and hepta-acetylated lipid A is strongly inflammatory, in contrast to tetra-acetylated lipid A [8, 9]. The pathogenic Salmonella LPS has the smooth oligosaccharide component and the hexa-acetylated lipid A. This assures that the host cells are strongly engaged and potent inflammatory response is mounted. Type I interferon production in response to TLR4 engagement occurs predominantly through a MyD88 independent, TRIF-dependent mechanism [10].
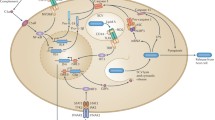
Recent studies have shown that Salmonella exploits this induction of a strong inflammatory response to promote its intracellular survival [11, 12]. CpG treatment of mice that normally resolve S. typhimurium infection resulted in host susceptibility [12]. This was due to the enhanced intracellular proliferation of Salmonella, which requires expression of the Salmonella pathogenicity island 2 (SPI-2) genes [12]. In another study, when TLR2-TLR4-TLR9 triple knock-out mice were infected with Salmonella, they survived better than combinations of double knock-outs of the same TLR members [11]. Again this was shown to operate through induction of SPI-2 genes, which were induced in response to TLR engagement. Activated TLR9 recruits MyD88, IRAK1, IRAK4, and TRAF6 to phosphorylate/activate IRF7, followed by IRF7 translocation in the nucleus where it can activate type I IFNs production [13]. This is summarized in Fig. 1.

Major induction pathways of type I IFNs by Salmonella and role of type I IFN during infection at the cellular level. Salmonella SPI-1 effectors induce its engulfment in a SCV or phagosome where the SPI-2 effectors get induced. Although many functions are described for SPI-2 effectors, it remains unclear whether they regulate type I IFN production. Once in the phagosome or SCV, activated TLR9 can relay signals to IRF7 to stimulate IRGs and the TLR5 similar to TLR4 via MyD88 pathway activates the NF-κB complex. LPS activated TLR4, signals through MyD88 or TRIF-dependent pathways. The MyD88 pathway, via activated NF-κB, leads to induction of proinflammatory cytokines and chemokines and the TRIF pathway leads to IRF3 activation and type I IFN production. The type I IFN produced then engages the IFNAR to induce production of several hundreds of interferon regulated genes or gamma-activated sequences (GAS) via autocrine loop. IKK inhibitor of NF kappa-B kinase, TRAF tumor necrosis factor receptor-associated factor
Type III Secretion System-Dependent Induction of Type I IFN
Salmonella infects various types of cells. While phagocytic cells such as macrophages and dendritic cells can rapidly phagocytose Salmonella, the non-phagocytic cells are infected through a type III secretion system (T3SS) encoded in the SPI-1 cluster of genes. The T3SSs are needle-like structures canonically used by bacteria to bridge bacterial cytoplasm with the host cytosol and translocate proteinaceous effector molecules, which in case of pathogenic bacteria subvert host cell signaling [14]. The SPI-1 induces host cell structures that promote engulfment of Salmonella and its intracellular translocation into vacuoles, termed Salmonella containing vacuoles, SCVs. Professional phagocytic cells don’t require SPI-1 to phagocytose Salmonella, and once intracellular, the host could potentially recognize other pathogen-associated molecular patterns (PAMPs) beside LPS. However, SPI-1 is active in the phagocytic cells as well. PrgJ, a capping protein? of the T3SS of SPI-1, gets removed from the needle structure of T3SS and enables secretion of Salmonella effectors. This allows Salmonella to engage the NLRC4 inflammasome [15]. Flagellin, which is expressed by Salmonella, and is needed for its virulence, serves as a signal for TLR5 and NLRC4 inflammasome engagement, which in turn leads to activation and production of proinflammatory cytokines [16]. The flagellum is evolutionary related to the T3SS machinery and in certain conditions can secrete proteins as well [17, 18]. By engaging the inflammasomes, the production of active IL-1β is maintained, which is able to positively feed into the type I IFN production by inhibiting the DUBA, deubiqutinase known to remove K63 ubiquitination of TRAF3 [19]. K63 ubiquitination of TRAF3 is a major modification required for IFN gene expression [20].
In a study that addressed the role of caspase-8 during Salmonella infection it was shown that caspase-8 is recruited to the inflammasome complex. This recruitment was shown to be specific to S. typhimurium infection and as part of that complex it contributed positively to IL-1β production [21]. The production of active IL-1β seems to be fine-tuned, as it is shown that SipB, a Salmonella SPI-1 effector protein, promotes its production [22]. Active IL-1β has many other functions, yet the IL-1R signaling by modulating TRAFs remains instrumental for type I IFN production [13, 20]. It is important to note that IL-1 signaling can also accelerate the degradation of IFNAR by activating kinases that add phospho-moiety to a so-called degron sequence within the IFNAR protein [23], therefore adding complexity to the role of IL-1 signaling in type I IFN production and signaling.
Microarray studies focused on the host response to Salmonella infection revealed that many genes are specifically activated. RAW24.7, a murine macrophage cell line infected by S. typhimurium, was assessed for gene expression. The following genes were found to be upregulated: MIP-1α, MIP-1β, MIP-2α, IL-1β, TNF receptor, CD40, IκBα, IκBβ, NF-E2, IRF1, and c-rel among many [24]. In a similar study it was shown that SPI-1 effectors exploit host pathways that are independent of TLR engagement. Many genes in uninfected control remained at same expression level as cells infected by SPI-1 mutant Salmonella strain [25]. In that same study STAT3, a transcriptional factor with pleiotropic effects was upregulated [25], which in cooperation with IRF1 regulated the production of IL-10 [26]. Indeed, IL-10 is an anti-inflammatory cytokine, which has been shown to promote the intracellular proliferation of Salmonella [27].
Role of Type I IFNs During Salmonella Infection
In various infectious disease models (e.g., Listeria, Mycobacteria, Trypanosoma, Candida), it has been shown that IFNAR-deficient mice display enhanced survival [28–31]. Similarly, IFNAR-deficient mice display enhanced survival during infection with virulent S. typhimurium [32]. It is conceivable that pathogens from different domains of life or classes have converged in utilizing mechanisms of subverting the host immune defenses, and the above-mentioned examples would reiterate the importance of type I IFN signaling in host–pathogen interactions. The complexity of interferon signaling pathways and its impact on Salmonella pathogenesis was further revealed in another study in which UBP43-deficient mice (alternatively known as USP18) were shown to have elevated type I interferon signaling, yet these mice were able to control Salmonella better in vivo, since the splenic bacterial burden was reduced in UBP43-deficient mice; however, there was no difference in host susceptibility between WT and UBP43-deficient mice [33, 34]. UBP43 is a member of the “Ubiquitin specific protease” family that cleaves ISG15, a ubiquitin-like posttranslational modification (PTM) of proteins, which appears to be dependent on IFN-signaling [35]. The mechanism behind the better control of Salmonella in UBP43-deficient mice was attributed to the sustained and hyperactive JAK-STAT1 signaling, as the failure to remove ISG15 from the JAK1 resulted in prolonged JAK1-STAT1 signaling [36]. Furthermore, UBP43-deficient mice displayed elevated expression of genes that are dependent on type I IFN signaling (ISGs), and were hypersensitive to LPS-induced septic shock [33]. While these results may appear to be at odds with the phenotype obtained in IFNAR-deficient mice, however, the UBP43 deficient mice display elevated inflammatory signaling in contrast to IFNAR-deficient mice. Elevated inflammatory signaling in UBP43-deficient mice may promote initial clearance of bacteria, but the overt inflammatory response may lead to fatality at a later time period. Work on Salmonella invasiveness after treatment with type I IFN, suggests that epithelial cells are less susceptible to invasion [37], and because of that impaired invasion it is argued that mice challenged intragastrically with Salmonella show enhanced survival if treated with type I IFNs [38].
Furthermore type I IFN signaling is implicated in the regulation of inflammasome activation, and stimulation of necrosome formation, both presently understood as distinct signaling complexes. Inflammasomes are protein complexes that enable activation of inflammatory caspases, which drive immune responses by stimulating the production of proinflammatory cytokines, and by inducing pyroptosis, a mechanism of proinflammatory cell death [39]. Work done on elucidating the mechanisms involved in inflammasome regulation by IFNAR signaling indicated that type I IFN inhibits the production of IL-1β, through regulation of the NLRP3, leading to reduced transcript levels of pro-IL-1β [40]. Yet still, during infection with gram-negative bacteria, type I IFN promotes IL-1β production by controlling caspase-11 activity [41], and most likely such duality is dependent on the amount of IFN-β.
Necrosome is a protein complex that when assembled leads the host cell to necroptosis, a proinflammatory mechanism of cell death. Typically it is induced by TNFα-TNFR1 interaction in the absence of apoptosis [42]. During S. typhimurium infection of macrophages it was shown that type I IFN signaling stimulates necrosome activation leading to necrotic cell death, where IFNAR KO bone marrow macrophages showed enhanced survival [32]. Type I IFN signaling is the critical check-point of necrosome activation in macrophages. During in vivo infection, IFNAR-deficient mice had more macrophages, which correlated to better control of Salmonella. Additionally, the abrogated cytokine signaling downstream of IFNAR can also be a contributing factor, as the pleiotropic effects of IFN signaling can modulate subsequent downstream cytokine and chemokine signaling. Necroptosis is induced by IFN-α/β and IFN-γ signaling pathways independent of death receptors signaling, but dependent on Protein kinase RNA-activated (PKR) and Fas-associated death domain (FADD) [43]. Further, even TNF-dependent necrosome activation appears to be dependent on type I IFN signaling (S. Sad, unpublished).
A hallmark of necroptosis is the release of damage associated molecular patterns (DAMPs) that can act as “secondary” ligands during host–pathogen interactions and can become major drivers of inflammatory responses, although their contribution is often neglected. Necrosome activation that is associated with Salmonella infection that is notorious for inducing host cell death generates overt pathology leading to adverse outcome. During infections by pathogens that are able to inhibit caspases, necroptosis can be regarded as a backup mechanism that initiates inflammatory cell death and alerts the immune system defenses. Specifically, in Salmonella infection, the outcome and progression are multifactorial and will not be only dependent on type I IFN signaling [44], yet the IFNAR-deficient mice show significantly reduced susceptibility to Salmonella infection [32].
Final Remarks
New pathways of type I IFN signaling have emerged that seem to indicate that the impact of type I IFN signaling may be highly dependent on the disease context [45]. The IFNAR KO mice have been used extensively in many studies and have revealed both the positive and negative role of type I IFN signaling. At the cellular level the role of type I IFN signaling is also complex. Resistance to LPS shock is mediated by ablation of type I IFN signaling, as IFNAR1 KO, but not the IFNAR2 KO, mice are resistant to LPS [45, 46]. Type I IFN appears to be a key mechanism that impacts inflammasome and necrosome activation, although the precise mechanistic details are lacking currently. These two distinct signaling complexes, inflammasome and necrosome, might have substantial cross-talk since both are controlled by type I IFN. Salmonella is a chronic intracellular pathogen, which results in persistent activation of immune response. It is therefore quite conceivable that type I IFN signaling plays a key role in this process, which results in a deleterious host outcome due to persistent pathology.
References
Broz P, Monack DM (2013) Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol 13:551–565
Iwasaki A, Medzhitov R (2010) Regulation of adaptive immunity by the innate immune system. Science 327:291–295
Huber M, Kalis C, Keck S, Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Beutler B, Galanos C, Freudenberg MA (2006) R-form LPS, the master key to the activation of TLR4/MD-2-positive cells. Eur J Immunol 36:701–711
Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, Freudenberg M, Beutler B (2005) CD14 is required for MyD88-independent LPS signaling. Nat Immunol 6:565–570
Nikaido H (2003) Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67:593–656
Wilkinson SG (1996) Bacterial lipopolysaccharides–themes and variations. Prog Lipid Res 35:283–343
Bowen WS, Minns LA, Johnson DA, Mitchell TC, Hutton MM, Evans JT (2012) Selective TRIF-dependent signaling by a synthetic toll-like receptor 4 agonist. Sci Signal 5:ra13
Janusch H, Brecker L, Lindner B, Alexander C, Gronow S, Heine H, Ulmer AJ, Rietschel ET, Zahringer U (2002) Structural and biological characterization of highly purified hepta-acyl lipid A present in the lipopolysaccharide of the Salmonella enterica sv. Minnesota Re deep rough mutant strain R595. J Endotoxin Res 8:343–356
Lapaque N, Takeuchi O, Corrales F, Akira S, Moriyon I, Howard JC, Gorvel JP (2006) Differential inductions of TNF-alpha and IGTP, IIGP by structurally diverse classic and non-classic lipopolysaccharides. Cell Microbiol 8:401–413
Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN (2002) TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat Immunol 3:392–398
Arpaia N, Godec J, Lau L, Sivick KE, McLaughlin LM, Jones MB, Dracheva T, Peterson SN, Monack DM, Barton GM (2011) TLR signaling is required for Salmonella typhimurium virulence. Cell 144:675–688
Wong CE, Sad S, Coombes BK (2009) Salmonella enterica serovar typhimurium exploits Toll-like receptor signaling during the host-pathogen interaction. Infect Immun 77:4750–4760
Gonzalez-Navajas JM, Lee J, David M, Raz E (2012) Immunomodulatory functions of type I interferons. Nat Rev Immunol 12:125–135
Cornelis GR (2006) The type III secretion injectisome. Nat Rev Microbiol 4:811–825
Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A (2010) Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A 107:3076–3080
Lage SL, Buzzo CL, Amaral EP, Matteucci KC, Massis LM, Icimoto MY, Carmona AK, D’Imperio Lima MR, Rodrigues MM, Ferreira LC, Amarante-Mendes GP, Bortoluci KR (2013) Cytosolic flagellin-induced lysosomal pathway regulates inflammasome-dependent and -independent macrophage responses. Proc Natl Acad Sci U S A 110:E3321–E3330
Karlinsey JE, Tanaka S, Bettenworth V, Yamaguchi S, Boos W, Aizawa SI, Hughes KT (2000) Completion of the hook-basal body complex of the Salmonella typhimurium flagellum is coupled to FlgM secretion and fliC transcription. Mol Microbiol 37:1220–1231
Paul K, Erhardt M, Hirano T, Blair DF, Hughes KT (2008) Energy source of flagellar type III secretion. Nature 451:489–492
Tseng PH, Matsuzawa A, Zhang W, Mino T, Vignali DA, Karin M (2010) Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat Immunol 11:70–75
Hacker H, Tseng PH, Karin M (2011) Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol 11:457–468
Man SM, Tourlomousis P, Hopkins L, Monie TP, Fitzgerald KA, Bryant CE (2013) Salmonella infection induces recruitment of caspase-8 to the inflammasome to modulate IL-1beta production. J Immunol 191:5239–5246
Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A (1999) The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc Natl Acad Sci U S A 96:2396–2401
Fuchs SY (2013) Hope and fear for interferon: the receptor-centric outlook on the future of interferon therapy. J Interferon Cytokine Res 33:211–225
Rosenberger CM, Scott MG, Gold MR, Hancock RE, Finlay BB (2000) Salmonella typhimurium infection and lipopolysaccharide stimulation induce similar changes in macrophage gene expression. J Immunol 164:5894–5904
Bruno VM, Hannemann S, Lara-Tejero M, Flavell RA, Kleinstein SH, Galan JE (2009) Salmonella typhimurium type III secretion effectors stimulate innate immune responses in cultured epithelial cells. PLoS Pathog 5:e1000538
Saraiva M, O’Garra A (2010) The regulation of IL-10 production by immune cells. Nat Rev Immunol 10:170–181
Nguyen T, Robinson N, Allison SE, Coombes BK, Sad S, Krishnan L (2013) IL-10 produced by trophoblast cells inhibits phagosome maturation leading to profound intracellular proliferation of Salmonella enterica typhimurium. Placenta 34:765–774
Chessler AD, Caradonna KL, Da’dara A, Burleigh BA (2011) Type I interferons increase host susceptibility to Trypanosoma cruzi infection. Infect Immun 79:2112–2119
Majer O, Bourgeois C, Zwolanek F, Lassnig C, Kerjaschki D, Mack M, Muller M, Kuchler K (2012) Type I interferons promote fatal immunopathology by regulating inflammatory monocytes and neutrophils during Candida infections. PLoS Pathog 8:e1002811
Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ, Barry C, Kaplan G (2005) Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J Interferon Cytokine Res 25:694–701
O’Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, Perry AK, Nguyen BO, Lane TF, Taniguchi T, Miller JF, Cheng G (2004) Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med 200:437–445
Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S (2012) Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar typhimurium. Nat Immunol 13:954–962
Kim KI, Malakhova OA, Hoebe K, Yan M, Beutler B, Zhang DE (2005) Enhanced antibacterial potential in UBP43-deficient mice against Salmonella typhimurium infection by up-regulating type I IFN signaling. J Immunol 175:847–854
Richer E, Yuki KE, Dauphinee SM, Lariviere L, Paquet M, Malo D (2011) Impact of Usp18 and IFN signaling in Salmonella-induced typhlitis. Genes Immun 12:531–543
Sgorbissa A, Brancolini C (2012) IFNs, ISGylation and cancer: Cui prodest? Cytokine Growth Factor Rev 23:307–314
Malakhova OA, Yan M, Malakhov MP, Yuan Y, Ritchie KJ, Kim KI, Peterson LF, Shuai K, Zhang DE (2003) Protein ISGylation modulates the JAK-STAT signaling pathway. Genes Dev 17:455–460
Bukholm G, Degre M (1983) Effect of human leukocyte interferon on invasiveness of Salmonella species in HEp-2 cell cultures. Infect Immun 42:1198–1202
Bukholm G, Berdal BP, Haug C, Degre M (1984) Mouse fibroblast interferon modifies Salmonella typhimurium infection in infant mice. Infect Immun 45:62–66
Lamkanfi M, Dixit VM (2012) Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol 28:137–161
Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P, Tschopp J (2011) Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34:213–223
Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA (2012) TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150:606–619
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11:700–714
Thapa RJ, Nogusa S, Chen P, Maki JL, Lerro A, Andrake M, Rall GF, Degterev A, Balachandran S (2013) Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci U S A 110:E3109–E3118
Ramos-Morales F (2012) Impact of Salmonella enterica type III secretion system effectors on the eukaryotic host cell. ISRN Cell Biol 2012:32
de Weerd NA, Vivian JP, Nguyen TK, Mangan NE, Gould JA, Braniff SJ, Zaker-Tabrizi L, Fung KY, Forster SC, Beddoe T, Reid HH, Rossjohn J, Hertzog PJ (2013) Structural basis of a unique interferon-beta signaling axis mediated via the receptor IFNAR. Nat Immunol 14:901–907
Mahieu T, Park JM, Revets H, Pasche B, Lengeling A, Staelens J, Wullaert A, Vanlaere I, Hochepied T, van Roy F, Karin M, Libert C (2006) The wild-derived inbred mouse strain SPRET/Ei is resistant to LPS and defective in IFN-beta production. Proc Natl Acad Sci U S A 103:2292–2297
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Shutinoski, B., Sad, S. (2014). The Detrimental Role of Type I Interferon Signaling During Infection with Salmonella typhimurium . In: Parker, D. (eds) Bacterial Activation of Type I Interferons. Springer, Cham. https://doi.org/10.1007/978-3-319-09498-4_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-09498-4_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-09497-7
Online ISBN: 978-3-319-09498-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)