
Abstract
Multidimensional time-resolved spectroscopy allows disentangling particular aspects of the molecular dynamics, which are normally hidden from linear techniques. In this chapter, we show how third- and fifth-order techniques using sub-20 fs pulses can be applied to address coherence and population dynamics in the excited states of biomolecules. In particular, broadband four wave mixing is combined with an initial pump pulse to promote population to the excited state. With this approach, it is possible to interrogate the potential surface of the excited and ground states during the excited state evolution with a time resolution better than 20 fs. Three general aspects of the excited state dynamics are discussed. (1) The assignment of vibrational coherence to the respective excited state potential is illustrated for retinal in solution and in the protein environment. By changing the excitation wavelength and comparing low- and high-frequency vibrational coherence content, it is shown that low-frequency modes are predominantly originated in the excited state, while high-frequency modes belong to the ground state. (2) The temporal resolution of dark electronic states in lycopene is investigated with pump-DFWM. Contrasting to lower-order techniques, pump-DFWM allows to snapshot the ultrafast population relaxation directly after the excitation of the S2 electronic state. (3) The evolution of the vibrational coherence in the excited state is demonstrated for β-carotene. This gives accurate information on the instantaneous frequency, populations and even anharmonicities of all relevant vibrational modes on the potential surface of the excited state.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
9.1 Introduction
The evolution of photochemical transformations has been in the focal point of ultrafast time-resolved spectroscopy since its advent [1–6]. In this regard, the ability to follow the population and coherence dynamics in the femtosecond time scale with high resolution, particularly in excited states, is a major pre-requisite to fully understand how structural changes in (bio-)chromophores take place [7–10].
Time-resolved spectroscopy of excited potential surfaces is, though, more challenging than the ground state spectroscopy due to two major aspects. The first one is the number of excited molecules contributing to the signal. Under normal excitation regimes (i.e. well below saturation), the number of excited molecules is much smaller than the number of molecules in the ground state. Contributions from the excited state may therefore be weak and difficult to detect. This can be specially complicated in (self-)heterodyne detection methods if ground and excited state contributions spectrally overlap. In methods using such kind of detection, the intensities of different contributions add linearly, which can obscure, or even totally cancel, weak signals. Such an effect can be observed e.g. in transient absorption (TA) when a negative signal like stimulated emission (SE) overlaps with an excited state absorption (ESA). This contrasts to background-free third-order methods, where all contributions will be detected with a much higher sensitivity to small variations of the dynamics in the excited state.
The second aspect is related to the time scale of processes in the excited state. After excitation, population and coherence in the higher electronic states are not in equilibrium but evolve in many (bio-)chromophores often on a very fast time scale. Such time scales range from hundreds of femtoseconds as found for the isomerization of protein-bound retinal [11] to just some tens of femtoseconds as observed for the internal conversion between excited states of carotenoids [12]. While ultrafast transient population evolution can in principle be resolved by using state-of-the-art sub-10 fs pulses [13], following the evolution of the vibrational coherence is not trivial. Not just the amplitude of oscillatory phenomena contains information on the deactivation of the excited state, but also the frequency and the phase may offer important insight. Nevertheless, determination of frequency and phase during an ultrafast relaxation process is demanding, which has received a lot of attention recently. It requires accurate analysis in order to avoid phase and frequency changes due to pure analysis artifacts [14, 15].
One way of addressing these central aspects in time-resolved spectroscopy is to increase the dimensionality of the nonlinear optical interaction. Multidimensional time-resolved spectroscopy employs additional ultrashort pulses in order to open a new spectral and/or temporal observation window, which is normally hidden from lower-order techniques [16]. For example, this allows probing a specific molecular response without the need of complex fitting algorithms or assumptions [17, 18]. In this chapter, we show how four wave mixing methods with three beams like coherent anti-Stokes Raman scattering (CARS) and transient grating (TG) can be combined with an additional initial pump beam to be selective to excited state dynamics in the condensed phase (Fig. 9.1). The so called pump-degenerate four wave mixing (pump-DFWM) technique [19, 20] consists of two interactions with the initial pump pulse to produce coherences and populations in the excited state, which will be probed by the consecutive four wave mixing sequence. In this sense, pump-DFWM can be thought of as a “pump-probe” method, where the probe is not a single pulse, but is a nonlinear probe sequence of three ultrashort pulses. Due to their bandwidths, the pulses of the DFWM sequence generate both CARS (and its Stokes counterpart CSRS) and TG contributions.

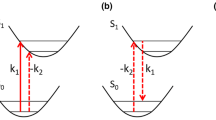
Interaction scheme of (a) pump-DFWM, (b) electronically resonant DFWM and (c) nonresonant DFWM. In pump-DFWM (a), the excited potential can be exclusively investigated by pre-exciting the ground state population with the initial pump. This is not the case when using DFWM only: Resonant DFWM (b) excites and probes dynamics in both potential surfaces, i.e., excited and ground state dynamics
Three fundamental aspects of the excited state dynamics found in several molecular systems are discussed using such nonlinear experiments. Firstly, the assignment of vibrational coherence to the respective electronic states is shown in the case of Retinal Protonated Schiff Base (RPSB) using DFWM (Fig. 9.1(b) and (c)) with sub-20 fs time resolution (Sect. 9.3.1). Tuning the excitation spectrum of the DFWM sequence is exploited with its high sensitivity to disentangle the origin of low- (<800 cm−1) and high-frequency (>800 cm−1) molecular modes. The second aspect is the detection of short-living dark electronic states, whose excitation via one-photon is forbidden. In Sect. 9.3.2, pump-DFWM (Fig. 9.1(a)) is applied to lycopene in order to unravel the presence of additional electronic states after the relaxation of the S2 state. The focus is the unique capability of pump-DFWM to detect additional relaxation pathways due to its higher dimensionality. The experimental results are further supported by a simulation in the response function formalism. In the last experimental Sect. 9.3.3, vibrational coherences in a vibrationally excited electronic state are addressed with pump-DFWM. In particular the vibrational evolution of high-frequency modes are resolved with a high time and spectral resolution. This is demonstrated for β-carotene during the internal conversion between S2 and S1.
9.2 Pump-Degenerate Four Wave Mixing
9.2.1 Signal Generation
Pump-DFWM is an ultrafast technique able to obtain the complete vibrational spectra as well as electronic population and coherence relaxation in excited state potentials [21, 22]. The excitation with the initial pump (Fig. 9.2(a)) generates population and vibrational coherence in the excited state, which will be probed by the nonlinear interactions with the DFWM sequence. Several delay times between the excitation pulses can be defined (Fig. 9.2(a)). The first one is the delay T between the initial pump pulse and the DFWM sequence. During this delay, population as well as vibrational coherence induced by the initial pump will relax. The second time interval is the delay τ 12 between the pump pulse and Stokes of the DFWM. During this time, as in pure DFWM, the electronic coherence between the participating states will evolve. This has been exploited by us to distinguish normal molecular modes from polarization beating [23]. The last time interval is the delay τ 23 between the pump/Stokes and probe pulses of the DFWM sequence.

(a) Scheme showing the respective delays between all beams. (b) Extended folded BOXCARS phase-matching scheme used in the (pump-)DFWM experiment
The higher dimensionality of pump-DFWM allows to follow the molecular dynamics in the excited PES with unprecedented time and spectral resolution (see also Sect. 9.2.2). The transient dynamics triggered by the initial pump pulse is selected and probed by the DFWM sequence. Therefore, each DFWM transient obtained at a given delay T after the excitation contains detailed information on transient populations and coherences [24–26]. The DFWM signal can be separated into two components, an oscillatory part related to the vibrational coherence and a non-oscillatory part. The amplitude of the whole DFWM scales with the electronic population squared of the excited state while the decay of the DFWM non-oscillatory signal gives information on the electronic population relaxation. The oscillatory signal contains information about the vibrational coherence like dephasing times, vibrational frequencies and phases of involved molecular Raman modes. This allows to build a snapshot of the structural changes during the relaxation of the chromophore and a precise picture of chemical transformations even in complex biomolecules (Fig. 9.3). Here it is interesting to understand the interplay between the time and spectral resolution [27]. Although the time resolution of such snapshots is a priori given by the delay T and the pulse durations (Fig. 9.2(a)), the measurement of each vibrational spectrum also modifies the time resolution. In order to measure a spectrum, a DFWM transient with a given length (delay τ 23) must be Fourier transformed. The longer the DFWM transient, the higher will be the spectral resolution, but at the same time, the lower will be the time resolution of the snapshot, and vice-versa. In other words, the interplay between spectral and time resolution in pump DFWM may be fine adjusted by the length of the transient measured during the delay τ 23.

Scheme of pump-DFWM in the excited potential surface and the generation of CARS (as well as CSRS) spectra during the relaxation. For each delay between the initial pump and the DFWM sequence (delay T), a DFWM transient is obtained by varying the delay τ 23. After Fourier transformation of each transient, snapshots of the vibrational spectra can be plotted during the relaxation on the excited potential surface
9.2.2 Setup Description
In our setup [21, 22, 28–31], the laser for the DFWM sequence is initially generated in a broadband noncollinear optical parametric amplifier (nc-OPA), which is pumped by a commercial femtosecond laser system (1 KHz, 300 μJ at 800 nm). After pulse compression in a prism compressor, typical pulse durations are about 11 and 15 fs depending on the central wavelength. After beam-splitting the nc-OPA output in three DFWM beams (pump, Stokes and probe beams), two delay stages are used to control the delay (Fig. 9.2(a)) between pump and Stokes (τ 12) and between Stokes and probe (τ 23). In general, the relative delay τ 23 is scanned with a step length of about 2–3 fs in order to obtain enough data sampling for high-frequency modes. All three beams are focused using the same concave mirror (f=25 cm) in a folded BOXCARS phase-matching geometry (Fig. 9.2(b)). Typical spot diameters are around 60–100 μm and the energies are 20 nJ for pump and Stokes and 15 nJ for the probe.
The combination of DFWM with an initial pump is straightforward. The IP pulse is generated in a second nc-OPA with similar pulse parameters as the DFWM nc-OPA. In such a way, the spectrum can be individually tuned from the DFWM’s nc-OPA, which is an important requirement for the detection of excited electronic states (see Sect. 9.2.3). The delay T between the IP and the DFWM sequence is controlled by an additional delay stage. The IP beam is focused using an additional concave mirror in an extended folded BOXCARS phase-matching geometry. This allows controlling individually the spot size of the IP beam. Typical IP pulse energies used in pump-DFWM experiments depend on several parameters like spectral overlap between excitation and absorption spectra, which will be discussed below. In general, in order to achieve a high signal-to-noise-ratio (SNR), energies in the range between 30–80 nJ are used.
The detection of the (pump-)DFWM signal is performed using two photomultipliers at two different wavelengths. An interferometric filter with a FWHM of 10 nm is used in front of each detector. In order to avoid signal attenuation by using a 50 % beam splitter, the signal beam is split with high-pass filters into two beams. In this case, the low-frequency spectral region is reflected to one detector while the high-frequency spectral region is transmitted to the other detector. This always allows one to measure two complementary wavelengths. The main drawback of this setup is that the signal amplitude at different detection wavelengths cannot be directly compared due to different detection parameters in each detector (e.g. photomultiplier amplification, filter transmission, etc.). This can be easily overcome by spectrally resolving the signal in a spectrometer and detecting the signal with a CCD camera.
9.2.3 Role of Spectral Overlap
A central point in the investigation of excited state dynamics with (pump-)DFWM is the spectral overlap between excitation pulses and the molecular absorption. In this regard, there are two important aspects how DFWM and pump-DFWM can be tuned in order to separate different signal contributions.
The first aspect can be illustrated by a comparison between DFWM and transient absorption (TA). Both techniques are third-order time-resolved methods. DFWM, however, uses three beams and homodyne detection, while TA uses two beams and self-heterodyne detection by the probe beam. In principle, both methods are able to generate vibrational coherence: In DFWM, the first two pulses excite two or more different vibrational levels. Similarly, in transient absorption vibrational levels lying within the bandwidth of the pump pulse are excited. Except for these similarities, DFWM presents additional features not found for e.g. TA. One of these features is the possibility of efficient non-resonant excitation (Fig. 9.1(c)). DFWM can efficiently generate vibrational modes with non-resonant spectra in comparison to TA, which clearly contains only ground state modes. This can be contrasted to resonant (or even near-resonant) excitation, which potentially contains both excited and ground state modes. By comparing both signals, it is possible to assign modes to their electronic potential.
The second aspect is related to the detection of excited state dynamics without any interference of ground state contributions. The spectrum tuning of initial pump and DFWM allows detecting exclusively excited state dynamics. This is performed by making the initial pump spectrum resonant (or near-resonant if the molecular absorption is not perfectly within the nc-OPA wavelength range) which leads to the excitation of the excited state as well as ground state vibrational manifold. The suppression of ground state contributions is achieved by the correct tuning of the DFWM spectrum: It must be tuned to the excited state absorption (ESA) or stimulated emission (SE) spectral regions. If the ESA and SE do not overlap with the ground state absorption, this leads to a pure excited electronic state signal. This can be easily understood since the DFWM signal is nonresonant with the ground state absorption and the DFWM signal scales with the transition dipole moment μ 8. Non-resonant transition dipole moments are several orders of magnitude smaller than resonant transition dipole moments, generating resonant signals 106 stronger than non-resonant signals [32, 33].
For partially or totally overlapping ESA/SE and ground state absorption, the pump-DFWM will intrinsically have ground state contributions, which can be, however, strongly reduced by changing the energy relation of the initial pump and the DFWM sequence. Third order techniques with homodyne detection like DFWM signal provide two control knobs which are critical in this respect: The signal depends (i) on the population difference squared between the involved states (Δn 2=(n e−n g)2) and (ii) on the intensity of each incoming beam (I pu I St I pr∼I 3). By decreasing the energy of the total DFWM sequence, the DFWM signal is automatically diminished. This overall decrease of the DFWM can be compensated by increasing the energy of the initial pump pulse, which also leads easily to a suppression of the ground state contributions. This can be easily understood since the population difference between the excited state and ground state diminishes.
9.3 Results and Discussion
In the following, three different applications will be presented. Firstly, pure DFWM is applied to RPSB in order to disentangle ground from excited state vibrational coherence. This first experiment is followed then by two experiments using pump DFWM applied to carotenoids, where the excited dynamics is exclusively investigated.
9.3.1 Assignment of Vibrational Coherence to Electronic States Using Pure DFWM
9.3.1.1 Introduction
Oscillations in the DFWM signal contains important information about vibrational coherence dynamics. In this context, a major issue of the signal analysis is the assignment of such modulations to specific electronic states of the sample molecule, since resonant excitation generates vibrational coherences in both ground as well as excited electronic states. In order to achieve this goal, spectral tuning of the excitation spectra together with spectral resolution of the signal light in DFWM experiments have previously been used for investigations on coherence dynamics in Bacteriorhodopsin (BR) and retinal protonated Schiff-bases (RPSB) [29, 30].
Particular interest resides in the characterization of wave packet dynamics in these samples because of the occurring photo-isomerization reactions which are important model reactions in terms of their biological relevance [3] as well as technical applications as photo-chemical switches [34]. The observed vibrational dynamics embrace a large energetic region ranging from around 100 cm−1 to more than 1500 cm−1. High-frequency modes (>800 cm−1) reflect single- and double-bond stretching or substituent wagging motion where the vibrational frequency is dependent on the specific structure of the chromophore and its environment. Contrary to that, low-frequency modes (<800 cm−1) represent delocalized motion from groups of atoms. By following the evolution of the vibrational frequencies accompanying, e.g. photo-induced chemical reactions, one may hope to directly identify the chemically relevant modes [14, 35]. In this context, ground state vibrational modes which are directly activated by the excitation pulses do generally not contain important information about the photo-induced reaction but mainly report on vibrational dephasing caused by solvent-solute interactions. Excited state vibrational coherences on the other hand, are more useful for following photo-induced structural evolution as such dynamics can take place directly in the excited state. Therefore, a clear separation between ground and excited state signal contributions is desirable.
9.3.1.2 Results
Excitation spectra for RPSB (Fig. 9.4(a)) were located in the red wing of the ground state absorption and centred at 510 nm (spectrum 1) and 570 nm (spectrum 2). Both spectra access different vibrational manifolds in the excited state, however, both have strong overlap with an intense ESA band (500–600 nm) [36, 37]. Spectrum 2 also covers the spectral region of such a excited state emission. Based on these spectral characteristics, both excitation spectra can induce ground as well as excited state coherences. Due to a reduced overlap with high-frequency excited state vibrational manifolds, spectrum 2 can only give rise to low-frequency excited state coherences.

(a) Absorption spectrum of all-trans RPSB in ethanol (black) together with the experimental excitation spectra (blue (1), orange (2)). (b) Absorption spectrum of BR (purple) in aqueous HEPES-buffer together with the experimental excitation spectra (orange (1) and red (2)) (Color figure online)
The excitation spectra for BR are tuned to cover the centre and the red part of the absorption spectrum. Similar to the RPSB experiments, this allows the different excitation spectra to access different vibrational manifolds in the excited state which then give rise to different vibrational coherence dynamics. Differing from the RPSB excitation, the excitation spectra used with BR partially overlap with two excited state absorption bands (ESA 1 (S4←S1) and ESA 2 (S3←S1) in Fig. 9.4(b)) of retinal in BR located at around 500 nm and around 650 nm, respectively. In particular, spectrum 1 overlaps with the red wing of ESA 1 and also with the blue wing of ESA 2. Excitation spectrum 2 overlaps only with the ESA 2.
Figures 9.5(a) and (b) show for RPSB that high- and low-frequency modes contribute to the total signal with different relative intensities compared to the non-oscillatory population dynamics. High relative contributions of high-frequency modes are observed for spectrum NR. For this excitation spectrum, no clear low-frequency modulations of RPSB are observed throughout the spectrally-resolved signal dynamics. The application of spectrum 1, on the other hand, reduces the relative intensity of high-frequency modes. In addition, intense low-frequency modulations appear in the dynamics. The low-frequency modulations are particularly strong for signal detection around the centre of the excitation spectrum.

Representative spectrally-resolved DFWM dynamics for RPSB (a) and (b) and BR (c) and (d). Black lines indicate the experimental data, red lines represent exponential fits to the population dynamics. Blue lines give residual oscillatory dynamics after subtraction of the exponential fit (Color figure online)
Figures 9.5(c) and (d) show similar transient data from the spectrally-resolved DFWM experiments on BR. Again, clear high- as well as low-frequency vibrational coherence dynamics can be discerned for the different excitation spectra. However, the modes contribute to the dynamics with different relative intensities between the non-oscillatory population dynamics and the vibrational coherence dynamics. For instance, high-frequency modes are observed with high relative intensity when BR is excited in the red wing of its ground state absorption whereas similar modes decrease in their relative intensities when the excitation spectrum is tuned towards the centre of the ground state absorption. Importantly, different observations are made for the low-frequency modes which most strongly contribute to the dynamics for excitation spectrum 1 while their relative contributions are only low for excitation with spectrum 2.
The described observations can clearly be visualized by calculating FFT spectra from the transient data of RPSB and BR after subtraction of a multi-exponential fit to the non-oscillatory population dynamics. For RPSB and BR samples mainly three and four high-frequency modes show strong intensities in the FFT spectra, respectively (Fig. 9.6). These high-frequency modes (at about 1565 cm−1, 1200 cm−1 and 1000 cm−1 for RPSB and 1525 cm−1, 1200 cm−1, 1155 cm−1 and 1010 cm−1 for BR) reflect mainly retinal chain single- and double-bond stretching modes [38]. The vibrational frequencies of the modes do not depend on the different excitation spectra and nor on the different spectral regions of signal detection. Only four BR additional bands in the energetic region between 800–1000 cm−1 appear in the FFT spectra for excitation with spectrum 1 and detection wavelengths on the blue side of the excitation spectrum (Fig. 9.6(d)). These modes reflect primarily out-of-plane wagging motion of retinal chain substituents [15, 38]. Such modes are absent in all FFT spectra for RPSB (note that the 885 cm−1 is a solvent mode from ethanol).

FFT spectra of RPSB ((a) and (b)) and BR ((c) and (d)) for selected excitation spectra. For RPSB, low-frequency modes only contribute to the signal for excitation spectrum 1 (b). For BR, low-frequency modes strongly increase in their relative intensities for spectrum 1 (d) in comparison to spectrum 2 (c)
For both RPSB as well as BR intense low-frequency modes can be discerned in the energetic region between 100–300 cm−1 (Fig. 9.6). For RPSB one broad dominant band is located at approximately 120 cm−1 only for spectrum 1. Contrasting to that, two clear bands are found for BR at 160 cm−1 and 210 cm−1 for both spectra 1 and 2. However, the contributions of these two modes are much stronger for spectrum 1.
9.3.1.3 Discussion
The protein-environment strongly reduces the lifetime of the S1 state of all-trans retinal in BR by nearly one order of magnitude compared to RPSB [36, 39, 40]. This effect has previously been discussed to originate from either electrostatic or steric interactions [14, 15, 36, 39–41]. It may thus be expected that band positions and dephasing time constants from vibrational coherence dynamics in both ground as well as excited electronic states will also be affected by the highly different environmental interactions in both molecules. The energetic positions of the most prominent high-frequency modes (Fig. 9.6) closely match the ground state vibrational frequencies of RPSB which are known from resonance Raman measurements [38]. Beside the energetic positions of the high frequency modes, the lack of changes in the vibrational frequencies and their lifetimes (not shown) [29, 30] when the different excitation spectra were used, helps to assign high-frequency modes to ground-state wave packet motion.
The experimentally observed dependence of the relative contributions of the high-frequency modes on the detection wavelength for a specific excitation spectrum exactly matches theoretical expectations for spectrally-resolved DFWM [29]. The highest relative contributions are expected on both blue- as well as red-detuned detection wavelengths while these contributions are at a minimal level near the centre of the excitation spectrum. The decreasing intensity of the high-frequency modes for spectrum 1 of RPSB (Fig. 9.6(b)) and spectrum 1 and blue-detuned detection wavelengths for BR (Fig. 9.6(b)) has, however, different origins [29]. The first origin is based on the fact that red-detuned excitation spectra, for which a large part of the applied spectral intensity is off-resonant (e.g. spectrum 2 for RPSB and spectrum 2 for BR), favour the excitation of ground state vibrational coherences over the induction of excited state population. The second effect is based on interference between ground and excited state response pathways which is due to the homodyne signal detection in DFWM: In spectral regions of intense ESA, the relative intensity of ground state dynamics is controlled by an interference term between ground state and excited state response pathways. The intensity of ground state dynamics is determined by the different ratios of transition dipole moments for ground state bleach and ESA.
In contrast to the high-frequency dynamics, the observed out-of-plane modes (only for BR, 800–1000 cm−1), which are observed only for excitation spectrum 1, exhibit negligible Franck-Condon activity [38]. For out-of-plane modes from the ground state, one would expect such dynamics to contribute to both excitation spectra for BR, which is, however, not observed. As the modes are observed with high relative intensity only in spectral regions of ESA 1 (500–570 nm) this indicates that the out-of-plane modes are detected through a probing pathway exploiting ESA transitions. In total, the above mentioned points show that the out-of-plane modes originate from excited state dynamics in agreement with earlier speculations on the basis of pump-probe experiments [15].
In this context, it is interesting to note that out-of-plane modes are clearly observable for excited state dynamics of BR while similar vibrational dynamics seem to be negligible for RPSB even for signal detection in spectral regions throughout the intense ESA band centred at 500 nm (Fig. 9.6). The importance of this observation concerns information about the excitation mechanism of these modes: For a direct excitation of the modes by the laser pulse in the excited state, these modes need to be normal modes of the all-trans retinal chromophore. However, retinal normal modes cannot simply be absent in case of RPSB in comparison to the BR dynamics. This, together with the clear dependence of the out-of-plane modes on the excitation wavelength, indicates that the activation of such modes in BR play an important role in the relaxation pathway of the excited state leading to double bond isomerization. This point will be discussed in more detail below in the context of the low-frequency modes.
Similar to the out-of-plane modes, the band positions of the low-frequency modes (Fig. 9.6) do not match with previously reported resonance Raman experiments of Schiff bases in BR or in solution [38]. Also for these modes the pronounced dependence of their observation on the excitation wavelengths together with very short lifetime of the low-frequency coherences for both RPSB and BR (<600 fs) indicates that the modes reflect excited state dynamics. In addition, these observations and conclusions are in perfect agreement with previous results for BR and RPSB in pump-probe experiments and time-resolved fluorescence experiments [39, 40]. The spectral region of observation of the low-frequency modes which is highly correlated to the spectral region of ESA in both RPSB and BR additionally substantiates that the modulations stem from excited state vibrational coherences. These observations extend the current picture of excited state wave packet dynamics of RPSB and BR by showing that the modes are observable in both spectral regions of excited state stimulated emission [39, 40] and also via multiple electronic transitions of ESA. Hence, the reported DFWM experiments resolve long standing inconsistencies for excited state dynamics of RPSB and BR: Previously, such excited state wave packet dynamics were only resolved for excited state emission [39, 40] for RPSB and only for the ESA 1 band in BR [15].
In the following we discuss the origin of the dependence of the low-frequency modes on the excitation and detection wavelength (Fig. 9.6). Low frequency modes in RPSB can only be observed with resonant excitation spectrum (spectrum 1 in Fig. 9.4(a)). For BR, low-frequency modes have a stronger relative intensity (when compared to high-frequency modes) for excitation with excess photon energy (spectrum 1 in Fig. 9.4(b)). The observation that the modulation intensity depends on the excitation wavelength indicates that the modulations are not directly activated by the excitation pulses.
This holds as long as the spectral shape of the ESA bands at the detection wavelengths of interest do not change significantly for the applied excitation spectra what has been demonstrated previously for RPSB [36]. An alternative activation mechanism of the low-frequency modes compared to a direct excitation through light-matter interaction can be given as coherent internal vibrational energy redistribution starting from directly excited high-frequency vibrational levels in the S1 state [40] (Fig. 9.7). Such an activation mechanism has been proposed for RPSB previously based on the need for a huge amount of excess vibrational energy (∼5000 cm−1) in order to observe the low-frequency modes [40]. However, our DFWM experiments reveal that such a threshold can only be on the order of a few hundreds of wavenumbers (200–500 cm−1). In the context of the activation of the low-frequency modes, it is important to note that very similar observations have been determined for the out-of-plane modes for BR (Fig. 9.6). This observation is indicative for a conclusion that both types of modes are acceptor modes for access vibrational energy deposition in the excited state and are therefore responsible for ultrafast relaxation possibly along the reaction coordinate. Their specific observation only for BR can be indicative for the conclusion that these modes are responsible for the much faster isomerization in the protein environment compared to the chromophore in solution and also for the much higher selectivity and quantum yield for photoproduct formation [42].

Proposed activation mechanism for low-frequency and out-of-plane excited state vibrational coherences in RPSB and BR. The initial intramolecular vibrational energy redistribution (IVR) takes place between Franck-Condon active modes and non-Franck-Condon active out-of-plane and low-frequency modes
9.3.2 Detection of Dark States
9.3.2.1 Introduction
The identification and characterization of dark electronic states is of the greatest challenges in spectroscopy of excited dynamics. Since these states cannot be excited directly, as the denotation ‘dark state’ already implies, identification of dark states on photochemical processes in biological systems is often difficult. Nonetheless, the processes that cannot be explained without the presence of dark states are numerous and the examples range from photo-damage of DNA [43] to light-harvesting [44]. In the latter case, a long standing and still ongoing debate is held on the possible participation of dark electronic states in the energy dissipation pathway in carotenoids. It is well known that the first bright electronic singlet state (S2) of these natural pigments that is excited via absorption of light in the green-blue region of the visible spectrum is not the lowest lying excited singlet state. The first excited singlet state S1 is indeed a very well characterized dark state, whose energy, electronic lifetime and vibrational spectra [12] have been determined in many spectroscopic investigations. However, several experimental findings such as the deviation of the S2 lifetime dependence on the conjugation length of the carotenoids from the energy gap law raise the question whether additional dark states between the S2 and S1 states exist and potentially participate in the relaxation pathway [45, 46]. Such states have already been predicted by theory [47] but are extremely difficult to detect spectroscopically, since they cannot even be excited via two-photon absorption, as is the case for the S1 state [48], and have very short lifetimes.
Applying pump-DFWM to lycopene, a carotenoid with 11 conjugated double bonds, we find direct evidence for the contribution of an additional electronic state in the rapid deactivation from the S2 to the S1 state [31]. This contribution manifests itself in a long-living signal that appears at short delay times T between the initial pump pulse and the DFWM sequence and it can be assigned to an additional dark state by numerical model simulations of lycopene’s pump-DFWM signal.
9.3.2.2 Results
The excitation spectra for the pump-DFWM experiments on lycopene are shown in Fig. 9.8 together with lycopene’s linear absorption spectrum. The spectrum of the initial pump pulse overlapped with the ground state absorption to the first bright electronic singlet state S2 (Fig. 9.9). The DFWM spectrum was centered at 600 nm corresponding to the S1–Sn excited state absorption.

Excitation spectra (DFWM: red; initial pump: blue) together with lycopene’s ground state absorption spectrum (black line) in THF (Color figure online)

Excitation scheme for the pump-DFWM experiments in lycopene
The pump-DFWM signal was detected at several different wavelengths ranging from the blue edge (560 nm) of the S1 absorption to the far red edge (640 nm) and is shown in Fig. 9.10 as 2D plots of the probe delay τ 23 against the initial pump delay T (see Fig. 9.2 for definition of delays).

Spectrally resolved pump-DFWM signal of lycopene at different detection wavelengths
The plots in Fig. 9.10 clearly show a slow rise of the signal along the T axis at small probe delays τ 23 for all detection wavelengths. This rise illustrates the flow of population from the initially excited S2 state into the S1 state where resonant DFWM takes place [12]. Exponential fitting of the rise gives a time constant of 140 fs. The oscillatory and non-oscillatory contributions of the DFWM signal emerge along the delay τ 23 between the DFWM probe pulse and the temporally overlapped pump and Stokes pulses. Along the τ 23-axis of Fig. 9.10, the expected exponential decay of the slowly varying component of the signal is visible for late initial pump delays (T>100 fs). In a narrow time window between T=10 fs and T=80 fs, however, a signal with a long lifetime in τ 23 (>2 ps) occurs. This component has a maximum at about T=40 fs and has larger amplitude at blue-detuned detection wavelengths (560 nm) than at red-detuned detection wavelengths (640 nm), compared to the rapidly decaying S1 signal at later T delays.
9.3.2.3 Discussion
The pump-DFWM signal of carotenoids for late initial pump delays T (∼800 fs) can easily be explained in the framework of the relaxation from the initially excited S2 state to the S1 state including the process of vibrational relaxation in the S1 state (see also the following Sect. 9.3.3). The interpretation of the long living signal at T=40 fs requires a more detailed discussion. The temporal evolution of this signal is striking since its very long lifetime in τ 23 (2.5 ps) and is paired with a rapid decay along the T-axis. The latter aspect reveals this signal stemming from a process taking place during the relaxation from S2 to S1. However, none of these two states can account for such a signal: At T=40 fs the S1 state has almost negligible population. Since the DFWM signal depends on the squared population difference (Δn 2) between the involved states, the S1 state can definitely be ruled out as source for such strong signal. On the other hand, the S2 state is instantaneously populated by the initial pump pulse so that any signal from this state should appear directly at time zero and not delayed by 40 fs.
In order to clarify the origin of this unique signal at early initial pump delays we performed numerical model simulations based on the Brownian oscillator model [31, 49]. Details for the simulations can be found in [31]. The simulation results are shown in Fig. 9.11. Note that the laser pulses in the simulations are approximated by δ-functions. The signal in Fig. 9.11 hence displays integration over all wavelengths and can be just qualitatively compared to the spectrally resolved experimental signals in Fig. 9.10. Nevertheless, the prominent features of the experimental data shown in Fig. 9.10 are well reproduced.

Simulated pump-DFWM signal of lycopene
In order to reproduce all features of the experimental pump-DFWM signal, several different models were tested [31]. Nevertheless, the only way to reproduce the long-living signal at T=40 fs in our simulations was to introduce a stimulated emission pumping DFWM (SEP-DFWM) [50] process between an additional state (X in Fig. 9.12) and a vibrationally hot ground state (hot-S0). This additional state is located energetically directly below the S2 state and is populated extremely rapidly with a time constant of 20 fs. The decay from S2 to X leads to the delay of 40 fs of the signal along the T-axis, and since X decays into the S1 state, the signal is vanished before significant S1 excited state absorption appears. The long lifetime in τ 23 on the other hand is due to the slow decay of the hot ground state (6 ps). The assignment of the signal to a hot ground state is supported by the temporal evolution of the vibrational modes (not shown here) [31]. All other tested models beginning with the simplest model considering only S2 and S1 over models including either SEP-DFWM between S2 and hot-S0 or relaxation via an additional ‘X state’ without SEP-DFWM up to models incorporating S2–S m absorption processes did not lead to a successful replication of the experimental signal [31].

Time constants for the relaxation pathway in lycopene underlying the model simulations
The relaxation scheme shown in Fig. 9.12 implies that the ‘X state’ corresponds to such an additional dark electronic state discussed in literature. However, X could as well be thought of as the approach of a Franck-Condon window for the S2–hot-S0 transition within the first 40 fs after excitation. Measurements in lycopene using different initial pump spectra showed no dependence of the temporal position of the long-living signal on the energy of the IP. Since a shift of the IP spectrum leads to excitation to another position on the S2 electronic hypersurface, this lack of dependence actually argues for X being an electronic state between S2 and S1 as already suggested in other experimental investigations [45, 46, 51, 52].
In our model the generally accepted S2 lifetime of 130 fs in lycopene, which is normally determined either by fluorescence or via the rise time of the S1 excited state absorption, is composed of the ultrafast relaxation from S2 to X (20 fs) and the X lifetime (110 fs). Similar time constants were found in sub-picosecond absorption spectroscopy in carotenoids with varying conjugation lengths [53]. In this study, the intermediate state in lycopene was identified to be of \(3\mathrm{A}_{\mathrm{g}}^{-}\) symmetry and lifetimes of 20 fs and 150 fs were determined for S2 and \(3\mathrm{A}_{\mathrm{g}}^{-}\), respectively. Further indication for the S2–S1 relaxation via an additional state was given in a recent femtosecond transient absorption study of lutein and β-carotene combined with quantum chemical calculations [46]. In this case, the existence of the intermediate state was inferred from the observation that the fluorescence spectra of these carotenoids differ from a direct mirror image of the absorption. The authors assigned a long lifetime to the S2 state (100–170 fs, depending on the solvent) and a shorter one (80 fs and 50 fs for lutein and β-carotene, respectively) to the intermediate state, determined by quantum chemical calculations as \(1\mathrm{B}_{\mathrm{u}}^{-}\). Another indirect hint for a dark state was found in femtosecond time-resolved absorption and Kerr-gate fluorescence experiments on different β-carotene homologues [52]. The behavior of the S2 relaxation rates depending on the conjugation length could only be explained by the existence of an additional state located between S2 and S1. Similar to studies on lutein and β-carotene [46], the longer lifetime of about 100 fs was assigned to the S2 state while the identical rise time of the S1 state was explained with a very fast S x –S1 relaxation (10 fs). The dark nature of the additional state in combination with the extremely rapid relaxation from S2 to S1 in carotenoids makes it very difficult to observe this state in spectroscopic studies using transient absorption and to differentiate between the time constants of the S2 state and the dark state. Using the method of pump-DFWM, however, we can distinguish between these two adjacent states and unambiguously assign of the longer lifetime of 110 fs to the dark state due to its intense stimulated emission that leads to a unique signal in this technique.
9.3.3 Vibrational Coherence Evolution in the Excited State
9.3.3.1 Introduction
Among the incoherent population relaxation (Sect. 9.3.2), the evolution of the ultrafast vibrational coherence in the excited state is an additional but very important aspect in understanding chemical transformations. Not just the frequency of oscillatory phenomena can give significant hints on the flow of a given reaction, but also how the frequencies change during the relaxation process may also help to re-construct the relaxation pathway and the participating molecular degrees of freedom. The high temporal and spectral resolution of pump-DFWM make this technique an ideal tool for the analysis of the shift of the vibrational frequencies in time. Another important aspect of the vibrational dynamics is the population time of individual modes. Rise and decay times of vibrational mode amplitudes present us a better picture on how modes are populated and depopulated and, sometimes, allow for the identification of vibrational modes specific for a given electronic state. In pump-DFWM, the evolution of the vibrational population can be measured by detecting the vibrational mode intensity for several initial pump delays T.
In this section, we apply pump-DFWM to resolve the evolution of the vibrational coherence in the excited states of carotenoids. Similar to lycopene investigated on the last section, the S1 state will be populated with excess vibrational energy, given the considerable difference in zero-point energies between S1 and S2. McCamant et al. [54] proposed a two-stage mechanism for internal vibrational redistribution (IVR) on S1 for β-carotene in solution. The authors employed femtosecond stimulated Raman scattering and explained their results without invoking any intermediate states between S1 and S2 in β-carotene. Rondonuwu et al. [55] however, investigated polyenes of similar conjugation length by sub-picosecond time-resolved Raman scattering. The authors suggested the following energetic pathway upon excitation of S2 (\(1\mathrm{B}_{\mathrm{u}}^{+}\)): \(1\mathrm{B}_{\mathrm{u}}^{+} \rightarrow 3\mathrm{A}_{\mathrm{g}}^{-} \rightarrow 1\mathrm{B}_{\mathrm{u}}^{-} \rightarrow 2\mathrm{A}_{\mathrm{g}}^{-} \rightarrow 1\mathrm{A}_{\mathrm{g}}^{-}\). Hence, the puzzle of carotenoid’s deactivation network is not resolved by either time dependent vibrational or transient absorption spectroscopy.
9.3.3.2 Results
A typical 2D transient data set for β-carotene is shown in Fig. 9.13. The non-oscillatory dynamics of the pump-DFWM signal along the T-axis in β-carotene show similar characteristics found for lycopene (Sect. 9.3.2). The long-lived signal at T-delays (T=100–130 fs) can be related to the same feature observed for lycopene at shorter T-delays. This contribution comes also from a vibrationally excited ground state (hot-S0), which is excited via a dark state X by the DFWM sequence via SEP-DFWM [50] directly after the relaxation of the S2 state. The longer vibrational dephasing time of this signal is additional evidence that this contribution does not come from the excited state, but must be related to a long time vibrational dynamics in the ground state.

(a) Typical 2D scan for β-carotene; (b) Selected DFWM transients for three values of delay T: 100 fs (blue), 490 fs (red) and 850 fs (black). The color used for the dotted lines in (a) correspond to the color of the respective transients in (b)
In Fig. 9.14, the Fourier transformation of the oscillatory contribution detected at the red wing of the absorption (λ det=610 nm) is shown. Two major vibrational contributions can be distinguished: (i) the vibration at 1140 cm−1 can be associated with the C–C single-bond stretch mode and (ii) the vibration at 1528 cm−1 is related to the C=C double bond stretch mode. Besides these two major contributions, several additional modes are observed. One of them, at 1765 cm−1, is a typical contribution which is associated with the C=C stretch mode characteristic for the S1 state [54]. This mode cannot be found for any other electronic state, dark or bright, in carotenoids. Other small contributions at 1368 cm−1, 1626 cm−1 and 1698 cm−1 were recently proposed to be related to the formation of dark states in lycopene (N=11) and spheroidene (N=10) [55]. The two peaks at about 915 cm−1 and 1045 cm−1 are modes of the solvent (THF).

Fast Fourier transform spectra of DFWM transients for three values of delay T (100, 490 and 850 fs). The colors used correspond to the same color coding used in Fig. 9.13
As shown in Fig. 9.15(a), the C–C stretch mode evolves from its corresponding wavenumber in the ground state (∼1160 cm−1) to a lower wavenumber in the excited state at 1140 cm−1 within the lifetime of the S2 state in THF (∼170 fs). After this time the frequency does not change during the population time of the S1 state. The C=C stretch mode evolves initially (Fig. 9.15(b)) similarly compared to the evolution of the C–C mode, i.e., the central wavenumber varies from its value in the ground state 1532 cm−1 down to 1522 cm−1 during the lifetime of the S2 state. After this time, different to the C–C mode dynamics, the wavenumber of the C=C mode drifts to 1528 cm−1 with a time scale of 260±20 fs.

Dependence of the central frequency of two vibrational modes of β-carotene on the delay time between initial pump and the DFWM sequence: (a) 1140 cm−1 C–C single-bond stretching, (b) 1528 cm−1 C=C double-bond stretching
9.3.3.3 Discussion
The evolution of the C=C stretch mode at 1528 cm−1 after T∼200 fs can be explained in terms of vibrational energy relaxation in the excited state. For earlier values of T, the molecule is still vibrationally excited. As T increases and the system relaxes, lower vibrational levels will be eventually populated and, due to vibrational anharmonicity, wavepackets generated by the DFWM sequence will show a higher vibrational energy. For β-carotene in THF the time constant associated with the relaxation of this mode is 260 fs. A similar behavior cannot be observed for the C–C vibrational mode. The absence of any evolution for the C–C mode at later T values and the significant variation of the central frequency of the C=C mode are two central features in understanding the internal conversion between S2 and S1. The frequency of strongly coupled vibrational modes shifts more than for modes which are weakly coupled. Therefore, it becomes clear that the C=C stretching mode is the strongest coupled mode in the early times during the internal conversion between S2 and S1. This assignment is in good agreement with other experimental observations [54]. A similar behavior is also present for the C=C mode at 1765 cm−1 (not shown), although the shift seems to be much smaller [21].
The excited state potential surface can be further mapped by plotting the peak area of each individual Raman mode (Fig. 9.16). The evolution of the area of a CARS/CSRS peak is directly related to the changes of population in the mode [49] and, therefore, indicates the relaxation pathway on the multidimensional PES. The peak area is calculated around the center wavenumber with 10 cm−1 width. The two main vibrational contributions to the signal at 1140 cm−1 and 1528 cm−1 rise almost instantaneously with a maximum value at 100–130 fs. At this time delay, as depicted in Fig. 9.13, the stimulated pump excitation (SEP) mechanism is present. After this delay the peak integral remains almost constant during the relaxation of the system. This merely confirms that the C–C and the C=C modes are present in hot-S0 as well as in the excited electronic states. The behavior of the other vibrational modes is different. As expected, the S1-specific C=C stretch mode at 1760 cm−1 has negligible magnitude for early values of T, but then increases with a time constant of 360±20 fs. The other modes at 1368 cm−1, 1628 cm−1 and 1698 cm−1 show a similar behavior with time constants of 320±20 fs, 500±30 fs and 350±20 fs, respectively. The last two modes show an additional fast rise which peaks at about 100 fs. This is explained by the absence of these modes in the ground state, generating a fast rising signal as soon as the system is excited and starts to relax.

Variation of selected FFT peaks depicted in Fig. 9.14. The integral of the peaks, was calculated with 10 cm−1 width around the given wavenumber and its value was normalized
The lack of a clear vibrational signature specific to a dark state in β-carotene is in agreement with the picture obtained for other carotenoids using pump-DFWM. The dark state observed between the S2 and the S1 state has a lifetime which is between 20–80 fs, depending on the carotenoid length. In the lifetime of such a dark state, the C–C or C=C modes will barely be able to oscillate two times before the electronic state has completely decayed, which will lead to a contribution very short in duration in the DFWM signal. Therefore, the signal of such a dark state will be hardly detectable with DFWM (as well as in TA or other any other third-order technique). This demonstrates the power of a pump DFWM: By using an initial pump pulse to excite the molecule, the presence of such ultrafast transient states can be clearly resolved in the deactivation.
9.4 Conclusions
Multidimensional time-resolved spectroscopy methods use additional laser-matter interactions to open new temporal and/or spectral observation windows. This allows observing molecular phenomena normally hidden from time-resolved techniques with low-dimensionality. We have exploited third- and fifth-order nonlinear methods based on four wave mixing to investigate the excited state of biomolecules. This was demonstrated to Schiff-bases in BR and solution as well as for different carotenoids in solution. With (pump-) DFWM, it is possible to resolve the activation and deactivation of Raman modes and electronic population, leading to a better understanding of the chemical changes, in particular in electronically excited states.
References
M. Dantus, M.J. Rosker, A.H. Zewail, Real-time femtosecond probing of transition-states in chemical-reactions. J. Chem. Phys. 87(4), 2395–2397 (1987)
J. Dobler et al., Excited-state reaction dynamics of bacteriorhodopsin studied by femtosecond spectroscopy. Chem. Phys. Lett. 144(2), 215–220 (1988)
R.A. Mathies et al., Direct observation of the femtosecond excited-state cis-trans isomerization in bacteriorhodopsin. Science 240(4853), 777–779 (1988)
P.F. Barbara, G.C. Walker, T.P. Smith, Vibrational-modes and the dynamic solvent effect in electron and proton-transfer. Science 256(5059), 975–981 (1992)
J.L. Herek et al., Femtosecond real-time probing of reactions. 9. Hydrogen-atom transfer. J. Chem. Phys. 97(12), 9046–9061 (1992)
K. Lenz et al., Resonance Raman and femtosecond absorption studies of vibrational-relaxation initiated by ultrafast intramolecular proton-transfer. Chem. Phys. Lett. 229(4–5), 340–346 (1994)
A.H. Zewail, Femtochemistry: recent progress in studies of dynamics and control of reactions and their transition states. J. Phys. Chem. 100(31), 12701–12724 (1996)
W. Fuss, W.E. Schmid, S.A. Trushin, Time-resolved dissociative intense-laser field ionization for probing dynamics: femtosecond photochemical ring opening of 1, 3-cyclohexadiene. J. Chem. Phys. 112(19), 8347–8362 (2000)
M. Dantus, Coherent nonlinear spectroscopy: from femtosecond dynamics to control. Annu. Rev. Phys. Chem. 52, 639 (2001)
W. Wohlleben et al., Coherent control for spectroscopy and manipulation of biological dynamics. ChemPhysChem 6(5), 850–857 (2005)
R.W. Schoenlein et al., The 1st step in vision—femtosecond isomerization of rhodopsin. Science 254(5030), 412–415 (1991)
T. Polívka, V. Sundström, Ultrafast dynamics of carotenoid excited states—from solution to natural and artificial systems. Chem. Rev. 104(4), 2021–2071 (2004)
D. Polli et al., Broadband pump-probe spectroscopy with sub-10-fs resolution for probing ultrafast internal conversion and coherent phonons in carotenoids. Chem. Phys. 350(1–3), 45–55 (2008)
T. Kobayashi, T. Saito, H. Ohtani, Real-time spectroscopy of transition states in bacteriorhodopsin during retinal isomerization. Nature 414(6863), 531–534 (2001)
A. Kahan et al., Following photoinduced dynamics in bacteriorhodopsin with 7-fs impulsive vibrational spectroscopy. J. Am. Chem. Soc. 129(3), 537–546 (2007)
S. Mukamel, Multidimensional femtosecond correlation spectroscopies of electronic and vibrational excitations. Annu. Rev. Phys. Chem. 51, 691–729 (2000)
W. Wohlleben et al., Multichannel carotenoid deactivation in photosynthetic light harvesting as identified by an evolutionary target analysis. Biophys. J. 85(1), 442–450 (2003)
I.H.M. Vanstokkum et al., Conformational dynamics of flexibly and semirigidly bridged electron donor-acceptor systems as revealed by spectrotemporal parameterization of fluorescence. J. Phys. Chem. 98(3), 852–866 (1994)
J. Oberle et al., Enhancement and subpicosecond dynamics of optical nonlinearities of excited-states—trans-stilbene in solution. Chem. Phys. Lett. 241(4), 281–289 (1995)
M. Motzkus, S. Pedersen, A.H. Zewail, Femtosecond real-time probing of reactions. 19. Nonlinear (DFWM) techniques for probing transition states of uni- and bimolecular reactions. J. Phys. Chem. 100(14), 5620–5633 (1996)
T. Buckup et al., Multidimensional spectroscopy of beta-carotene: vibrational cooling in the excited state. Arch. Biochem. Biophys. 483(2), 219–223 (2009)
J. Hauer, T. Buckup, M. Motzkus, Pump-degenerate four wave mixing as a technique for analyzing structural and electronic evolution: multidimensional time-resolved dynamics near a conical intersection. J. Phys. Chem. A 111(42), 10517–10529 (2007)
J.P. Kraack, M. Motzkus, T. Buckup, Selective nonlinear response preparation using femtosecond spectrally resolved four-wave-mixing. J. Chem. Phys. 135, 224505 (2011)
T. Joo, A.C. Albrecht, Electronic dephasing studies of molecules in solution at room-temperature by femtosecond degenerate 4-wave-mixing. Chem. Phys. 176(1), 233–247 (1993)
T.H. Joo, A.C. Albrecht, Vibrational frequencies and dephasing times in excited electronic states by femtosecond time-resolved 4-wave-mixing. Chem. Phys. 173(1), 17–26 (1993)
B.I. Grimberg et al., Ultrafast nonlinear spectroscopic techniques in the gas phase and their density matrix representation. J. Phys. Chem. A 106(5), 697–718 (2002)
S. Mukamel, J.D. Biggs, Communication: Comment on the effective temporal and spectral resolution of impulsive stimulated Raman signals. J. Chem. Phys. 134(16) (2011)
T. Hornung, H. Skenderovic, M. Motzkus, Observation of all-trans-beta-carotene wavepacket motion on the electronic ground and excited dark state using degenerate four-wave mixing (DFWM) and pump-DFWM. Chem. Phys. Lett. 402(4–6), 283–288 (2005)
J.P. Kraack et al., Ground- and excited-state vibrational coherence dynamics in bacteriorhodopsin probed with degenerate four-wave-mixing experiments. ChemPhysChem 12(10), 1851–1859 (2011)
J.P. Kraack, T. Buckup, M. Motzkus, Vibrational analysis of ground and excited electronic states of all-trans retinal protonated Schiff-bases. Phys. Chem. Chem. Phys. 13, 21402–21410 (2011)
M.S. Marek, T. Buckup, M. Motzkus, Direct observation of a dark state in lycopene using pump-DFWM. J. Phys. Chem. B 115(25), 8328–8337 (2011)
G. Beadie et al., Towards a FAST-CARS anthrax detector: analysis of cars generation from DPA. J. Mod. Opt. 51(16–18), 2627–2635 (2004)
M. Mehendale et al., All-ultraviolet time-resolved coherent anti-Stokes Raman scattering. Opt. Lett. 31(2), 256–258 (2006)
N. Hampp, Bacteriorhodopsin as a photochromic retinal protein for optical memories. Chem. Rev. 100(5), 1755–1776 (2000)
I. Iwakura, A. Yabushita, T. Kobayashi, Observation of transition state in Raman triggered oxidation of chloroform in the ground state by real-time vibrational spectroscopy. Chem. Phys. Lett. 457(4–6), 421–426 (2008)
O. Bismuth et al., Photochemical dynamics of all-trans retinal protonated Schiff-base in solution: excitation wavelength dependence. Chem. Phys. 341(1–3), 267–275 (2007)
B. Loevsky et al., A new spectral window on retinal protein photochemistry. J. Am. Chem. Soc. 133(6), 1626–1629 (2011)
A.B. Myers, R.A. Harris, R.A. Mathies, Resonance Raman excitation profiles of bacteriorhodopsin. J. Chem. Phys. 79(2), 603–613 (1983)
B.X. Hou et al., Comparing photoinduced vibrational coherences in bacteriorhodopsin and in native and locked retinal protonated Schiff bases. Chem. Phys. Lett. 381(5–6), 549–555 (2003)
G. Zgrablic, S. Haacke, M. Chergui, Vibrational coherences of the protonated Schiff base of all-trans retinal in solution. Chem. Phys. 338(2–3), 168–174 (2007)
J. Léonard et al., Functional electric field changes in photoactivated proteins revealed by ultrafast Stark spectroscopy of the Trp residues. Proc. Natl. Acad. Sci. USA 106(19), 7718–7723 (2009)
S.P. Balashov et al., Quantum yield ratio of the forward and back light reactions of bacteriorhodopsin at low-temperature and photosteady-state concentration of the bathoproduct-k. Photochem. Photobiol. 54(6), 955–961 (1991)
I. Vaya et al., Fluorescence of natural DNA: from the femtosecond to the nanosecond time scales. J. Am. Chem. Soc. 132(34), 11834–11835 (2010)
R. Nakamura et al., Dark excited states of carotenoid regulated by bacteriochlorophyll in photosynthetic light harvesting. J. Phys. Chem. B (2011)
Y. Koyama et al., Excited-state dynamics of overlapped optically-allowed 1B(u)(+) and optically-forbidden 1B(u)(−) or 3A(g)(−) vibronic levels of carotenoids: possible roles in the light-harvesting function. Int. J. Mol. Sci. 11(4), 1888–1929 (2010)
E. Ostroumov et al., Electronic coherence provides a direct proof for energy-level crossing in photoexcited lutein and beta-carotene. Phys. Rev. Lett. 103(10) (2009)
P. Tavan, K. Schulten, The low-lying electronic excitations in long polyenes: a PPP-MRD-CL study. J. Chem. Phys. 85(11), 6602–6609 (1986)
P.J. Walla et al., Excited-state kinetics of the carotenoid S-1 state in LHC II and two-photon excitation spectra of lutein and beta-carotene in solution: efficient car S-1 → Chl electronic energy transfer via hot S-1 states? J. Phys. Chem. A 106(10), 1909–1916 (2002)
S. Mukamel, Principles of Nonlinear Optical Spectroscopy (Oxford University Press, New York, 1995), p. 543
P.H. Vaccaro, Advanced Series in Physical Chemistry: Molecular Dynamics and Spectroscopy by Stimulated Emission Pumping (World Scientific, New York, 1994)
G. Cerullo et al., Photosynthetic light harvesting by carotenoids: detection of an intermediate excited state. Science 298(5602), 2395–2398 (2002)
D. Kosumi et al., The dependence of the ultrafast relaxation kinetics of the S-2 and S-1 states in beta-carotene homologs and lycopene on conjugation length studied by femtosecond time-resolved absorption and Kerr-gate fluorescence spectroscopies. J. Chem. Phys. 130(21) (2009)
R. Fujii et al., Two different pathways of internal conversion in carotenoids depending on the length of the conjugated chain. Chem. Phys. Lett. 369(1–2), 165–172 (2003)
D.W. McCamant, P. Kukura, R.A. Mathies, Femtosecond time-resolved stimulated Raman spectroscopy: application to the ultrafast internal conversion in beta-carotene. J. Phys. Chem. A 107(40), 8208–8214 (2003)
F.S. Rondonuwu et al., Singlet internal conversion processes in the order of IBu → 3A(g) (−) → IBu → 2A(g) (−) → IA (g) (−) in all-trans-spheroidene and lycopene as revealed by subpicosecond time-resolved Raman spectroscopy. Chem. Phys. Lett. 429(1–3), 234–238 (2006)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Buckup, T., Kraack, J.P., Marek, M.S., Motzkus, M. (2014). On the Investigation of Excited State Dynamics with (Pump-)Degenerate Four Wave Mixing. In: de Nalda, R., Bañares, L. (eds) Ultrafast Phenomena in Molecular Sciences. Springer Series in Chemical Physics, vol 107. Springer, Cham. https://doi.org/10.1007/978-3-319-02051-8_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-02051-8_9
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-02050-1
Online ISBN: 978-3-319-02051-8
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)