
Abstract
The immune system functions to protect the host from pathogens. To counter host defense mechanisms, pathogens have developed unique strategies to evade detection or restrict host immune responses. Programmed cell death is a major contributor to the multiple host responses that help to eliminate infected cells for obligate intracellular pathogens like viruses. Initiation of programmed cell death pathways during the early stages of viral infections is critical for organismal survival as it restricts the virus from replicating and serves to drive antiviral inflammation immune recruitment through the release of damage-associated molecular patterns (DAMPs) from the dying cell. Necroptosis has been implicated as a critical programmed cell death pathway in a diverse set of diseases and pathological conditions including acute viral infections. This cell death pathway occurs when certain host sensors are triggered leading to the downstream induction of mixed-lineage kinase domain-like protein (MLKL). MLKL induction leads to cytoplasmic membrane disruption and subsequent cellular destruction with the release of DAMPs. As the role of this cell death pathway in human disease becomes apparent, methods identifying necroptosis patterns and outcomes will need to be further developed. Here, we discuss advances in our understanding of how viruses counteract necroptosis, methods to quantify the pathway, its effects on viral pathogenesis, and its impact on cellular signaling.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Programmed cell death
- Necroptosis
- Poxviruses
- Herpesviruses
- Influenza viruses
- E3L
- Z-nucleic acid binding protein
- vIRA
Introduction
Viruses are obligate intracellular parasites that depend on host cell machinery for replication. As viruses evolve and increase pathogenicity, infected hosts counter these changes by developing immune strategies that protect the host. Mammalian host immune systems, in particular, have evolved defense mechanisms that increase resistance to infection as well as strategies to recognize viruses and target infected cells for elimination. In a perpetual arms race, the viral pathogens respond to these host defense mechanisms by developing gene products that antagonize or allow the virus to evade detection via the immune system. While the strategies used to subvert innate immunity are recognized as crucial to ensuring the propagation and transmission of infections, our full understanding of their role in disease pathogenesis remains limited. Part of what researchers have been able to discover is that many viruses manage to overcome the full impact of a host antiviral state, involving the interferon (IFN) family and other inflammatory cytokines, by counteracting pathogen pattern recognition receptors (PRRs) and downstream signaling cascades (Pestka et al. 2004; Amarante-Mendes et al. 2018). Without employing these pathogenic strategies, elements produced from viral infections typically are recognized by PRRs which trigger an inflammatory response causing the upregulation of inflammatory cytokines along with programmed cell death pathways. Programmed cell death of infected cells has long been recognized as a highly effective mechanism to restrict pathogens by eliminating host machinery needed for viral replication and subsequent spread. The critical process has been exemplified by the abundance of virus-encoded inhibitors that have been found to suppress programmed cell death pathways(Mocarski et al. 2015; Jorgensen et al. 2017; Koehler and Jacobs 2021; Guo et al. 2022a). Here we discuss a specific type of programmed cell death known as necroptosis.
Necroptosis is part of a subgroup of programmed cell death known as necrosis. Necrosis was previously believed to be a dysregulated or accidental type of cell death that results from a toxic insult or physical stressors. Now it has become recognized to include a multitude of distinct regulated pathways with similar morphologies including necroptosis, pyroptosis, ferroptosis, pyronecrosis, parthanatos, oxytosis, and NETosis. Of these morphologies, necroptosis is characterized by a gain in cell volume, swelling of organelles, plasma membrane rupture, and subsequent loss of intracellular contents (Berghe et al. 2014; Green and Llambi 2015; Cotsmire et al. 2021). Unlike apoptosis and pyroptosis, it is independent of caspases (Vercammen et al. 1998; Holler et al. 2000) and generated from death receptor activation (TNF superfamily) due to ligand sensing by PPRs along with innate sensors. Necroptosis has also been implicated to contribute to numerous non-infectious pathological medical conditions including Alzheimer’s disease, Parkinson’s disease, age-related macular degeneration, skin autoimmunity/inflammation, tumor invasion to chemotherapeutics during colorectal cancer treatment, and many more (Bonnet et al. 2011; Moriwaki et al. 2015; Hanus et al. 2015; Bozec et al. 2016; Zhang et al. 2017). Non-infectious diseases and their relationship to necroptosis have been reviewed at length previously in “Necroptosis in the Pathophysiology of Disease” (Khoury et al. 2020). Since the pathway is also so critical for the early control of both DNA and RNA viruses, this work will focus on necroptosis as it relates to viral pathogens. We will explore the viruses that induce necroptosis, the strategies viruses employ to disrupt it, and the ways this death pathway has been studied.
Necroptosis Pathway and Players
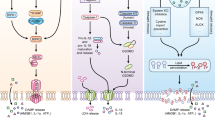
Programmed cell death is a genetically regulated process leading to the death of cells that was synonymous with apoptosis for some time. However, over the past several years, an array of alternative programmed cell death pathways have been identified and established as important regulators of the innate immune response to viral infections. Through these discoveries, the field of virus-induced, programmed cellular death rapidly evolved along with apoptosis, necroptosis, and pyroptosis, representing key pathways involved in the innate response to intracellular pathogens. Out of these pathways, necroptosis is characterized as an inflammatory form of programmed cell death believed to function as an alternative to extrinsic apoptosis, observed when caspases are inhibited (Vercammen et al. 1998; Li and Beg 2000; Holler et al. 2000; Mocarski et al. 2011). Activation of necroptosis leads to a signal cascade dependent on the receptor-interacting serine/threonine protein kinase (RIPK)3 and the downstream pseudokinase executer, MLKL (Sun et al. 2002; Murphy et al. 2013; Rodriguez et al. 2016) (Fig. 2.1). Because necroptosis is a caspase-independent programmed cell death, it is thought to play a critical role in the host defense mechanism against viruses that encode anti-apoptotic genes (Mocarski et al. 2011, 2015; Koehler and Jacobs 2021).

Necroptosis pathway and the three adaptors. Necroptosis is a form of programmed cell death that is initiated in response to various stimuli, such as viral infection. The signaling pathway of necroptosis involves three major adaptor proteins, namely receptor-interacting protein kinase 1 (RIPK1), TIR domain-containing adapter-inducing interferon-β (TRIF), and Z-DNA binding protein 1 (ZBP1). RIPK1: Various stimuli such as tumor necrosis factor (TNF) binding to TNF receptor 1 (TNFR1) activate RIPK1. RIPK1 has a death domain that enables it to interact with other death domain-containing proteins. When caspase-8 is inhibited, RIPK1 undergoes autophosphorylation and recruits other proteins to form a complex called the necrosome. TRIF: The second adaptor protein involved in the necroptosis pathway is TRIF. TRIF is a signaling protein that plays a role in the immune response to viral infection through the activation of TLR3. When TRIF is activated by a viral infection, it recruits RIPK1 to form a complex that leads to necroptosis. ZBP1: The third adaptor protein involved in the necroptosis pathway is ZBP1. ZBP1 is a cytoplasmic Z-RNA sensor. When ZBP1 recognizes Z-RNA, it undergoes a conformational change and recruits RIPK3 to form a complex that leads to necroptosis. Activation of any of these three adaptors results in the recruitment and activation of RIPK3. Activated RIPK3 then phosphorylates and activates MLKL, which translocates to the plasma membrane, where it oligomerizes and forms pores, leading to necroptotic cell death
Much of our current understanding of the necroptosis pathway arises from studies investigating TNF-α signaling in the presence of a caspase inhibitor (de Almagro et al. 2017). Through these studies, it is now known that binding of a TNF-α homotrimer with TNF-R1 promotes the formation of a membrane-associated signaling complex termed complex I through scaffolding of TRADD, RIPK1, TRAF2, and cIAP1/cIAP2 (Li et al. 2012; Chen et al. 2019). Formation of complex I leads to two distinct outcomes depending on cosigning and other cellular factors. In cellular conditions that promote the polyubiquitination of RIPK1 via cIAP1/cIAP2 and LUBAC, complex I drives pro-survival signaling via NFκB and MAP kinase (O’Donnell et al. 2007; Declercq et al. 2009; Dikic et al. 2009). In contrast, when RIPK1 undergoes deubiquitination, complex I dissociates from the membrane and becomes cytoplasmic in order to recruit FADD to form complex II which favors death signaling (Li et al. 2012). by inducing the dimerization and activation of caspase 8 causing apoptosis. This activation of caspase 8 not only initiates extrinsic apoptosis but also precludes necroptosis since it cleaves essential necroptotic mediators including RIPK1, RIPK3, and CYLD (Oberst et al. 2011). However, in situations where caspase activity is restricted, necroptosis proteins are not inactivated by caspase 8, but instead, RIPK1 recruits RIPK3 to complex II, thereby forming the necrosome. This can occur with viral caspase inhibitors like serpin or chemical inhibitors such as zVAD-fmk. This interaction of RIPK1 with RIPK3 drives large amyloid-like complexes via RIP homotypic interaction motifs (RHIMs) (Li et al. 2012). The combination of blocking caspase 8 activity and the formation of the necrosome by RIPK1 recruitment of RIPK3 is the molecular switch that turns cell death from apoptosis to necroptosis. Necrosome formation then results in the recruitment of MLKL via its interaction with RIPK3 which causes MLKL to be phosphorylated to its active state by the kinase activity of RIPK3 (Wang et al. 2014a; Rodriguez et al. 2016). Activation of MLKL leads to its oligomerization and localization to the plasma membrane causing a disruption of the membrane (Murphy et al. 2013; Dondelinger et al. 2014; Dovey et al. 2018; Samson et al. 2020). While the precise mechanism of this disruption is not fully understood, current evidence suggests that it may involve one of two mechanisms. These mechanisms involve either the activation of cation channels causing an alteration of cellular osmotic pressures leading to a membrane disruption or MLKL pore formation causing a direct breakdown of the membrane. Regardless of the mechanism, subsequent rupturing of the membrane then results in the release of intracellular DAMPs that drive local inflammatory processes.
While the cytokine-driven pathway in which RIPK1 activates RIPK3 has been fundamental to defining the general necroptosis pathway (Sun et al. 2002, 2012; Polykratis et al. 2014; Moriwaki et al. 2015; de Almagro et al. 2017), two other proteins are known to activate RIPK3 through RHIM domain interactions to cause necroptotic cell death. TICAM1, also known as TRIF, is one of these proteins that activate RIPK3 after undergoing interactions with TLR3 or TLR4 (Kaiser et al. 2008, 2013; He et al. 2011). These toll-like receptors initiate the process after being activated through receptor–ligand interactions. TLR3 is activated by dsRNA known to be present in many viral replication processes, while TLR4 recognizes LPS which is a common gram-negative bacterial virulence factor. The other RHIM domain-containing protein that can activate RIPK3 is ZBP1, formerly called DAI and DLM1. ZBP1 triggers necroptotic cell death after sensing Z-form nucleic acids (Z-NA) released during viral infections (Zhang et al. 2020; Koehler et al. 2021).
Overall, RIPK3 can be activated to initiate necroptotic cell death through RHIM domain interactions with RIPK1, TRIF, or ZBP1. Regardless of the trigger, activation of RIPK3 and subsequent MLKL phosphorylation lead to a rupturing of the plasma membrane and release of pro-inflammatory DAMPs which drives a local inflammatory response.
Necroptosis Inhibitors
Many necroptotic inhibitors have been developed over the past several years that target different components of the cell death pathway including RIPK1, RIPK3, and MLKL (Fig. 2.2). Notably, RIPK1 inhibitors include necrostatin (Nec)-1 and GSK2982772. Nec-1 is a tryptophan-based inhibitor that works by stabilizing RIPK1 into its inactive form (Degterev et al. 2013). RIPK1 inhibitors: the most studied and well-characterized RIPK1 inhibitors are kinase inhibitors that bind to the ATP-binding pocket of the kinase domain and block its activity. GSK’963, for example, has been shown to inhibit RIPK1 kinase activity and prevent the formation of the necrosome complex, which is composed of RIPK1, RIPK3, and MLKL (Berger et al. 2015). Other RIPK1 inhibitors, such as Nec-1s and Nec-1h, have been shown to stabilize RIPK1 in a non-phosphorylated state, thereby preventing its recruitment to the necrosome (Degterev et al. 2013; Cao and Mu 2021). A derivative of Nec-1, Nec-1s, has also been developed and shows increased specificity for RIPK1 binding along with better stabilization of RIPK1 into its inactive conformation compared to Nec-1. While both of these compounds have been utilized to study a multitude of disease processes, poor pharmacokinetic properties have made their potential limited.

The chemical inhibitors of necroptosis and their targets. The necroptosis signaling pathway involves a series of protein–protein interactions and post-translational modifications that ultimately lead to the activation of the executioner protein, MLKL, and subsequent cell death. The signaling pathway of necroptosis involves three major adaptor proteins, namely RIPK1, TRIF, and ZBP1. Chemical inhibitors can target specific upstream adaptors of RIPK3, RIPK3, or the downstream executer MLKL. This figure summarizes the chemical inhibitors and their specific targets
RIPK3 inhibitors include dabrafenib and other small-molecule inhibitors (Martens et al. 2020). Dabrafenib has been traditionally used in the treatment of melanoma where it acts as a B-RAF inhibitor (Rheault et al. 2013). Recently though, it has also been found to compete for an ATP-binding site on RIPK3, thereby inhibiting RIPK3’s kinase capability (Li et al. 2014). This compound has been implemented in studies using human hepatocytes where it has shown promise in reducing acetaminophen-induced necrosis. GSK’840, GSK’843, and GSK’872 are also effective RIPK3 inhibitors but have been found to cause a dose-dependent induction of apoptosis when administered alone which has made their use limited (Mandal et al. 2014). Compounds that inhibit both RIPK1 and RIPK3 have been studied as well. The FDA-approved chemotherapeutic drugs pazopanib and ponatinib inhibit both of these components of the necroptotic pathway but are unlikely to have broad implications due to their associated cardiotoxicity. GSK’074 is another dual inhibitor recently developed that has shown promise in inhibiting necroptosis while potentially being less cardiotoxic based on early mouse models (Zhou et al. 2019; Chen et al. 2022).
MLKL is a critical downstream effector of necroptosis, a programmed form of cell death that is implicated in various pathological conditions (Murphy 2020). MLKL oligomerizes and translocates to the plasma membrane, where it forms pores that disrupt cellular membranes and lead to cell death. Chemical inhibitors of MLKL have been developed as potential therapies for necroptosis-related diseases. One such inhibitor necrosulfonamide has been shown to bind to a different site on MLKL and block its translocation to the plasma membrane, preventing the execution of necroptosis (Dondelinger et al. 2014; Su et al. 2014). These MLKL inhibitors have demonstrated efficacy in various disease models, including myocardial infarction, ischemic stroke, and acute pancreatitis, suggesting their potential as novel therapies for necroptosis-related disorders (Dondelinger et al. 2014; Zhang et al. 2017, 2022). However, further research is needed to optimize their efficacy and safety in humans. Another MLKL inhibitor that has shown promise as a potential therapeutic agent is NSA (necrosulfonamide-α), which is a derivative of necrosulfonamide. NSA binds specifically to the MLKL protein and inhibits its oligomerization and translocation to the plasma membrane, thereby preventing necroptosis. In addition to its potent anti-necroptotic activity, NSA has also been shown to have anti-inflammatory effects in various disease models, including colitis and sepsis (Jiao et al. 2020). These findings suggest that NSA could be a promising therapeutic agent for necroptosis-related disorders with an added anti-inflammatory benefit. Further preclinical studies and clinical trials will be needed to assess the efficacy and safety of NSA in humans.
In summary, many necroptotic inhibitors have been developed that target a variety of critical components of the cell death pathway. The complexity of signaling involved in necroptosis has contributed to the pros and cons associated with each inhibitor choice currently available. Challenges include unwanted signaling overlap with the apoptotic death pathway, a lack of target specificity, and unwanted tissue toxicity. While these challenges do represent hurdles in working to develop necroptosis-based therapeutics, current progress suggests that continued research into this intricate cell death pathway could yield significant insights into treating a multitude of disease processes in the future.
In Vitro Necroptosis in Cultured Cells
Necroptotic death was very difficult to identify and study for several years since only a few specific cell lines actually had the necessary machinery to elicit necroptosis as a result of epigenetic regulation (Koo et al. 2015). Of these necroptotic-capable cells, the L929 and SVEC4-10 lines have been utilized the most due to ease of use and long-established protocols. HT29 cells are used frequently as well but need to have ZBP1 artificially expressed due to the limited presence of the protein in the line. Other necroptotic-capable lines include 3T3-SA, BMDM, HMEC-1, IMR-90, J2, MDM, MEF, U937, and THP-1 cells. Of these, the THP-1 line has been found to have variable necroptotic expression, based on differentiation status. Other lines that do not have the necessary necroptotic machinery have been adapted to have necroptotic capabilities through either transduction or ectopic expression of missing components. These lines include NIH3T3, HEK293T, HF, and HT29 cells. A full list of cell lines utilized to study necroptotic death can be found in Table 2.1. This is not an exclusive list as not all cells have been evaluated. However, prior to initiating studies, there should be a careful evaluation of a culture systems ability to undergo necroptosis and the suitability for the specific virus.
In Vivo Models of Necroptosis
Concerns over the complicated delivery of chemical necroptotic inhibitors with potential off-target effects have led to the development of a multitude of genetically modified in vivo models to study the cell death pathway. Many of these genetically modified models have worked to alter necroptotic steps involved in the RIPK1-RIPK3-MLKL and ZBP1-RIPK3-MLKL pathways. A list of necroptosis-deficient mice and the genetic targets utilized to study the role of necroptotic death in viral pathogenesis can be found in Table 2.2.
The RIPK1–/– mice generated have not been able to be implemented in studying necroptosis since they demonstrate perinatal lethality due to apoptosis and RIPK1-independent necroptosis (Berger et al. 2014; Polykratis et al. 2014). As such, RIPK1 knock-in models have been developed in which the native RIPK1 gene is replaced with a genetically modified version that alters the kinase capabilities of RIPK1 making it catalytically inactive. Ripk1K45A and Ripk1D138N/D138N are two of these catalytically inactive RIPK1 knock-in models that produce viable mice while inhibiting RIPK1-dependent necroptosis (Berger et al. 2014; Polykratis et al. 2014). This suggests that the kinase activity of RIPK1 is needed for a necroptotic response, but not necessary for embryonic development. A RIPK1 scaffold mechanism has been proposed which suggests that RIPK1 prevents ZBP1-induced necroptotic death. It is interesting to also note that RIPK1 knockout mice can be rescued when crossed with RIPK3/Caspase-8 double knockout mice or ZBP1–/– mice which suggests RIPK1’s dual role in both apoptotic and necroptotic pathways. Domain-specific ZBP1 mutants have been developed but have not been applied to viral research, but would likely give significant insights to advance our understanding of virus-induced necroptosis in vivo.
While these animal models are useful in studying RIPK1-induced necroptosis, they are not able to prevent all forms of necroptosis such as those involved in RIPK3 activation via TRIP or ZBP1. Consequently, catalytically inactive RIPK3 knock-in mice have also been developed (Newton et al. 2014). While these lines can prevent necroptotic death, they are subject to death by RIPK3-mediated apoptosis, evident in influenza A viral and HSV models (Nogusa et al. 2016b; Kuriakose et al. 2016; Guo et al. 2022b). This suggests that RIPK3 also has a dual role in both apoptotic and necroptotic pathways. The dual necroptotic and apoptotic roles both RIPK1 and RIPK3 play help to represent the complexity of these regulators involved with multiple immunologic responses. Due to this complexity, MLKL-knockout mice have also been produced and are currently the best option for studying the impact of necroptotic cell death in various diseases (Wu et al. 2013; Moerke et al. 2019; Tovey Crutchfield et al. 2021; Cao et al. 2022). This is evident in studies showing intact pro-inflammatory cytokine, NF-kB, and MAPK signaling post-LPS or TNF treatment in MLKL–/– mouse models immune to necroptotic death (Wu et al. 2013).
Other in vivo models have looked at TLR-induced necroptosis through TLR3–/– mice or Z-NA-induced necroptosis through ZBP1–/– mice. In TLR3–/– mice, TLR3 ligand sensing of dsRNA cannot occur to activate TRIF (Chen et al. 2021; Kaiser et al. 2013). Research utilizing this model has shown that TLR3–/– mice infected with various viruses show greater viral loads and disease progression compared to controls (Vercammen et al. 2008; Zhang et al. 2013; Perales-Linares and Navas-Martin 2013). This is interesting since no viral infection has been able to produce TLR3-dependent necroptosis in vitro with the pathway only being studied with synthetic RNAs designed to mimic viral nucleic acids (Kaiser et al. 2013). Future research will need to delve into whether or not certain viruses actually utilize this pathway to stimulate necroptotic death in vitro. ZBP1–/– mice on the other hand have been able to produce evidence of necroptotic cell death when infected with various viruses that correlates with in vitro studies (Thapa et al. 2016; Koehler et al. 2017; Upton et al. 2019). In these mice, ZBP1 cannot sense Z-NA released during viral infections to stimulate necroptotic death. Since many viruses have developed inhibitors to counteract necroptotic cell death, work utilizing this model will be interesting to follow as it could lead to the development of novel therapeutics designed to treat a variety of viral infections.
Cellular Patterns and Methods to Evaluate Necroptosis
Since cell death pathways are complex in nature and share a multitude of components with one another, distinctly identifying necroptotic processes from other cell death pathways has been challenging. Therefore, it comes as no surprise that necroptosis identification tends to require a multifaceted approach that assesses many different interactions within the pathway. Additionally, in the setting of viral infections, it is important to note that viruses have been found to inhibit necroptosis at different levels which makes it critical to assess which component a particular virus of interest may be targeted to inhibit the pathway. This is especially important in the development of therapeutics which require specific targets to be efficacious.
Metabolic Viability Assays
Metabolic viability assays are an important method to evaluate cell death that utilizes markers of mitochondrial activity to detect changes manifested in viable or nonviable cells. Here we describe three assays that can be used to measure metabolic changes in cells exposed to viral infections:
ATP Assay
Adenosine triphosphate (ATP) is an organic, energy-carrying molecule that is essential to fueling many cellular processes. Produced in the mitochondria by metabolically active cells, ATP can be assayed to determine the changes in cell viability with nonviable cells showing diminished ATP levels from a lack of production. While not specific to only necroptotic death, diminished ATP levels help us to broadly determine if some type of cellular demise is taking place for a given disease process. Most commonly this is done by using a bioluminescent assay that uses the two-part firefly luciferase reaction. Lysing cells to release intracellular ATP in the presence of a luciferin enzyme and substrates can effectively measure the concentrations of cellular ATP. The available ATP in the lysate is used to adenylate luciferin forming luciferyl-adenylate. In the second step of the reaction, oxidative decarboxylation of luciferyl-adenylate forms an excited oxyluciferin. When this oxyluciferin returns to the ground state, light is emitted which can be measured by a luminometer. The measurement of emitted light from this reaction can be correlated to the amount of ATP in the sample. In utilizing this technique, it is important to note that in cases of programmed cell death, ATP breakdown is delayed from death execution steps and may produce a false negative limiting the use as an immediate indicator of a necroptotic death. This makes using appropriate time points essential when using the technique to study necroptosis. Additionally, pairing the assay with small-molecule or genetic modification approaches can verify if the cell death events observed are in fact a result of necroptosis.
MTT Assay
The MTT assay is a very common colorimetric assay to measure cell viability or cytotoxicity in viral infections. Viable, metabolically active cells produce mitochondrial dehydrogenases that can reduce MTT, a yellow tetrazolium salt, to form purple formazan crystals. Cells that are no longer metabolically active or producing mitochondrial dehydrogenases are dead or dying and these cannot complete the reduction reaction to generate the purple formazan. The purple color correlates to the number of metabolically active cells and the intensity of the color can be spectrophotometrically measured: the darker the color, the greater the number of viable cells.
Resazurin Assay
Similar to the MTT assay, the resazurin assay is also an indicator dye assay that measures cell viability based on the reduction of blue resazurin by mitochondrial reductases to red fluorescent resorufin. Cells that are dead or dying will not have the necessary reductases to convert the blue resazurin to red resorufin. The measurement of the resorufin produced is proportional to the number of viable cells in the sample, the greater red fluorescence signal corresponds to an increase in the number of metabolically active cells, and, conversely, more blue fluorescence indicates more cell death, including cells undergoing necroptosis.
These cell viability assays are not specific indicators of necroptosis, but they are indicators of cell death that can be paired with other assays to determine if necroptosis is occurring. Additionally, these assays are not accurate indicators of the timing of cell death, and they do require careful control of cell number and an understanding that states or conditions that cause metabolic senescence should be avoided.
Membrane Integrity Assays
Cell membrane integrity assays are crucial to assessing virus-induced necroptotic cell death, the hallmark of which is cell swelling and rupture due to plasma membrane damage.
LDH Assay
The lactate dehydrogenase (LDH) assay uses LDH to measure a loss in cell membrane integrity. In viable or uninfected cells, the LDH enzyme is present only inside the cells, but infection or other cytotoxic insults can degrade the plasma membrane and allow the release of this enzyme into the media. In a damaged cell, LDH catalyzes the reduction of NAD+ to NADH and the oxidation of lactate to pyruvate. In the second step, the NADH is used to catalyze the reduction of an indicator dye, like luciferin or resazurin. The resulting amount of fluorescence or color measured is an indicator of membrane integrity loss. This assay is not a specific measure of necroptosis but should be used with other assays such as inhibitors or genetic knockouts to verify that an increase in LDH levels is the result of necroptosis.
Membrane Impermeant DNA Dyes
A simple method for quantifying cell death is to use DNA-binding fluorescent dyes that are impermeant to healthy cell membranes. These fluorescent nucleic acid-binding dyes can penetrate the compromised membranes of dead or dying cells and the fluorescence of the nucleic acid-bound dye can be measured using live cell imaging. This method gives a real-time measurement of membrane permeability to help establish the timing of signaling events in cells that are dying. This is an effective tool to use for cells undergoing necroptosis where the pathway results in pore formation and the dyes can access the DNA and can be utilized as a real-time measurement of loss of membrane integrity in live cell imaging to give kinetic insights into death (Upton et al. 2019). Many dye systems are available including SYTOX from Invitrogen and Incucyte Cytotox Dyes from Sartorius.
Necroptosis Morphology
Various imaging techniques have been implemented to assess the morphological characteristics of cells undergoing necroptosis. These techniques have been used to show evidence of typical characteristics of cells undergoing necroptotic death, including a gain in cell volume, swelling of organelles, and a rupturing of the plasma membrane (Newton et al. 2014; Cotsmire et al. 2021) (Fig. 2.3). Since this process of cellular swelling with subsequent membrane rupture generates fairly fragile specimens, techniques that rely on fixation like transmission microscopy and confocal imaging have been unsuccessful. However, techniques that work by freezing the specimen in place before fixation such as cryo-electron microscopy have shown moderate success (Daley-Bauer et al. 2017). Additionally, utilizing necroptotic inhibitors cells deficient in necroptotic machinery that halt cell death has allowed more traditional imaging techniques to observe activation of the necroptosis pathway for unaffected proteins at discrete time points. Ultimately though, given the quick onset and fragility of the necroptosis process, live cell imaging has proven to be the most successful in studying the pathway and provides critical kinetic information (Koehler et al. 2017, 2021; Upton et al. 2019). This technique allows a researcher to observe the necroptotic process in real time to differentiate morphological manifestations from those observed in other cellular death pathways.

Diagram of common morphology of a virus-infected cell undergoing necroptosis. During necroptosis, cells undergo a series of morphological changes that ultimately lead to cell death. At the early stage of necroptosis, cells undergo swelling and rounding, followed by the formation of blebs on the cell surface. The blebs are caused by the disintegration of the plasma membrane and the release of cytoplasmic contents. The cells also exhibit a loss of plasma membrane integrity, which leads to the leakage of intracellular contents into the extracellular space. As the process progresses, the cells become progressively more swollen and eventually burst open, releasing their contents into the extracellular environment. This final stage of necroptosis is characterized by the formation of large membrane pores, which allow the influx of water and ions into the cell and lead to osmotic lysis. Overall, the morphology of cells undergoing necroptosis is characterized by swelling, membrane blebbing, loss of membrane integrity, and osmotic lysis. These changes are distinct from other forms of programmed cell death, such as apoptosis, which is characterized by shrinkage, condensation of chromatin, and fragmentation of the nucleus
Immuno-techniques
Immuno-techniques utilized to study necroptosis rely on antibody–antigen interactions to isolate and characterize various components pertinent to the necroptotic pathway. These techniques include immunohistochemistry, immunoprecipitation, and western blots.
Immunohistochemistry
This technique utilizes chromogen-tagged antibodies that attach to antigens on critical necroptotic components. After allowing time for these antibodies to adhere to their respective antigens, a substrate is added that reacts with the tagged antibody complex to produce a color change. This color change correlates to the amount of a particular necroptotic component present. While this technique makes theoretical sense for studying the pathway, antibody cross-reactivity has made it difficult to assess if the changes observed are accurate. As such, the technique has not been utilized too much in present studies but has potential for future research endeavors as more specific antibodies are being developed.
Immunoprecipitation
These techniques work by precipitating out necroptotic components of interest using immobilized antibodies. Direct immunoprecipitation techniques involve antibodies immobilized on density beads that can directly bind to antigens of interest. Indirect immunoprecipitation techniques are performed by exposing a solution containing necroptotic components of interest to antibodies directed toward specific antigens on these components. After allowing time for these antibodies to bind to their antigens, beads designed to capture antibodies are added to the solution and bind to the antibody–antigen complexes. After necroptotic components of interest have been immobilized by either technique, the remaining solution is washed away. This allows these components to be isolated for further analysis with other techniques such as immunoblotting or proteomic analysis. This approach is useful in determining which necroptotic components are being affected by various disease processes and protein interactions that propagate necroptosis signaling. Careful consideration should be taken to minimize off-targeting effects of non-specific antibody interactions. The use and development of engendered cell lines that express epitope-tagged necroptotic proteins has drastically mitigated the off-targeting effects observed and facilitated key advances in our understanding of necroptosis signaling (Sridharan et al. 2017; Upton et al. 2019).
Western Blots
Western blots are common experiments used to study various cell signaling events including necroptosis protein activation states. These experiments work by separating different proteins of interest by size with denaturing gel electrophoresis. This allows for the identification and semi-quantification of the levels of proteins in a given sample. A multitude of antibodies have been developed that specifically target activated or phosphorylated necroptotic proteins including MLKL and RIPK3 which have been used to document necroptosis signaling in virus-infected cells. This helps to garner evidence that a particular component of interest is present and activated by controlling for both size and phosphorylation state.
The Viruses That Activate or Inhibit Necroptosis
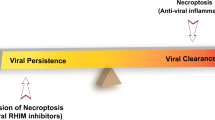
Much like apoptosis, necroptosis plays out as a cell-autonomous innate immune mechanism to kill virus-infected cells and cut short productive infection. This potent host defense mechanism limits viral replication and spread by eliminating infected cells (Koehler et al. 2017; Upton et al. 2019). The significance of necroptosis as a host defense mechanism has been reinforced by the identification of virus-encoded inhibitors that block specific steps in the pathway (Fig. 2.4). These viral inhibitors sustain infection allowing the virus to replicate and spread. A series of reports demonstrated the significance of necroptosis in restricting viral pathogenesis and how ZBP1 is a critical mediator of virus-induced necroptosis observed during innate immune sensing of viruses in both DNA and RNA viruses.

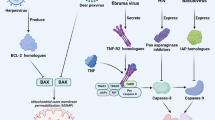
Viral inhibitors of necroptosis and their cellular targets. Crm (light purple) proteins are found in orthopoxviruses but are best described in the cowpox virus. These proteins are competitive inhibitors of TNF receptor signaling and prevent RIPK1-mediated necroptosis. ICP6(blue) is the HSV1 and HSV2 RHIM-containing inhibitor that targets host RHIM interactions of adaptors and RIPK3. M45(Red) are found in MCMV and are an RHIM-containing protein similar to ICP6 in HSV. M45 blocks necroptosis by preventing RHIM interactions of the adaptor proteins and RIPK3, thereby blocking all three pathways of necroptosis. vIR (green) is found is poxviruses and functions to enhance the turnover of RIPK3 preventing it from activating MLKL. vMLKL is a truncated MLKL homologue and functions to restrict necroptosis by acting as a decoy preventing host MLKL from getting phosphorylated. E3(dark purple) is a Z-RNA competitive inhibitor found in orthopoxviruses and prevents the activation of ZBP1. HCMV has an unidentified inhibitor of MLKL (burgundy)
DNA Viruses
Herpesviruses
Herpesviruses are a large family of double-stranded DNA viruses. The Herpesviridae family consists of more than 100 viruses, nine of which primarily infect humans where infection and replication take place in the epithelial cells and some of which can cause lifelong latent infections. Necroptosis studies utilizing herpes viruses have included investigations with murine cytomegalovirus (MCMV), human cytomegalovirus (HCMV), herpes simplex virus 1 (HSV1), and herpes simplex virus 2 (HSV2).
Murine Cytomegalovirus
MCMV is a species-specific double-stranded DNA virus that is a natural pathogen in wild mice populations. While MCMV infection triggers necroptotic cell death, MCMV has also evolved viral inhibitors to counteract the host defense mechanisms to ensure viral replication. Early evaluation of a mutant MCMV transposon library identified M45 as an important gene for endothelial and macrophage cell survival in vitro and viral replication (Brune et al. 2001; Lembo et al. 2004). Delving further to elucidate M45-encoded protein function, the sequence was analyzed and found to contain an N-terminal RHIM domain facilitating binding to RHIM-containing receptor-interacting kinases, RIPK1 and RIPK3 (Kaiser et al. 2008). To examine the impact of this protein on necroptosis, cells were infected with recombinant MCMV engineered to have a nonfunctional M45 RHIM domain by mutations of four amino acids in the RHIM sequence (Kaiser et al. 2008). Cells infected with the M45-deficient MCMV (M45MutRHIM) had decreased cell viability compared to wild-type M45. The M45-encoded viral inhibitor of RIP activation (vIRA) was found to impede the RIPK1 and RIPK3 complex from forming via RHIM domains. To evaluate this disruption, the roles of these receptor interaction kinases were individually examined. Treatment of cells with RIPK1-specific inhibitor Necrostatin-1 did not reveal any decrease in cell survival (Upton et al. 2010a), suggesting a RIPK1-independent mechanism. The independence from RIPK1-mediated necroptosis was confirmed in MEFs from mice deficient in RIPK1 where these mice were also insensitive to necroptosis induction (Upton et al. 2010a). vIRA inhibition of RIPK3-dependent necroptosis was confirmed through various knockout studies. M45mutRHIM-infected cells expressing reduced RIPK3 via shRNAs were not susceptible to necroptosis and had no reduced cell viability. Additionally, the mutant M45mutRHIM virus did not demonstrate viral replication in the wild-type animal; however, in the RIPK3 knockout, the wild-type and mutant virus had comparable levels of replication confirming the importance of RIPK3 for the inhibition of necroptosis via vIRA (Upton et al. 2010a). This was the first time that a RHIM domain-containing protein other than RIPK1 was found to be capable of stimulating necroptosis through RIPK3 (Nailwal and Chan 2019).
More recent studies identified that ZBP1 interacts with RIPK3 and that this complex is the target for the MCMV vIRA necroptosis inhibitor (Upton et al. 2012). Similar to experiments with RIPK3, M45mutRHIM infection of ZBP1-deficient cells and mice resulted in viral replication of the mutant virus (Upton et al. 2012). These data implicate the vIRA in preventing the ZBP1 RHIM from binding and activating RIP3, halting the ZBP1/RIPK3/MLKL necroptotic pathway and protecting the infected cell, allowing it to continue replication and pathogenesis (Upton et al. 2012). Further studies identified viral transport factor immediate early protein 3 (IE3) as a pivotal player in ZBP1-dependent necroptosis in MCMV infection. Recombinant viruses for wild-type MCMV and M45mutRHIM were generated that contained an IE3 protein fused to the C-terminus (Sridharan et al. 2017). Under normal conditions, the addition of the destabilizing domain triggers rapid proteosomal degradation. Chemical ligand Shield-1 stabilizes the destabilization domain and allows the transcriptional functions to persist. Results from these studies demonstrated that M45mutRHIM-infected cells would undergo ZBP1-mediated necroptosis in the presence of Shield-1 but would be resistant to necroptotic death in its absence. This showed that the early transcription of viral RNA via IE3 is required for necroptosis and is the ligand for ZBP1-mediated necroptosis. One last interesting MCMV study found that when MCMV’s viral inhibitor of caspase 8-induced apoptosis (vICA) is altered to be nonfunctional, cells show evidence of RIPK3, MLKL, and caspase 8 activation. This showed that apoptotic and necroptotic processes could occur simultaneously.
Human Cytomegalovirus
HCMV studies reveal significant species-specific differences in the human virus compared to the murine strain. HCMV functions to block necroptosis after the phosphorylation of RIPK3 and MLKL. This resistance to necroptotic death is dependent on viral transcriptional processes regulated by the viral protein IE1; however, necroptosis-resistant cells still show evidence of MLKL phosphorylation (Omoto et al. 2015). It has been shown that the HCMV UL36 protein is a key player in inhibiting necroptosis by interacting with MLKL and inducing its demise (Fletcher-Etherington et al. 2020). UL36, a dual cell death pathway inhibitor also able to inhibit apoptosis by binding caspase 8, is capable of inhibiting necroptosis in a species-specific manner and does not inhibit necroptosis in murine cells.
Herpes Simplex Viruses
Studies investigating herpes simplex viruses (HSV1 and HSV2) have also found that these viruses are able to inhibit necroptosis in virus-infected human cells (Guo et al. 2015, 2018). This mechanism of cell death inhibition is similar to that of MCMV, where vIRA blocks the necrosome formation by RHIM interaction of RIPK3 and prevents further execution of the necroptotic pathway through RIPK3 and MLKL (Upton et al. 2010a, b, 2012). In HSV1 infection of human cells, ICP6 acts as an inhibitor that contains an N-terminal RHIM domain allowing it to competitively bind and sequester RIPK3 from the other interacting proteins, blocking necrosome formation and inhibiting the activation of RIPK3/MLKL-dependent necroptosis. Strangely enough, ICP6 has been found to induce necroptotic death in HSV1-infected murine cells through interactions of the RHIM domains on ICP6 and RIPK3 (Wang et al. 2014b). Later studies revealed HSV1 infection triggers necroptosis through the ZBP1/RIPK3-dependent pathway. Evaluation of ZBP1-deficient MEFs and mouse cell lines found the ZBP1-deficient cells resistant to necroptosis, confirming the importance of ZBP1 in HSV1 infection in mouse cells (Guo et al. 2018). Evaluation of MLKL phosphorylation during WT and ICP6 RHIM mutant virus infection revealed that the ICP6 mutant virus promotes the formation of a necrosome-like complex involving ZBP1/RIPK3/MLKL to promote MLKL phosphorylation and necroptosis in mouse cells (Guo et al. 2018). To analyze if ZBP1 also contributes to the initiation of necroptosis in human cells, ZBP1 expressing HT29 and ZBP1 deficient cells were infected with HSV1 ICP6 RHIM mutant virus. As in the mouse cells, the ZBP1-expressing human cells showed an induction of cell death, but the ZBP1-deficient cells did not. Using various inhibitors of necroptosis pathway players, RIPK3 and MLKL and caspase inhibitor zVAD, it was determined that the HSV ICP6 RHIM mutant virus-induced cell death through ZBP1/RIPK3/MLKL-mediated necroptosis. WT HSV1 infection did not induce cell death in the human cells, showing that the ICP6 RHIM was required for HSV1 necroptosis inhibition in its natural host species. These studies highlight species-specific differences in how viral inhibitors act on cells, suggesting a viral restriction in non-natural host infection. In addition to necroptotic inhibition in human cells, ICP6 has demonstrated an ability to bind to and inhibit caspase 8 (Langelier et al. 2002; Esaki et al. 2010; Dufour et al. 2011). This means that the viral protein ICP6 can inhibit both necroptosis and apoptosis in human cells.
Poxviruses
Poxviruses are a double-stranded DNA family of viruses. Most notable among these viruses may be the Orthopoxvirus genus due to the infamy of the variola virus (smallpox virus). The Orthopoxvirus genus includes vaccinia virus (VACV), cowpox virus (CPXV), Mpox (formerly monkeypox), and ectromelia (ECTV). Like other dsDNA viruses, this family encodes numerous accessory virulence factors that inhibit cell death.
Vaccinia virus (VACV) is the prototypic model for orthopoxvirus research and the poxvirus strain utilized in smallpox vaccinations. Early VACV studies identified a novel necrotic cell death pathway that eliminated virus infected cells. Researchers found that VACV encodes a caspase-8 inhibitor known as CrmA-like ortholog B13R (aka Spi2), which blocks caspase-dependent cell death pathways and sensitizes infected cells for this necrotic death pathway through TNF signaling (Li and Beg 2000). TNF signaling in VACV infection was further clarified using VACV-infected cells that are normally sensitive to necroptosis. RIPK1-deficient cells showed a reduction in necrosis, confirming RIPK1 as an essential player in the pathway. Further, TNFR-2 was also shown to be essential for this pathway in an in vivo infection by creating TNFR-2-deficient mice and observing reduced inflammation in the liver (Chan et al. 2003). RNA interference screens identified RIPK3 as another pivotal player in the necroptosis pathway. Wild-type and RIP3K-deficient cells were infected with VACV showing the deficient cells resistant to TNF-α-induced necroptosis. As with the RIPK1-deficient mice, RIPK3-deficient mice infected with VACV showed much less necrosis and liver inflammation than infected wild-type mice (Cho et al. 2009), showing that RIPK3 is part of the host defense mechanism protecting against VACV. Together, during VACV infection, these two protein kinases form a RIPK1–RIPK3 necrosome complex through RHIM interactions, RIPK1 thereby activating RIPK3 to phosphorylate and subsequently activate MLKL to execute necroptosis (Sun et al. 2002, 2012; Rodriguez et al. 2016). Later studies would look at another RHIM-containing stimulator of necroptosis known as ZBP1. In addition to two RHIM domains (RHIM-A and RHIM-B), this protein contains two Z-NA (Zα) binding domains located at the N-terminus. These Z-NA binding domains can be triggered by Z-NA released during VACV infections. Once triggered, type I IFN-signaling increases along with further ZBP1 expression. Utilizing RHIM domain interactions, ZBP1 can then instigate necroptotic cell death via RIPK3/MLKL activation. This necroptotic pathway is not observed in wild-type VACV infections, however, since the virus also encodes a protein with a Zα-binding domain known as E3 which serves to inhibit necroptosis (Koehler et al. 2017).
Studies examining how VACV utilizes E3 to prevent necroptotic death have produced some interesting findings. In vivo studies have shown that mice infected with a mutant VACV that encodes a truncated E3L lacking a Zα-binding domain did not exhibit severe pathology comparable to mice infected with the wt virus. However, pathogenicity was restored in both ZBP1–/– and RIPK3–/– mice. Additionally, follow-up in vitro studies demonstrated that knocking out the Zα-binding domains of ZBP1 was sufficient for increasing viral pathogenesis. The culmination of these and other studies provided evidence that VACV’s E3 is a competitive inhibitor of ZBP1/RIPK3/MLKL-induced necroptosis (Koehler et al. 2021). This provides another example of how viruses have co-evolved with us to evade our innate immune system.
Another virus that utilizes necroptotic inhibitors is cowpoxvirus (CPXV). Cowpox is known to block caspase activation by CrmA (homologue of VACV B13R) making the cells susceptible to necroptosis by triggering RIPK1/RIPK3 (Kettle et al. 1997; Li and Beg 2000; Chan et al. 2003). Most poxviruses, like VACV, also encode various amounts of TNF inhibitors to block signaling to restrict TNF-induced necroptosis. CPXV has been found to encode many inhibitors: CrmB, CrmC, CrmD, and CrmE (Cunnion 1999). Additionally, CPXV has been found to produce a CD30 homologue with exclusive TNFSF8 binding whose role in pathogenesis is still unknown (Panus et al. 2002). Aside from these TNF inhibitors, a targeted small interfering RNA (siRNA) screen was able to identify a novel inhibitor of RIPK3 generated by CPXV termed viral inducer of RIPK3 degradation (vIRD) (Liu et al. 2021). vIRD binds to RIPK3, causing RIPK3 ubiquitination and subsequent proteasome degradation as detected in western blots. Mice infected with mutant CPXV in which vIRD had been deleted showed significant decreases in many pathogenic processes including mortality. These pathogenic processes were able to be reversed in mice deficient in RIPK3 and MLKL. This evidence shows vIRD was able to inhibit all known forms of necroptotic cell death by directly targeting RIPK3 for degradation.
Other poxviruses have been co-opted to study the vIRD inhibitor including VACV, ectromelia virus (ECTV), and leporipoxvirus myxoma virus (MYXV). VACV encodes a defective vIRD that has been truncated. When a functional version of vIRD is integrated into VACV, mice infected with the mutant virus show increased viral titers. MYXV, which also lacks a functional vIRD, has been shown to be more pathogenic in RIPK3-deficient hosts, which makes sense since these hosts would have resistance to necroptotic death. ECTV, which also has an intact vIRD ortholog, induced RIPK3 degradation and resistance to TNF-induced necroptosis (Liu et al. 2021). ECTV, however, does not fit the typical necroptosis story where the crucial players, RIPK3 and MLKL, do not seem to play the same role in controlling viral infections (Montoya et al. 2023). Studying these inhibitors and comparing them to other viruses can help us to understand more about how viruses have continued to evolve survival mechanisms within their hosts.
Another method used in some poxviruses to inhibit necroptotic death is to mimic viral proteins. BeAn58058 and Cotia poxviruses encode proteins with high enough homology to competitively bind and sequester RIPK3 and out-compete host MLKL and disrupt necroptosis (Petrie et al. 2019). These mimic proteins have been identified in several clades of poxviruses, including avipoxviruses and leporipoxviruses.
RNA Viruses
Influenza A
Influenza A (IAV) is a negative, enveloped single-stranded RNA virus from the Orthomyxoviridae family that causes seasonal and sometimes severe respiratory tract infections in humans worldwide. Current and commonly circulating subtypes of influenza are A(H1N1) and A(H3N2). IAV infection typically affects human lung epithelial cells and triggers cell death. IAV was originally thought to mainly activate the apoptotic cell death pathway. After the discovery of RIPK3 as a crucial factor for cell death in IAV infections, it was recognized that the cell types most commonly used in viral and medical research were not appropriate to study cell death pathways for IAV infection because they did not express RIPK3 (Nogusa et al. 2016a).
Research utilizing the influenza A virus P8 strain model has helped to illuminate the dual role ZBP1 and RIPK3 play in both apoptosis and necroptosis. Early on, it was found that a ZBP1/RIPK3-dependent signaling axis induced cell death in human lung epithelial cells and MEFs following IAV infection. MEFs from RIPK3-deficient mice were infected with PR8 and found to be resistant to cell death and cytopathic effects, whereas wild-type mice showed susceptibility to the infection resulting in cell death (Nogusa et al. 2016a). However, MEFs generated to be deficient in MLKL, the executioner of necroptotic death, were determined to be just as susceptible as wild types to cell death (Nogusa et al. 2016b). This suggested that ZBP1 was capable of stimulating a non-necroptotic form of cell death post-IAV infection. Further genetic analyses revealed the absence of MLKL-activated ZBP1 and could trigger apoptosis in IAV-infected MEFs. Consequently, it was found that both the apoptosis regulator Fadd and the necroptosis executor MLKL needed to be knocked out in order to rescue IAV-infected MEF survival. Looking further into this ZBP1-stimulated apoptotic pathway demonstrated some interesting findings. After ZBP1 is activated post-IAV infection, RIPK3 is needed for the early onset of apoptosis, but not required at later time points. Additionally, the kinase activity of RIPK3 has been shown to be needed for necroptotic but not apoptotic cell death. As such, catalytically inactive RIPK3-expressing mice are subject to early caspase-8-dependent apoptosis, while those deficient in RIPK3 altogether (RIPK3–/–) are subject to later onset apoptotic cell death.
Additional studies verified that IAV was able to generate Z-RNA and activate ZBP1 to initiate the activation of RIPK3 and cell death pathways (Zhang et al. 2020). By transfecting labeled antiserum to Z-NA, it was found that the antiserum co-localized with Z-RNAs in the nucleus of infected cells. This Z-RNA was then shown to co-localize with ZBP1 and activate RIPK3 death signaling culminating in cell death.
While program cell death can be advantageous as a host defense mechanism, necroptosis, by way of its inflammatory nature, can also have severe pathogenic disadvantages when triggered by IAV infection. This was evaluated by examining MLKL-, ZBP1-, and RIPK3-deficient animals after IAV infection. Again, MLKL did not show any increased lethality or viral load, while ZPB1–/– and RIPK3–/– mice had significantly higher pathogenesis than their wild-type counterparts (Zhang et al. 2020).
Reovirus
Reoviruses are non-enveloped RNA viruses with ten linear double-stranded genomic segments (Danthi et al. 2013). Four main serotypes of mammalian ReoV have been identified. These serotypes are represented by prototype strains Type 1 Lang (T1L), Type 2 Jones (T2J), Type 3 Dearing (T3D), and Type 4 Ndelle (T4N) (Attoui et al. 2001).
It was previously believed that the primary PCD pathway triggered by ReoV infection was apoptosis. However, recent studies of T3D-infected L929 cells demonstrated that necroptosis is an important innate mechanism triggered in response to infection. L929 cells infected with the T3D strain underwent a rapid induction of death even in the presence of a caspase inhibitor (Berger and Danthi 2013). The cell death was characterized to be necroptosis as a result of loss of membrane integrity that was inhibited by necrostatins. A follow-up study then demonstrated that IFN-β production, cellular incorporation of viral genomic RNA, and de novo synthesis of viral dsRNA were required to induce necroptosis (Berger et al. 2017). Together, these results suggest that incoming genomic RNA is detected by RLRs in the cytoplasm of the infected cell, which then signals via the adaptor protein MAVS to produce IFN-β followed by the synthesis of viral RNA establishing a pathway to upregulate and detect the likely ligand of dsRNA. Additionally, it was found that knockdown of the dsRNA binding protein σ3 resulted in enhanced necroptosis which was attributed to RLR-mediated increase in IFN-β production (Roebke et al. 2020). Further support of multifactorial death requirements is indicated by the enhanced rate of necroptosis when the ReoV μ1 protein is knocked down, which is associated with an increase of accumulated progeny dsRNA and viral protein synthesis. The specific mechanism of necroptosis induction by ReoV remains unclear. One proposed mechanism is the downregulation of the cIAP family of E3 ubiquitin ligase ReoV (Kominsky et al. 2002). cIAPs are known to polyubiquinate RIPK1 and RIPK3 resulting in kinase inhibition and therefore the decreased cIAP could result in more RIPK1 and RIPK3 and thus more necroptosis (Annibaldi et al. 2018). This model, however, does not address the need for de novo dsRNA synthesis required for necroptosis activation (Jiffry et al. 2021). It is also unclear if the induction of necroptosis, as in many other virus models, contributes to viral clearance or pathogenesis. RIPK3 deficiency does result in an increase in ReoV progeny virions suggesting potential for a protective mechanism indicative of a potentially protective role for necroptosis in ReoV clearance (Yue and Shatkin 1997; Berger et al. 2017). Both cultured BMDMs and L929 fibroblasts undergo necroptosis within a few days post-infection (DeBiasi et al. 2004; Xing et al. 2016; Gummersheimer and Danthi 2020). It remains unclear if ReoV-induced necroptosis occurs within biologically relevant cell types in the gut, CNS, or heart and, furthermore, whether Reo-mediated necroptosis is protective or deleterious due to inflammation during other RNA viral infections like IAV (Balachandran and Rall 2020). More studies are warranted to further clarify the role of necroptosis in ReoV infections and its role in enhancing or restricting pathogenesis.
Respiratory Syncytial Virus
Human respiratory syncytial virus (RSV) is a nonsegmented negative-stranded RNA virus from the Paramyxoviridae family (Collins et al. 2013). This enveloped virus is a significant cause of acute bronchiolitis in infants, the outcomes and severity of which are associated with necroptosis. It is hypothesized that infants suffering acute RSV infection may later be predisposed to respiratory complications, such as asthma and wheezing, later in life. While much of the data is clinical, reports of RSV-triggered necroptosis are relevant to the focus of this chapter. Early RSV necroptosis studies used selective inhibitors of necroptosis pathway players to determine that RSV infection activated the necroptotic pathway RIPK1/RIPK3/MLKL (Muraro et al. 2018; Simpson et al. 2020). Further studies demonstrated that RSV infection may involve TNF-mediated macrophage necroptosis which can cause increased lung damage during RSV infections. This is supported by findings showing that Tnfr1–/– mice demonstrate decreased numbers of necrotic macrophages in lung tissue with less RIPK3 and MLKL expression, while elevated levels of TNF in RSV-infected infants correlate with increased disease severity (Santos et al. 2021). RSV-infected mice demonstrate an increased expression of activated RIPK1 and MLKL with no increase in activated caspase-3 expression which correlates with neutrophilic inflammation and airway epithelial cell sloughing (Simpson et al. 2020). When MLKL or RIPK1 were taken out of the equation using pharmacological inhibitors, inflammation and airway remodeling were prevented and there was a decreased viral load. Furthermore, limiting necroptosis in RSV infection in neonatal mice was associated with decreased viral or allergen-triggered asthma later on in life. These findings argue that necroptotic processes stimulated by RSV infection are actually harmful by increasing viral pathogenesis and making those who become infected more susceptible to developing asthma. This is important to note in the development of future therapeutics as researchers will need to weigh the benefits versus harms of utilizing an inflammatory form of cell death in necroptosis.
SARS-CoV-2
SARS-CoV-2 is a positive single-stranded RNA virus. SARS-CoV-2 infection has been shown to induce a strong inflammatory response that results in severe lung disease in critically ill patients. SARS-CoV-2 infection in lung epithelial cells revealed activation of NF-κB, upregulation of inflammatory cytokines, upregulation of ZBP1, and significant cell death. It has been reported that SARS-CoV-2 triggers the production of viral Z-RNA and initiates the ZBP1-RIPK3-MLKL necroptosis pathway. In ZBP1 CRISPR knockout epithelial cells, a reduced MLKL phosphorylation confirmed the role of necroptosis. Additionally, a significant reduction in inflammatory cytokines released in the ZBP1-deficient cells. This showed that SARS-CoV-2 infection activates necroptosis and induces pro-inflammatory signaling (Li et al. 2023). It was shown that ZBP1 deficiency in mice also reduced immune cell infiltration and lung damage, demonstrating that in the mouse model, ZBP1 is important for SARS-CoV-2 pathogenesis (Li et al. 2023). Further down the pathway, RIPK3 knockdown cells did not appear to impact viral load, but a significant reduction of inflammatory cytokines and chemokines was demonstrated. This depletion of MLKL did not reduce the inflammatory response. Inhibition of the RIPK3 kinase activity also did not appear to impact the inflammatory response but only the necroptosis activation; thus, the scaffolding of RIPK3 was determined to be crucial for triggering the inflammatory response resulting in severe lung damage.
Flaviviruses
Flaviviruses are a family of positive, single-stranded, enveloped RNA viruses. This genus of viruses includes mosquito-transmitted viruses such as yellow fever, Japanese encephalitis, Dengue fever, West Nile, and Zika virus (Pierson and Diamond 2020). Ticks are also responsible for some flavivirus transmission, causing encephalitis and hemorrhagic fever viruses. These include Palm Creek virus, Parramatta River virus, Kyasanur Forest disease, Alkhurma disease, and Omsk hemorrhagic fever.
Zika virus (ZIKV) is a flavivirus transmitted by mosquitoes that can cause various neurological outcomes in humans. ZIKV infection in pregnancy can result in the virus crossing the placenta resulting in severe fetal abnormalities like microcephaly (Coyne and Lazear 2016; Costa and Ko 2018). Infection in adulthood can also result in neurological injury but is rare (Mehta et al. 2018). Early studies conducted found that neurons infected with ZIKV activated a cell-restricting pathway. Using mice deficient in the crucial necroptotic players, ZBP1, RIPK3, or the RIPK1 kinase activity, researchers found that the incidence of paresis to be rapidly increased during CNS infection demonstrating the protective role of necroptosis in neuron ZIKV infection. After activation of the ZBP1 necroptosis pathway, instead of completing necroptosis, RIPK signaling in ZIKV-infected neurons causes them to enter a metabolic state that suppresses viral genome replication (Daniels et al. 2019). This change in metabolic state is evidenced by the identification of IRG1 production of itaconate that inhibits succinate dehydrogenase activity in the mitochondria, restricting viral replication (Daniels et al. 2019). Intracranially infected mice deficient in IRG1 had a greater viral burden, but injection of itaconate rescued the IRG1-deficient mice from this viral burden. Later studies, however, found evidence of necroptosis occurring in ZIKV-infected human astrocytes and resulted in a protective effect on glial cells. This is of interest since astrocytes are known to support neurons in many ways including providing key components to fuel neuronal metabolism. It is important to also note that the necroptotic cell death observed was RIPK1 independent and associated with an upregulation of ZBP1 (Wen et al. 2021). While these are some fascinating findings, ultimately, studies using ZIKV-infected MLKL-knockout mice have shown no differences in mortality compared to wild types infected with the virus which suggests that necroptosis does not play a major role in pathogenesis (Daniels et al. 2019).
Necroptotic processes in West Nile virus (WNV) models have not been thoroughly studied. What has been demonstrated is that microarray analysis of brain tissues harvested from WNV-infected mice shows an upregulation of necroptotic genes (Peng and Wang 2019). While these genes are upregulated, other in vivo studies have shown that RIPK3-deficient mice had increased mortality after WNV infection; however, in mice deficient in MLKL, the executioner of necroptosis, no impact on mortality was observed. These data suggest RIPK3 is involved in coordinating other immune responses including chemokine expression and lymphocyte recruitment (Daniels et al. 2017). This further demonstrates the complexity of key necroptotic components and the multiple roles RIPK3 plays in the host defense.
Rotavirus
Rotavirus (RV) is a double-stranded RNA virus of the Reoviridae family and a leading cause of gastroenteritis in young children. Rotavirus infection mainly impacts enterocyte cells in the small intestine where infection concludes in cell death. RV induces necroptosis and apoptosis, the latter being well characterized. In early RV-induced necroptosis studies, RV was demonstrated to trigger MLKL phosphorylation, oligomerization of MLKL, and translocation to the plasma membrane for pore formation (Mukhopadhyay et al. 2022) and shown to act through RIPK1-dependent pathway. Interestingly, a cooperative element between apoptosis and necroptosis appears to be a part of RV infection. When necroptosis is inhibited, apoptosis is activated and cell viability is also increased, resulting in an antiviral state where infected cells are phagocytized and RV replication reduces. When apoptosis is restricted in vivo, necroptosis becomes the death pathway of choice and a proviral response occurs. As a consequence of necroptosis, infected cells burst and the viral contents are released to promote infection (Soliman et al. 2022).
Conclusion
Viruses have invested substantial resources in the inhibition of programmed cell death by the host, highlighting the importance of cell death in the arms race between hosts and pathogens. While caspase-dependent cell death is traditionally thought of as the default host cell death pathway, numerous virus-encoded gene products have been identified that inhibit caspase-dependent cell death. However, inhibition of caspase-dependent cell death sensitizes cells to MLKL-dependent necroptotic cell death. Thus, viruses have evolved multiple mechanisms to inhibit necroptotic cell death that can be engaged to gain further understanding of the consequences of unleashing or restricting this death pathway. Viral inhibitors target RIPK3 degradation or redistribution, ZBP1/DAI antagonist, restriction of RHIM activation, TNFR activation, and MLKL antagonist. Antagonism of necroptosis is important for poxviral resistance to interferon, both in cells in culture and in animal models, and for pathogenesis in animal models. Thus, inhibition of necroptosis appears to be an important means for viruses to maintain an edge in the arms race between viruses and their hosts.
Abbreviations
- AIM2:
-
Absent in melanoma 2
- ATP:
-
Adenosine triphosphate
- DAMPs:
-
Damage-associated molecular patterns
- DNA:
-
Deoxy-ribose nucleic acid
- dsRNA:
-
Double-stranded RNA
- hpi:
-
Hours post-infection
- IFN:
-
Type I interferon
- ISG:
-
IFN-stimulated gene
- LPS:
-
Lipopolysaccharide
- MEF:
-
Mouse embryo fibroblasts
- MLKL:
-
Mixed-lineage kinase-like
- PAMPs:
-
Pathogen-associated molecular patterns
- PKR:
-
Double-stranded RNA-dependent protein kinase
- PRRs:
-
Pattern recognition receptors
- RHIM:
-
RIP homotypic interaction motif
- RIPK:
-
Receptor-interacting protein kinase
- RNase:
-
Ribonuclease
- serpins:
-
Serine protease inhibitors
- ssRNA:
-
Single-stranded ribonucleic acid
- TNF:
-
Tumor necrosis factor
- TRIF/TICAM-1:
-
TIR domain-containing adaptor inducing interferon-β/TIR domain-containing adaptor molecule 1
- VACV:
-
Vaccinia virus
- wt:
-
Wild type
- Za:
-
Z-nucleic acid binding domain
- ZBP1:
-
Z-nucleic acid binding protein 1
- Z-NA:
-
Z-form nucleic acid
References
Amarante-Mendes GP, Adjemian S, Branco LM et al (2018) Pattern recognition receptors and the host cell death molecular machinery. Front Immunol 9:2379
Annibaldi A, Wicky John S, Vanden Berghe T et al (2018) Ubiquitin-mediated regulation of RIPK1 kinase activity independent of IKK and MK2. Mol Cell 69:566–580.e5. https://doi.org/10.1016/j.molcel.2018.01.027
Attoui H, Biagini P, Stirling J et al (2001) Sequence characterization of Ndelle virus genome segments 1, 5, 7, 8, and 10: evidence for reassignment to the genus Orthoreovirus, family Reoviridae. Biochem Biophys Res Commun 287:583–588. https://doi.org/10.1006/bbrc.2001.5612
Balachandran S, Rall GF (2020) Benefits and perils of necroptosis in influenza virus infection. J Virol 94:e01101-19. https://doi.org/10.1128/JVI.01101-19
Berger AK, Danthi P (2013) Reovirus activates a caspase-independent cell death pathway. mBio 4:e00178-00113. https://doi.org/10.1128/mBio.00178-13
Berger SB, Kasparcova V, Hoffman S et al (2014) RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol Baltim Md 1950 192:5476–5480. https://doi.org/10.4049/jimmunol.1400499
Berger SB, Harris P, Nagilla R et al (2015) Characterization of GSK’963: a structurally distinct, potent and selective inhibitor of RIP1 kinase. Cell Death Discov 1:15009. https://doi.org/10.1038/cddiscovery.2015.9
Berger AK, Hiller BE, Thete D et al (2017) Viral RNA at two stages of reovirus infection is required for the induction of necroptosis. J Virol 91:e02404-16. https://doi.org/10.1128/JVI.02404-16
Berghe TV, Linkermann A, Jouan-Lanhouet S et al (2014) Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15:135–147. https://doi.org/10.1038/nrm3737
Bonnet MC, Preukschat D, Welz P-S et al (2011) The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity 35:572–582. https://doi.org/10.1016/j.immuni.2011.08.014
Bozec D, Iuga AC, Roda G et al (2016) Critical function of the necroptosis adaptor RIPK3 in protecting from intestinal tumorigenesis. Oncotarget 7:46384–46400. https://doi.org/10.18632/oncotarget.10135
Brune W, Ménard C, Heesemann J, Koszinowski UH (2001) A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science 291:303–305. https://doi.org/10.1126/science.291.5502.303
Cao L, Mu W (2021) Necrostatin-1 and necroptosis inhibition: Pathophysiology and therapeutic implications. Pharmacol Res 163:105297. https://doi.org/10.1016/j.phrs.2020.105297
Cao T, Ni R, Ding W et al (2022) MLKL-mediated necroptosis is a target for cardiac protection in mouse models of type-1 diabetes. Cardiovasc Diabetol 21:165. https://doi.org/10.1186/s12933-022-01602-9
Chan FK-M, Shisler J, Bixby JG et al (2003) A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem 278:51613–51621. https://doi.org/10.1074/jbc.M305633200
Chen J, Kos R, Garssen J, Redegeld F (2019) Molecular insights into the mechanism of necroptosis: the necrosome as a potential therapeutic target. Cells 8:1486. https://doi.org/10.3390/cells8121486
Chen Y, Lin J, Zhao Y et al (2021) Toll-like receptor 3 (TLR3) regulation mechanisms and roles in antiviral innate immune responses. J Zhejiang Univ Sci B 22:609–632. https://doi.org/10.1631/jzus.B2000808
Chen L, Zhang X, Ou Y et al (2022) Advances in RIPK1 kinase inhibitors. Front Pharmacol 13:976435
Cho Y, Challa S, Moquin D et al (2009) Phosphorylation-driven assembly of RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137:1112–1123. https://doi.org/10.1016/j.cell.2009.05.037
Collins PL, Fearns R, Graham BS (2013) Respiratory syncytial virus: virology, reverse genetics, and pathogenesis of disease. Curr Top Microbiol Immunol 372:3–38. https://doi.org/10.1007/978-3-642-38919-1_1
Costa F, Ko AI (2018) Zika virus and microcephaly: where do we go from here? Lancet Infect Dis 18:236–237. https://doi.org/10.1016/S1473-3099(17)30697-7
Cotsmire SM, Szczerba M, Jacobs BL (2021) Detecting necroptosis in virus-infected cells. Methods Mol Biol Clifton NJ 2225:199–216. https://doi.org/10.1007/978-1-0716-1012-1_11
Coyne CB, Lazear HM (2016) Zika virus—reigniting the TORCH. Nat Rev Microbiol 14:707–715. https://doi.org/10.1038/nrmicro.2016.125
Cunnion KM (1999) Tumor necrosis factor receptors encoded by poxviruses. Mol Genet Metab 67:278–282. https://doi.org/10.1006/mgme.1999.2878
Daley-Bauer LP, Roback L, Crosby LN et al (2017) Mouse cytomegalovirus M36 and M45 death suppressors cooperate to prevent inflammation resulting from antiviral programmed cell death pathways. Proc Natl Acad Sci U S A 114:E2786–E2795. https://doi.org/10.1073/pnas.1616829114
Daniels BP, Snyder AG, Olsen TM et al (2017) RIPK3 restricts viral pathogenesis via cell death-independent neuroinflammation. Cell 169:301–313.e11. https://doi.org/10.1016/j.cell.2017.03.011
Daniels BP, Kofman SB, Smith JR et al (2019) The nucleotide sensor ZBP1 and kinase RIPK3 induce the enzyme IRG1 to promote an antiviral metabolic state in neurons. Immunity 50:64–76.e4. https://doi.org/10.1016/j.immuni.2018.11.017
Danthi P, Holm GH, Stehle T, Dermody TS (2013) Reovirus receptors, cell entry, and proapoptotic signaling. In: Pöhlmann S, Simmons G (eds) Viral entry into host cells. Springer, New York, pp 42–71
de Almagro MC, Goncharov T, Izrael-Tomasevic A et al (2017) Coordinated ubiquitination and phosphorylation of RIP1 regulates necroptotic cell death. Cell Death Differ 24:26–37. https://doi.org/10.1038/cdd.2016.78
DeBiasi RL, Robinson BA, Sherry B et al (2004) Caspase inhibition protects against reovirus-induced myocardial injury in vitro and in vivo. J Virol 78:11040–11050. https://doi.org/10.1128/JVI.78.20.11040-11050.2004
Declercq W, Vanden Berghe T, Vandenabeele P (2009) RIP kinases at the crossroads of cell death and survival. Cell 138:229–232. https://doi.org/10.1016/j.cell.2009.07.006
Degterev A, Maki JL, Yuan J (2013) Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ 20:366. https://doi.org/10.1038/cdd.2012.133
Dikic I, Wakatsuki S, Walters KJ (2009) Ubiquitin-binding domains—from structures to functions. Nat Rev Mol Cell Biol 10:659–671. https://doi.org/10.1038/nrm2767
Dondelinger Y, Declercq W, Montessuit S et al (2014) MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 7:971–981. https://doi.org/10.1016/j.celrep.2014.04.026
Dovey CM, Diep J, Clarke BP et al (2018) MLKL requires the inositol phosphate code to execute necroptosis. Mol Cell 70:936–948.e7. https://doi.org/10.1016/j.molcel.2018.05.010
Dufour F, Bertrand L, Pearson A et al (2011) The ribonucleotide reductase R1 subunits of herpes simplex virus 1 and 2 protect cells against poly(I · C)-induced apoptosis. J Virol 85:8689–8701. https://doi.org/10.1128/JVI.00362-11
Esaki S, Goshima F, Katsumi S et al (2010) Apoptosis induction after herpes simplex virus infection differs according to cell type in vivo. Arch Virol 155:1235–1245. https://doi.org/10.1007/s00705-010-0712-2
Fletcher-Etherington A, Nobre L, Nightingale K et al (2020) Human cytomegalovirus protein pUL36: A dual cell death pathway inhibitor. Proc Natl Acad Sci U S A 117:18771–18779. https://doi.org/10.1073/pnas.2001887117
Green DR, Llambi F (2015) Cell death signaling. Cold Spring Harb Perspect Biol 7:a006080. https://doi.org/10.1101/cshperspect.a006080
Gummersheimer SL, Danthi P (2020) Reovirus core proteins λ1 and σ2 promote stability of disassembly intermediates and influence early replication events. J Virol 94:e00491-20. https://doi.org/10.1128/JVI.00491-20
Guo H, Omoto S, Harris PA et al (2015) Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 17:243–251. https://doi.org/10.1016/j.chom.2015.01.003
Guo H, Gilley RP, Fisher A et al (2018) Species-independent contribution of ZBP1/DAI/DLM-1-triggered necroptosis in host defense against HSV1. Cell Death Dis 9:816. https://doi.org/10.1038/s41419-018-0868-3
Guo H, Koehler HS, Dix RD, Mocarski ES (2022a) Programmed cell death-dependent host defense in ocular herpes simplex virus infection. Front Microbiol 13:869064. https://doi.org/10.3389/fmicb.2022.869064
Guo H, Koehler HS, Mocarski ES, Dix RD (2022b) RIPK3 and caspase 8 collaborate to limit herpes simplex encephalitis. PLoS Pathog 18:e1010857. https://doi.org/10.1371/journal.ppat.1010857
Hanus J, Anderson C, Wang S (2015) RPE necroptosis in response to oxidative stress and in AMD. Ageing Res Rev 24:286–298. https://doi.org/10.1016/j.arr.2015.09.002
He S, Liang Y, Shao F, Wang X (2011) Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A 108:20054–20059. https://doi.org/10.1073/pnas.1116302108
Holler N, Zaru R, Micheau O et al (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1:489–495. https://doi.org/10.1038/82732
Jiao J, Wang Y, Ren P et al (2020) Necrosulfonamide ameliorates neurological impairment in spinal cord injury by improving antioxidative capacity. Front Pharmacol 10:1538
Jiffry J, Thavornwatanayong T, Rao D et al (2021) Oncolytic reovirus (pelareorep) induces autophagy in KRAS-mutated colorectal cancer. Clin Cancer Res Off J Am Assoc Cancer Res 27:865–876. https://doi.org/10.1158/1078-0432.CCR-20-2385
Jorgensen I, Rayamajhi M, Miao EA (2017) Programmed cell death as a defence against infection. Nat Rev Immunol 17:151–164. https://doi.org/10.1038/nri.2016.147
Kaiser WJ, Upton JW, Mocarski ES (2008) Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol Baltim Md 1950 181:6427–6434. https://doi.org/10.4049/jimmunol.181.9.6427
Kaiser WJ, Sridharan H, Huang C et al (2013) Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem 288:31268–31279. https://doi.org/10.1074/jbc.M113.462341
Kettle S, Alcamí A, Khanna A et al (1997) Vaccinia virus serpin B13R (SPI-2) inhibits interleukin-1beta-converting enzyme and protects virus-infected cells from TNF- and Fas-mediated apoptosis, but does not prevent IL-1beta-induced fever. J Gen Virol 78(Pt 3):677–685. https://doi.org/10.1099/0022-1317-78-3-677
Khoury MK, Gupta K, Franco SR, Liu B (2020) Necroptosis in the pathophysiology of disease. Am J Pathol 190:272–285. https://doi.org/10.1016/j.ajpath.2019.10.012
Koehler HS, Jacobs BL (2021) Subversion of programed cell death by poxviruses. Curr Top Microbiol Immunol. https://doi.org/10.1007/82_2020_229
Koehler H, Cotsmire S, Langland J et al (2017) Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc Natl Acad Sci U S A 114:11506–11511. https://doi.org/10.1073/pnas.1700999114
Koehler H, Cotsmire S, Zhang T et al (2021) Vaccinia virus E3 prevents sensing of Z-RNA to block ZBP1-dependent necroptosis. Cell Host Microbe 29:1266–1276.e5. https://doi.org/10.1016/j.chom.2021.05.009
Kominsky DJ, Bickel RJ, Tyler KL (2002) Reovirus-induced apoptosis requires mitochondrial release of Smac/DIABLO and involves reduction of cellular inhibitor of apoptosis protein levels. J Virol 76:11414–11424. https://doi.org/10.1128/jvi.76.22.11414-11424.2002
Koo G-B, Morgan MJ, Lee D-G et al (2015) Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res 25:707–725. https://doi.org/10.1038/cr.2015.56
Kuriakose T, Man SM, Malireddi RKS et al (2016) ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol 1:aag2045. https://doi.org/10.1126/sciimmunol.aag2045
Langelier Y, Bergeron S, Chabaud S et al (2002) The R1 subunit of herpes simplex virus ribonucleotide reductase protects cells against apoptosis at, or upstream of, caspase-8 activation. J Gen Virol 83:2779–2789. https://doi.org/10.1099/0022-1317-83-11-2779
Lembo D, Donalisio M, Hofer A et al (2004) The ribonucleotide reductase R1 homolog of murine cytomegalovirus is not a functional enzyme subunit but is required for pathogenesis. J Virol 78:4278–4288. https://doi.org/10.1128/jvi.78.8.4278-4288.2004
Li M, Beg AA (2000) Induction of necrotic-like cell death by tumor necrosis factor alpha and caspase inhibitors: novel mechanism for killing virus-infected cells. J Virol 74:7470–7477. https://doi.org/10.1128/jvi.74.16.7470-7477.2000
Li J, McQuade T, Siemer AB et al (2012) The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150:339–350. https://doi.org/10.1016/j.cell.2012.06.019
Li J-X, Feng J-M, Wang Y et al (2014) The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis 5:e1278. https://doi.org/10.1038/cddis.2014.241
Li S, Zhang Y, Guan Z et al (2023) SARS-CoV-2 Z-RNA activates the ZBP1-RIPK3 pathway to promote virus-induced inflammatory responses. Cell Res 33:201–214. https://doi.org/10.1038/s41422-022-00775-y
Liu Z, Nailwal H, Rector J et al (2021) A class of viral inducer of degradation of the necroptosis adaptor RIPK3 regulates virus-induced inflammation. Immunity 54:247–258.e7. https://doi.org/10.1016/j.immuni.2020.11.020
Mandal P, Berger SB, Pillay S et al (2014) RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell 56:481–495. https://doi.org/10.1016/j.molcel.2014.10.021
Martens S, Hofmans S, Declercq W et al (2020) Inhibitors targeting RIPK1/RIPK3: old and new drugs. Trends Pharmacol Sci 41:209–224. https://doi.org/10.1016/j.tips.2020.01.002
Mehta R, Soares CN, Medialdea-Carrera R et al (2018) The spectrum of neurological disease associated with Zika and chikungunya viruses in adults in Rio de Janeiro, Brazil: A case series. PLoS Negl Trop Dis 12:e0006212. https://doi.org/10.1371/journal.pntd.0006212
Mocarski ES, Upton JW, Kaiser WJ (2011) Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat Rev Immunol 12:79–88. https://doi.org/10.1038/nri3131
Mocarski ES, Guo H, Kaiser WJ (2015) Necroptosis: The Trojan horse in cell autonomous antiviral host defense. Virology 479–480:160–166. https://doi.org/10.1016/j.virol.2015.03.016
Moerke C, Bleibaum F, Kunzendorf U, Krautwald S (2019) Combined knockout of RIPK3 and MLKL reveals unexpected outcome in tissue injury and inflammation. Front Cell Dev Biol 7:19. https://doi.org/10.3389/fcell.2019.00019
Montoya B, Knudson CJ, Melo-Silva CR et al (2023) Resistance to poxvirus lethality does not require the necroptosis proteins RIPK3 or MLKL. J Virol 97:e01945-22. https://doi.org/10.1128/jvi.01945-22
Moriwaki K, Bertin J, Gough PJ et al (2015) Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis 6:e1636. https://doi.org/10.1038/cddis.2015.16
Mukhopadhyay U, Patra U, Chandra P et al (2022) Rotavirus activates MLKL-mediated host cellular necroptosis concomitantly with apoptosis to facilitate dissemination of viral progeny. Mol Microbiol 117:818–836. https://doi.org/10.1111/mmi.14874
Muraro SP, De Souza GF, Gallo SW et al (2018) Respiratory syncytial virus induces the classical ROS-dependent NETosis through PAD-4 and necroptosis pathways activation. Sci Rep 8:14166. https://doi.org/10.1038/s41598-018-32576-y
Murphy JM (2020) The killer pseudokinase mixed lineage kinase domain-like protein (MLKL). Cold Spring Harb Perspect Biol 12:a036376. https://doi.org/10.1101/cshperspect.a036376
Murphy JM, Czabotar PE, Hildebrand JM et al (2013) The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39:443–453. https://doi.org/10.1016/j.immuni.2013.06.018
Nailwal H, Chan FK-M (2019) Necroptosis in anti-viral inflammation. Cell Death Differ 26:4–13. https://doi.org/10.1038/s41418-018-0172-x
Newton K, Dugger DL, Wickliffe KE et al (2014) Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343:1357–1360. https://doi.org/10.1126/science.1249361
Nogusa S, Slifker MJ, Ingram JP et al (2016a) RIPK3 is largely dispensable for RIG-I-like receptor- and type I interferon-driven transcriptional responses to influenza A virus in murine fibroblasts. PLoS One 11:e0158774. https://doi.org/10.1371/journal.pone.0158774
Nogusa S, Thapa RJ, Dillon CP et al (2016b) RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe 20:13–24. https://doi.org/10.1016/j.chom.2016.05.011
Oberst A, Dillon CP, Weinlich R et al (2011) Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471:363–367. https://doi.org/10.1038/nature09852
O’Donnell MA, Legarda-Addison D, Skountzos P et al (2007) Ubiquitination of RIP1 regulates an NF-kappaB-independent cell-death switch in TNF signaling. Curr Biol CB 17:418–424. https://doi.org/10.1016/j.cub.2007.01.027
Omoto S, Guo H, Talekar GR et al (2015) Suppression of RIP3-dependent necroptosis by human cytomegalovirus. J Biol Chem 290:11635–11648. https://doi.org/10.1074/jbc.M115.646042
Panus JF, Smith CA, Ray CA et al (2002) Cowpox virus encodes a fifth member of the tumor necrosis factor receptor family: A soluble, secreted CD30 homologue. Proc Natl Acad Sci U S A 99:8348–8353. https://doi.org/10.1073/pnas.122238599
Peng B-H, Wang T (2019) West Nile virus induced cell death in the central nervous system. Pathog Basel Switz 8:215. https://doi.org/10.3390/pathogens8040215
Perales-Linares R, Navas-Martin S (2013) Toll-like receptor 3 in viral pathogenesis: friend or foe? Immunology 140:153–167. https://doi.org/10.1111/imm.12143
Pestka S, Krause CD, Walter MR (2004) Interferons, interferon-like cytokines, and their receptors. Immunol Rev 202:8–32. https://doi.org/10.1111/j.0105-2896.2004.00204.x
Petrie EJ, Sandow JJ, Lehmann WIL et al (2019) Viral mlkl homologs subvert necroptotic cell death by sequestering cellular RIPK3. Cell Rep 28:3309–3319.e5. https://doi.org/10.1016/j.celrep.2019.08.055
Pierson TC, Diamond MS (2020) The continued threat of emerging flaviviruses. Nat Microbiol 5:796–812. https://doi.org/10.1038/s41564-020-0714-0
Polykratis A, Hermance N, Zelic M et al (2014) Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol Baltim Md 1950 193:1539–1543. https://doi.org/10.4049/jimmunol.1400590
Rheault TR, Stellwagen JC, Adjabeng GM et al (2013) Discovery of dabrafenib: a selective inhibitor of Raf kinases with antitumor activity against B-Raf-driven tumors. ACS Med Chem Lett 4:358–362. https://doi.org/10.1021/ml4000063
Rodriguez DA, Weinlich R, Brown S et al (2016) Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ 23:76–88. https://doi.org/10.1038/cdd.2015.70
Roebke KE, Guo Y, Parker JSL, Danthi P (2020) Reovirus σ3 protein limits interferon expression and cell death induction. J Virol 94:e01485-20. https://doi.org/10.1128/JVI.01485-20
Samson AL, Zhang Y, Geoghegan ND et al (2020) MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat Commun 11:3151. https://doi.org/10.1038/s41467-020-16887-1
Santos LD, Antunes KH, Muraro SP et al (2021) TNF-mediated alveolar macrophage necroptosis drives disease pathogenesis during respiratory syncytial virus infection. Eur Respir J 57. https://doi.org/10.1183/13993003.03764-2020
Simpson J, Loh Z, Ullah MA et al (2020) Respiratory syncytial virus infection promotes necroptosis and HMGB1 release by airway epithelial cells. Am J Respir Crit Care Med 201:1358–1371. https://doi.org/10.1164/rccm.201906-1149OC
Soliman M, Seo J-Y, Baek Y-B et al (2022) Opposite effects of apoptotic and necroptotic cellular pathways on rotavirus replication. J Virol 96:e0122221. https://doi.org/10.1128/JVI.01222-21
Sridharan H, Ragan KB, Guo H et al (2017) Murine cytomegalovirus IE3-dependent transcription is required for DAI/ZBP1-mediated necroptosis. EMBO Rep 18:1429–1441. https://doi.org/10.15252/embr.201743947
Su L, Quade B, Wang H et al (2014) A plug release mechanism for membrane permeation by MLKL. Structure 22:1489–1500. https://doi.org/10.1016/j.str.2014.07.014
Sun X, Yin J, Starovasnik MA et al (2002) Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem 277:9505–9511. https://doi.org/10.1074/jbc.M109488200
Sun L, Wang H, Wang Z et al (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148:213–227. https://doi.org/10.1016/j.cell.2011.11.031
Thapa RJ, Ingram JP, Ragan KB et al (2016) DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 20:674–681. https://doi.org/10.1016/j.chom.2016.09.014
Tovey Crutchfield EC, Garnish SE, Hildebrand JM (2021) The role of the key effector of necroptotic cell death, MLKL, in mouse models of disease. Biomolecules 11:803. https://doi.org/10.3390/biom11060803
Upton JW, Kaiser WJ, Mocarski ES (2010a) Pathogen subversion of RIP3-dependent necrosis. Cell Host Microbe 7:302–313. https://doi.org/10.1016/j.chom.2010.03.006
Upton JW, Kaiser WJ, Mocarski ES (2010b) Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 7:302–313. https://doi.org/10.1016/j.chom.2010.03.006
Upton JW, Kaiser WJ, Mocarski ES (2012) DAI complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11:290–297. https://doi.org/10.1016/j.chom.2012.01.016
Upton JW, Kaiser WJ, Mocarski ES (2019) DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 26:564. https://doi.org/10.1016/j.chom.2019.09.004
Vercammen D, Beyaert R, Denecker G et al (1998) Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 187:1477–1485. https://doi.org/10.1084/jem.187.9.1477
Vercammen E, Staal J, Beyaert R (2008) Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clin Microbiol Rev 21:13–25. https://doi.org/10.1128/CMR.00022-07
Wang H, Sun L, Su L et al (2014a) Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 54:133–146. https://doi.org/10.1016/j.molcel.2014.03.003
Wang X, Li Y, Liu S et al (2014b) Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci U S A 111:15438–15443. https://doi.org/10.1073/pnas.1412767111
Wen C, Yu Y, Gao C et al (2021) RIPK3-dependent necroptosis is induced and restricts viral replication in human astrocytes infected with Zika virus. Front Cell Infect Microbiol 11:637710. https://doi.org/10.3389/fcimb.2021.637710
Wu J, Huang Z, Ren J et al (2013) Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res 23:994–1006. https://doi.org/10.1038/cr.2013.91
Xing J, Weng L, Yuan B et al (2016) Identification of a role for TRIM29 in the control of innate immunity in the respiratory tract. Nat Immunol 17:1373–1380. https://doi.org/10.1038/ni.3580
Yue Z, Shatkin AJ (1997) Double-stranded RNA-dependent protein kinase (PKR) is regulated by reovirus structural proteins. Virology 234:364–371. https://doi.org/10.1006/viro.1997.8664
Zhang S-Y, Herman M, Ciancanelli MJ et al (2013) TLR3 immunity to infection in mice and humans. Curr Opin Immunol 25:19–33. https://doi.org/10.1016/j.coi.2012.11.001
Zhang S, Tang M-B, Luo H-Y et al (2017) Necroptosis in neurodegenerative diseases: a potential therapeutic target. Cell Death Dis 8:e2905. https://doi.org/10.1038/cddis.2017.286
Zhang T, Yin C, Boyd DF et al (2020) Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell 180:1115–1129.e13. https://doi.org/10.1016/j.cell.2020.02.050
Zhang X, Zhang Y, Wang F et al (2022) Necrosulfonamide alleviates acute brain injury of intracerebral hemorrhage via inhibiting inflammation and necroptosis. Front Mol Neurosci 15:916249. https://doi.org/10.3389/fnmol.2022.916249
Zhou T, Wang Q, Phan N et al (2019) Identification of a novel class of RIP1/RIP3 dual inhibitors that impede cell death and inflammation in mouse abdominal aortic aneurysm models. Cell Death Dis 10:226. https://doi.org/10.1038/s41419-019-1468-6
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Lawson, C.A., Titus, D.J., Koehler, H.S. (2023). Approaches to Evaluating Necroptosis in Virus-Infected Cells. In: Vijayakrishnan, S., Jiu, Y., Harris, J.R. (eds) Virus Infected Cells. Subcellular Biochemistry, vol 106. Springer, Cham. https://doi.org/10.1007/978-3-031-40086-5_2
Download citation
DOI: https://doi.org/10.1007/978-3-031-40086-5_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-40085-8
Online ISBN: 978-3-031-40086-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)