
Abstract
Nanostructured dielectric materials are a special class of high-quality electronic-grade materials having confinement of at least one of its dimensions at the nanoscale for advanced nanoelectronics, nanodevice, optical, and many other potential applications. Nanodielectric materials became significantly important with the progression of the miniaturization of device dimensions. Currently, transistor fabrication technology has seen many advancements reaching almost atomic scales that inculcate very high quality and ultrathin gate dielectric layers. High capacitance nanodielectric gate materials are extremely important in high-throughput thin-film transistors (TFTs) for potentially hysteresis-free and low-voltage action. Moreover, organic–inorganic hybrid nanodielectric materials are the most-sought-after for flexible electronics on account of mechanical bendability. Organic nanodielectric materials are highly desired for advanced low-voltage operating carbon nanotube (CNT)-based TFTs and for complementary logic gates applications. Another fact is the ease of deposition of dielectric solution over plastic substrates for flexible nanoelectronic applications. Other categories of nanodielectric materials include carbon black composited with polymers to form nanocomposites, silica aerogel, epoxy microcomposites enhanced with nanosized clays, crystalline dielectric materials, and nanometric dendrimers. Next-generation applications can be classified into increased stress, higher temperature, stress-grading, and surface modification submissions. Engineered nanodielectric layers are of paramount importance for self-cleaning or easy-to-clean surfaces for outdoor applications in cars and windows, for example. The main feature of a high static contact angle provides for a nonwetting surface. Next-generation highly efficient insulation materials are the need of the hour that is free from unambiguous dielectric breakdown character including surface flashover between the dielectric surface and vacuum or surrounding environment. Nanoscale fillers, nanodielectric materials, and nanoparticles have been synthesized and worked on for state-of-the-art applications. This chapter is a concise effort to present information about nanosized dielectric materials, their synthesis methods, and their state-of-the-art applications.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
3.1 Introduction
The term “nanodielectrics” entails a multicomponent nanostructure possessing dielectric properties that are engineered in a way such that it is at least one of the dimensions is confined in the nanoregime typically less than 100 nm [1,2,3,4]. Fréchette [1] points out two different definitions of nanodielectrics as follows, “Nanodielectrics are those materials composed of multicomponent dielectrics confined in nanodimensions, so that its dielectric properties are altered as compared to the bulk phase”. “Nanodielectric can be defined as materials that exhibit altered properties due to nanoconfinement within 100 nm of at least one of its dimensions”. This definition is closer to a specific feature of nanostructured materials showing modified novel properties at the nanoscale.
Michel F. Fréchette states in the IEEE Electrical Insulation Magazine that he coined the word nanodielectrics entailing a combination of nanotechnology and dielectrics [1]. This concept of nanostructured dielectric materials came to the forefront in form of published literature after 2001. In the first experimental report, current–voltage characteristics were markedly modified by the use of a nano-additives-based mixed compound in the case of a ZnO varistor.
Nanodielectric materials exhibit completely distinguished performance as exemplified by the above-stated definitions and should not be confused with micron-sized agglomerated structures possessing distinct properties due to the inclusion of defects or even mild enhancement in dielectric properties. In addition to the chief feature of nanoconfinement in form of a nanoparticle, nanocrystal, nanocomposites, nanopowders, or mixed nanomaterials, the fundamental process should allow for the dielectric behavior to prevail. Michel F. Fréchette recognizes four different types of nanodielectrics that are nanoclusters of varying aspect-ratios, multilayered nanostructured dielectric films, nanocoating of only a few nanometers thickness, and nanophases. In a sense, self-assembly lies at the core of the development of a nanodielectric material along with the potential of erecting a structure based on building blocks and interfaces.
Nanodielectric materials can be utilized in a range of applications that includes space charge suppression, high-density energy storage, nonlinear field-grading applications, high thermal conductivity applications, and even in advanced applications in biomedical systems [1, 4,5,6,7]. Nanodielectric materials with nonlinear field-grading properties mean that the dielectric properties can be varied as a function of the electric field-induced stress [5]. Information acquired as a result of nonlinear field grading can be used in the distribution of electric fields over the dielectric surfaces homogeneously. This is particularly useful in the prevention of the formation of electric stress zones in high-voltage applications. Sometimes, nonlinear field grading can be performed intentionally to induce nonlinear electrical characteristics by incorporation of nanofillers into the polymer matrix. Nonlinear field-grading characteristics are also useful for outdoor high-voltage applications relying on high-voltages. Another state-of-the-art application of nanodielectrics is in integrated circuitry (IC) which has scaled down to as low as 3 nm node in the fabrication of next-generation devices. As a result, highly advanced nanodielectric materials are the need of the hour for effective electrical and thermal insulation as more heat per volume will be generated. Additionally, super-advanced nanodielectrics as gate materials are required to avoid electron tunneling phenomenon due to confinement of thickness in the 1–3 nm range [8,9,10]. This is highly desirable to control the increment of open-circuit current or leakage current because of the operation of nanodevices in the high-voltage regime. The development of thinner insulation material with much enhanced dielectric strength will ensure significantly reduced dielectric layer material for the same voltage level in a much more compact integrated circuitry nearing approximately atomic dimensions for a single node. Nanofillers-polymer composite materials are significantly useful because of low costs, flexible features, high breakdown strength, and high permittivity. Further, the incorporation of inorganic nanomaterials in a polymer base avoids not only the incorporation of space charges but accumulation as well. Space charges are detrimental to the local electric field distribution that degrades the efficiency of the involved polymer as a function of time under high voltages of the direct current operation. Enis Tuncer and Isidor Sauers have reviewed the nanoparticle as fillers in a polymer matrix along with their applications in a detailed manner [11]. Dielectric materials are of prime commercial value in the sense that lesser insulation material for the same voltage level with a positively modified breakdown can result in the development of much more efficient, cost-effective, and weight-reduced applications including power capacitors, transformers, and even power cables. Voltage endurance is an important parameter in the case of solid insulation materials that depict the time taken in a breakdown on the application of an electric field that is lower in magnitude in comparison with the stress generated due to a field leading to instant failure. Nanodielectrics can also improve direct current (DC)-based transmission cable systems. Nanodielectric material systems can improve mechanical features in form of tensile strength, elongation at the point of fracture, hardness, resistance to wettability and temperature variations, and electrical properties in form of enhancement of tolerance level against electrical arcing and reduced dielectric permittivities. Nanocomposite-enameled wires were subjected to repetitive surge voltage in a study forwarded by Okuba et al. and Hayakawa and Okubo which estimated a breakdown lifetime longer by a factor of 1000 as compared to bulk-phased enameled wires under similar surge voltage application [12]. Nanodielectrics can also be promising for high-temperature dielectric applications that include capacitors, compact transformers, and generators. Nanodielectrics are also under consideration for use in cryogenic applications with a focus on thermoplastics, such as polyvinyl alcohol (PVA), polymethyl methacrylate (PMMA), or even a thermoset. A remarkable feature is that high permittivity particles on compositing with low permittivity surfactant produce a material that possesses low dielectric permittivity.
For insulation applications, nanodielectrics must possess sound features of high electric strength, effective relative permittivity along with low dielectric loss (δ) of the tailored material medium as per the application, and high tolerance level against material erosion, high stability of the optimized nanostructure design for prolonged periods in a reduced nano-entropy configuration, stable behavior as a function of temperature, or even on prolonged hours of operation including effective mechanical properties. Effective relative permittivity along with low dielectric loss (δ) need to be engineered in a nanodielectric material according to the requirements. In terms of commercial applications, the switchover from the current module of bulk insulation materials to nanodimensional materials with a brand-new set of properties is a giant step. All original equipment manufacturers (OEM) are fearful of making a changeover because of dreadful commercial implications. The important features of nanodielectric materials are inclusive of highly efficient voltage endurance, high fidelity against discharge-driven deformations, superior dielectric behavior with distention for intrinsic space charge collection, and increment in alternating current (AC) breakdown strength with positive consequences of enhancing the operational electric load that become significant at high temperatures under application of divergent fields along with direct voltage. This can ensure a highly concise mechanical design of electric machines along with thermal features.
3.1.1 Theory of Dielectric Materials
Dielectrics belong to the broader class of materials that possess the property of polarizability under the application of an electric field. The formation of dipolar structures allows the containment of an electrostatic field within them [13]. Insulators are also dielectric materials that prevent the flow of leakage currents or open-circuit currents in devices. Dielectrics have become extremely important in integrated circuitry (IC) since shrinking of device architectures to almost atomic scales (1–3 nm node) has compelled the development of ultrathin high-k dielectric materials that can not only provide effective gating but prevent leakage currents as well. Under the effect of an electric field, positive charges are along the direction of the electric field (E) while negative charges are aligned in a direction opposite to the direction of the electric field (E). In this process, dielectric material remains neutral as a whole, although, electrons get displaced from their mean vibrating positions in length scales smaller than atomic sizes. No significant charge displacement is observed at the macro-scale similar to the conductors to cause higher conductivities. G. C. Psarras has provided details of the theory of dielectrics in his book chapter on “Fundamentals of Dielectric Theories” in a very nice manner [13].
Polarization happens due to the distribution of charged species in a dielectric with a vector pointing from the midpoint of negatively charged species toward the midpoint of positively charged species. The developed electric dipole moment (μ) due to the absolute value of positive and negative charges with distance r between the two centers is given as follows,
The total dipole moment can be calculated by summing the above product over a range of different dipoles. Polarization (P) is defined as the total electric moment per unit volume (V) of the dielectric material, which is given as,
Polarization is an important parameter that practically resembles charge surface density with units of C m−2 and dimensions as [L−2TI]. Polarization can be defined as the average sum of n electric dipole moments contained in a unit volume which can be expressed as,
In an isotropic material, under the effect of zero electric fields (E = 0), permanent electric dipole moments are arranged randomly. On application of an E, permanent electric moments are along the direction of E, while induced dipole moments (μind) are also developed as outlined above. Hence, dipole moment as a whole is expressed as,
And, in that case, total polarization is,
The coefficient of polarization or polarizability (α) is an important dielectric parameter for dielectrics that give the ability of atoms or molecules to get polarized.
In the case of linear dielectrics, polarizability can be defined as the total dipole moment developed in a dielectric under the application of an external electric field (E) or better total dipole moment produced per unit of electric field (E).
Hence,
In the above equation, E is the acting electric field and not the field applied externally. Polarizability possesses dimensions of volume and can be estimated for dielectric materials with different types of atoms.
Electric displacement vector (\(\vec{\user2{D}}\)) with \({\varvec{\varepsilon}}_{0} = 8.854\user2{*}10^{ - 12} \user2{ F}/{\varvec{m}}\) is expressed as follows,
Noticeable point is that polarization relates to induced charges over edge surfaces of the sample in a parallel plate capacitor filled with a dielectric material, electric displacement pertains to independent charges at the electrode surfaces, while electric field vector \(({\mathbf{D}}\)) depends on both independent and induced charges.
The relative dielectric constant (\({\varvec{\varepsilon}}\)) of a material is defined as follows,
Relative dielectric constant (\(\varepsilon\)) in terms of electric displacement vector \(({\varvec{D}}\)) is expressed as,
Here, \(\frac{{\mathbf{P}}}{{{{\varvec{\upvarepsilon}}}_{0} {\mathbf{E}}}}\) is electric susceptibility that provides the degree of induced electric polarization due to the application of an electric field.
Dielectrics can be classified into polar and nonpolar dielectrics. Polar dielectrics possess permanent electric molecular dipoles since the line joining the midpoints of positive and negative charge carriers is not along a single direction. Permanent molecular dipole moments persist even in the absence of the application of an electric field. The essence lies in the asymmetrical nature of the molecules constituting molecular dielectrics with the value of dipole moment increasing on increasing the difference in electronegativity of the constituent atoms. On the other hand, symmetrical molecules in nonpolar molecular dielectrics are aligned, so that the midpoints of positive and negative charge carriers coincide to cause a loss of nonsymmetrical nature. Molecular dipole moments tend to be oriented in an externally applied electric field. Perfect molecular orientation happens when the imposed force due to an externally applied field overcomes the thermal energy of molecules. Dipoles tend toward incremental alignment with an increase in the strength of the external electric field while the ordering becomes randomized on increasing the temperature based on additionally imparted thermal energy. Atoms do not possess permanent electric dipole moments because of being spherically symmetric. However, under the effect of an externally applied field, their electron clouds acquire certain orientations leading to the development of induced dipole moments. There may be interactions happening between permanent electric moments and induced dipoles that may onset a distortion in the perfect alignment of dipoles in the direction of an electric field.
Technically, polarization can be classified into distortional and orientational polarizability.
-
(i)
Distortional polarizability is also known as deformational polarizability. Distortion polarizability can be classified commonly into electronic and atomic or ionic polarizability based on relative deformations or distortions of electronic charges or between ions generated due to an externally applied electric field. It is to be noted that electronic polarizability keeps on increasing as a function of the increment in the number of electrons. The genesis lies in the fact that electrons in outer shells of atoms with many electrons experience a lesser degree of binding forces with nuclei imparting significant contribution to α-values. Atomic or ionic polarizability is a consequence of displacements of atoms or ions from their equilibrium positions under the effect of an externally applied electric field. Atomic or ionic polarizability progresses much slower in comparison with electronic polarization due to the higher mass of ions or atoms than electrons. However, both electronic and atomic polarizability has low relaxation times making them “fast” processes. Another important parameter is that deformational polarization has no dependence on temperature, and hence, no energy loss is reflected as a function of temperature.
-
(ii)
Orientational polarizability is also called as dipolar polarizability. Permanent electric dipole moment is exhibited due to structural molecules or groups in polar dielectric materials. Randomly oriented dipoles get perfectly aligned in the direction of an externally applied electric field. In the structural configuration of solids, the relative displacement of charges with localized attachment to atoms or molecules leads to distortional and orientational polarizability. Space charge is an important element in dielectrics that portray additional charge carriers moving short distances across the dielectric and imparting overall polarization and leading to an enhancement in the capacitance of dielectric material. An important characteristic is a constriction in movement or traveling paths due to trapping at the grain boundaries or interfaces in the dielectric material. Space charge contribution can be significant that becomes indistinct or approximately impossible to filter out from other polarizability contributions. Hence, space charge or interfacial polarizability (α) appears as a fourth term in the total polarizability of a material in addition to electronic, atomic, or ionic and orientational polarizabilities. As pointed out previously, polarization attempts to overcome thermal energy-based contribution to kinetic energy (K.E.) of molecules and hence is a function of the applied temperature.
Determination of the static value of dielectric permittivity or dielectric constant as well as polarization is a direct function of dipole moments of a polar dielectric. Peter J. W. Debye adopted Langevin’s theory of statistic orientation to derive an analytical explanation of permanent dipole moments of a polar dielectric material. As pointed out in this theory, the cumulative effect of two opposing actions generated due to an externally applied electric field that aligns dipoles in preferred orientations and thermal agitation-driven randomization of dipoles results in a statistical equilibrium. Here, the definite rotation of molecules as well as their corresponding relative interactions are neglected in the derivation of statistical equilibrium. The assumption is based on factors that include very slowly varying externally applied electric field to achieve polarizations of all different types in an equilibrium state, permanent dipole moment (μ) is free from effects of temperature as well as applied fields, dipole axes can adopt any alignment for the direction of externally applied field in an initial phase, Boltzmann statistical distribution of permanent electric dipole moments in a direction to that of the applied field, and isotropic nature of the dielectric material. Based on above-mentioned assumptions, Debye formulated the theory of dielectrics by doing away with complexities resulting in dielectric materials due to close interactions of molecules attributed to dense packing fractions.
3.1.2 Nanodielectrics
The development of nanodielectric technology relies heavily on morphological features of nanomaterials that can acquire granular microstructure, one-dimensional (1D) nanofiber, and two-dimensional (2D) nanoplatelets. Such morphologically varying nanostructures can be used as fillers for spatial distribution in a polymer matrix creating an interface of two distinguished materials. This interfacial zone has a strong bearing on the overall properties reflected by these typified polymer-nanoparticles mixed nanocomposites. Advanced tailoring of nanodielectric properties is possible by suitable modifications in the microstructure of the nanoparticle along with tuning of filler percentage and spatial distribution in the polymer matrix. Zhong et al. have reviewed polymer-nanoparticle composite materials as nanodielectrics in detail [6].
Nanodielectric materials are mainly of two types nanostructured ceramics and nanocomposites. The use of nanocrystalline materials as starting precursor material leads to the development of a highly efficient final product. A simplistic approach is to mingle nanostructured particles in a polymer matrix for the preparation of polymer nanocomposites. However, highly efficient approaches need to be followed for the development of nanometric dielectric materials. Nanodielectric particles of very small sizes show an agglomeration tendency. The great interest in nanodielectric materials lies in the large surface area available in nanosized surfaces, better functionalization opportunities offering high loading sites, and highly modified properties in contrast to their bulk counterparts. Spontaneous dielectric polarization as a function of the externally applied electric field happens in nanodielectric materials [14]. Nanosized dipoles are formed in the nanostructured dielectric material at an instant under an external electric force. Generally, the electric polarization of perfect insulator materials is termed dielectric polarization. Dielectric polarization can be explained based on the following three mechanisms of electronic, atomic or ionic, and orientational polarization,
-
(i)
Spatial shift of electron cloud about the nuclei that induce the formation of atomic dipoles developing electronic polarization (Pe),
-
(ii)
Dislocation of various atomic nuclei from their initial relative positions in an inverse space to induce molecular dipoles developing atomic or ionic polarization (Pa),
-
(iii)
Alignment of permanent molecular dipoles under the influence of an electric field force to develop orientational polarization (Pμ).
The resultant dielectric polarization vector (\(\vec{\user2{P}}\)) is the actual sum of all the above-mentioned three contributing polarization vectors.
Electronic polarization and atomic polarization as well as depolarization are very fast processes in dielectric materials in comparison with orientational polarization as well as disorientation polarization.
Engineering of nanodielectric materials requires suitable tailoring of nanostructure shape and sizes, morphological features, design of structures with appropriate aspect ratios, and relevant interfacial modifications. Nanofillers can be suitably divided into classes of 0D, 1D, 2D, and 3D types with each specific type of nanofiller improving on the particular properties of mechanical, electrical, thermal, and optical properties. Quantum dots belong to the category of 0D quantum confined nanofillers. Similarly, nanoshells, nanolaminas, and nanoplates belong to 1D confined nanofillers. Nanotubes, nanowires, and nanofibers are 2D confined nanostructures. Nanosized SiO2 forms a good example of 3D confined quantum structures.
Polymer nanocomposites (PNCs) can be synthesized by the application of three major PNC components of the polymer matrix, nanomodified particles along with transitional constituents, such as surfactants and intercalants [15,16,17,18,19,20]. A range of polymers is available in the commercial market that can be exploited as a matrix for the synthesis of PNCs. Different polymers including polyvinyl alcohol (PVA) [21, 22], polyethylene glycol (PEG) [23], and even di-block copolymers (poly-styrene-poly-ethylene oxide) [24, 25] can be used for the preparation of PNCs. Polymers need to be processed with nanomodified particles and transitional constituents carefully in a thermal environment below their glass transition temperature (Tg). Earlier thermosets were used frequently for PNCs which were subsequently replaced by thermoplastics [5, 26,27,28]. Polyolefins (POs) belong to the category of thermoplastic polymers possessing intricate organic molecular structures with relatively low melting points (M.P), semi-crystalline, cost-effective, less resistant to degradation against UV-radiation, mechanical breakage, and oxidation chemical means along with the temperature of processing not to exceed 250 °C [28]. Clay-containing PNCs can be synthesized by reactive, solution, and melt-compounding methods. The reactive method involves in situ polymerization technology, solution method is focused on creating a dispersion of organoclay in a solution dissolved with polymer, while melt-compounding consists of formation by compounders [28]. Clay platelets as well as polymers are of the separate chemical constitution with polymers being hydrophobic and largely immiscible with hydrophilic clays, mostly silicates. Compatibilization is a two-step process forming a diffused ionic layer in a region surrounding the clay platelet along with the dispersion of extra ionic bunches in the material medium [28]. The interface between clay and matrix offers a range of complex reactions. Organic molecules are physisorbed followed by solidification resulting in enhancement of overall net solid content. Significant enhancement of electric modulus along with barrier characteristics due to totaling of a small quantity of effectively distributed clay particles is caused because of increment in solid content at the interface. This may also halt the UV-driven degradation process. The inclusion of clay particles enhances the degree of crystallinity along with providing efficient nucleation sites. An important parameter is the optimization of type and quantity of intercalant as well as compatibilizer that may cover the clay particles effectively and halt effective nucleation. The inclusion of an optimized content of clay in PNC is an important parameter that shows the most effective performance in the 2–4 wt% range of organoclay. The degradation of clays may be driven catalytically by transition metal ions as contaminants. Organoclay does not have a sound effect on dielectric properties when added in small quantities but has a pronounced effect on mechanical as well as barrier characteristics.
3.1.3 Interface Chemistry
Interface chemistry plays a major role in each of the steps involved in the synthesis methodology of nanocomposites. Glass transition temperature (Tg) in polymers is a classical illustration of the involved interfacial chemistry in nanocomposites [29]. Simulation or modeling or even experimental study of interfacial chemistry-related characteristics is an important factor that can be tailored effectively by varying simulation or experimental conditions. A plethora of literature has been presented on interface chemistry in nanocomposites. Alcoutlabi and McKenna concluded in their report that the present theoretical accounts to explain Tg are not sufficient to describe the distinct features at the nanoscale [28, 30]. The defect diffusion model inculcated factors of concentration of defects, percolation fraction, defect lattice geometry correlation length, and defect–defect interaction enthalpy [28]. This model on nanoconfined Tg was presented by Bendler et al. and focused on a calculable association between the percolation ratio obtained by fixed to transportable areas and Tg [31]. Lewis et al. have pointed out that nanocomposite presents an interface between nanofiller and polymer matrix and it has nothing to do with a simplistic two-component phasic system [32, 33]. As a result, the incorporation of a minimum of a single interphase region becomes of prime importance to get a nanoproduct in which structure–property relationships and involved chemistries become distinct in contrast to the two initial bulk phases that react primarily to produce the nanocomposite. A significant distinction of this system is that nanoparticles possess the enormously exposed surface area to contribute to the interphase region in a nanocomposite.
Complex permittivity (ε) is an important parameter and dielectric response to externally applied AC electric fields studied in terms of complex permittivity. Complex permittivity consists of a real part (ε’) and an imaginary part (ε'’). The real part of complex permittivity (ε’) entails energy stored in a dielectric material, while the imaginary part of complex permittivity (ε'’) talks about different mechanisms that correlate to the dissipation of energy. Tanaka gave an initial modeling theory that considered a nanocomposite formed by the dispersion of nanoparticles in a polymer matrix. In a model based on this approach as shown in Fig. 3.1, nanoparticles are encircled by three different layers of separate structural geometry covered by a diffuse Gouy–Chapman charge layer [34]. In the core lies the nanoparticle, while other layers lie one above the other as shown in the model given below. Adjacent two layers close to the core nanoparticle in the model, consisting of immobilized species that bear a strong interacting relationship with their inner neighbor, are covalently attached to it. An enhanced free volume including chain mobility lies at the core of this system confined in an area between the immobilized shells and undisturbed matrix summing up with other factors to generate a distinct effect on the dielectric nature of the formed three-phase system.

Schematic representation of the interfacial model for core–shell structured nanoparticles for advanced nanodielectric materials [34]
The rule of mixing the composite material consisting of two systems A and B portrays that the properties of the finally derived composite material will lie in between the values shown by precursor materials A and B.
Raetzke and Kindersberger have discussed the impact of interphase in terms of fraction based on the diameter of the particle and thickness of interphase signifying the role of physical or even chemical bonding at the interphase, acting radius, and correlated material properties [29]. As pointed out above, nanoparticles as fillers possess a surrounding area covered with polymer forming the interphase. This surrounding layer around the nanoparticles or interphase has a thickness typically in the range of 0.5–10 nm. Polymeric chains contained in the surrounding matrix are bound to the particle surface by physical or chemical bonds that may possess orientations as shown in Fig. 3.2a and b. The interphase structure depends on a range of parameters that include filler or type of nanoparticles, surface treatment meted-out, type of matrix material, and even the crystalline or semi-crystalline nature of interphase that plays a deciding role. Properties of interphase are completely different from surrounding matrix material because of distinct interphase structure and typical bond formation with the particle surface.

Schematic representation of a nanofiller surrounded by polymer matrix in such that a polymer chains are arranged parallel to the nanofiller surface, and b polymeric chains oriented radially about the nanofiller surface [29]
Attachment of polymeric chains over nanofiller surfaces happens by chemical bonds that depend on polar or nonpolar characteristics of the two bonding species. An interesting factor is the conversion of surface groups from nonpolar to polar characteristics due to surface treatment. Ehrenstein has pointed out that chemical bonds of polymeric chains possess bond energies between 40 and 800 kJ mol−1 with bond lengths typically lying in the range of 0.075 to 3 nm [35]. On the other hand, bond energies in physical bonds are typically between 0.2 and 25 kJ mol−1 for bond lengths between 0.5 and 0.8 nm.
An explanation of interphase phenomena in terms of a multicore model entails involving all three layers, so that the contribution from interphase becomes significant along with nanofiller surfaces impacting the polymeric chains. Different nanofiller concentrations will produce interphase made up of different fractions. For the establishment of a simplistic explanation, it is pertinent to consider the three separate layers as a single interphase layer possessing thickness, j. Bond lengths and atomic diameters are the parameters important for the determination of thickness j of interphase in the multicore model. Typically, the first layer possesses a thickness in the range of 1–1.5 nm in which polymeric functional groups are chemically bonded to the nanofiller surfaces, and consists of contribution from nanofiller surfaces as well, with a typical value of 0.4 nm. The thickness of the initially attached organic layer in a tight-binding approach lies in the range of ~0.3 nm–1.0 nm, which typically is dependent upon the type of polymer. The second layer has polymeric chains attached to nanofiller surfaces or the first layer as per the multicore model. In the second layer, the sterical arrangement of polymeric functional groups is affected mainly and facilitates an ordered arrangement. In the parallel arrangement of polymeric chains about the nanofiller surface, as shown in Fig. 3.2a, the little number of functional groups constitute mere thickness of the second layer that is dependent on smaller bond lengths of H2-bonds as well as dipole bonds. It signifies that the thickness of the second layer depends on the degree of strength of the bonds. The thickness of the interphase layer can be estimated by the diameter of polymeric chains typically in the range of ~0.3 nm to 1.0 nm along with bond lengths between 0.5 and 0.8 nm. A typical thickness between 1.5 and 8 nm prevails for about 2–4 entangled polymeric chains in the second layer. In the case of radial attachment of polymeric groups to the nanofiller surface, as shown in Fig. 3.2b, the second layer can adopt higher thicknesses based on polymeric chain lengths.
In contrast to the first and second polymer layers, the third layer is affected by the orientation of polymeric chains in the second layer, and usually, the thickness is between 1 and 3 nm. With all the above three-layered structure considerations, the thickness (j) of interphase lies in the range of 3–12 nm.
Interphase structure greatly impacts the mechanical, dielectric, electrical and thermal conductivity, and prevention of material degradation. The mobility of polymeric chains affects dielectric properties, such as dielectric loss (tan δ). On increasing interphase fractions, the permittivity of dielectric material can show drastic changes.
Raetzke and Kindersberger have given a simplified model for the estimation of interphase faction at different fractions of nanofillers [29]. Assumptions for this model are outlined below,
-
(i)
All particles are assumed to be spherical and uncapped in interphase having nonvariable thickness j.
-
(ii)
All particles are assumed to be equal in size with a constant diameter “d”.
-
(iii)
Homogeneous dispersion of particles happens in dispersing medium or in the matrix.
In a cubic configuration with homogeneous dispersion, as shown in Fig. 3.3a, eight particles share each corner in the cubic geometry along with one particle at the body center of the cubic structure. Let the center-to-center distance of the two closest lying particle centers be r0. This type of particle arrangement in a cubic configuration can accommodate a maximum nanofiller concentration of 68%.

a For homogeneous dispersions of particles, a cubic geometry can be assumed for a perfect arrangement of particles such that eight particles share each corner of the cube similar to the cubic crystal geometry in face-centered cubic (fcc) configuration. This can be described as the fundamental geometry of the arrangement of particles that are dispersed homogeneously in a polymer matrix. In addition, a body-centered cubic (bcc)-type configurational structure is also visible such that a particle is placed at the center of the body of the cube, so that a maximum of 68% of nanofiller concentration by volume can be accommodated [29]. b Nonoverlapping particles are represented schematically here since nanofiller particles are placed at considerable distances attributed to low nanofiller concentrations used in the formation of nanocomposites. As a result, interphases of closely-lying particles do not partake in the formation of common regions. Interphase fraction is supposed to increase linearly with an increment in nanofiller concentration [29]
Estimation of interphase fraction or fj can be performed by knowing about nanofiller concentration Vf, while interphase thickness or j is another important parameter. Four different conditions can be considered in this regard,
-
(i)
$$\frac{2}{\sqrt 3 }\left( {{\varvec{d}} + 2{\varvec{j}}} \right) \le \user2{ r}_{0}$$(3.10)
At low concentrations of nanofillers, interparticle distance is significantly high, and hence, no overlapping of neighboring interphases happens. Considering spherical volumes to be Vd and Vd+2j for diameters d and d + 2j, respectively, let the interphase volume be represented as Vj, then,
Now, the estimation of interphase fraction fi to nanofiller concentration f can be given as,
-
(ii)
$$\left( {{\varvec{d}} + 2{\varvec{j}}} \right)\user2{ } \le \user2{ r}_{0} < \frac{2}{\sqrt 3 }\left( {{\varvec{d}} + 2{\varvec{j}}} \right)$$(3.14)
In this range, interphase fraction enhances with nanofiller concentration, so that closely lying interphases overlap.
-
(iii)
$$\frac{2\surd 2}{3}\left( {{\varvec{d}} + 2{\varvec{j}}} \right) < {\varvec{r}}_{0} < \left( {{\varvec{d}} + 2{\varvec{j}}} \right)$$(3.16)
High overlapping of closely lying interphases happens in the above-specified range. Under this condition, interphase concentration reduces because nanofillers begin to shift toward closely lying interphases with an increment in nanofiller concentration (Fig. 3.4).

Schematic representation of the diagonal element of fundamental geometry of particles reflecting overlapping of interphases under the third condition [29]
-
(iv)
$${\varvec{r}}_{0} < \frac{2\surd 2}{3}\left( {{\varvec{d}} + 2{\varvec{j}}} \right)$$(3.18)
If interphase fraction dominates in the overall structural configuration, the effect of incremental nanofiller concentration leads to a decremental interphase fraction (Fig. 3.5).

Schematic representation of the interphase-based formation of the complete polymeric layer [29]
3.2 Synthesis, Microstructural, and Dielectric Characterization of Nanodielectrics
Different techniques employed for nanoparticle synthesis are also used for the preparation of dielectric nanoscale materials. This is inclusive of simple solution techniques, co-precipitation methods, simple mixing of nanoparticles in a polymer environment for the formation of polymer nanocomposites preparation of nanoparticles under vacuum, hydrothermal synthesis, sol–gel method, sonochemical synthesis of nanoparticles, microwave-assisted synthesis, template-based synthesis approach, and application of biological templates for synthesis of nanoparticles and physical methods that include pulsed laser ablation.
3.2.1 Simple Solution Techniques for Nanodielectric Particles
Solution-based mixing of nanofillers with polymers in a solution along with the melt-compounding method in the context of broader mechanical mixing of two components is a well-established process. Mechanical mixing is a common process in industrial production that entails the management of a diverse physical system to produce a resulting homogeneous system in the configuration of unit operation. Mass or heat transfer happens due to the mechanical agitation of multiple phases. In the simple solution chemistry or mechanical mixing method, polymers are usually dissolved in a solution followed by the dispersion of nanofillers in this polymer solution to synthesize nanocomposites in the solution. This nanocomposite solution is subjected to evaporation to get solidified nanocomposites in a mixed configuration. Shear forces in the simple solution chemistry method are of a much lower degree due to the imposition of mechanical agitation of a lesser degree. This is usually performed by placing the mixed components solution on a mechanical stirrer under constant magnetic stirring under the effect of temperatures if so required. However, the applied temperature must be kept below its Tg to avoid the transformation of the polymeric phase. Due to this fact, external parameters play a vital role in the dispersion of nanofillers in the polymer solution, such as surface loading of nanoparticles, surface modification by organic molecules, and presence of binders. The advantage of this method involves high control over the produced nanocomposites since constituents can be mixed in desired percentages and mechanical mixing. Another benefit is to have better control over the shape, size, and morphological features of nanoparticles that are produced separately as one of the constituents. Ajay Vasudeo Rane and others have given details of clay-polymer nanocomposites prepared by mechanical mixing of polymers with both pristine and modified clays [36]. Solid–solid phase mixing and solid–liquid phase mixings are the stereotyped methods in the formation of clay-polymer nanocomposites. Solid–solid mixing in this case is further classified into convective and intensive mixing. Convective mixing generates a random state eventually with the potential risk of demixing constituents due to segregation caused by variations in shape, size, and density. Convective mixing is not suitable for the mechanical mixing of materials possessing cohesive nature mostly exercised in the case of very finely divided particles or even wet compounds. Convective or transportation forces are of a very mild nature that is not sufficient to overcome relatively strong cohesive forces acting among particles. As a result, energy-based mixing or impact employment or application of shear stress may be followed to negate the effect of cohesive interparticle forces. Four different synthesis methods of clay-polymer nanocomposites include in situ template synthesis, solution intercalation, in situ intercalative polymerization, and melt intercalation [36]. Melt intercalation in conjunction with a shear mixer is the preferred method for the synthesis of clay-polymer nanocomposites based on thermoplastics and elastomers. Solution intercalation is applied for the synthesis of clay-polymer nanocomposites in which different thermosets, thermoplastics, and elastomers based on solubility parameters are dissolved in the specified solvents followed by intercalation with solvent-dissolved nanoclay particles. At the industrial scale, mechanical mixing is performed in batches inline or dynamic mixing with mixers possessing the capability to operate at 1500 or 1800 rpm.
Ren et al. have compared the dielectric properties of pristine polyetherimide (PEI) polymeric materials with those of polyetherimide polymer composited with hafnium oxide (HfO2) nanoparticles synthesized mainly by the solution casting method [37]. Solutions were cast over glass slides followed by drying at a suitable temperature for a specific period to evaporate the solvent. Dried solution casted films were peeled-off and dried again to obtain 9 to 12 μm thick films.
Nanocomposites enhanced with HfO2 nanoparticles show an improved dielectric constant along with a reduction in high-field current density. A difference in morphological features of the pristine PEI and PEI-HfO2 nanocomposites was observed from scanning electron micrographs (SEM) as shown in Fig. 3.6.

SEM images of pristine PEI and HfO2-enhanced nanocomposites in PEI matrix [37]. Morphological changes are visible at the microscale of 5 μm with a network formation in the HfO2-enhanced matrix in comparison with a rather larger layered-type formation in pristine PEI composites [37]. Reprinted with permission from ref. [37]. Copyright (2021) (Elsevier)
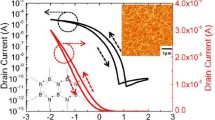
For pristine PEI and PEI/HfO2 nanocomposites with various nanofiller contents, a comparison of D-E loops was performed at E = 300 MV.m−1 as shown in Fig. 3.3a and b. It was inferred that electric displacement (D) is steadily improved as a function of the induction of HfO2 nanofillers in the PEI matrix along with more than 95% efficiency of the charge–discharge cycle. At nanofiller contents of 1 vol%, 3 vol%, 5 vol%, 7 vol%, and 9 vol%, D-value increased to 1.03 × 10–2, 1.07 × 10–2, 1.11 × 10–2, 1.14 × 10–2, and 1.22 × 10–2 C m−2 for nanocomposites from 9.77 × 10–3 C m−2 of pristine PEI at 300 MV m−1.Thus, the D-value reflects a trend similar to that of dielectric constant since D changes in a linear manner as a function of \({\varvec{\varepsilon}}_{{\mathbf{r}}} \left( {{\varvec{D}} = {\varvec{\varepsilon}}_{{\mathbf{0}}} {\varvec{\varepsilon}}_{{\mathbf{r}}} {\varvec{E}}} \right)\) in the case of linear dielectrics Fig. 3.7.

a D-E loops of pristine PEI and PEI/HfO2 nanocomposites derived at E = 300 MV m−1. Inset shows the Dmax and charge–discharge efficiency in % of pristine PEI and PEI/HfO2 nanocomposites as a function of nanofiller content at E = 300 MV m−1. b Maximum discharged energy density (J cm−3) and leakage current density (A cm−2) were derived as a function of nanofiller content in vol% for pristine PEI and PEI/HfO2 nanocomposites [37]. Reprinted with permission from ref. [37]. Copyright (2021) (Elsevier)
Figure 3.8 shows a schematic of the PEI/HfO2 nanocomposites with nanoparticles at the core and first, second, and third layers as the bonding, bound, and loose layers typically in a Guoy–Chapman electrical double-layer configuration [37]. Band diagram with HOMO and LUMO bands can also be seen along with this core–shell nanoparticle interfacial model. Charge incorporation from electrodes and charge transport including charge trapping and de-trapping in pristine PEI and PEI/HfO2 nanocomposites can be seen in Fig. 3.10. High degree of thermal and electrical stimulation prevails only at high temperatures and electric fields, respectively, and hence, conduction mechanisms based on charge density in pristine PEI are significantly boosted to overcome the potential barrier conveniently. However, an opposite mechanism happens in PEI/HfO2 nanocomposites, and charge carriers are trapped in holes or positive vacancies formed at the interface of HfO2 nanofillers and polymer matrix. Insertion of wide band gap HfO2 dielectric nanofillers impedes the electrical conduction mechanism as documented by significantly reduced leakage current density.

Band diagrams for the core–shell nanoparticle interfacial model along with HOMO and LUMO bands. Evac is the vacuum energy level, EF is the Fermi energy level, EC is the conduction band, EV is the valence band, EAf is electron affinity, and BGF is the band gap of nanofiller, while EAm and BGm are electron affinity and bandgap of the polymer matrix, respectively [37]. Reprinted with permission from ref. [37]. Copyright (2021) (Elsevier)
3.2.2 Co-precipitation Method
Co-precipitation is a synthesis method in which compounds soluble in a solution are abolished during the precipitation process [38]. More than one compounds become soluble instantly to make happen precipitation when dissolved in a single solution phase. However, the degree of contamination becomes of a very high degree. Generally, co-precipitation is the process of bringing down involved materials that are dissolved in normal conditions. Precipitation can be understood as the process of formation of insoluble solid-phase particles from a super-saturated solution on the dissolution of a substance. Precipitant is the chemical reagent that causes the formation of the insoluble solid phase.
Precipitation happens in a much faster manner in a strongly super-saturated solution. The super-saturated solution has its concentration exceeding its solubility owed to the formation of a mixture of solvents, evaporation of solvents, and associated temperature changes. Precipitation involves the development of a solid phase that forms an interface with the constituent solution. Interface chemistry gets involved here inculcating free energy connected with the dissolution reaction including relative surface energy formation between the two phases. Free energy of dissolution reaction enhances the degree of randomness and hence entropy of the system in an exothermic or endothermic reaction mechanism. The nicety of the co-precipitation method is that no precipitate product will be obtained in case no appropriate nucleation sites are found associated with a lesser favorable energy.
On the other hand, post-precipitation is a method in which precipitation of the unwanted phase happens initially after the precipitation of the required compound. The degree of contamination is low. On this formed precipitate, a precipitation layer is formed over this initially formed layer.
Co-precipitation usually involves the instant happening of nucleation or beginning of precipitate formation, growth of solid phase, and coarsening along with agglomeration in the solid content. Regarding the formation of nanocomposites, Ostwald ripening, and other secondary processes tailor the shape, size, morphological features, and associated properties of the resulting products. Co-precipitation of nanocomposites is a widely used process for the formation of metals from aqueous solutions or obtained from nonaqueous solutions by reduction, electrochemical reduction, formation of oxides from aqueous and nonaqueous solutions, or even microwave-sonication-assisted co-precipitation methods. Some of the benefits of co-precipitation in the formation of nanocomposites are fast synthesis procedures along with control of shape, size and composition, modification of particle surfaces along with homogeneous character, offering synthesis facility at low temperatures, energy efficiency, and no use of organic solvents as a medium. Disadvantages are inclusive of nonapplicability to noncharged species, taking a bit of time to synthesize, associated irregularity in batch-to-batch production, the inclusion of impurities, and nonconformity with reactants that possess highly varying precipitation rates.
Vadivel et al. formed magnetic nanosized particles of chromium (Cr)-displaced cobalt ferrite (CoFe2-xCrxO4) at different Cr concentrations of 0.0, 0.1, 0.2, and 0.3 by co-precipitation followed by annealing for 3 h at T = 600 °C [39]. Multicomponent particles were single phasic in a cubic spinel geometry with an average crystallite size of 15–23 nm. Dielectric studies revealed that both dielectric constant (ε) and dielectric loss (ε) were analyzed to be higher in comparison with the pristine CoFe2O4 nanoparticles. Fluorescence spectral analysis displayed UV emissions in the strong red zone as well as in the weak blue zone. Figure 3.9a and b show the variation of dielectric constant (εʹ) and dielectric loss (εʹʹ) as a function of frequency in various Cr-substituted CoFe2O4 compounds. It can be seen that the dielectric constant (εʹ) increases at reduced f-values.


Variation of dielectric constant (εʹ) as a function of logarithmic (f) at different temperatures of 40, 60, 80, 100, 120, 140, 160, 180, and 200 °C is shown for (a) CoFe2O4, (b) CoFe1.9Cr0.1O4, (c) CoFe1.8Cr0.2O4, and (d) CoFe1.7Cr0.3O4 nanoparticles in Fig. 3.10a–d. This is observable that the dielectric constant (εʹ) increases from 8.0 × 103 to ~ 1 × 104 at room temperatures on increasing Cr from 0.0 to 0.2 and 0.3. The effect of temperature is that the value of the dielectric constant (εʹ) increases with increasing temperature. Dielectric loss (εʹʹ) for the four samples under the same operating temperatures is shown in Fig. 3.11a–d. Dielectric loss (εʹʹ) at room temperature for the four samples shows erratic behavior with value decreasing from ~ 1.4 × 104 to 8.0 × 103 in the case of CoFe1.9Cr0.1O4, again rising to 1.0 × 104 for CoFe1.8Cr0.2O4, and finally rising to 2.0 × 104 for CoFe1.7Cr0.3O4. However, all the samples show comparatively higher values of dielectric loss for the operational temperature of 200 °C.

Krishna et al. used chemical co-precipitation for the synthesis of lanthanum oxide nanoparticles by employing organic and biological capping agents [40]. The organic capping agent was ethylene-diamine-tetra-acetic acid (EDTA) and the biological capping agent was starch and deoxy-ribo-nucleic-acid (DNA). Lanthanum oxide nanoparticles are obtained by annealing the carbonates at temperatures of 450 to 700 °C. Dielectric permittivity was estimated from drawing plots of capacitance as a function of applied frequencies for the two operational temperatures. Dielectric permittivity was found to possess enhanced values at low operational frequencies and reduced values at high operational frequencies. Under AC conduction, maximum AC conductivity is achieved at high operational frequencies.
Joshi et al. co-precipitated nickel ferrite nanoparticles of single-phase cubic spinel crystal geometry in a space group of Fd3m possessing an average crystallite size of 8 to 20 nm based on annealing temperature [41]. Strong temperature dependence was observed at all the operational frequencies for dielectric permittivity, dielectric loss, and AC conductivity measurements. Frail polaron hopping was discovered between Fe3+/Fe2+ ions for dominant ac-conduction. Optical bandgap was estimated to lie in the range of 1.27 eV to 1.47 eV for NiFe2O4 by UV–visible diffuse spectra and established as an indirect bandgap material. Dielectric constant (εʹ) and dielectric loss (tanδ) were studied at room temperature as shown in Fig. 3.12. Dielectric constant showed frequency dispersion in the lower frequency range that became constant nature for frequencies beyond 10 kHz. The dielectric constant value fell sharply in the low-frequency region and was found to increase gradually at higher operational frequencies. Electronic, ionic, and orientational polarizations as well as space charge were the reasons for high dielectric constant values in the region of low frequencies. Space charge polarization was the dominating factor at high temperatures and low frequencies.

Dielectric constant (εʹ) and dielectric loss (δ) were also drawn as a function of temperature for the sample calcined at 550 °C for 2 h as shown in Fig. 3.13. Curves were acquired at different operational frequencies of 10 kHz, 25 kHz, 50 kHz, 75 kHz, 100 kHz, and 1 MHz. The normal behavior of magnetic semiconductor ferrite shows an initial increment in dielectric constant as a function of temperature. The formation of free localized dipoles increases the dielectric constant (εʹ) due to supplemental thermal energy. These free localized dipoles tend to line up along a direction in which the field is applied. Enhancement in temperature causes the creation of lattice phonons that undergo interaction with electrons for electron–phonon scattering.

Figure 3.14 shows the variation of AC conductivity σac with angular frequency at different temperatures of 40, 60, 80, 100, 120, 140, 160, 180, and 200 °C [41]. It can be observed that σac shows dispersion characteristics at higher operating frequencies with a variable gradient. Eventually, all σac curves meet each other on higher operating frequencies. This shows the presence of several temperature induced as well as multiple relaxation processes. This electrical conduction can be expressed by a phenomenal expression in which the hopping of charge carriers is the major mechanism. This relationship is called Jonscher’s universal power law,

Here, K is a constant that depends on temperature, s occupies a value between 0 and 1, and ω is the angular frequency of the AC field. The value of s was found to increase as a function of temperature which can be explained based on small polaron hopping.
3.2.3 Hydrothermal Process
Hydrothermal synthesis involves chemical reactions of precursor substances kept in an air-tight sealed reactor heated to create temperatures and pressures above the normal atmospheric ranges [42]. When a reaction takes place at elevated temperatures and pressures in an organic solvent, it is more commonly referred to as solvothermal synthesis.
The advantage of the hydrothermal synthesis method is that it can be used to dissolve most of the materials in an appropriate solvent by thermal treatment and creating pressures in the vicinity of the critical point. This synthesis method is an effective method for the production of metastable or intermediate-phase compounds. A range of flexibility is offered in this synthesis method by tailoring parameters of reaction time, temperature, and change of surfactant, solvent, and precursors. Another benefit is to obtain products with reduced M.P., a tendency to undergo pyrolysis, and possessing high vapor pressures. A high disadvantage of the hydrothermal process is that it does not offer any flexibility to observe and study reaction mechanisms, rates, and other parameters in situ because of the happening of reactions under high pressures in the sealed reactor.
Köseoğlu et al. hydrothermally synthesized Mn0.2Ni0.8Fe2O4 nanoparticles assisted with polyethylene glycol (PEG) [43]. Temperature-dependent properties as a function of frequency (1 Hz to 3 MHz) showed dielectric dispersion that is based on Koop’s theory. Koop’s theory relies on the Maxwell–Wagner model focused on homogeneous double structure with reference to interfacial polarization [44].
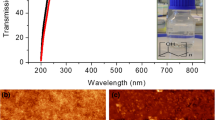
Jiang et al. have shed light on nanoscale BaTiO3 as well as polymer-BaTiO3 nanocomposites with tailored properties in form of a review [45]. Polymer-based nanocomposites of BaTiO3 have several advantages over pristine BaTiO3 nanocrystals. Hydrothermal as well as solvothermal processes of BaTiO3 form the core of this detailed review article. In a work performed by Phromviyo et al. Ag nanoparticles were hydrothermally synthesized by applying Aloe vera-plant extracted solution in form of a surface stabilizer and reducing agent to produce Ag@Ale-NPs [46]. This was followed by the preparation of Ag@Ale-NPs/PVDF polymer nanocomposites by liquid-phase backed dispersion as well as hot-pressing techniques. Modified surfaces of Ag@Ale-NPs were confirmed by X-ray photoelectron spectroscopy (XPS). Dielectric properties were studied at two different filler compositions of 0.18 and 0.22 in volume fractions. At the volume fraction of 0.18, high dielectric permittivity (ε) of ≈92.5 along with highly reduced tangential loss of 0.049 was discovered at 1 kHz and room temperature. On increasing the nanoparticle volume fraction to 0.22, a substantially higher value of ε of ≈257.2 became evident along with a low loss of tanδ ≈0.26. Interfacial polarization along with the micro capacitance effect in the PVDF matrix was the factors responsible for outstanding dielectric properties of the synthesized Ag@Ale-NPs/PVDF polymer nanocomposites.
SEM images of surface-modified nanoparticles of Ag@Ale-NPs by reacting Aloe vera extract in a reactor for 3 h, 6 h, and 12 h in hydrothermal synthesis is shown in Fig. 3.15 [46].

Morphological features along with particle size distributions of a 164 nm with S.D. = 56, b 228 nm with S.D. = 62, and c 303 nm with S.D. = 90 as visible in SEM images of surface-modified Ag@Ale-NPs prepared with the help of Aloe vera plant extracts reacted for three different times of 3 h, 6 h, and 12 h [46]. Reprinted with permission from ref. [46], Copyright (2018) (Elsevier)
Microstructure of nanocomposites was also observed by SEM imaging in a largely fractured cross-sectional area for filler volume fractions of fAg@Ale = 0.08 and fAg@Ale = 0.22 [46]. Microstructure consisted of random distribution of Ag@Ale-NPsin a surrounding PVDF polymer matrix in a cluster configuration more specifically on increasing filler volume fraction. Cluster configuration indicated the high tendency of Ag@Ale-NPs to agglomerate due to interparticle cohesive forces. Information on agglomeration is essential for establishing a correlation between microstructure and derived dielectric propertiesFig. 3.16.

The tangential dielectric loss was estimated to change from 0.039 for a filler volume fraction of 0.22 to 0.049 with a filler volume fraction of 0.18 under an operational frequency of 1 kHz. Cluster formation was detected for the filler volume fraction of 0.18 with each of the clusters separated by a polymer layer of PVDF resulting in mild enhancement of the percolation threshold. However, the presence of a barrier polymer layer prevented the formation of weak conductive links between different conductive clusters. Hence, dielectric tangential loss (tanδ) at lower frequencies was maintained at lower values. Lower operating frequencies are an approximation to direct current (DC) conductivities. Free electrons generated from conductive clusters tend to assemble at the interface of the PVDF barrier layer between different loose nanoparticle agglomerates. This gives rise to the effect of interfacial polarization. Low tangential loss (tan δ) of polymeric nanocomposites was ascribed to micro capacitor formation because of two close-lying nanoparticles clusters acting as microelectrodes along the direction of the applied electric field separated by a dielectric polymer barrier layer (Fig. 3.17).

3.2.4 Sol–gel
Sol–gel method involves the formation of a sol or solution that slowly but eventually progresses in form of a diphasic system reflecting a behavior similar to gels [47]. Gel-resembling diphasic system consists of a solid as well as a liquid phase. Sol–gel-derived particles can possess surface morphological features in the geometry of discrete or distinct particles and extend to more regular networks of polymeric chains. Sol–gel method possesses the capability to produce ultrafine divided distinct particles in ceramic powders of single as well as multicomponent constitution at the nanometer scale. Metal nanoparticles are usually derived by the sol–gel process by mixing the precursor substances in the liquid phase, formation of stable sol that is normally transparent by hydrolysis and polycondensation reactions, and further aging of sols leads to the development of a 3D structural network followed by drying, calcination, and sintering.
Advantages of sol–gel are inclusive of the (i) formation of a homogeneous product at relatively low temperatures, (ii) better control over shape, size, porosity, and (iii) metal-oxide or ceramic particles in a polymer matrix form an α-β matrix for enhanced applications, and (iv) suitable methodology for the formation of multicomponent particles with the possibility to include dopant atoms.
Yadav et al. used a honey-mediated sol–gel combustion method for the synthesis of fcc-spinel ferrite CoFe2O4 nanoparticles subsequently calcined at 500, 700, 900, and 1100 °C [48]. The dielectric constant (ε) of CoFe2O4 nanoparticles was plotted as a function of frequency, and it decreased with increment in frequency. At lower operating frequencies that are nearly identical to DC conductivity, ε-values lie on the higher side depicting effects associated with space charges, ionic charges, and interfacial polarization. Contrarily, ε-values became practically independent of applied frequencies because formed electric dipoles were not capable of catching up with the fast-varying electric field lines as a function of time. Again, Koop’s theory is of paramount importance here as outlined above relying on dielectric formation in which two heterogeneous medium layers of the Maxwell–Wagner type are considered. The dielectric module consists of insulating grain boundaries separating the conductive grains in a granular or polycrystalline or multicrystalline configuration. At low operational frequencies, grain boundaries play a major role as compared to the grain itself, so that ε-values become effectively high. Moreover, the hopping mechanism of charge carriers takes place, so that charge carriers pile up at the insulating boundary because of high resistances and thus producing polarization. This polarization or accumulation of charge carriers is not observed at higher operating frequencies since the charge carriers can not pursue the AC electric field at higher frequencies. Dielectric constant varied as a function of cationic distribution depending on granular size and type of microstructure. Dielectric loss suffers from a lag or phase difference in polarization about the AC electric field. This may be caused due to microstructural attributes, such as grain boundaries, imperfections, defects or dislocations, or distortions in unit cell lattice volume. The hopping frequency whereby becomes equal to the frequency of the applied AC field, and oscillating ions begin receiving maximized electrical energy. This is reflected in form of a power loss peak in the plot of tan δ versus frequency (f). The mathematical expression for maximum tangential loss in dielectric materials is given as,
The mathematical relationship between relaxation time (τ) and jumping probability per unit time (p) is given as,
Debye relaxation is observed when the rate of hopping of jumping probability per unit time becomes equal to ac-frequency.
Vasudevan et al. applied the sol–gel method for the synthesis of poly(vinyl pyrrolidone) (PVP) doped with varying concentrations of titanium dioxide (TiO2) to obtain PVP-TiO2 nanocomposites [49]. Dielectric properties were estimated for the nanocomposites in the range of operational frequencies of 1 kHz to 2 MHz in the vicinity of room temperature. The dielectric constant and tangential loss were estimated to be reduced at high operational frequencies of AC conduction. Conductivity values were 10–7 to 10–8 S cm−1, which decreased steeply on increasing wt% of TiO2 due to enhancement in randomness or reduction of homogeneity in microstructure pattern in the whole volume of the nanocomposite.
Singh et al. used the sol–gel method for the preparation of polysulfone-ZnO nanocomposites and studied frequency-dependent and temperature-dependent dielectric properties [50]. Dipolar polarization was solely deciphered to be responsible for the observed dielectric behavior, while interfacial polarization was partly. Dielectric relaxation was studied in terms of electric modulus to observe the effects of electrode polarization and resolving low-frequency relaxation mechanisms. Figures 3.9 and 3.10 show the real (Mʹ) and imaginary (Mʹʹ) parts of complex electric modulus of pure polysulfone and ZnO-loaded polysulfone nanocomposites at different operating temperatures of 30, 60, 90, and 120 °C (Fig. 3.18).

Mathematical equations for the calculation of real and imaginary parts of electric modulus are given as,
It can be observed from Fig. 3.19 that the electric modulus of pristine and ZnO-loaded nanocomposites shows an increasing trend as a function of increasing frequencies at different operational temperatures. However, the imaginary part (M”) of pristine and ZnO-loaded nanocomposites do not show any peak in the curve at an operating temperature of 30 °C. Observance of sharp peaks at operational temperatures of 90 °C and 120 °C is representative of dielectric relaxation at higher operational frequencies reflecting very quick crystalline relaxation propagating with retentive symmetries. At higher temperatures of 120 °C, peaks of imaginary parts (M”) of dielectric relaxation shift toward higher frequencies. Good broadening of peaks is observed for M” at 90 °C for different loading wt% of nanofillers. However, the peak became sharp and peak-broadening decreased at 120 °C of operational temperature reflecting Maxwell–Wagner–Sillers (MWS) type crystalline relaxation in any asymmetric mode. Further, it was inferred that the peak reduced in a vertical orientation on increasing the wt% of nanofillers.

Figure 3.20 shows the variation of AC conductivity (σAC) in S m−1 of the pristine PSF and ZnO-loaded nanocomposites at mass% of (i) 3%, (ii) 5%, and (iii) 10% at the specific frequencies of 20 kHz, 50 kHz, and 95 kHz derived at two different temperatures of 30 °C and 180 °C.

It can be observed from Fig. 3.20 that the σAC value is 3.61 × 10–7 S m−1 for pristine PSF, while it becomes 5.55 × 10–7 S m−1 for 10 mass% ZnO-loaded PSF at f = 20 kHz and T = 30 °C showing an increment of 53.74%. At T = 180 °C, σAC value was 1.56 × 10–5 S m−1 for pristine PSF which became 2.03 × 10–5 S m−1 for 10 mass % ZnO-loaded PSF at the same f = 20 kHz showing an increment of ~30%. It can also be observed easily that σAC value increased by an order of 100 as reflected by determined σAC values on switching T = 30 °C to T = 180 °C. This is close to that of a semiconducting type of behavior with ZnO itself being a wide-bandgap material. An increment in σAC values at T = 180 °C indicates that more free charge carriers become available at higher temperatures due to enhancement in kinetic energy (K.E.) because of an increased supply of thermal energy. Another reason is the reduced interparticle distance and increased probability of much-enhanced contacts developing higher probabilities of charge carrier transfer. This mechanism happens at different nanofiller concentrations to develop overlapped interfacial regions inside the nanocomposites. Dispersion of nanofillers in the polymer matrix is an important factor in the crossover of interfacial regions in the nanocomposites that leads to the formation of percolation of charge carriers in weakly conducting chains.
3.3 Conclusion
Nanodielectric materials offer countless opportunities for high-end applications based on nanoelectronics, nanosensors, and high-voltage-based electrical transmission devices as well as improvement in the performance of ceramic-based applications. Dispersion of nanofillers (spherically shaped nanoparticles, nanowires, nanotubes, etc.) in a matrix, usually a polymer, produces a nanocomposite material with much-enhanced properties. An interfacial region is formed at the boundary of the matrix and particle surface that possesses modified properties in comparison with both matrix and nanofiller. The design of nanodielectrics gives high consideration to properties of interphase in addition to that of nanofillers and matrix since interphase fraction is the substantial fraction in composite material. A major problem is that the majority of the peer-reviewed research reports do not include quantification of nanofiller dispersions. Further, concerning industrial applications, advanced research on optimized concentration of nanofillers in a matrix is required along with prolonged usage of derived nanocomposite materials in several cycles. In addition, the huge volume of consistent and reliable datasets is needed that can be applied to prepare models in a rather empirical approach along with providing due consideration to breakdown as well as recovered state after prolonged use in several cycles, a multiscale model to include parameters related to nanofiller type and functionalization, matrix, along with conditions of processing, and, last but not the least, involved costing as it has straight industrial consequences.
References
Frèchette MF (2013) What are nanodielectrics ? IEEE ElectrInsul Mag 29:8–11
IEEE (2010) Nanodielectrics: a panacea solving all electrical insulation problems? 10th IEEE Intl Conf Solid Dielectr 1–29
FrèchetteM F, Reed CW, Sedding H (2006) Progress, understanding and challenges in the field of nanodielectrics. IEEJ Trans Fund Mater 126:1031–1043
Stevens GC, Vaughan AS (2013) Nanodielectrics and their role in power transmission applications. In: Melhem Z (ed) Electricity transmission, distribution and storage systems, pp 206–241
Cao Y, Irwin PC, Younsi K (2004) The future of nanodielectrics in the electrical power industry. IEEE Trans Dielectr Electr Insul 11:797–807
ZhongS-L D-M, ZhouW-Y C-W (2018) Past and future on nanodielectrics. IET Nanodielectr 1:41–47
Bhugra VS (2020) Advanced magnetic and dielectric nanomaterials, Ph.D. Thesis, Victoria University of Wellington, pp 1–214
Marks T, Ye P, Facchetti A, Lu G, Lin H (2007) High performance field effect transistors with self-assembled nanodielectrics. US Patent (US2007/0284629 A1), pp 1–10
Stallings K, Smith J, Chen Y, Zeng L, Wang B, di Carlo G, Bedzyk MJ, Facchetti A, Marks TJ (2021) Self-assembled nanodielectrics for solution-processed top-gated amorphous IGZO thin film transistors. ACS Appl Mater Interf 13:15399–15408
Lee S, Han H, Kim C-H (2020) Nanodielectrics approaches to low-voltage organic transistors and circuits. Eur Phys J Appl Phys 91:20201(1–8)
Tuncer E, Sauers I (2010) Industrial applications perspective of nanodielectrics. In: Nelson J (ed) Dielectric polymer nanocomposites. Springer, Boston, MA
Okubo H, Nakamura Y, Inano H, Hayakawa N, Hiroshima S, Hirose T, Hamaguchi M (2007) Lifetime characteristics of nanocomposite enameled wire under surge voltage application. Ann Rep Conf Electr Insul Dielect Phenom 1–4
Psarras GC (2018) Fundamentals of dielectric theories. In: Dang Z-M (ed) Dielectric polymer materials for high-density energy storage. Elsevier
Jiang S, Jin L, Hou H, Zhang L (2019) Polymer-based nanocomposites with high dielectric permittivity. In: Polymer-based multifunctional nanocomposites and their applications, pp 201–243
Singh M, Apata IE, Samant S, Wu W, Tawade BV, Pradhan N, Raghavan D, Karim A (2021) Nanoscale strategies to enhance the energy storage capacity of polymer dielectric capacitors: review of recent advances. Poly Rev 62:211–260
Luo H, Wang F, Guo R, Zhang D, He G, Chen S, Wang Q (2022) Progress on polymer dielectrics for electrostatic capacitors application. Adv Sci 9:2202438(1–25)
Balaraman AA, Dutta S (2022) Inorganic dielectric materials for energy storage applications: a review. J Phys D: Appl Phys 55:183002(1–38)
Hassan YA, Hu H (2020) Current status of polymer nanocomposite dielectrics for high-temperature applications. Compos Part A: Appl Sci Manufact 138:106064(1–24)
Pandey JC, Singh M (2021) Dielectric polymer nanocomposites: past advances and future prospects in electrical insulation perspective. SPE Poly 2:236–256
Yuan J, Yao S, Poulin P (2016) Dielectric constant of polymer composites and the routes to high-k or low-k nanocomposite materials. In: Huang X, Zhi C (eds) Polymer nanocomposites. Springer, Cham
Reddy PL, Deshmukh K, Chidambaram K, Nazeer Ali MM, Sadasivuni KK, Kumar YR, Lakshmipathy R, Khadheer Pasha SK (2019) Dielectric properties of polyvinyl alcohol (PVA) nanocomposites filled with green synthesized zinc sulphide (ZnS) nanoparticles. J Mater Sci: Mater Elect 30:4676–4687
Roy AS, Gupta S, Sindhu S, ParveenA RPC (2013) Dielectric properties of novel PVA/ZnO hybrid nanocomposite films. Comp Part B: Engg 47:314–319
Li Y, Bi X, Wang S, Zhan Y, Liu H-Y, Mai Y-W, Liao C, Lu Z, Liao Y (2020) Core-shell structured polyethylene glycol functionalized graphene for energy-storage polymer dielectrics: combined mechanical and dielectric performances. Comp Sci Technol 199:108341(1–9)
Mao H, You Y, Tong L (2018) Dielectric properties of di-block copolymers containing a poly-arylene ether nitrile block and a poly-arylene ether ketone block. J Mater Sci: Mater Electr 29:3127–3134
Samant SP, Grabowski CA, Kisslinger K, Yager KG, Yuan G, Satija SK, Durstock MF, Raghavan D, Karim A (2016) Directed self-assembly of block copolymers for high breakdown strength polymer film capacitors. ACS Appl Mater Inter 8:7966–7976
Tanaka T, Imai T (2013) Advances in nanodielectric materials over the past 50 years. IEEE ElectrInsul Mag 29:10–23
Andritsch TM (2010) Epoxy based nanodielectrics for high voltage DC applications—synthesis, dielectric properties and space charge dynamics, Ph.D. Thesis, Delft University of Technology
International Science Nanodielectrics (2010) International conference on solid dielectrics, Postdam Germany, pp 1–29
Raetzke S, Kindersberger J (2006) The effect of interphase structures in nanodielectrics. IEEJ Trans Fund Mater 126:1044–1049
Alcoutlabi M, McKenna GB (2005) Effects of confinement on material behaviour at the nanometre size scale. J Phys: Condens Matt 17:R461–R524
Bendler JT, Fontanella JJ, Shlesinger MF, Wintersgill MC (2009) The defect diffusion model and the glass transition in nanoscale and bulk films. J Comput Theor Nanosci 6:1–5
Lewis TJ (2004) Interfaces are the dominant feature of dielectrics at the nanometric level. IEEE Trans Dielect Electr Insul 11:739–753
Lewis TJ (2005) Interfaces: nanometric dielectrics. J Phys D Appl Phys 38:202–212
Tanaka T (2005) Dielectric nanocomposites with insulating properties. IEEE Trans Dielect Electr Insul 12:914–928
Ehrenstein GW (1999) Polymer-Werkstoffe, Carl Hanser Verlag 2 Auflage
Rane AV, Kanny K, Abitha VK, Patil SS, Thomas S (2017) Clay-polymer composites: design of clay polymer nanocomposite by mixing. In: Jlassi K, Chehimi MM, Thomas S (eds) Clay polymer nanocomposites, pp 113–144
Ren L, Yang L, Zhang S, Li H, Zhou Y, Ai D, Xie Z, Zhao X, Peng Z, Liao R, Wang Q (2021) Largely enhanced dielectric properties of polymer composites with HfO2 nanoparticles for high-temperature film capacitors. Comp Sci Technol 201:108528(1–32)
Kolthoff IM (1932) Theory of coprecipitation: the formation and properties of crystalline precipitates. J Phys Chem 36:860–881
Vadivel M, Ramesh Babu R, Sethuraman K, Ramamurthi K, Arivanandhan M (2014) Synthesis, structural, dielectric, magnetic and optical properties of Cr substituted CoFe2O4 nanoparticles by coprecipitation method. J MagnMagn Mater 362:122–129
Krishna PGA, Tharayil NJ (2019) Dielectric properties of lanthanum oxide nanoparticle synthesized using chemical coprecipitation method. AIP Conf Proc 2162:020079(1–9)
Joshi S, Kumar M, Chhoker S, Srivastava G, Jewariya M, Singh VN (2014) Structural, magnetic, dielectric and optical properties of nickel ferrite nanoparticles synthesized by coprecipitation method. J Mol Struct 1076:55–62
Yang G, Park S-J (2019) Conventional and microwave hydrothermal synthesis and application of functional materials: a review. Mater 12:1177(1–18)
Köseoğlu Y, Bay M, Tan M, Baykal A, Sözeri H, Topkaya R, Akdoğan N (2011) Magnetic and dielectric properties of Mn0.2Ni0.8Fe2O4 nanoparticles synthesized by PEG-assisted hydrothermal method. J Nanopart Res 13:2235–2244
Farea AMM, Kumar S, Batoo KM, Yousef A, Lee CG, Alimuddin (2008) Structure and electrical properties of Co0.5CdxFe2.5-xO4 ferrites 464:361–369
Jiang B, Iocozzia J, Zhao L, Zhang H, Harn Y-W, Chen Y, Lin Z (2019) Barium titanate at the nanoscale: controlled synthesis and dielectric and ferroelectric properties. Chem Soc Revs 48:1194–1228
Phromviyo N, Chanlek N, Thongbai P, Maensiri S (2018) Enhanced dielectric permittivity with retaining low loss in poly(vinylidene fluoride) by incorporating with Ag nanoparticles synthesized via hydrothermal method. Appl Surf Sci 446:59–65
Kumar A, Yadav N, Bhatt M, Mishra NK, Chaudhary P, Singh R (2015) Sol–gel derived nanomaterials and it’s applications: a review. Res J Chem Sci 5:1–8
Yadav RS, Kuřitka I, Vilcakova J, Havlica J, Masilko J, Kalina L, Tkacz J, Švec J, Enev V, Hajdùchovà M (2017) Impact of grain size and structural changes on magnetic, dielectric, electrical, impedance and modulus spectroscopic characteristics of CoFe2O4 nanoparticles synthesized by honey mediated sol-gel combustion method. Adv Nat Sci: Nanosci. Nanotechnol 8:045002(1–14)
Vasudevan P, Thomas S, Arunkumar KV, Karthika S, Unnikrishnan NV (2015) Synthesis and dielectric studies of poly(vinyl pyrrolidone)/titanium dioxide nanocomposites. IOP Conf Ser: Mater Sci Engg 73:012015
Singh PK, Gaur MS, Chauhan RS (2013) Dielectric properties of sol–gel synthesized polysulfone-ZnO nanocomposites. J Therm Analy Calor 111:743–751
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2024 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Dwivedi, S. (2024). Synthesis, Microstructural and Dielectric Characterization of Nanodielectrics. In: Moharana, S., Gregory, D.H., Mahaling, R.N. (eds) Emerging Nanodielectric Materials for Energy Storage. Nanostructure Science and Technology. Springer, Cham. https://doi.org/10.1007/978-3-031-40938-7_3
Download citation
DOI: https://doi.org/10.1007/978-3-031-40938-7_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-40937-0
Online ISBN: 978-3-031-40938-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)