
Abstract
From initially overlooked as peculiarities of uncertain biological importance or rare isoforms generated as a result of splicing errors to commonly regarded as relevant regulatory molecules, circular RNAs (circRNAs) now represent an extensively studied subgroup of non-coding RNAs (ncRNAs). The structural uniqueness of these endogenous biomolecules confers resistance to exonuclease activity, resulting in higher stability than observed in their linear counterparts, which alongside their reported function of gene expression regulators, predisposes them to potential use as robust biomarkers and/or powerful therapeutic targets. During the last decade, we have witnessed a boom in circRNA research deciphering various mechanisms of their biogenesis and an in-depth description of their biological functions, including miRNA and protein sponging, yet regulation of these events remains unclear. Despite recent advances in high-throughput genomic technology and novel bioinformatic approaches allowing circRNAs characterization in detail, analyzing these molecules continues to be challenging, leaving multiple biological knowledge gaps unsolved.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
Circular RNAs (circRNAs) are long, covalently closed single-stranded molecules. Albeit still included in the sizeable non-coding RNA (ncRNA) family, their function as a template for peptides has also been reported multiple times to date. These RNAs, generated during an event called “backsplicing” and widely expressed across the eukaryotic tree of life, are increasingly acknowledged by the scientific community owing to their unique properties and powerful mode of action.
Although not generated during backsplicing, first circRNAs were identified in the middle of the 70 s as circular genomes in plant viroids. A few years later, the detection of first mammalian circRNAs extracted from cytoplasmic fraction of HeLa cells using electron microscopy was reported. In the breakthrough year, 2013, a sequence conservation study by Memczak et al. (2013) proved that circRNAs could act as post-transcriptional regulators. In the same month, other relevant studies were published, confirming the biological importance of circRNAs (Jeck et al. 2013). Hansen et al. (2013) were the first to report a functional analysis of naturally expressed circRNA and elucidated the now widely accepted role of circRNAs demonstrating that circRNAs could function as microRNA (miRNA) sponges to specifically bind a miRNA molecule and prevent it from binding to its target, thereby affecting the regulation of gene expression. Until the publication of crucial papers reporting circRNA regulatory properties, these molecules were considered functionless byproducts of aberrant splicing events.
In recent years, aberrant expression of circRNAs has been associated with a wide range of pathological conditions. According to various studies, it plays a crucial role in the pathogenesis and progression of various disorders, including for example cancer, neurodegenerative diseases, or cardiovascular diseases. Due to the circRNA loop structure with close ends unavailable for ribonucleases, these molecules are presumably more stable than their linear counterparts (Jeck et al. 2013), and, additionally, they are usually expressed in a tissue- or cell-type-specific manner (Memczak et al. 2013; Salzman et al. 2013). Therefore, they have been proposed as potential reliable prognostic or diagnostic biomarkers or therapeutic targets and have been extensively explored.
With the development of high-throughput sequencing technologies and dedicated bioinformatic approaches, the biology of circRNAs and their molecular and cellular functions have been gradually revealed, leading to the common understanding that these molecules represent key players in various molecular biological processes. However, primarily due to their unusual loop structure lacking free 5’ and 3'-ends-ends, there are several reasons why the true nature of circRNA has long escaped the attention of scientists worldwide. When employing standard bioinformatic pipelines for high-throughput sequencing data analysis, any reads that span back-splice junctions (BSJs) will be discarded because they do not align to the linear reference genome. Nevertheless, there are now countless bioinformatics approaches for processing sequencing data (Szabo and Salzman 2016). Further, the lack of a poly(A) end must be considered when preparing sequencing complementary DNA (cDNA) libraries, as poly(A) selection is used in many protocols in the ribosomal RNA (rRNA) removal step (Pandey et al. 2019). Moreover, circRNAs are only a minority of total RNAs and must be purified before downstream analyses (Xiao and Wilusz 2019). Likewise, it is necessary to design modified primers when using RT-qPCR (reverse transcription followed by quantitative polymerase chain reaction) detection since primers designed according to the linear genome will not distinguish the linear RNA from the circRNA. CircRNAs need to be actively searched for, and standard molecular biology and bioinformatics approaches must be explicitly modified, thus, research on these unique molecules remains substantially challenging.
2 Biogenesis and CircRNA Subtypes
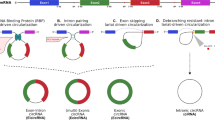
Although the complete mechanism of biogenesis and its regulation remain elusive, general knowledge of circRNA origins has improved considerably in recent years. During canonical splicing, a splice donor, an upstream 5’ splice site, binds to a splice acceptor, a downstream 3’ splice site, across an intron to discard it. Nevertheless, many pre-mRNAs can undergo an alternative splicing, such as backsplicing, during which circRNAs are generated (Fig. 1).

Mechanisms of circRNA biogenesis
CircRNAs are derived from canonical splice sites (Jeck et al. 2013; Memczak et al. 2013) and are reported to be dependent on the splicing machinery, which usually is not efficient enough to form a linear RNA (Jeck et al. 2013; Ashwal-Fluss et al. 2014). Therefore, circRNAs transcription can compete with canonical pre-mRNA (precursor messenger RNA) splicing and affects its rate (Ashwal-Fluss et al. 2014). On the other hand, authors of a study on Drosophila melanogaster observed an increase in circRNA levels when depleted splicing factors (Kramer et al. 2015). Another study on the same organism cells demonstrated that depletion of U2 small nuclear ribonucleoprotein components leading to spliceosome inhibition increases the ratio of circRNAs to linear RNAs (Liang et al. 2017a). These findings suggest that when pre-mRNA processing is inhibited, emerging RNA can be guided into alternative pathways that promote backsplicing.
During spliceosome-mediated backsplicing, the downstream splice-donor site and the upstream splice-acceptor site on the same pre-mRNA must be brought into proximity by looping of flanking intron sequence to be ligated (Kramer et al. 2015). This event gives rise to the most abundant and well-studied circRNAs, which tend to have long introns flanking the exons involved in backsplicing, exonic circRNAs (ecircRNAs). Besides, these molecules are often derived from genes with highly active promoters. More seldom, if the circle retains introns, exon–intron circRNAs (EIciRNAs) are generated (Li et al. 2015). Loop structure formation may be enabled by various mechanisms, including base pairing in the presence of inverted repetitive sequences (such as Alu elements) in flanking introns (Jeck et al. 2013), dimerization of RNA-binding proteins that bind to specific motifs located in flanking introns, such as quaking (Conn et al. 2015), muscleblind (Ashwal-Fluss et al. 2014), or FUS (Errichelli et al. 2017), or the presence of non-repetitive complementary sequences (Liang and Wilusz 2014). Furthermore, data from a study by Kramer et al. indicate that the production of circRNAs can be controlled by multiple hnRNP (heterogeneous nuclear ribonucleoprotein) and SR (serine-arginine) proteins, cis-acting elements and trans-acting splicing factors, acting in a combinatorial manner (Kramer et al. 2015).
Alternatively, if the lariat structure generated throughout pre-mRNA splicing avoids linearization, is cleaved from the nascent mRNA, maintains a circular form, and circumvents subsequent degradation, it can lead to the formation of circular intronic RNAs (ciRNAs) by so-called lariat-driven circularization. Above all, circRNAs composed of exons that are not surrounded by suitable complementary sequences in the pre-mRNA can arise by the process of exon skipping (Zhang et al. 2013). Additionally, tRNA intronic circular (tricRNAs) produced during metazoan tRNA splicing have been described (Lu et al. 2015). Chu et al. (2018) have even recently summarized 10 different types of circRNA.
During backsplicing (a), introns are excised from the pre-mRNA, and exons are covalently linked. The presence of long flanking introns, inverted repeat elements (such as Alu elements), or trans-acting RNA binding proteins (RBPs) supports the backsplicing. Base pairing between inverted Alu elements or the dimerization of RBPs guides a downstream splice-donor site (SD) into immediate proximity with an upstream splice-acceptor site (SA). An upstream branch point (BP) attacks a downstream SD leading to attacking an upstream SA, consequently forming exone-intron circRNAs (EIcircRNAs) or internally spliced exonic circRNAs (ecircRNAs). In the exon-skipping event (b), a lariat containing both introns and one or more exons forms during canonical splicing of the primary transcript and is cleaved from the molecule. The ends of the exons are brought to proximity allowing backsplicing to take place, giving rise to an ecircRNA or EIcircRNA. Moreover, circRNAs may arise from intronic lariat precursors escaping the debranching step of canonical linear splicing resulting in circular intronic RNA (ciRNA) (c). BSJ, back-splice junction; circRNA, circular RNA; pre-mRNA, precursor messenger RNA.
3 Export and Turnover
Following biogenesis, EcircRNAs are, for the most part, exported to the cytoplasm (Jeck et al. 2013; Memczak et al. 2013), whereas EIciRNAs and ciRNAs are found in the nucleus (Zhang et al. 2013; Li et al. 2015). Although the export is not fully understood, a recent study correlated the mechanism with the molecular length (Huang et al. 2018), providing the first findings on circRNAs export from the nucleus. The study indicated that ATP-dependent RNA helicase DDX39A and the spliceosome RNA helicase DDX39B are involved in this process. In addition to the cytoplasm, circRNAs can be exported into the extracellular space encapsulated in exosomes and enriched according to the cell of origin (Sun et al. 2021).
Albeit pathways behind circRNA turnover in vivo have just recently begun to be more perspicuous, much remains unknown. Due to the lack of free 5’ and 3’ ends and the resultant resistance to exonucleases, circRNAs are characterized as remarkably stable molecules. This fact allows some circRNAs to accumulate to levels that may exceed the linear form (Jeck et al. 2013; Memczak et al. 2013). Nevertheless, they are prone to degradation by circulating endonucleases (Park et al. 2019) or cleavage mediated by miRNAs, as shown in Vitis vinifera L. (Gao et al. 2019). Degradation in a manner dependent on a highly complementary miR-671 binding site has been observed in ciRS-7 (also known as CDR1as), a reported sponge for miR-7. The bond results in circRNA cleavage activation by the protein Argonaute 2 (AGO2). It has been shown that the regulatory circuit involves the long- intergenic ncRNA (lincRNA) Cyrano binding and targeting miR-7 for degradation leading to indirect modulation of the ciRS-7 degradation by miR-671. This event can be supported by miR-7 drafting the miR-67 silencing complex to the circRNA or by yet unexplained mechanism (Kleaveland et al. 2018).
As for endonucleases, which have been linked to circRNA turnover at a more general level, data indicate that ribonuclease P (RNase P) is able to degrade m6A-modified circRNAs (Park et al. 2019) and, additionally, circRNAs are globally cleaved by 2-5A-dependent ribonuclease (Rnase L) during viral infection (Liu et al. 2019).
4 Biological Functions
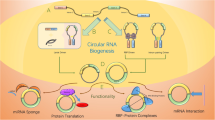
Experimental evidence suggests that circRNAs function as significant cell biology and pathophysiology regulators. It has been reported that these molecules are able to modulate gene expression at both transcriptional and post-transcriptional levels (Fig. 2).

Overview of circRNA functions
4.1 MiRNA Sponging
Although a myriad of circRNAs has been identified to date, only a minor fraction of this group has been assigned a specific biological function, with the majority being miRNA sponging in the cytoplasm (Hansen et al. 2013; Memczak et al. 2013). The process represents the inhibition of the miRNA function by binding target miRNA complementary sequences directly or indirectly to the specific circRNA “sponge”, leading to increased expression of genes that would be silenced by the RNA interference mechanism in the absence of circRNA (Wang et al. 2016). Moreover, it has been reported that most of the circRNAs act as competitive endogenous RNAs (ceRNAs), which may modulate miRNA action by binding to miRNA response elements (MREs) (Mitra et al. 2018).
Probably the best-examined circRNA, ciRS-7, contains over 70 binding sites for miR-7 in humans and more than 60 conserved binding sites across 32 vertebrates and is abundantly expressed in various tissue types, suggesting its ability to regulate the expression of miR-7 target genes (Hansen et al. 2013; Memczak et al. 2013). An in vivo functional experiment conducted on mice demonstrated that removal of the ciRS-7 locus from the genome resulted in the downregulation of miR-7 expression (Piwecka et al. 2017), while, on the contrary, other findings indicated a negative correlation between ciRS-7 and miR-7 expression (Weng et al. 2017).
Another circRNA, circABCB1O, has been identified to sponge miR-1271 resulting in carcinogenesis promotion in breast cancer cells through the bioinformatically predicted circABCB10/miR-1271 axis (Liang et al. 2017b). CircHIPK3, derived from the homeodomain interacting protein kinase 3 gene, has been reported multiple times to participate in tumorigenesis and cancer progression, and promoted, for example, gallbladder cancer cell growth by sponging miR-124 (Kai et al. 2018). Also, circ-Foxo3, derived from the forkhead box O3 gene, allegedly repressed cell proliferation and cell cycle progression by sponging specific miRNAs that regulate the production of Foxo3 mRNA (Du et al. 2016). Interestingly, it has been hypothesized that higher circRNA levels may work in cooperation to sponge many miRNAs (Bachmayr-Heyda et al. 2015). Nonetheless, this claim has not been verified experimentally.
However, this effect is presumably intrinsic only to some circRNAs. Militello et al. (2017) concluded that investigated circRNAs do not significantly bind miRNAs, so it is impossible to ascribe this function across the board to all circRNAs. For many circRNAs, their abundance is also limiting in this regard, i.e., the fact that there are not enough molecules present in the cell for efficient sponging of the relevant miRNAs.
4.2 CircRNA–Protein Interaction
Based on the available cross-linking immunoprecipitation datasets, some circRNAs are suggested to interact with RBPs in various manners. Besides the interaction with proteins during their own biogenesis, as mentioned previously, circRNAs have been investigated as protein sponges or decoys regulating RBP-dependent functions (Ashwal-Fluss et al. 2014; Abdelmohsen et al. 2017), protein scaffolds providing the formation of the enzyme–substrate complex (Du et al. 2016; Zeng et al. 2017), protein function enhancers (Zhang et al. 2013; Li et al. 2015), or recruiting proteins to a specific place of need (Chen et al. 2018). The emerging role of circRNA–protein interactions has been recently covered in a comprehensive review by Das et al. (2021).
In human cervical carcinoma HeLa cells, for example, circPABPN1 binds to RBP Hu-antigen R (HuR), a protein influencing gene expression programs and hence cellular phenotypes. This leads to the suppression of the nuclear poly(A) binding protein 1 (PABPN1) translation (Abdelmohsen et al. 2017). Holdt et al. (2016) demonstrated that circular antisense non-coding RNA in the INK4 locus (circANRIL), generally expressed at higher levels than the linear isoform, acts atheroprotectively by regulating rRNA maturation and atherogenesis pathways. The circRNA binds to pescadillo homolog 1 (PES1), an essential 60S-preribosomal assembly factor, effectuating the diminishment of pre-rRNA processing and ribosome biogenesis in vascular smooth muscle cells and macrophages. Consequently, circANRIL induces nucleolar stress and p53 activation, resulting in the possible removal of hyperproliferative cells from atherosclerotic plaques.
The previously acknowledged study of Du et al. (2016) reported, for the first time, a circRNA serving as a protein scaffold. In murine fibroblasts, circ-Foxo3 interacts with p-21- and cyclin-dependent kinase 2 (CDK2). The formation of the circ-FOxo3-p21-CDK2 ternary complex prevents the CDK2 from its function, subsequently resulting in cell cycle arrest. In another study, a circRNA highly expressed in neonatal human cardiac tissue, circ-Amotl1, binds to both 3-phosphoinositide-dependent protein kinase 1 (PDK1), leading to the PDK1-dependent phosphorylation of AKT1 and its subsequent nuclear translocation. In a mouse model, this cascade was reported to have a cardioprotective function (Zeng et al. 2017). Moreover, circRNAs can modulate the host genes transcription by controlling the initiation and elongation of RNA polymerase II (RNAP II) (Zhang et al. 2013). Further PAR-CLIP analysis showed that EIcircRNAs co-immunoprecipitate with RNAP II and can modulate transcription (Li et al. 2015).
CircRNAs have been hypothesized to have a protein-recruiting function. It was confirmed for circRNA FECR1, which coordinates the regulation of DNA methylating and demethylating enzymes and consequently represents an upstream controller of breast cancer growth. FECR1 recruits TET1 to the promoter region of FLI1, its host gene promoting tumor growth in solid tumors and acting as an oncogenic driver in hemato-oncological diseases, which leads to the demethylation of CpG sites and active transcription (Chen et al. 2018).
4.3 Translation of CircRNAs
Although circRNAs are commonly classified as ncRNAs, some of these molecules are confirmed and thousands are predicted to include a putative open reading frame (ORF) with an upstream IRES, thus, serving as a template for peptide synthesis. For linear mRNAs, translation depends on the presence of the 5’ cap and poly(A) end, which are absent in circRNA. However, as demonstrated with engineered circRNAs with m6A RNA modification incorporated (Wang and Wang 2015), or in the process of translation using the internal ribosome entry site (IRES), which allows binding of ribosome independently of the 5′ cap, has been observed. In human cells, the sequences identified as IRES contain a modified N6-methyladenosine (Yang et al. 2017), the most abundant base modification of RNA.
Albeit the functional relevance of most circRNA-originated peptides remains elusive, they are often shortened versions of the canonical protein lacking crucial domains to fulfill their original function and allegedly might compete with their full-length counterparts. They may naturally act in various manners, such as decoys, modulators of alternative protein complexes. For example, FBXW-185aa (Yang et al. 2018), PINT87aa (Zhang et al. 2018a, b), and SHPRH-146aa (Zhang et al. 2018a) (peptides derived namely from circ-FBXW7, circPINTexon2, and circ-SHPRH) are all suggested to work as tumor suppressors in glioblastoma. Moreover, recent findings of a novel study suggested a pervasive translation of circRNAs, providing profound implications in translation control (Fan et al. 2022).
Peptides of circRNA origin can be expressed under conditions unusual to the canonical protein, such as in times of cellular stress, or function in different cellular compartments to the canonical protein, therefore function as regulated product within the cell (Legnini et al. 2017; Pamudurti et al. 2017). The translation of circ-ZNF609, a circRNA derived from zinc finger protein 609 gene, or m6A-containing circRNAs were confirmed significantly increased upon heat shock proteins in human cells using advanced mass spectrometry-based analysis and overexpression of circRNA plasmids (Legnini et al. 2017; Yang et al. 2017). Circ-ZNF609 contains an ORF spanning from the start codon, as well as the linear counterpart, and terminating at an in-frame STOP codon, created upon circularization. The molecule was shown to control myoblast proliferation specifically (Legnini et al. 2017). Additionally, it was found that after starvation of D. melanogaster circ-Mbl-peptide was produced and stabilized (Pamudurti et al. 2017).
5 Profiling and Analysis of CircRNAs
As circRNAs have increasingly fallen under the radar of many researchers, the demand for molecular biology and bioinformatics methods to investigate these molecules with precision and depth has also increased. Current advances in high-throughput genomic technology and bioinformatic strategies allow circRNAs characterization in precise detail, yet there are many experimental hardships associated specifically with circRNA research. A set of guidelines for circRNA studies on the authors’ experience in detail has been recently proposed (Nielsen et al. 2022).
5.1 Purification and Enrichment of CircRNAs
Expression of circRNAs has been observed in many cell types and tissues (Rahimi et al. 2021), and detectable levels have also been identified in various liquid biopsies (Wang et al. 2022). Besides, their higher stability compared to linear RNAs may favor them when analyzing partially degraded samples. These molecules can be isolated with various commercially available kits designed for total RNA isolation, nonetheless, their low abundance in total RNA, estimated at 1%, from biological material is problematic (Salzman et al. 2013). Apparently, this relates mainly to rapidly proliferating cells where circRNAs have less time to accumulate to the detection limit (Bachmayr-Heyda et al. 2015).
As the first step of circRNA profiling, total RNA isolation, quantity, and quality control are recommended (Nielsen et al. 2022). For relatively low-abundant circRNAs, a subsequent enrichment step prior to cDNA library preparation is crucial. The most widespread strategy is incorporating RNase R treatment into the procedure. This enzyme hydrolyzes linear RNA in the 3'-5’ direction, leaving circular and lariat structures intact. However, Xiao and Wilusz (2019) showed that RNAs with highly structured 3’ ends, such as snRNAs and histone mRNAs, resist RNase R treatment. Authors also found that RNase R stalls in the structure of many mRNAs, particularly in G-rich sequences that are referred to as G-quadruplexes. It was demonstrated that standard protocols involving RNase R could fail to digest > 20% of all highly expressed linear RNAs. An improved protocol for a more efficient method of RNase R treatment resolved these difficulties by adding a polyadenylation step, modifying the composition of the reaction buffer, and prolonging incubation time. In the same year, an approach called RPAD (RNase R Treatment, Polyadenylation, and Poly(A) + RNA Depletion) was reported, which includes an extra rRNA depletion step. It claims to drastically deplete linear RNAs leading to the isolation of highly pure circRNAs from total RNA pools (Pandey et al. 2019), ready as an input for the cDNA library preparation procedure and subsequent bioinformatic analysis.
5.2 Bioinformatic Approaches on CircRNAs and Their Global Profiling
Characterization and accurate quantification of circRNA is critical, but it is still a challenging research problem. Linear RNA and circRNA are co-expressed, and the presence of the BSJ is the only unique feature of circRNAs that can distinguish them from linear RNA. To precisely quantify circRNA expression levels using RNA-seq, it is crucial to perform deep sequencing with longer reads (>100 nucleotides) (Nielsen et al. 2022). Sequencing coverage not only increases sensitivity but also leads to higher false-positive rates. Fortunately, the false positive rate of BSJs can be reduced by adding the circRNA enrichment step during library preparation, determining the amount of circRNA in enriched vs. non-enriched samples, and setting a strict threshold for circRNA read counts (Szabo and Salzman 2016).
Most circRNA detection algorithms support both single-end (SE) and paired-end (PE) data analysis. Using PE sequencing leads to higher sensitivity and, in some cases, even higher specificity, but it is not always a possible approach due to its higher price (Szabo and Salzman 2016). CircRNA identification tools first recognize fusion junction sites and then use different filters to detect the corresponding circRNA. These tools can be simply classified as annotation-dependent and independent (Ye et al. 2017). Tools requiring gene annotation can only be used in an organism with well-annotated genes, such as a mouse or human (Memczak et al. 2013). De novo prediction algorithms are not dependent on gene annotation and show promising results but with a higher rate of false positives. Finally, combining several detection tools with different analytical approaches is strongly endorsed to diminish false positive results (Hansen et al. 2016).
Short-read sequencing platforms cannot detect the internal arrangement of exons located more than 100–150 nucleotides adjacent to the BSJ of circRNAs. In recent studies, Oxford Nanopore Technology long-read sequencing has been used to characterize circRNA isoforms and discovered novel exons and microexons. A relatively low read count limits the long-read sequencing methods compared to second-generation PE sequencing (Rahimi et al. 2021). According to the results, nanopore long-read sequencing detected more circRNAs than short-read sequencing. Results suggest that long-read sequencing could determine expression levels of circRNAs that are not detected by short-read sequencing technology. The data show that highly expressed circRNAs are generally observed by both long-read and short-read sequencing methods. In addition, each method also detects many lower expressed circRNAs not discovered by the other technique (Rahimi et al. 2021). The length of the sequenced fragments was proved to be a significant factor affecting the efficiency of circRNA recognition by the CIRI-long tool (Zhang et al. 2021).
Interestingly, a study of scRNAseq (small conditional RNA sequencing) detected ~ 90% of specifically expressed circRNAs in fewer than ten cells in both human and mouse samples, suggesting further difficulties in detecting circRNAs by bulk RNA-seq techniques (Wu et al. 2022). Over of circRNA profiling is shown in Fig. 3.

Overview of circRNA profiling
5.3 PCR-Based Analyses
As there is a vast resemblance between circRNA and linear RNA sequences and, furthermore, enzymatic procedures used during library preparation for high-throughput sequencing may produce false-positive BSJs, it is necessary to perform the experimental validation (Nielsen et al. 2022). Since conventional RT-qPCR cannot distinguish precisely between circular and linear variants, the method must be modified. Among the most used approaches is RT-qPCR with divergent primers allowing the generation of BSJ amplicons (Fig. 4) followed or not by gel electrophoresis and/or Sanger sequencing (Abdelmohsen et al. 2017). Subsequent Sanger sequencing can further verify the junction sequence and may be able to reveal the complete sequence of the studied circRNA. Frequently, RT-qPCR must be preceded by RNase R treatment, especially when detecting low-abundant circRNAs. Recently, a comprehensive protocol for circRNA quantification levels in cells using digital droplet PCR (dd-PCR) was reported (Das et al. 2022).

Illustration of the complementary sequence of primers and the direction of replication on circular and linear molecules
Particular attention should be paid to the RT step. Most RT enzymes exhibit template-switching activity and can probably contribute significantly to false positives in the detection of circRNAs (Tang et al. 2018). Other authors observed continuous circumnavigation of the single circRNA molecule, producing concatemeric cDNAs as a consequence of RT by either random primers or a gene-specific primer. Therefore, each of these cDNA molecules provided multiple priming targets for the RT-qPCR primer pairs, resulting in a false increase in circRNA levels. This difficulty can be minimized by combining PCR detection with hybridization-based methods such as, for example, northern blotting or microarrays, or in situ hybridization-based microscopy (Nielsen et al. 2022).
6 CircRNAs as Potential Biomarkers in Human Cancers
Since highly stable circRNAs are implicated in various diseases, including cancer, their biomarker potential in diagnosis or disease development monitoring has been considered. Additionally, they are currently being explored as potential therapeutic targets. Aberrant expression of some circRNAs in cells may be associated with their phenotypic changes, such as angiogenesis, reduced apoptosis, or the ability to invade and migrate, characteristics that can lead to their uncontrolled proliferation and/or metastasizing. Recently, a comprehensive review outlining circRNA modulation of cancer hallmarks and molecular pathways, resulting in cancer progression support and metastasizing, has been published (Yarmishyn et al. 2022). Although only several are highlighted in this chapter, numerous studies have focused on circRNA involvement in the pathophysiology of many diseases.
A focus on circRNA expression aberrations in papillary thyroid carcinoma has revealed several tissue circRNAs that could be exploited as biomarkers or therapeutic targets. Increased expression of circ_0058124 was observed, which silencing in vitro reduced the viability of tumor cells, their ability to generate colony formation and migration, and led to increased apoptosis. In the mouse model, silencing prevented circ_0058124 tumor growth (Liu et al. 2020). Similar properties were also revealed for circFOXM1, the blocking of which prevented tumor growth in vitro and in vivo. Its increased expression correlated with larger tumor size, higher grade, and the occurrence of lymph node metastases (Ye et al. 2020).
In circ_0016788, higher expression was observed in hepatocellular carcinoma tissue than in non-tumor tissue and was determined to be an independent factor predicting overall patient survival (Cheng et al. 2020). As an opposite example, lower expression of hsa_circ_0078602 in tumor tissue correlated with poor prognosis for patient survival, as demonstrated by Kou et al. (2019).
Hsa_circ_0004585 was reported to have significantly increased expression in tumor tissues of colorectal cancer patients. Its expression was positively correlated with tumor size, suggesting the molecule’s involvement in the carcinogenesis process. In addition, hsa_circ_0004585 was also detected in peripheral blood and therefore has the potential to be used as a non-invasive diagnostic biomarker (Tian et al. 2019). Another promising diagnostic biomarker could be hsa_circ_0001696, expressed significantly higher in tumor tissue than in healthy tissue. The results of the research conducted by Li et al. (2021) suggest that decreased expression of hsa_circ_0001696 leads to an increase in the number of colonies and tumor cell migration promotion.
Zhou et al. (2021) found increased levels of circPARP4 in glioblastoma (GBM) tissue and showed that its presence promotes tumor cell division, migration, and invasiveness. An experiment in a mouse model showed that silencing circPARP4 led to significant tumor shrinkage. In contrast, lower expression of circ_0001946 in GBM cells led to increased invasiveness and proliferation of tumor cells and reduced apoptosis. This effect was further confirmed in an in vivo experiment in a mouse model, where increased expression of circ_0001946 reduced migration, invasiveness, and tumor proliferation (Li and Diao 2019). The authors of these studies suggest that the molecules under investigation have potential use in targeted therapy of GBM.
The involvement of specific circRNAs in pathophysiological cellular processes has been demonstrated not only in other cancers lung cancer but also in other types of diseases, such as Alzheimer’s disease (Dube et al. 2019) or cardiovascular diseases (Wang et al. 2016).
7 Perspective
In the last decade, countless new insights have been gained about circRNA structure, biogenesis, properties, and functions. Moreover, the involvement of these unique molecules in the pathophysiology of many diseases, consisting of their ability to regulate gene expression by various mechanisms, has been described. Recent years have also seen significant improvements in the capabilities of molecular biological and bioinformatic approaches for circRNA study in detail. Despite all this, however, the field remains challenging. It awaits further remarkable discoveries that will move us closer to a complete understanding of circRNAs and, most importantly, harnessing their potential in clinical practice and improving the well-being of patients.
References
Abdelmohsen K, Panda AC, Munk R et al (2017) Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol 14:361–369
Ashwal-Fluss R, Meyer M, Pamudurti NR et al (2014) circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 56:55–66
Bachmayr-Heyda A, Reiner AT, Auer K et al (2015) Correlation of circular RNA abundance with proliferation–exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep 5:8057
Chen N, Zhao G, Yan X et al (2018) A novel FLI1 exonic circular RNA promotes metastasis in breast cancer by coordinately regulating TET1 and DNMT1. Genome Biol 19:218
Cheng F, Wang L, Zhang J (2020) Circular RNA 0016788 displays as a biomarker for tumor progression and poor prognosis in surgical hepatocellular carcinoma patients. J Clin Lab Anal 34:e23300
Chu Q, Bai P, Zhu X et al (2018) Characteristics of plant circular RNAs. Brief Bioinform 21:135–143
Conn SJ, Pillman KA, Toubia J et al (2015) The RNA binding protein quaking regulates formation of circRNAs. Cell 160:1125–1134
Das A, Das D, Panda AC (2022) Quantification of Circular RNAs Using Digital Droplet PCR. J Vis Exp 187
Das A, Sinha T, Shyamal S, Panda AC (2021) Emerging Role of Circular RNA–Protein Interactions. Noncoding RNA 7:48
Du WW, Yang W, Liu E et al (2016) Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res 44:2846–2858
Dube U, Del-Aguila JL, Li Z et al (2019) An atlas of cortical circular RNA expression in Alzheimer disease brains demonstrates clinical and pathological associations. Nat Neurosci 22:1903–1912
Errichelli L, Dini Modigliani S, Laneve P et al (2017) FUS affects circular RNA expression in murine embryonic stem cell-derived motor neurons. Nat Commun 8:14741
Fan X, Yang Y, Chen C et al (2022) Pervasive translation of circular RNAs driven by short IRES-like elements. Nat Commun 13:1–15
Gao Z, Li J, Luo M et al (2019) Characterization and Cloning of Grape Circular RNAs Identified the Cold Resistance-Related Vv-circATS1. Plant Physiol 180:966–985
Hansen TB, Jensen TI, Clausen BH et al (2013) Natural RNA circles function as efficient microRNA sponges. Nature 495:384–388
Hansen TB, Venø MT, Damgaard CK et al (2016) Comparison of circular RNA prediction tools. Nucleic Acids Res 44:e58
Huang C, Liang D, Tatomer DC et al (2018) A length-dependent evolutionarily conserved pathway controls nuclear export of circular RNAs. Genes Dev 32:639–644
Jeck WR, Sorrentino JA, Wang K et al (2013) Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19:141–157
Kai D, Yannian L, Yitian C et al (2018) Circular RNA HIPK3 promotes gallbladder cancer cell growth by sponging microRNA-124. Biochem Biophys Res Commun 503:863–869
Kleaveland B, Shi CY, Stefano J et al (2018) A Network of Noncoding Regulatory RNAs Acts in the Mammalian Brain. Cell 174:350-362.e17
Kou P, Zhang C, Lin J et al (2019) Circular RNA hsa_circ_0078602 may have potential as a prognostic biomarker for patients with hepatocellular carcinoma. Oncol Lett 17:2091–2098
Kramer MC, Liang D, Tatomer DC et al (2015) Combinatorial control of Drosophila circular RNA expression by intronic repeats, hnRNPs, and SR proteins. Genes Dev 29:2168–2182
Legnini I, di Timoteo G, Rossi F et al (2017) Circ-ZNF609 Is a Circular RNA that Can Be Translated and Functions in Myogenesis. Mol Cell 66:22-37.e9
Li PF, Zhang ZX, Yuan X et al (2021) Hsa_circ_0001696 modulates cell proliferation and migration in colorectal cancer. Oncol Lett 21:154
Li X, Diao H (2019) Circular RNA circ_0001946 acts as a competing endogenous RNA to inhibit glioblastoma progression by modulating miR-671-5p and CDR1. J Cell Physiol 234:13807–13819
Li Z, Huang C, Bao C et al (2015) Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol 22:256–264
Liang D, Tatomer DC, Luo Z et al (2017a) The output of protein-coding genes shifts to circular RNAs when the pre-mRNA processing machinery is limiting. Mol Cell 68:940
Liang D, Wilusz JE (2014) Short intronic repeat sequences facilitate circular RNA production. Genes Dev 28:2233–2247
Liang HF, Zhang XZ, Liu BG et al (2017b) Circular RNA circ-ABCB10 promotes breast cancer proliferation and progression through sponging miR-1271. Am J Cancer Res 7:1566
Liu CX, Li X, Nan F et al (2019) Structure and Degradation of Circular RNAs Regulate PKR Activation in Innate Immunity. Cell 177:865-880.e21
Liu L, Yan C, Tao S et al (2020) Circ_0058124 Aggravates the Progression of Papillary Thyroid Carcinoma by Activating LMO4 Expression via Targeting miR-370-3p. Cancer Manag Res 12:9459–9470
Lu Z, Filonov GS, Noto JJ et al (2015) Metazoan tRNA introns generate stable circular RNAs in vivo. RNA 21:1554–1565
Memczak S, Jens M, Elefsinioti A et al (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495:333–338
Militello G, Weirick T, John D et al (2017) Screening and validation of lncRNAs and circRNAs as miRNA sponges. Brief Bioinform 18:780–788
Mitra A, Pfeifer K, Park KS (2018) Circular RNAs and competing endogenous RNA (ceRNA) networks. Transl Cancer Res 7:S624–S628
Nielsen AF, Bindereif A, Bozzoni I et al (2022) Best practice standards for circular RNA research. Nat Methods 19:1208–1220
Pamudurti NR, Bartok O, Jens M et al (2017) Translation of CircRNAs. Mol Cell 66:9-21.e7
Pandey PR, Rout PK, Das A et al (2019) RPAD (RNase R treatment, polyadenylation, and poly(A)+ RNA depletion) method to isolate highly pure circular RNA. Methods 155:41–48
Park OH, Ha H, Lee Y et al (2019) Endoribonucleolytic Cleavage of m6A-Containing RNAs by RNase P/MRP Complex. Mol Cell 74:494-507.e8
Piwecka M, Glažar P, Hernandez-Miranda LR et al (2017) Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 357:eaam8526
Rahimi K, Venø MT, Dupont DM et al (2021) Nanopore sequencing of brain-derived full-length circRNAs reveals circRNA-specific exon usage, intron retention and microexons. Nat Commun 12:4825
Salzman J, Chen RE, Olsen MN et al (2013) Cell-type specific features of circular RNA expression. PLoS Genet 9:e100377
Sun R, Liu W, Zhao Y et al (2021) Exosomal circRNA as a novel potential therapeutic target for multiple myeloma-related myocardial damage. Cancer Cell Int 21:311
Szabo L, Salzman J (2016) Detecting circular RNAs: bioinformatic and experimental challenges. Nat Rev Genet 17:679–692
Tang C, Yu T, Xie Y et al (2018) Template switching causes artificial junction formation and false identification of circular RNAs. bioRxiv 259556
Tian J, Xi X, Wang J et al (2019) CircRNA hsa_circ_0004585 as a potential biomarker for colorectal cancer. Cancer Manag Res 11:5413–5423
Wang K, Long B, Liu F et al (2016) A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J 37:2602a–2611a
Wang Y, Wang Z (2015) Efficient backsplicing produces translatable circular mRNAs. RNA 21:172–179
Wang Z, Yu R, Chen X et al (2022) Clinical utility of cerebrospinal fluid-derived circular RNAs in lung adenocarcinoma patients with brain metastases. J Transl Med 20:74
Weng W, Wei Q, Toden S et al (2017) Circular RNA ciRS-7-A Promising Prognostic Biomarker and a Potential Therapeutic Target in Colorectal Cancer. Clin Cancer Res 23:3918–3928
Wu W, Zhang J, Cao X et al (2022) Exploring the cellular landscape of circular RNAs using full-length single-cell RNA sequencing. Nat Commun 13:3242
Xiao MS, Wilusz JE (2019) An improved method for circular RNA purification using RNase R that efficiently removes linear RNAs containing G-quadruplexes or structured 3’ ends. Nucleic Acids Res 47:8755–8769. https://doi.org/10.1093/NAR/GKZ576
Yang Y, Fan X, Mao M et al (2017) Extensive translation of circular RNAs driven by N6-methyladenosine. Cell Res 27:626–641
Yang Y, Gao X, Zhang M et al (2018) Novel Role of FBXW7 Circular RNA in Repressing Glioma Tumorigenesis. J Natl Cancer Inst 110:304–315
Yarmishyn AA, Ishola AA, Chen CY et al (2022) Circular RNAs Modulate Cancer Hallmark and Molecular Pathways to Support Cancer Progression and Metastasis. Cancers (basel) 14:862
Ye CY, Zhang X, Chu Q et al (2017) Full-length sequence assembly reveals circular RNAs with diverse non-GT/AG splicing signals in rice. RNA Biol 14:1055–1063
Ye M, Hou H, Shen M et al (2020) Circular RNA circFOXM1 Plays a Role in Papillary Thyroid Carcinoma by Sponging miR-1179 and Regulating HMGB1 Expression. Mol Ther Nucleic Acids 19:741–750
Zeng Y, Du WW, Wu Y et al (2017) A Circular RNA Binds To and Activates AKT Phosphorylation and Nuclear Localization Reducing Apoptosis and Enhancing Cardiac Repair. Theranostics 7:3842–3855
Zhang J, Hou L, Zuo Z et al (2021) Comprehensive profiling of circular RNAs with nanopore sequencing and CIRI-long. Nat Biotechnol 39:836–845
Zhang M, Huang N, Yang X et al (2018a) A novel protein encoded by the circular form of the SHPRH gene suppresses glioma tumorigenesis. Oncogene 37:1805–1814
Zhang M, Zhao K, Xu X et al (2018b) A peptide encoded by circular form of LINC-PINT suppresses oncogenic transcriptional elongation in glioblastoma. Nat Commun 9:4475
Zhang Y, Zhang XO, Chen T et al (2013) Circular intronic long noncoding RNAs. Mol Cell 51:792–806
Zhou J, Wang H, Hong F et al (2021) CircularRNA circPARP4 promotes glioblastoma progression through sponging miR-125a-5p and regulating FUT4. Am J Cancer Res 11:138
Acknowledgements
The project National Institute for Cancer Research (Programme EXCELES, ID Project Number LX22NPO5102)—Funded by the European Union—Next Generation EU.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Ruckova, M., Al Tukmachi, D., Slaby, O. (2023). Biology of Circular RNAs and Methodological Approaches to Their Study. In: Barciszewski, J. (eds) RNA Structure and Function. RNA Technologies, vol 14. Springer, Cham. https://doi.org/10.1007/978-3-031-36390-0_15
Download citation
DOI: https://doi.org/10.1007/978-3-031-36390-0_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-36389-4
Online ISBN: 978-3-031-36390-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)