
Summary
Mycobacterium tuberculosis and M. leprae are causative microorganisms for tuberculosis and leprosy, respectively. In contrast to avirulent mycobacteria, which are effectively eliminated by the host immune system, M. tuberculosis and M. leprae can persist and cause diseases in infected individuals. It is unclear how the pathogens survive in the presence of cell-mediated immune responses. Recent studies have revealed that while essential for controlling viral replication, type I interferons (IFNs) also impairs the host control of intracellular bacteria. This chapter overviews recent discoveries, obtained from animal and human studies, on the role of type I IFN in regulating immune response and resistance to mycobacterial infection. The proposed mechanisms explaining how the classic phagosomal pathogens trigger type I IFN production in macrophages as well as how the cytokines may interfere with protective immune responses and regulate disease outcome are also discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Systemic Lupus Erythematosus Patient
- Mycobacterial Infection
- Lepromatous Leprosy
- Phagosomal Maturation
- Phagosomal Membrane
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Mycobacterial Infection and Tuberculosis
Mycobacteria are slow growing, facultative intracellular bacilli that primarily reside in macrophages. The Mycobacterium genus comprises more than 100 different species. Among them are the pathogenic species Mycobacterium tuberculosis and M. leprae, causing tuberculosis (TB) and leprosy, respectively. This chapter will discuss mainly on the role of type I interferons (IFNs) in M. tuberculosis infection, which is the focus of the majority of recent studies. TB is one of the major infectious diseases worldwide [1]. Two billion people are infected with M. tuberculosis, 10 % of whom will eventually develop active TB disease [2]. Annually, more than eight million people develop TB, which is responsible for over 1.3 million deaths, figures still grossly underestimated due to failures in reporting and detection [1, 3]. Once considered to be on its way to extinction, M. tuberculosis is posing a significant threat to global health due to the emergence of multi-drug resistant strains [3]. M. bovis Bacillus Calmette–Guérin (BCG), the only TB vaccine available for humans, is ineffective in protecting adults against pulmonary TB [4]. Therefore understanding the immune responses to the pathogen may lead to improved vaccination and therapy.
Host Immune Response to M. tuberculosis
Host control of M. tuberculosis infection in both humans and mice depends on cell-mediated immunity [5, 6]. Interestingly however, despite the development of an adaptive immune response, some bacilli resist killing and survive within macrophages in granulomas [7, 8]. Mycobacterial granulomas are typically composed of lymphocytes and infected macrophages. The former cell population is thought to provide cytokine mediators necessary for macrophage activation and restriction of intracellular growth of mycobacteria [6, 9]. Importantly, defects in lymphocyte recruitment and retention or effector function during chronic infection can lead to a breakdown of immunity and result in progressive infection [10, 11].
Resistance to mycobacteria is critically dependent on the T helper 1 (Th1) response [5, 6]. Thus, patients and animals deficient in IFN-γ, IL-12, STAT1, or T-cells show significantly increased susceptibility to mycobacterial infections [12]. In addition to CD4+ lymphocytes, natural killer (NK) cells also contribute to cytokine production in M. tuberculosis infection [13]. Critically, host control of M. tuberculosis infection requires intact IFN-γ receptor signaling in both hematopoietic and non-hematopoietic components [14]. It is believed that the key effector function of IFN-γ is to activate infected macrophages to produce antimicrobial mediators, such as nitric oxide and p47 immunity-related GTPases [15]. But, emerging evidence from recent studies indicates that this long-held concept may represent an over-simplified view. For example, in addition to impaired bacterial control, M. tuberculosis-infected Ifng –/– mice show severe pulmonary pathology associated with dramatically increased accumulation of neutrophils [13, 16]. Together, these findings suggest that IFN-γ plays a broader role in inflammation and infection beyond its proposed function in bacterial killing.
Mycobacterial Virulence Mechanisms
Following infection, avirulent mycobacteria are effectively cleared by host defence mechanisms and are unable to persist in the host. In contrast, virulent mycobacteria establish persistent infection in the infected host. The ability of M. tuberculosis to avoid host antimicrobial strategies is well documented [17–21]. One strategy involves the action of the M. tuberculosis mannose-capped lipoarabinomannan that is incorporated into lipid rafts of the plasma membrane, thereby executing arrest in phagosomal maturation [22–25]. The bacterium is known to block fusion of lysosomes as well as inhibit phagosomal acidification [26]. This prevents the activation of a number of pH-dependent antimicrobial compounds, such as maturation of cathepsin D [22], which are required for destroying intracellular bacteria.
It has been assumed that because M. tuberculosis is broadly equipped to combat phagosomal maturation via inhibition of Golgi-trafficking, phagosome acidification and lysosomal fusion, that an operational phagosome would be effective in clearing infection [27]. However, the identification of bacterial strategies that allow the survival of M. tuberculosis mutants in fully mature phagosomes challenges this assumption. These strategies include the ability to deactivate reactive oxygen species (ROS) and protect against NOS2 damage [8]. Thus, it is unlikely that M. tuberculosis causes definitive arrest of phagosomal maturation, but rather delays it [28]. This may be a temporary measure to allow the bacteria to adapt and initiate transcription in response to the intracellular environment [28]. Recent studies suggest that rather than using the mycobacterial phagosome as a replicative niche, as traditionally believed, the mycobacterially-altered phagosomes act as a preparation and “waiting room” for escape of the bacteria into the cytosol [29]. This suggestion has significant ramifications, both for bacterial survival and the host defence mechanisms involved.
In a seminal study, van der Wel et al. [30] report that virulent M. tuberculosis but not heat-killed mycobacteria or vaccine strain M. bovis BCG are present within the cytosol of macrophages 2 days after infection. This translocation is dependent upon the bacterial secretion system, early secretory antigenic target 6 system 1 (ESX-1) apparatus transcribed by the region of difference-1 (RD-1) genes that are present only in virulent mycobacteria including M. tuberculosis [31, 32]. However, interestingly, permeabilization of the phagosomal membrane and cytosolic access to bacterial pathogen-associated molecular patterns (PAMPs) may occur within hours of infection under certain conditions [28], long before complete translocation of bacilli into the cytosol occurs. This may allow bacterial components access to the cytosol and avoid their sensing by endosomal Toll-like receptors (TLRs) [33]. Indeed, M. tuberculosis β-lactamase catalytic activity occurs in the cytosol progressively, from less than 2 days post infection [29]. These observations suggest that release of bacterial products precedes complete escape of the pathogen. Regardless of the sequence of events, this “phagosomal escape” represents a newly characterized virulence mechanism of M. tuberculosis, although whether RD-1 mediates this purported partial permeabilization of the membrane [34] or allows complete translocation of the bacilli into the cytosol [31] remains unclear. Finally, this phagosomal-cytosolic access hypothesis is further supported by studies demonstrating the ability of virulent (RD-1 competent) M. tuberculosis to prime CD8+ T cell responses [35] and activate the inflammasome [36, 37] since both processes require the access of microbial products to cytosolic immune pathways.
Type I IFN Production and IFN Signature
In the case of mycobacterial infections, type I IFNs are produced in vitro by M. tuberculosis-infected murine macrophages [38, 39], as well as human monocyte derived macrophages [17], dendritic cells [40], and differentiated monocytic THP-1 cells [41]. Importantly type I IFNs are induced during infection with virulent but not avirulent mycobacteria such as with M. bovis BCG [17, 21, 42], indicating that type I IFN induction is unique to virulent mycobacteria.
In mice, type I IFNs and their inducible genes are detected in M. tuberculosis-infected tissues [21, 43]. Similarly, in humans IFN-β is expressed in leprosy skin lesions [44]. Although type I IFN cytokine genes are undetectable in peripheral blood of infected human subjects, a large set of IFN-inducible genes are readily detected in the same cells. In a seminal study, Berry et al. [45] identified the presence of a 393 transcript gene signature in whole blood of active TB patients. Further analysis revealed that 86-transcripts can distinguish active TB from other types of inflammatory conditions, such as systemic lupus erythematosus (SLE), which is known to be associated with an enhanced type I IFN gene signature [46–48]. Interestingly, 10–25 % of latently infected subjects also displayed the IFN signature [45]. Considering that approximately 10 % of latent M. tuberculosis-infected individuals will eventually develop active TB in their lifetime, it would be interesting to examine whether the IFN gene signature detected in latently infected individuals could be predictive for disease progression.
This type I IFN-induced gene signature, which includes the transcription factors IRF1, IRF7, Oct-1, and proteins of the STAT family, has been confirmed in patients with active TB in multiple recent reports [49–52]. Importantly, these transcriptional profiles are also observed in experimental models of M. tuberculosis infection in mice and in vitro in human cell lines [49, 50], providing an avenue to investigate these observations in more detail at a functional level. A study examining the expression of interferon regulated genes (IRGs) in cattle infected with M. bovis (the causative agent of bovine TB) found an increase in type I and type II IFN regulated genes such as CXCL10, STAT1, IFI16, IRF7, and OAS1 [53], suggesting that similar virulence mechanisms may be conserved across mycobacterial species.
Leprosy is another major human mycobacterial disease in which a type I IFN gene signature is associated with severe disease outcome. Leprosy has traditionally been classified into two major types; tuberculoid and lepromatous. Historically, the self-healing tuberculoid leprosy is believed to be associated with Th1 responses whereas disseminated lepromatous leprosy, which is characterized by uncontrolled infection and increased tissue pathology, is driven by a strong Th2 response. Interestingly, a recent study demonstrated that lepromatous leprosy is associated with an IFN-β-dependent gene signature [44], providing a novel mechanism for uncontrolled bacterial growth associated with this severe form of the disease. Together, studies in both M. tuberculosis and M. leprae infections have clearly established that the presence of an IFN-inducible gene signature is associated with disease progression and more severe clinical manifestations, although the issue as to whether the observed gene signature is driven by type I, type II, or both remains unclear.
One of the major challenges in TB management is to effectively distinguish latent infection from active disease and to monitor treatment efficacy. To date, no laboratory test is available for these purposes. Therefore, the identification of the whole blood IFN signature associated with active TB disease has generated considerable interest in the clinical and basic TB research communities because of its potential in diagnosing and monitoring TB disease progression [54, 55]. However, the practical impact of the discovery remains unclear, as multiple recent studies have demonstrated that the IFN-inducible gene signature is also associated with other diseases. For example, melioidosis, a disease caused by the intracellular bacterium Burkholderia pseudomallei, also contains the 86-gene IFN signature [52]. Another example is sarcoidosis, a lung disease associated with pulmonary lesions similar to TB, in which the blood IFN gene signature has 80 % overlap with that of active TB patients enrolled in the same study [51] and in the study published by Berry et al. These data suggest that while the IFN signature is associated with acute inflammatory conditions it is not specific for mycobacterial infection and TB.
Regulation of Host Resistance to Mycobacterial Infection by Type I IFN Signaling
The hypervirulence of clinical isolates of M. tuberculosis correlates with the enhanced synthesis of endogenous type I IFN. Mice deficient in the receptor for these cytokines display significantly reduced bacterial loads when chronically infected with M. tuberculosis [21, 38, 56]. More recently, mice deficient in interferon regulatory factor 3 (IRF3), a key molecule required for type I IFN production, were shown to display significantly increased survival and decreased bacterial load relative to their wild-type counterparts following M. tuberculosis infection [34]. Interestingly, in the absence of IFN-γ signaling, type I IFNs play a protective role in M. tuberculosis infection as mice deficient in both Ifngr and Ifnar display increased pathology and mortality than Ifngr single deficient mice [57]. Therefore, it appears the detrimental effect of type I IFN is dependent on the presence of an IFN-γ response to M. tuberculosis.
Conversely, M. tuberculosis-infected mice with elevated type I IFN levels show reduced survival and increased bacillary burden compared to control mice. Intranasal administration of poly:ICLC, a compound that stimulates high-level production of type I IFN, exacerbates pulmonary TB in wild-type but not in type I IFN receptor deficient mice [58]. In addition, increased mycobacterial burden is reported in a mouse model of influenza A virus and M. tuberculosis co-infection [59]. Importantly, the impaired host resistance is dependent on type I IFN signaling, suggesting that concurrent viral infection can exacerbate TB by triggering type I IFN production.
Compared to animal studies, the effect of type I IFN on resistance to mycobacterial infection in humans is less clear. Increased incidence of TB disease has been reported in patients with active autoimmune diseases or on treatment with recombinant IFN-α. For example, it is established that type I IFNs play a key role in disease pathogenesis of SLE and an IFN-inducible gene signature is observed in patients with severe disease [46–48, 60, 61]. In this case, increased incidence of TB disease in SLE patients has been reported [62–64]. However, the causal role for SLE in TB disease progression is difficult to establish, as SLE patients are also more susceptible to other bacterial infections [65, 66]. This may be a consequence of the immunosuppressant therapy administered to SLE patients, rather than a direct exacerbation of TB by type I IFN signaling. It would be informative to examine whether TB/SLE patients displaying the IFN gene signature can benefit from combination therapy with the IFN-α blocking antibody currently being evaluated in clinical trials [67].
Type I IFN therapy is used frequently for the treatment of multiple sclerosis, hepatitis C virus, and a number of cancers [68]. The effect of type I IFN therapy on M. tuberculosis infection remains controversial, since both beneficial and detrimental effects have been reported [69–74]. In a number of cases administration of IFN-α alone or in combination with standard anti-mycobacterial regimen enhanced mycobacterial control in TB patients. In one study, aerosolized IFN-α lead to earlier resolution of infection and was associated with lower levels of IL-1β, IL-6, and TNF-α in bronchoalveolar lavage fluid [75]. In a second case, inclusion of IFN-β to a four drug regimen of mycobacterial antibiotics resulted in rapid resolution of a previously difficult-to-treat infection [76]. Together, these conflicting clinical data make it difficult to conclude the effect of type I IFNs on M. tuberculosis infection and TB disease in humans, although it is plausible that type I IFNs are detrimental to M. tuberculosis control in certain circumstances but beneficial in others.
Innate Sensing for Type I IFN Induction
Mycobacteria are complex microorganisms that primarily interact with cells of the phagocyte lineage (macrophages and dendritic cells). Recognition of mycobacterial products by surface and cytosolic pathogen recognition receptors (PRRs) leads to the activation of multiple innate pathways.
Cell surface sensing of M. tuberculosis 19 kDa lipoprotein occurs through TLR2 in association with TLR1 or TLR6 [77, 78]. However, these TLRs lack TRIF signaling adaptors and are unable to transduce signals culminating in IRF3 activation and IFN production [79]. In infected DCs, sensing of mycobacterial unmethylated CpG-DNA by TLR9 in the endosomal compartment is inhibited by the mycobacteria [80]. Consequently, TLR signaling is not necessary for M. tuberculosis-mediated induction of type I IFNs [19, 21, 42, 81, 82].
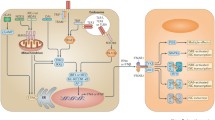
Virulent mycobacteria capable of causing damage to the phagosomal membrane can trigger type I IFN production in infected macrophages, suggesting that cytosolic sensing could be responsible for mycobacterium-induced type I IFN production. Damage to the phagosomal membrane is accomplished by the ESX-1 secretion system [21]. Known cytosolic receptors for muropeptides, components of the bacterial cell wall, are the nucleotide-binding oligomerization domain-containing protein (NOD) receptors [42, 43, 83]. In M. tuberculosis infection, bacterially derived N-glycolyl-muramyl dipeptides (MDPs) in the cytosol have been reported to activate the NOD2 sensor, which leads to the induction of type I IFN production in macrophages [42, 84] along with other functions in the innate and adaptive response [85]. In this model, NOD2 recognition of M. tuberculosis MDP triggers the ubiquitination of the receptor-interacting protein kinase 2 (RIP2), which subsequently activates TANK binding kinase 1 (TBK1) to stimulate IRF5 leading to the transcription of type I IFN genes (Fig. 1).

The postulated mechanisms underlying the induction and function of type I IFN in mycobacterial infection. Phagosomal damage caused by ESX-1, a mycobacterial secretion system present only in virulent mycobacteria, and subsequent cytosolic translocation of microbial products initiates the innate cascades for type I IFN production in mycobacterial-infected macrophages. Although both avirulent and virulent mycobacteria can activate NF-κB (not depicted) and MAPK pathways through surface expressed pattern recognition receptors, only virulent mycobacteria activate cytosolic innate mechanisms leading to type I IFN production. It is postulated that mycobacterial MDP recognized by NOD2 triggers a TBK1 and IRF5-dependent pathway (1) whereas mycobacterial double-stranded DNA activates a STING, TBK1, and IRF3-dependent mechanism (2). Both pathways are not effectively triggered by BCG or other RD1-deficient mycobacteria, which lack the ESX-1 secretion system. Finally, activation of the TPL-2/ERK pathway is capable of limiting M. tuberculosis induced type I IFN production (3) (indicated in red line). The production of type I IFN has been associated with increased susceptibility to mycobacterial infection. The cytokines inhibit known antimicrobial effector mechanisms, such as IFN-γ-induced MHC class II upregulation, IFN-γ receptor expression and IL-1β production. Type I IFNs also induce IL-10 and SOCS1 to suppress host protective Th1 response to the pathogen. Finally, over-production of type I IFN exacerbates M. tuberculosis infection by recruiting immature myeloid cells that are incompetent in killing intracellular bacteria
However, a distinct sensing mechanism has been proposed recently. Cox and colleagues have demonstrated that induction of type I IFN genes in murine cells requires the activation of the Stimulator of IFN genes (STING) by cytosolic mycobacterial products and subsequent phosphorylation of TBK1 and IRF3 [34]. Due to the fact that the pathway plays a key role in sensing bacterial DNA [86–89], the finding implies that mycobacterial DNA is a cytosolic PAMP for type I IFN induction. Indeed, M. tuberculosis DNA is detected in the cytosolic fraction of infected macrophage lysates [34].
Cytosolic cyclic-di-GMP is a molecule unique to bacterial but not mammalian cells [82]. Due to its bacterial specificity, c-di-GMP represents an important target for innate immune recognition, and has been shown to induce potent activation of cytosolic pathways [90]. Listeria monocytogenes c-di-AMP is known to induce type I IFN production [91] by binding to the cytosolic DNA sensor STING [92, 93]. However, unexpectedly, mycobacterial c-di-GMP and c-di-AMP are not involved in stimulating type I IFN production in infected murine macrophages [34] although mutations in c-di-GMP signaling within mycobacteria has been linked to impaired infectivity of M. tuberculosis [94, 95].
Function of Type I IFN in Mycobacterial Infection
While the mechanisms of action by which type I IFN increase mycobacterial virulence are still being investigated, some studies have provided important insights as to how the cytokines negatively affect anti-mycobacterial immunity (summarized in Fig. 1).
Recent work has demonstrated that type I IFNs regulate the production of IL-1β, a critical cytokine in host resistance to M. tuberculosis infection. Novikov et al. [17] demonstrate that exogenous and M. tuberculosis-induced type I IFNs are able to suppress IL1B gene transcription in human macrophages. Although the exact molecular mechanism responsible for the suppression has yet to be defined, a separate study found that the type I IFN-dependent IL-1β inhibition could be partially restored by blocking IL-10 activity [96]. In addition, IFN-β-dependent IL-10 suppresses IFN-γ-dependent antimicrobial mechanisms in M. leprae-infected human macrophages and is associated with the development of lepromatous leprosy [44]. IL-10 is an anti-inflammatory cytokine known to inhibit Th1 responses and macrophage antimicrobial effector functions. Importantly, the cytokine has been shown to exacerbate murine mycobacterial infection in some settings [97–99]. It is, therefore, possible that IL-10 induction by type I IFNs is one of the general mechanisms contributing to the pro-bacterial effect of type I IFNs.
In addition to inducing IL-10, type I IFNs upregulate negative regulators of IFN signaling including suppressor of cytokine signaling 1 (SOCS1) [100, 101]. Socs1 gene expression is elevated in infection with M. tuberculosis strains, particularly those of high virulence and associated with high IFN-α/β stimulating activity [39]. SOCS molecules are also found to be increased in active human TB cases and appear to correlate with disease severity [102, 103]. Experimentally, mouse macrophages deficient in type I IFN receptor demonstrated reduced Socs1 gene induction after mycobacterial infection [104]. Socs1 deficient macrophages displayed reduced bacterial numbers compared to wild-type macrophages and, importantly, this effect was dependent on the inhibition of IFN-γ signaling thereby providing a mechanism by which type I IFNs suppress the antimicrobial activities of IFN-γ [104]. In addition, it is well established that type I IFNs can down-regulate the expression of IFN-γ receptor [105, 106]. This down-regulation has also been observed in M. tuberculosis infection when infected mice were treated with the synthetic type I IFN inducer poly:ICLC [58]. Limiting the expression of cell surface IFN-γ receptor would likely impair IFN-γ-dependent effector functions in macrophages, such as induction of NOS2.
While multiple mechanisms have been postulated to explain the pro-bacterial function of type I IFNs in mycobacterial infection in vitro, the exact function of the cytokines in vivo is poorly understood. Type I IFN has been shown to down-regulate the Th1 response in one study [38] but not others [56, 58]. Interestingly, the detrimental effect of poly:ICLC treatment in M. tuberculosis-infected mice is linked to the dramatically increased accumulation of immature inflammatory monocytes [107]. Flow cytometric sorting experiments reveal that immature myeloid cells harbor significantly more bacteria than their mature counterparts. Therefore, type I IFNs may contribute to host susceptibility to M. tuberculosis infection by supplying a niche for mycobacterial growth and survival. The mechanisms that regulate type I IFN production in vivo are also not well defined. However, a recent study demonstrated that mice deficient in tumor progression locus-2 (TPL-2) show increased levels of IFN-β in the serum and bacillary loads in lungs compared to wild-type controls following infection with L. monocytogenes and M. tuberculosis. It is postulated that the activation of TPL-2 and extracellular signal-regulated kinase 1/2 (ERK1/2) pathways, possibly by TLR signals, prevents excessive production of type I IFN during the infections [108].
Concluding Remarks
Increasing evidence suggests that type I IFNs negatively regulate host resistance to intracellular pathogens including mycobacteria. There is an urgent need for a better understanding of the cytokines’ biological functions in infection, as well as identification of the host and mycobacterial factors required for the cytokine production. While the question as to whether an IFN-inducible gene signature will assist in identifying TB cases and latently infected individuals who are at high risk of developing active disease needs to be carefully examined in longitudinal studies, it is clear that type I IFN inducing agents should be used with caution in people with mycobacterial infection. Finally, it would be interesting to examine whether therapeutic blockade of some components of the type I IFN signaling pathway could promote bacterial clearance and reduce TB reactivation and transmission in humans.
References
World Health Organisation (2012) Global tuberculosis report 2012. WHO Press, Geneva
Zumla A, Raviglione M, Hafner R, von Reyn CF (2013) Tuberculosis. N Engl J Med 368:745–755
Dias HM, Falzon D, Fitzpatrick C, Floyd K, Glaziou P, Hiatt T, Lienhardt C, Nguyen L, Sismanidis C, Timimi H, Uplekar M, van Gemert W, Zignol M (2012) Global tuberculosis report. World Health Organisation, Geneva
Kaufmann SH, Hussey G, Lambert PH (2010) New vaccines for tuberculosis. Lancet 375:2110–2119
Cooper AM (2009) Cell-mediated immune responses in tuberculosis. Annu Rev Immunol 27:393–422
Philips JA, Ernst JD (2012) Tuberculosis pathogenesis and immunity. Annu Rev Pathol 7:353–384
Cosma CL, Sherman DR, Ramakrishnan L (2003) The secret lives of the pathogenic mycobacteria. Annu Rev Microbiol 57:641–676
Monack DM, Mueller A, Falkow S (2004) Persistent bacterial infections: the interface of the pathogen and the host immune system. Nat Rev Microbiol 2:747–765
Saunders BM, Britton WJ (2007) Life and death in the granuloma: immunopathology of tuberculosis. Immunol Cell Biol 85:103–111
Feng CG, Jankovic D, Kullberg M, Cheever A, Scanga CA, Hieny S, Caspar P, Yap GS, Sher A (2005) Maintenance of pulmonary Th1 effector function in chronic tuberculosis requires persistent IL-12 production. J Immunol 174:4185–4192
Feng CG, Britton WJ, Palendira U, Groat NL, Briscoe H, Bean AG (2000) Up-regulation of VCAM-1 and differential expansion of beta integrin-expressing T lymphocytes are associated with immunity to pulmonary Mycobacterium tuberculosis infection. J Immunol 164:4853–4860
Rosenzweig SD, Holland SM (2005) Defects in the interferon-gamma and interleukin-12 pathways. Immunol Rev 203:38–47
Feng CG, Kaviratne M, Rothfuchs AG, Cheever A, Hieny S, Young HA, Wynn TA, Sher A (2006) NK cell-derived IFN-gamma differentially regulates innate resistance and neutrophil response in T cell-deficient hosts infected with Mycobacterium tuberculosis. J Immunol 177:7086–7093
Desvignes L, Ernst JD (2009) Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity 31:974–985
Taylor GA, Feng CG, Sher A (2007) Control of IFN-γ-mediated host resistance to intracellular pathogens by immunity-related GTPases (p47 GTPases). Microbes Infect 9:1644–1651
Nandi B, Behar SM (2011) Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. J Exp Med 208:2251–2262
Novikov A, Cardone M, Thompson R, Shenderov K, Kirschman KD, Mayer-Barber KD, Myers TG, Rabin RL, Trinchieri G, Sher A, Feng CG (2011) Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1beta production in human macrophages. J Immunol 187:2540–2547
Koo IC, Wang C, Raghavan S, Morisaki JH, Cox JS, Brown EJ (2008) ESX-1-dependent cytolysis in lysosome secretion and inflammasome activation during mycobacterial infection. Cell Microbiol 10:1866–1878
Shi R, Otomo K, Yamada H, Tatsumi T, Sugawara I (2006) Temperature-mediated heteroduplex analysis for the detection of drug-resistant gene mutations in clinical isolates of Mycobacterium tuberculosis by denaturing HPLC, SURVEYOR nuclease. Microbes Infect 8:128–135
Kurenuma T, Kawamura I, Hara H, Uchiyama R, Daim S, Dewamitta SR, Sakai S, Tsuchiya K, Nomura T, Mitsuyama M (2009) The RD1 locus in the Mycobacterium tuberculosis genome contributes to activation of caspase-1 via induction of potassium ion efflux in infected macrophages. Infect Immun 77:3992–4001
Stanley SA, Johndrow JE, Manzanillo P, Cox JS (2007) The type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol 178:3143–3152
Fratti RA, Chua J, Vergne I, Deretic V (2003) Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc Natl Acad Sci U S A 100:5437–5442
Vergne I, Chua J, Deretic V (2003) Tuberculosis toxin blocking phagosome maturation inhibits a novel Ca2+/calmodulin-PI3K hVPS34 cascade. J Exp Med 198:653–659
Kang PB, Azad AK, Torrelles JB, Kaufman TM, Beharka A, Tibesar E, DesJardin LE, Schlesinger LS (2005) The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J Exp Med 202:987–999
Welin A, Winberg ME, Abdalla H, Sarndahl E, Rasmusson B, Stendahl O, Lerm M (2008) Incorporation of Mycobacterium tuberculosis lipoarabinomannan into macrophage membrane rafts is a prerequisite for the phagosomal maturation block. Infect Immun 76:2882–2887
Loeuillet C, Martinon F, Perez C, Munoz M, Thome M, Meylan PR (2006) Mycobacterium tuberculosis subverts innate immunity to evade specific effectors. J Immunol 177:6245–6255
van Crevel R, Ottenhoff TH, van der Meer JW (2002) Innate immunity to Mycobacterium tuberculosis. Clin Microbiol Rev 15:294–309
Welin A, Lerm M (2012) Inside or outside the phagosome? The controversy of the intracellular localization of Mycobacterium tuberculosis. Tuberculosis (Edinb) 92:113–120
Simeone R, Bobard A, Lippmann J, Bitter W, Majlessi L, Brosch R, Enninga J (2012) Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog 8:e1002507
van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, Brenner M, Peters PJ (2007) M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129:1287–1298
Houben D, Demangel C, van Ingen J, Perez J, Baldeon L, Abdallah AM, Caleechurn L, Bottai D, van Zon M, de Punder K, van der Laan T, Kant A, Bossers-de Vries R, Willemsen P, Bitter W, van Soolingen D, Brosch R, van der Wel N, Peters PJ (2012) ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol 14:1287–1298
Abdallah AM, Bestebroer J, Savage ND, de Punder K, van Zon M, Wilson L, Korbee CJ, van der Sar AM, Ottenhoff TH, van der Wel NN, Bitter W, Peters PJ (2011) Mycobacterial secretion systems ESX-1 and ESX-5 play distinct roles in host cell death and inflammasome activation. J Immunol 187:4744–4753
Burdette DL, Vance RE (2013) STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol 14:19–26
Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS (2012) Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 11:469–480
Winau F, Weber S, Sad S, de Diego J, Hoops SL, Breiden B, Sandhoff K, Brinkmann V, Kaufmann SH, Schaible UE (2006) Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity 24:105–117
Saiga H, Kitada S, Shimada Y, Kamiyama N, Okuyama M, Makino M, Yamamoto M, Takeda K (2012) Critical role of AIM2 in Mycobacterium tuberculosis infection. Int Immunol 24:637–644
Yang Y, Zhou X, Kouadir M, Shi F, Ding T, Liu C, Liu J, Wang M, Yang L, Yin X, Zhao D (2013) The AIM2 inflammasome is involved in macrophage activation during infection with virulent Mycobacterium bovis strain. J Infect Dis 208:1849–1858
Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, Barry CE 3rd, Freedman VH, Kaplan G (2001) Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc Natl Acad Sci U S A 98:5752–5757
Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ, Barry C, Kaplan G (2005) Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J Interferon Cytokine Res 25:694–701
Remoli ME, Giacomini E, Lutfalla G, Dondi E, Orefici G, Battistini A, Uze G, Pellegrini S, Coccia EM (2002) Selective expression of type I IFN genes in human dendritic cells infected with Mycobacterium tuberculosis. J Immunol 169:366–374
Weiden M, Tanaka N, Qiao Y, Zhao BY, Honda Y, Nakata K, Canova A, Levy DE, Rom WN, Pine R (2000) Differentiation of monocytes to macrophages switches the Mycobacterium tuberculosis effect on HIV-1 replication from stimulation to inhibition: modulation of interferon response and CCAAT/enhancer binding protein beta expression. J Immunol 165:2028–2039
Pandey AK, Yang Y, Jiang Z, Fortune SM, Coulombe F, Behr MA, Fitzgerald KA, Sassetti CM, Kelliher MA (2009) NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog 5:e1000500
Trinchieri G (2010) Type I interferon: friend or foe? J Exp Med 207:2053–2063
Teles RM, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, Komisopoulou E, Kelly-Scumpia K, Chun R, Iyer SS, Sarno EN, Rea TH, Hewison M, Adams JS, Popper SJ, Relman DA, Stenger S, Bloom BR, Cheng G, Modlin RL (2013) Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science 339:1448–1453
Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O’Garra A (2010) An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466:973–977
Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK (2005) Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum 52:1491–1503
Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW (2003) Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A 100:2610–2615
Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V (2003) Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 197:711–723
Ottenhoff TH, Dass RH, Yang N, Zhang MM, Wong HE, Sahiratmadja E, Khor CC, Alisjahbana B, van Crevel R, Marzuki S, Seielstad M, van de Vosse E, Hibberd ML (2012) Genome-wide expression profiling identifies type 1 interferon response pathways in active tuberculosis. PLoS One 7:e45839
Wu K, Dong D, Fang H, Levillain F, Jin W, Mei J, Gicquel B, Du Y, Wang K, Gao Q, Neyrolles O, Zhang J (2012) An interferon-related signature in the transcriptional core response of human macrophages to Mycobacterium tuberculosis infection. PLoS One 7:e38367
Maertzdorf J, Weiner J 3rd, Mollenkopf HJ, Network TB, Bauer T, Prasse A, Muller-Quernheim J, Kaufmann SH (2012) Common patterns and disease-related signatures in tuberculosis and sarcoidosis. Proc Natl Acad Sci U S A 109:7853–7858
Koh GC, Schreiber MF, Bautista R, Maude RR, Dunachie S, Limmathurotsakul D, Day NP, Dougan G, Peacock SJ (2013) Host responses to melioidosis and tuberculosis are both dominated by interferon-mediated signaling. PLoS One 8:e54961
Wang J, Zhou X, Pan B, Wang H, Shi F, Gan W, Yang L, Yin X, Xu B, Zhao D (2013) Expression pattern of interferon-inducible transcriptional genes in neutrophils during bovine tuberculosis infection. DNA Cell Biol 32:480–486
Berry MP, Blankley S, Graham CM, Bloom CI, O’Garra A (2013) Systems approaches to studying the immune response in tuberculosis. Curr Opin Immunol 25:579–587
Bloom CI, Graham CM, Berry MP, Wilkinson KA, Oni T, Rozakeas F, Xu Z, Rossello-Urgell J, Chaussabel D, Banchereau J, Pascual V, Lipman M, Wilkinson RJ, O’Garra A (2012) Detectable changes in the blood transcriptome are present after two weeks of antituberculosis therapy. PLoS One 7:e46191
Ordway D, Henao-Tamayo M, Harton M, Palanisamy G, Troudt J, Shanley C, Basaraba RJ, Orme IM (2007) The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J Immunol 179:522–531
Desvignes L, Wolf AJ, Ernst JD (2012) Dynamic roles of type I and type II IFNs in early infection with Mycobacterium tuberculosis. J Immunol 188:6205–6215
Antonelli LR, Gigliotti Rothfuchs A, Goncalves R, Roffe E, Cheever AW, Bafica A, Salazar AM, Feng CG, Sher A (2010) Intranasal poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J Clin Invest 120:1674–1682
Redford PS, Mayer-Barber KD, McNab FW, Stavropoulos E, Wack A, Sher A, O’Garra A (2014) Influenza A virus impairs control of Mycobacterium tuberculosis coinfection through a type I interferon receptor-dependent pathway. J Infect Dis 209:270–274
Bengtsson AA, Sturfelt G, Truedsson L, Blomberg J, Alm G, Vallin H, Ronnblom L (2000) Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus 9:664–671
Dall’era MC, Cardarelli PM, Preston BT, Witte A, Davis JC Jr (2005) Type I interferon correlates with serological and clinical manifestations of SLE. Ann Rheum Dis 64:1692–1697
Erdozain JG, Ruiz-Irastorza G, Egurbide MV, Martinez-Berriotxoa A, Aguirre C (2006) High risk of tuberculosis in systemic lupus erythematosus? Lupus 15:232–235
Prabu V, Agrawal S (2010) Systemic lupus erythematosus and tuberculosis: a review of complex interactions of complicated diseases. J Postgrad Med 56:244–250
Sayarlioglu M, Inanc M, Kamali S, Cefle A, Karaman O, Gul A, Ocal L, Aral O, Konice M (2004) Tuberculosis in Turkish patients with systemic lupus erythematosus: increased frequency of extrapulmonary localization. Lupus 13:274–278
Mitchell SR, Nguyen PQ, Katz P (1990) Increased risk of neisserial infections in systemic lupus erythematosus. Semin Arthritis Rheum 20:174–184
Zandman-Goddard G, Shoenfeld Y (2003) SLE and infections. Clin Rev Allergy Immunol 25:29–40
Yao Y, Richman L, Higgs BW, Morehouse CA, de los Reyes M, Brohawn P, Zhang J, White B, Coyle AJ, Kiener PA, Jallal B (2009) Neutralization of interferon-alpha/beta-inducible genes and downstream effect in a phase I trial of an anti-interferon-alpha monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum 60:1785–1796
Pestka S, Krause CD, Walter MR (2004) Interferons, interferon-like cytokines, and their receptors. Immunol Rev 202:8–32
Sabbatani S, Manfredi R, Marinacci G, Pavoni M, Cristoni L, Chiodo F (2006) Reactivation of severe, acute pulmonary tuberculosis during treatment with pegylated interferon-alpha and ribavirin for chronic HCV hepatitis. Scand J Infect Dis 38:205–208
Belkahla N, Kchir H, Maamouri N, Ouerghi H, Hariz FB, Chouaib S, Chaabouni H, Mami NB (2010) [Reactivation of tuberculosis during dual therapy with pegylated interferon and ribavirin for chronic hepatitis C]. Rev Med Interne 31:e1–3
Farah R, Awad J (2007) The association of interferon with the development of pulmonary tuberculosis. Int J Clin Pharmacol Ther 45:598–600
Telesca C, Angelico M, Piccolo P, Nosotti L, Morrone A, Longhi C, Carbone M, Baiocchi L (2007) Interferon-alpha treatment of hepatitis D induces tuberculosis exacerbation in an immigrant. J Infect 54:e223–e226
Perez-Elias MJ, Garcia-San Miguel L, Gonzalez Garcia J, Montes Ramirez ML, Muriel A, Machin-Lazaro JM, Martinez-Baltanas A, Zamora F, Moreno A, Martin-Davila P, Quereda C, Gomez-Mampaso E, Moreno S (2009) Tuberculosis complicating hepatitis C treatment in HIV-infected patients. Clin Infect Dis 48:e82–e85
Tsai MC, Lin MC, Hung CH (2009) Successful antiviral and antituberculosis treatment with pegylated interferon-alfa and ribavirin in a chronic hepatitis C patient with pulmonary tuberculosis. J Formos Med Assoc 108:746–750
Giosue S, Casarini M, Alemanno L, Galluccio G, Mattia P, Pedicelli G, Rebek L, Bisetti A, Ameglio F (1998) Effects of aerosolized interferon-alpha in patients with pulmonary tuberculosis. Am J Respir Crit Care Med 158:1156–1162
Zarogoulidis P, Kioumis I, Papanas N, Manika K, Kontakiotis T, Papagianis A, Zarogoulidis K (2012) The effect of combination IFN-alpha-2a with usual antituberculosis chemotherapy in non-responding tuberculosis and diabetes mellitus: a case report and review of the literature. J Chemother 24:173–177
Pecora ND, Gehring AJ, Canaday DH, Boom WH, Harding CV (2006) Mycobacterium tuberculosis LprA is a lipoprotein agonist of TLR2 that regulates innate immunity and APC function. J Immunol 177:422–429
Akira S, Takeda K (2004) Toll-like receptor signalling. Nat Rev Immunol 4:499–511
Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S (2003) Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301:640–643
Simmons DP, Canaday DH, Liu Y, Li Q, Huang A, Boom WH, Harding CV (2010) Mycobacterium tuberculosis and TLR2 agonists inhibit induction of type I IFN and class I MHC antigen cross processing by TLR9. J Immunol 185:2405–2415
Park JH, Kim YG, McDonald C, Kanneganti TD, Hasegawa M, Body-Malapel M, Inohara N, Nunez G (2007) RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol 178:2380–2386
Monroe KM, McWhirter SM, Vance RE (2010) Induction of type I interferons by bacteria. Cell Microbiol 12:881–890
Watanabe T, Asano N, Fichtner-Feigl S, Gorelick PL, Tsuji Y, Matsumoto Y, Chiba T, Fuss IJ, Kitani A, Strober W (2010) NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J Clin Invest 120:1645–1662
Leber JH, Crimmins GT, Raghavan S, Meyer-Morse NP, Cox JS, Portnoy DA (2008) Distinct TLR- and NLR-mediated transcriptional responses to an intracellular pathogen. PLoS Pathog 4:e6
Divangahi M, Mostowy S, Coulombe F, Kozak R, Guillot L, Veyrier F, Kobayashi KS, Flavell RA, Gros P, Behr MA (2008) NOD2-deficient mice have impaired resistance to Mycobacterium tuberculosis infection through defective innate and adaptive immunity. J Immunol 181:7157–7165
Watson RO, Manzanillo PS, Cox JS (2012) Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150:803–815
Ishikawa H, Ma Z, Barber GN (2009) STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792
Ishikawa H, Barber GN (2011) The STING pathway and regulation of innate immune signaling in response to DNA pathogens. Cell Mol Life Sci 68:1157–1165
Barber GN (2011) Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr Opin Immunol 23:10–20
Karaolis DK, Means TK, Yang D, Takahashi M, Yoshimura T, Muraille E, Philpott D, Schroeder JT, Hyodo M, Hayakawa Y, Talbot BG, Brouillette E, Malouin F (2007) Bacterial c-di-GMP is an immunostimulatory molecule. J Immunol 178:2171–2181
McWhirter SM, Barbalat R, Monroe KM, Fontana MF, Hyodo M, Joncker NT, Ishii KJ, Akira S, Colonna M, Chen ZJ, Fitzgerald KA, Hayakawa Y, Vance RE (2009) A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J Exp Med 206:1899–1911
Woodward JJ, Iavarone AT, Portnoy DA (2010) c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328:1703–1705
Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE (2011) STING is a direct innate immune sensor of cyclic di-GMP. Nature 478:515–518
Stewart GR, Patel J, Robertson BD, Rae A, Young DB (2005) Mycobacterial mutants with defective control of phagosomal acidification. PLoS Pathog 1:269–278
Cui T, He Z (2012) C-di-GMP signaling and implications for pathogenesis of Mycobacterium tuberculosis. Chin Sci Bull 57:4387–4393
Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, Oland S, Gordon S, Sher A (2011) Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity 35:1023–1034
Beamer GL, Flaherty DK, Assogba BD, Stromberg P, Gonzalez-Juarrero M, de Waal Malefyt R, Vesosky B, Turner J (2008) Interleukin-10 promotes Mycobacterium tuberculosis disease progression in CBA/J mice. J Immunol 181:5545–5550
Redford PS, Boonstra A, Read S, Pitt J, Graham C, Stavropoulos E, Bancroft GJ, O’Garra A (2010) Enhanced protection to Mycobacterium tuberculosis infection in IL-10-deficient mice is accompanied by early and enhanced Th1 responses in the lung. Eur J Immunol 40:2200–2210
Redford PS, Murray PJ, O’Garra A (2011) The role of IL-10 in immune regulation during M. tuberculosis infection. Mucosal Immunol 4:261–270
Piganis RA, De Weerd NA, Gould JA, Schindler CW, Mansell A, Nicholson SE, Hertzog PJ (2011) Suppressor of cytokine signaling (SOCS) 1 inhibits type I interferon (IFN) signaling via the interferon alpha receptor (IFNAR1)-associated tyrosine kinase Tyk2. J Biol Chem 286:33811–33818
Fenner JE, Starr R, Cornish AL, Zhang JG, Metcalf D, Schreiber RD, Sheehan K, Hilton DJ, Alexander WS, Hertzog PJ (2006) Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nat Immunol 7:33–39
Almeida AS, Lago PM, Boechat N, Huard RC, Lazzarini LC, Santos AR, Nociari M, Zhu H, Perez-Sweeney BM, Bang H, Ni Q, Huang J, Gibson AL, Flores VC, Pecanha LR, Kritski AL, Lapa e Silva JR, Ho JL (2009) Tuberculosis is associated with a down-modulatory lung immune response that impairs Th1-type immunity. J Immunol 183:718–731
Masood KI, Rottenberg ME, Salahuddin N, Irfan M, Rao N, Carow B, Islam M, Hussain R, Hasan Z (2013) Expression of M. tuberculosis-induced suppressor of cytokine signaling (SOCS) 1, SOCS3, FoxP3 and secretion of IL-6 associates with differing clinical severity of tuberculosis. BMC Infect Dis 13:13
Carow B, Ye X, Gavier-Widen D, Bhuju S, Oehlmann W, Singh M, Skold M, Ignatowicz L, Yoshimura A, Wigzell H, Rottenberg ME (2011) Silencing suppressor of cytokine signaling-1 (SOCS1) in macrophages improves Mycobacterium tuberculosis control in an interferon-gamma (IFN-gamma)-dependent manner. J Biol Chem 286:26873–26887
Kearney SJ, Delgado C, Eshleman EM, Hill KK, O’Connor BP, Lenz LL (2013) Type I IFNs downregulate myeloid cell IFN-gamma receptor by inducing recruitment of an early growth response 3/NGFI-A binding protein 1 complex that silences Ifngr1 transcription. J Immunol 191:3384–3392
Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL (2010) Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J Exp Med 207:327–337
Bozza PT, Bai X, Feldman NE, Chmura K, Ovrutsky AR, Su W-L, Griffin L, Pyeon D, McGibney MT, Strand MJ, Numata M, Murakami S, Gaido L, Honda JR, Kinney WH, Oberley-Deegan RE, Voelker DR, Ordway DJ, Chan ED (2013) Inhibition of nuclear factor-kappa B activation decreases survival of Mycobacterium tuberculosis in human macrophages. PLoS One 8:e61925
McNab FW, Ewbank J, Rajsbaum R, Stavropoulos E, Martirosyan A, Redford PS, Wu X, Graham CM, Saraiva M, Tsichlis P, Chaussabel D, Ley SC, O’Garra A (2013) TPL-2-ERK1/2 signaling promotes host resistance against intracellular bacterial infection by negative regulation of type I IFN production. J Immunol 191:1732–1743
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Stifter, S.A., Coleman, M.C., Feng, C.G. (2014). Regulation of Host Response to Mycobacteria by Type I Interferons. In: Parker, D. (eds) Bacterial Activation of Type I Interferons. Springer, Cham. https://doi.org/10.1007/978-3-319-09498-4_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-09498-4_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-09497-7
Online ISBN: 978-3-319-09498-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)