
Abstract
Is the time ripe for microbiome to be levered in clinical hepatology? Is the preventive, predictive, and personalized medicine (3PM/PPPM) realm offering a framework for just an intellectual exercise in the world of natura cogitans, or can we already predict, personalize, and prevent liver diseases by targeting microbiome? Can the knowledge be extrapolated to other branches of clinical medicine? In the following, these and other questions are approached from the perspective of a clinical hepatology enrouted in a resource-aware healthcare setting—an environment not usually perceived as being conducive to the translation of ideas from the bench to bedside. And that is just it: to include microbiome into mindsets possessed by 3PM irrespective of the setting. This chapter is offered to the readers who hesitate at the front of the door of perception thinking that microbiome is not for them and that they can do as well without it.
Microbiome science seems to be entering medicine with the new millennium but Hippocrateses “All disease origins in the gut” and ancient’s China “Yellow soup for the soul” could well be its predecessors. However, there is no doubt that the advances in molecular biology and computer science enabled us to look beyond the Petri dishes and discover the microbiome’s unknown universe. Trillions of microbes outnumber body cells and genes by the factors of 1.3 and one-two orders, respectively. Ninety-five percent of diseases have their pathophysiology linked to a deranged microbiome which—weighing more than the brain, became the heaviest organ of the body. Fascination with these numbers notwithstanding, it is now clear that what matters more is the microbiome composition, function, nutrients, predators, metabolites, and interactions. We know that the “healthy microbiome” is a relatively stable ecosystem entrenched in a baby gut as a relatively distinct enterotype by the means of mode of birth, mother’s touch, and early-life events. With its abundance and diversity, it resembles a rainforest. Its metabolism helps the whole body thrive from the very first breastfeeding. Healthy microbiome keeps the integrity of host–environment barriers steadfast, systemic inflammation low, and liver resilience high. When dynamics underlying the microbiome composition and function change, dysbiosis ensues. This state is epitomized by a desert losing the abundance of species and their diversity. Dysbiosis can be induced by factors such as hyperhygienic lifestyle, low physical activity, highly processed food, alcohol, antibiotics, stress, etc. Dysbiosis is known or a proposed causative factor of a host of liver diseases, from hepatitis to cirrhosis, acute -on-chronic liver failure, and cancer. Many liver syndromes are preceded by a relatively specific patterns of dysbiosis which are possible targets for prevention, prediction, and personalization. Obesity, non-alcoholic fatty liver disease, alcohol use disorder, alcohol-associated liver disease, ACLF, hepatic encephalopathy, and autoimmune liver disease - to name but a few. As for the domain of 3PM in liver diseases, huge endeavors are in place on the platforms of consortia such as MicrobPredict and alike.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Aim of the Chapter
Thanks to the exponential acceleration of a microbiome science (MS), in the near future we are about to witness the technological singularity. At the explosion, the realms of the predictive, preventive, and personalized medicine (3PM/PPPM) and microbiome research are bound to gravitate to the mysterium coniunctionis. Our chapter is aimed at providing the reader with an outlook on the particular topics related to the role of microbiome in liver diseases which belong to the top-ten causes of a global burden of morbidity, mortality, and cancer. And to emphasize until now hidden potential of microbiome analysis and healthy microbiome promotion in the practical application of 3PM as well as to stimulate further research expanding the visions of the next-generation healthcare.
2 Introduction
Microbes rule the world. It is that simple [1]
Microbial ecosystem is the oldest, richest, and most diverse living ecosystem on the planet hard-wired to all its vital processes [2, 3]. The approximately two-hundred thousand years old humankind of our kind with some 10,000 generations has evolved in an unimaginably diverse niche of microbes (our 719 billionth cousins-predecessors)—whose presence around-, on-, and in the body has become the necessary condition for survival [4]. Therefore, microbes in general are not to be considered enemies, but allies. Healthy microbiome implies healthy individual in the manner akin to the known statement “Mens sana in corpore sano” (originally from the first century AD, a Latin phrase by Roman poet Juvenal, translated in English as “a healthy mind in a healthy body”). Healthy microbiome is abundant, rich, diverse, and resilient—an ecosystem similar to a rainforest [5, 6]. Being at the same time an organ of the body and external environment, the microbiome of human being—holobiome, represents more than half the body’s cells and 99% of its DNA. The main difference from the human genome is that the microbiome can be changed—consciously for 3PM, or unconsciously by just living in the modern world. Manipulation of microbiome for prediction, prevention, and personalization has become the main area of interest of the modern hepatology. Inhabitants of the Western and westernized world are living amidst the microbiome diversity crisis. Their microbiome is like the ecosystem of a desert – deprived of diversity, richness, and resilience [5]. Human microbiome diversity – the necessary condition for a good health – has co-evolved with humans over the same two hundred thousand years, but at a mutational pace of 20 min per one microbial generation. They were living in harmony with the human genome (open for a genetic change at a pace of 20 years per one generation) until recently. The microbiome extinction coincides with the modernization of our society lasting roughly five human generations (but 2.5 million microbial generations); therefore, it is of no surprise that human genome has been caught absolutely unprepared [5]. The resultant dysbiosis-associated microbiome/genome functional mismatch is the root cause behind the chronic endotoxemia and low-grade inflammation leading to a pandemic of a non-communicable diseases (NCD) including chronic liver diseases (CLD) [7]. Gut is by far the greatest, the most diverse, and the most health-influential of the human body microbiomes and, if not stated otherwise, is referred to in this chapter. Provided healthy gut microbiome is the body’s intelligence agency responsible for peaceful handling of an external affairs (stressors and diet), liver - via the gut-liver axis - is its closest internal affairs proxy.
3 Microbiome in Historical Context and State of the Art
Apart from the Ancient China’s Yellow soup for the soul—which was in all the probability the first mention of a fecal microbial transplantation (FMT) for the gut–brain axis in the history of medicine, as well as of the Ancient Greece’s “Let your food be thy medicine” (Rephrased by J. F. Cryan to “Let food for your microbes be your medicine“)—which was probably the first record on a prebiotic nature of the food, the modern microbiome science (MS) has begun with the technology: Antony van Loewenhoek’s first-ever use of the microscope in 1683 displayed his own oral microbes, some still moving (“animalcules were in such enormous numbers that all the water... seemed to be alive”). More recently, the Nobel laureate of 1908 Elie Metchnikoff (then at the Pasteur’s Institute), after leaving macrophages to their own, begun to study longevity and, as described in his book The Prolongation of Life, noticed that the oldest inhabitants of the Parsa consumed a noticeably more lactic bacteria from a fermented food. Strachan and Bloch have laid foundations to the hygienic hypothesis of a global tsunami of NCD’s but, the really new era of MS exploded (according to the PubMed statistics) some 15 years ago as a consequence of advances in molecular biology and computer science. An enormous speed of—as Susan D. Lynch put it – “the enabling tools to interrogate microbial dark matter,” begun with the discoveries of microbiome’s: (1) composition via its genetic structure sequenced by 16S sRNA technology, followed by microbiome’s, (2) functional capacity (by the shotgun metagenomics), (3) gene expression (metatranscriptomics / RNA), (4) protein catalytic function (metaproteomics), and (5) metabolic activity of molecules (metabolomics) [4, 8,9,10]. Clearly, the real MS revolution is coming now—with the microbiome structure being currently linked with its function measured by molecular inputs and outputs. This has laid the foundations for brand-new areas of research such as foodomics, personalized diets, microbiome-pharmacogenetics, phage therapy, etc. Microbiome output interacts with receptors on the nearby and distant host cells (such as GPR, FFAR, PRR, TLR, LRR, RIG-1, CTLR, CB1, FXR, TGR5), and with nerve-endings of (e.g.) vagus nerve, and create a communication web with the distant organs and systems of the body; Emeran Mayer coined the term connectome [11, 12]. Its various extensions are called gut–liver axis, gut–brain axis, gut–muscle axis, etc. [13].
4 Understanding Microbiome Taxonomy and Function
The term microbiome was introduced by Joshua Lederberg in 2001 as a community of commensal, symbiotic, and pathogenic microorganisms within a body space or other environment. In this chapter, we use it as an umbrella term.
All microorganisms are given a name based on taxonomical rank-based classification. In the currently accepted scientific Classification of Life, there are three domains of microorganisms: the Eukaryotes, Bacteria, and Archaea. Within each domain, a several level species classifications can be found. Organized in a descending scale the domain level is followed by kingdom, division/phyla, class, subclass, order, suborder, family, genus, and species; in addition, species can involve several strains [14, 15]. In the scientific classification established by Carl von Linné, each distinct species is assigned to a genus using a two-part binary name (for example Escherichia coli). In 1987, Carl Woese divided the Eubacteria into 11 divisions based on 16S ribosomal RNA (SSU) sequences, which are—with several additions—still used today [16]. In the gut so far, dozens of bacterial phyla have been identified of which Firmicutes, Bacteroidota, Proteobacteria, Actinobacteria, and Verrucomicrobia are the most abundant; from the perspective of abundance, the first two phyla represent 90% of the microbiome [15]. Other phyla include Fusobacteria, Chloroflexi, Flavobacteria, Sphingobacteria, Planctomycetes, Cyanobacteria, Thermomicrobia, Xenobacteria, Aquificae, Chlorobia, and Chrysogenetes. The most abundant of species are Enterobacterium rectale, Bacteroides vulgatus, and Escherichia coli. Beneficial are species known for their symbiotic metabolic properties (see below) such as Akkermansia muciniphila, Roseburia spp, short-chain fatty acids-producing Bifidobacteria, Lactobacillus spp., etc. [14, 17].
The gut microbiome contains all the genomes of microbes inhabiting the gut including bacteria, archaea, viruses, and fungi [18]. Spurious is the categorization of phages, viruses, plasmids, prions, viroids, and free DNA. The term microbiome, as it was originally postulated, includes not only the community of the microorganisms, but also their “theater of activity” [19]. The latter involves the whole spectrum of molecules produced by the microorganisms, including their structural elements (nucleic acids, proteins, lipids, polysaccharides), metabolites (signaling molecules, toxins, organic, and inorganic molecules), and molecules produced by coexisting hosts and structured by the surrounding environmental conditions [20]. The term microbiome is also sometimes confused with the metagenome. Metagenome, however, is clearly defined as a collection of genomes and genes from the members of a microbiota. Microbiome - personal like a fingerprint and in adulthood relatively stable - can be characterized by bacterial clusters, grouped by some (but not all) authors to a three enterotypes: I. dominated by Bacteroides, II. by Prevotella, III. by Ruminococcus [21, 22]. Microbial community composition defined by the metagenome of a single sample can be characterized by its alpha-diversity (α-diversity), a numeric value summarizing the structure of the community, with respect to its richness, evenness, or both. Alpha-diversity belongs to the most validated metagenomic marker of gastrointestinal health. The loss of diversity has been linked to severity of a multitude of diseases. There is not yet a gold standard regarding α-diversity measures, even though the number of species (or Operational Taxonomic Units) and the Shannon diversity index are the two most widely used. The measure of similarity or dissimilarity of two communities is defined by beta diversity, which quantifies the (dis-)-similarities between communities or samples. Statistical or geometry approaches such as Bray-Curtis, Jaccard, and Jensen-Shannon divergence calculate such distances by counting the overlapped components, as well as an analysis of variance (PERMANOVA) or of similarity (ANOSIM). Finally, we can calculate gamma-diversity as the total observed richness of all samples within a habitat.
When reading MS literature in hepatology, it is of uppermost importance to be aware of which taxa are discussed or compared with which. Qin et al. (using HMP database) were the first to describe alterations in microbiome typical for liver cirrhosis, Bajaj et al. have created the numeric good–to–bad taxa ratio called cirrhosis dysbiosis ratio, and Schnabl et al. shifted the attention to the microbiome’s function and precision phage therapy—all considered the pioneering endeavors opening hepatology for 3PM/PPPM [14, 23, 24].
A healthy microbiome–host interface as photographed by the group from the Stanford is associated with a several liver health-sustaining features [4, 25,26,27,28,29,30]. The first, as mentioned above, is that the microbiome is rich in the number and abundance of symbiotic microbial species and has low proportion of pathogenic microorganisms; this should produce a health-sustaining metabolic output, leading to eubiosis, thick mucus, and tightly sealed epithelium not penetrable to bacteria and their products such as PAMPs and DAMPs (Pathogen-Associated Molecular Patterns, and Damage-Associated Molecullar Patterns) [11, 15, 23, 27, 31,32,33,34,35,36,37,38,39,40,41,42,43,44]. On the one side, the healthy microbiome’s output should be rich in the liver health-sustaining molecules or microvesicles such as: short-chain fatty acids (SCFA, butyrate, propionate, acetate); vitamins; secondary bile acids; endocannabinoids and other lipids; aryl-hydrocarbon receptor ligands such as tryptophan; psychoactive substances (called by Anderson, Cryan and Dinan psychobiotics); enterosynes; biotransformed medical drugs (PD 1—programmed cell death protein 1-based immunotherapy, digoxin, acetaminophen); etc. Of note, health-sustaining microbiome metabolic output includes also microbe-associated molecular patterns (MAMP) and pathogen-associated molecular patterns (PAMP), of which the lipopolysaccharide (LPS) is the prototype as it is needed in low levels for the proper immune function, but is harmful in higher levels. As regards the biotransformation of drugs by microbiome, relevant to hepatology are the microbiome-dependent liver toxicity of paracetamol and the possibility to overcome PD 1 resistance of tumors by FMT [45, 46]. On the other hand, healthy microbiome metabolism keeps under control the levels of pro-inflammatory cytokines and toxins such as trimethylamine N-oxide (TMAO); fructoselysine; imidazole propionate (IMP); paracetamol; etc.
5 Microbiome and the Liver
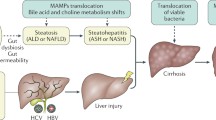
With its 10–100 trillion of symbiotic cells, up to 1500 species, dozens of millions of genes and weight up to 1.5–2 kg, gut microbiome is the richest, the most diverse and the most influential part of a holobiome [11]. Although we will not focus on the fungal microbiome and virome, their content of up to 1013 and 1015 microorganisms, respectively, is no less remarkable and certainly worth further research in hepatology [24, 47, 48]. The vertical microbiome gradient mirrors the health-sustaining abundances of microbes down the gastrointestinal tract from an oral cavity which contains 1011 bacteria, through stomach with 107, jejunum 107, ileum 1011, up to colon with 1014 bacteria, respectively [49]. The other features connected with the vertical gradient are the luminal pH (relevant also in the horizontal gradient), transit-time (more than ten-times longer in the colon than in the small intestine), oxygen pressure, etc. [11, 13]. The two most important examples of a vertical microbiome gradient breakdown in hepatology are a small intestinal bacterial overgrowth (SIBO) and oralization of gut microbiome [50,51,52]. Liver health-promoting horizontal microbiome gradient refers to the different concentrations of hydrogen (pH), oxygen, and symbionts / pathogens close to the gut mucosa as compared to the center of the gut lumen [53]. There is no doubt that a liver-health promoting microbiome is an ecosystem rich, diverse, and resilient like a rainforest and CLD-associated dysbiosis is more like a desert with a low diversity, low abundance of symbiotic species, overgrowth of pathogens, distorted gradients, and low resilience to changes [54,55,56,57].
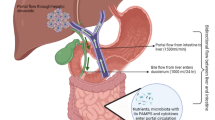
The gut–liver axis originally introduced in the 80s has been recently recognized as the key pathogenetic component in-, and a potential therapeutic target of-, virtually all the liver diseases [13, 58,59,60]. The anatomy of the axis is composed of an afferent and efferent limb, represented by the portal vein, and biliary tree, respectively. Between the two limbs lie the liver and the gut - the latter composed of microbiome and the complex intestinal barrier [61]. The healthy microbiome helps to maintain an unimpeded integrity of a gut barrier which, above many other tasks protects its intimate anatomical and functional proxy—the liver [28, 62, 63]. The barrier is made up of several interacting layers: (1) luminal microbiome with pathogens located far from mucus/mucosa; (2) the tightly sealed intestinal epithelium with a protective layer of (3) mucus; and (4) the sub-mucosal cells of the immune, lymphatic, nervous and blood systems [64]. Via portal circulation of 1 L/min and connectome, liver is the proxy encountering all the microorganisms and molecules traversing the gut barrier [65]. Although microbiome is considered an organ of the body, it is also an ecosystem representing the outer environment. These Janus-like properties are crucial for leveraging microbiome potential for 3PM purposes in hepatology. To approximate the merit of a mucus layer—one of the sine qua non’s of gut barrier - Erica Sonnenburg uses the proverb “Good fences make good neighbors.” Intestinal mucus prevents bacterial adhesion and translocation into the intestine and is composed of two parts. One, adjacent to the intestinal mucosa and called “the de-militarized zone“ for its lack of bacteria, is rich in peptidoglycans produced by Paneth cells and immunoglobulin A (IgA) by plasmatic cells [13], [60]. The second, which is in the direct contact with the luminal microbiome, is the glycocalyx produced by the goblet cells. Very rare exceptions notwithstanding, properly functioning mucus layer does not allow bacteria in the portal circulation and properly functioning microbiome governs the mucus barrier. Contact between the gut microbiome and the liver under physiological circumstances is thus relied only on the so-called postbiotics—products of microbiome. Any thinning and/or disturbed functionality of the mucus layer is the key component of the so-called leaky gut syndrome which has been proposed as the core pathophysiological mechanisms behind chronic liver diseases [66]. The most important global cause of thinning of a mucus layer is its consumption by dysbiotic bacteria; epitomized by Sonnenburgs as “hungry microbes eat you,” lack of dietary fiber which resists absorption in proximal gut causes starvation of colonic microbiome which then turns to mucus as the preferred source of substrates and energy [62]. Penetration of endotoxins such as LPS through the leaky gut results [67] in activation of the inflammatory process and inflammasomes. Moreover, dysbiosis induces MALT via regulatory T lymphocytes (Treg) and Th17 to the synthesis of transforming growth factor beta 1 (TGF β1), interleukin 17 (IL17), which regulate adipogenesis [35] and inflammation by Foxp3+Tregs processes—thus contributing to the development of liver inflammation and steatosis. Tight junctions, integral part of the barrier can be loosened by direct effect of alcohol metabolism, high-fat diet, and dysbiosis, which further accentuate leaky gut syndrome and close-up the vicious circle leading to progressive CLD [68]. Vertical microbiome gradient derangement, contributing to a leaky gut syndrome is characterized by the small intestinal bacterial overgrowth, and oralization of gut microbiome – both driven majorly by changes in bile acids [50, 69,70,71].
In summary, the proposed cascade of events, leading to the tsunami of CLDs has been primarily driven by the Western lifestyle-induced extinction of microbiome diversity which has taken place over just the few last generations of human evolution and therefore could not be followed by an adaptation on the side of the human host genome [5]. This disequilibrium leads to the leaky gut, translocation of bacteria and toxins to the portal blood, creating an inflamed intrahepatic milieu leading to an attack to the liver parenchyma by the reactive oxygen species, inflammatory molecules, and toxic metabolites; established is the state of chronic metabolic endotoxemia, impaired metabolic homeostasis, liver steatosis, inflammation, and fibrosis [13, 60, 72].
6 Liver Diseases and the Microbiome
6.1 General Considerations
In the Western and westernized world, the burden of CLDs has been increasing and this trend is predicted to continue [73,74,75]. The main drivers behind the tsunami are how we move, eat, drink, think, feel, and what we consider important and true; with a resulting 1.5 billion global cases of CLD, caused in the West mostly by ALD and NAFLD, accompanied by an autoimmune syndromes and hepatitis C [69, 70, 76, 77]. The individual and societal toll is mostly associated with the acute decompensation of cirrhosis (AD) and the syndrome of an acute-on-chronic liver failure (ACLF) [78,79,80]. Before AD/ACLF, the usual timeframe of CLD’s progression spans over twenty plus years, leaving plenty of room for a 3PM intervention. Most, if not all, of the CLDs are to a certain extent pathogenetically linked with dysbiosis; however, in ALD, NAFLD, autoimmune etiologies, and in cirrhotic stage of all the etiologies, the dysbiosis is considered the key pathogenetic component [13, 80,81,82,83,84]. It is important to acknowledge that, the chicken-egg puzzle of what is damaged first—microbiome, or liver, has not yet been solved.
In any case, microbiome has become decimated and hostile by the lack of dietary fiber and gastric acid, long-term racism of sugar, tribism of ethanol, and genocide of antibiotics, xenobiotics, and psychopathic hygiene [5]. Deprived of a citizenship, multiculturalism, livelihood, manna and soma, microbiome fires weapons such as LPS, PAMPs, DAMPs, toxic bile acid cocktails, cytolysin, candidalysin, and kynurenine [14, 70, 85]. The liver receives the blow and responds with an inflammatory cytokines and more toxic bile acid cocktails; it becomes stuffed by inflammatory cells and, as a consequence, hepatocytes cease to function or die [13, 59, 68].
There is also a chaos on the border: The intestinal mucus layer has been demolished by chemicals and eaten by gut bacteria - hungry due to the lack of dietary fiber; immune cells, mediators, and reactive oxygen species are scattered widely and gut pathogens have broken the gut barrier and translocate to the host portal blood to attack the liver. the liver is injured again and responds back again. And then the gut again. Up until cirrhosis stage of CLD will have evolved and, provided no effective therapy intervenes, cirrhosis decompensates, and other failing organs step in: kidneys; brain; coagulation; lungs. And, while before the decompensation patients might have had mild or no symptoms, at the stage of AD/ACLF, they are hospitalized, often on ICU with a dramatically reduced short-term survival [70, 78, 80, 86]. The difficult task to predict and prevent decompensation or to personalize its management is being undertaken by the Microb-Predict and other consortia and laboratories and scientists; on the other hand, the core concept of 3PM/PPPM is to react to these trends and therefore, it has been integrating ACLD and MS to its nucleus for transcription, translation, and action.
6.2 Alcohol-Associated Liver Disease
In addition to the general mechanisms behind the public health—scale domino of dysbiosis—leaky gut—risk of CLD, one in five people older than 15 years also drink alcohol [87, 88]. In the region of authors, the situation is even worse as the alcohol-associated liver disease (ALD; if not stated otherwise, ALD serves as an umbrella term, encompassing all the syndrome’s subtypes) is the leading cause of liver-related mortality, liver transplantation, liver morbidity, and cost to society [89, 90]. Most of the heavy drinkers will develop steatosis and, in at least one-third of them, it will progress to steatohepatitis [91,92,93]. However, to explain that “only” 8–20% of heavy drinkers will develop cirrhosis is the call for research into a genetic and enviromental (aggravating or protective) co-factors, of which one of the most promising is the microbiome [85, 94,95,96].
The main research questions in this regard are (1) can microbiome explain the extreme spectrum of ALD phenotypes in comparable drinkers; (2) can microbiome be used for the prediction of prognosis; and (3) for preventive and/or therapeutic interventions? [97, 98]. The spectrum of ALD is extreme: on the one hand, there are individuals with alcohol use disorder (AUD) who drink regularly harmful doses and have steatosis with minimal or no liver disease; and, on the other hand, many individuals drinking the same daily doses will develop severe alcoholic hepatitis (SAH), progressive ALD, cirrhosis, AD/ACLF, hepatocellular carcinoma (HCC), and are transplanted or die [87]. The one special entity, SAH, can develop on the top of almost any stage of ALD (albeit most of patients have cirrhosis), has no durably effective therapy and up to 50% 90-day mortality [93, 99].
In the landmark study from the Perlemuter group, researchers were able to determine the specific dysbiosis which was associated with the severity of ALD and to transmit ALD by transplanting this microbiome to animals [100]. This was the proof-of-concept that alcohol drives liver disease by hijacking microbiome, its metabolites (PAMPs, beta-glucan, bile acids, low indoles, and SCFA), and gut barrier/tight junctions, as recently reviewed elsewhere [94, 101]. Alcohol-associated dysbiosis concerns all the main domains—bacteria, fungi, and viruses. As for the bacterial dysbiosis, it has been shown that, patients with AUD and AH have dysbiosis with gradually decreasing beta diversity and Shannon alpha-diversity as compared to healthy non-alcohol-drinking controls [97, 102]. However, severity of AH was not predicted by microbiome analysis [103]. There were several taxa which were associated with the severity of SAH, prognosis, and response to therapy, e.g., increased Veillonella and decreased Prevotella. However, the most striking feature of AH-associated dysbiosis was orders-of-magnitude higher abundance of Enterococcus faecalis in AH as compared to both AUD and healthy controls; surprisingly enough, this feature did not correlate with the clinical outcome until the subjects with E. faecalis were further analyzed for strains producing the toxin, cytolysin. Then there was a gradual increase in cytolysin-positive strains along the cohorts (controls—AUD—AH) and this time the presence of cytolysin–positive E. faecalis was strongly associated with mortality (p<0.0001). Absolutely fascinating example of personalized/precision approach is to target these cytolysin-producing strains with phages - as already shown with C. crescentus and E. faecalis phages in experiment [102].
Taking into account that the effective therapy for SAH is an unmet need, it is of little surprise that the clinical research focused on new therapies targeting microbiome [98, 104, 105]. The first study by Philips et al. has shown improved survival in patients with SAH not previously responding to standard of care, if they were administered FMT from healthy donors via the upper gastrointestinal tract for eight days (p = 0.018 vs. historical controls) [106]. More studies with FMT are needed and, according to the clinicaltrials.gov, several are ongoing (one of them at the institutions of the authors—NCT58806). A cautionary note is needed regarding FMT, as drug-resistant bacteria such as E. coli and viruses such as Monkeypox can be transmitted [107, 108]. Promising piece of evidence for a predictive, preventive and personalized potential of certain gut microbial taxa is the case of Akkermansia muciniphila as a marker, predictor and therapeutic agent in AH [109]. Yet another way of addressing microbiome in ALD and AH/SAH are antibiotics [98]. A recent study has shown a promising alleviation of dysbiosis after therapy with rifaximin [97]. Moreover, the interesting 3PM aspect of this multicentric study was that baseline microbiome signature was able to predict prognosis and response to therapy with rifaximin.
Very recent research from the Schnabl group has shed light on up to now rarely scrutinized virome and fungal microbiome in three cohorts (non-alcoholic controls, patients with AUD, and patients with AH). In the study on virome, authors have shown graded alterations along the three cohorts, with the most remarkable increases in AH patients of the Shannon diversity and of mammalian viruses such as Parvoviridae, Circoviridae, and Herpesviridae (especially EBV); moreover, results were corelated with the severity of liver disease as reflected in the model for end-stage liver disease (MELD) score [110]. In another set of three studies, they provided evidence of a graded fungal overgrowth in patients with AUD and AH as compared to non-drinking controls [111,112,113]. The most significant overgrown fungus was candida albicans. As to the fungal diversities, beta diversity was not able to discriminate between AUD and AH, but was clearly distinctive of healthy controls; Shannon alpha-diversity was highest in AH patients and lowest in controls (similar to viral-, and at a difference with bacterial diversity). One of the conclusions was that fungal overgrowth is more dependent on the alcohol intake than on the stage of ALD.
As the crucial determinants of pathogenesis and mortality in ALD are microbiome and long-term abstinence, respectively, it is logical to attempt to address them simultaneously. This has been done by Bajaj et al. in their landmark phase 1 randomized study on patients with AUD addressing craving and AUD outcomes by single-dose FMT [114]. The FMT enema was selected in the OpenBiome for enrichment with Lachnospiraceae and Ruminococcaceae. Patients in active arm post-FMT had increased Shannon diversity, increased abundance of SCFA-producing Roseburia, Alistipes, and Odoribacter—usually decreased in ALD and cirrhosis, and reduced craving and AUD-related events.
6.3 Non-Alcoholic Fatty Liver Disease (NAFLD)
With the global prevalence of 25–30%, NAFLD (currently in the process of re-naming to metabolic-associated fatty liver disease—MAFLD and, in 2023 to steatotic liver disease - MASLD) is the most common etiology of CLD and, as a consequence of a pandemic of diabesity, it is the fastest growing indication for liver transplantation [76, 115, 116]. Similar to ALD, NAFLD encompasses a spectrum with only 10–20% of patients progressing to cirrhosis via non-alcoholic steatohepatitis with fibrosis over at least 10 years—the interval open for 3PM intervention [117, 118]. And, also similar to ALD, microbiome is one of the prime suspects modulating the phenotype toward benign or progressive disease or HCC [101, 119, 120]. As NAFLD is considered the liver manifestation of metabolic syndrome, transfer of obesity by FMT in animals has been taken as a proof-of-concept that NAFLD pathophysiology may be driven by dysbiosis [121, 122].
Obesity plus inactivity with sarcopenia, and insulin resistance-mediated delivery of free fatty acids from adipose tissue to the liver are the main factors leading to the first hit in the multiple-hit hypothesis of NAFLD. In the meantime, obesity is - according to some - associated with certain enterotypes, and certain microbiome signatures are associated with metabolic endotoxemia, low-grade inflammation, oxidative stress, endogenous alcohol production and various other “second and further hits,” to the already steatotic liver; this cascade of events leads to a progression of NAFLD to NASH, cirrhosis, liver failure, and/or HCC [120, 123,124,125]. NAFLD-associated dysbiosis is characterized by reduced SCFA producing Firmicutes, Ruminococcaceae, Prevotella, and Faecalibacterium and over-abundance of Bacteroides, Ruminococcus, Proteobacteria, and Enterobacteriaceae—the latter linked with the production of alcohol, the pathophysiological step toward NAFLD known as autobrewery [120, 126, 127].
However, microbial and metabolical signatures typical for NAFLD and its progression are less well characterized and more controversial than in ALD[59, 101]. Apart from autobrewery mechanism of liver injury, over-abundant gram-negative bacteria increase levels of LPS which inflames the gut and makes it leaky, activates inflammasome in the liver and, recruit macrophages to the adipose tissue; at the same time, depleted taxa produce less SCFA with their positive functions left lacking, which closes the vicious circle of a leaky gut, translocation, inflammation, metabolic derangement, and liver injury [128, 129]. Crucial in development and progression of NAFLD and NASH is the interplay between bile acids, FXR, FGF 19, and microbiome; dysbiosis leads to a skewed bile acid signaling with downstream effect on fibrogenesis [130,131,132].
Outlier between the usual microbiome-based pathophysiological concepts is the relationship between SCFA-producing bacteria and progression of NAFLD [101]. While SCFA are in general, as well as in other liver diseases, considered beneficial molecules and their producers a beneficial members of microbiome—usually associated with less inflammation, better energy metabolism, satiety, better gut barrier and a good liver prognosis, in NAFLD the associations tend to differ [103, 133]: Higher stool SCFA (and their producers, such as several Roseburia species and Faecalibacterium prausnitzii) of animals and patients with NAFLD associates with a more progressive disease, more inflammation, more fibrosis, and worse prognosis [134].
The foundations of a modern predictive and personalized medicine in a wider NAFLD realm were laid by the landmark study by Zeevi et al. [135]. Authors have shown that the main determinant of a metabolic response to a defined meal (the primordial pathomechanism in NAFLD) was the microbiome. They postulated the possibility of a microbiome analysis-based personalized nutritional intervention in a foreseeable future. A more recent follow-ups on this line of research are personalized approach to a weight loss, tailored according to host–microbiome characteristics; and the meta-analysis showing that, Lactobacillus supplementation positively impacts on glycemic and lipid indices [136, 137].
Twenty-one studies (considerably heterogeneous) were scrutinized in a meta-analysis of the first-generation probiotics and synbiotics in NAFLD patients; improvements in liver enzymes, steatosis, and liver stiffness were found but personalized recommendations on the certain type of biotic for certain patients/phenotypes of NAFLD could not be drawn [138]. As stated above, Lactobacillus supplementation had a positive impact on glycemic and lipid indices [136, 137]. Moreover, specific strains of Faecalibacterium prausnitzii were found to regulate microbiome and improve NAFLD in mice [139]. Currently, there are no ongoing studies with the next-generation biotics such as phages, or engineered bacteria in NAFLD/NASH. One outstanding exception is the domain of microbiome-bile acid signaling, where the focus of recent interest has been the FXR/FGF-19 pathway; obeticholic acid and engineered FGF 19 analogue are the studied molecules, with biopsy-proven NASH the indication. Of interest in this regard is as of now unpublished finding from GwangPyo Ko group of a reduced liver steatosis by a cell-free supernatant (a postbiotic [P9]) of Akkermansia muciniphila via GLP-1 pathway.
FMT has been formally investigated in three studies on a metabolic syndrome and two in NAFLD; while awaiting more data, experts doubt that FMT without a causal long-term lifestyle intervention could lead to a sustained benefit [101, 140, 141].
6.4 Autoimmune Diseases and the Microbiome
Autoimmune diseases of the liver comprise three major diseases - autoimmune hepatitis (AIH) with a prevalence of 0.5–1 cases per 100,000 inhabitants [142], primary biliary cholangitis (PBC) occurring in 20–40 cases per 100,000, and primary sclerosing cholangitis (PSC) with 6–10 cases per 100,000 in the Caucasian population[143].
In general, the pathogenesis of autoimmune liver diseases is not completely understood. Recent data from the genome-wide association studies and the multi-omic (metagenomic and metabolomic) studies of the microbiome have underlined some potential mechanisms by which the microbiome could play a role in the development of autoimmunity [144, 145]: a unique pattern of genetic susceptibility to immune system recognition of antigens with various HLA haplotypes, a unique succession of changes in the microbiome during immune system maturation leading to selective immune tolerance to various antigens encountered in the environment, the state of mucosal homeostasis balancing a pro-inflammatory and gut barrier disturbing microbiota and their metabolites with anti-inflammatory and gut barrier promoting processes, a liver immune system homeostasis balancing the immune response to microbial antigens, metabolites, and signaling molecules reaching the liver from the gut by promoting either anti-inflammatory or pro-inflammatory state, a toxic effect of various food additives, industrial or household pollutants disrupting the mucosal or liver immune system homeostasis.
6.5 Autoimmune Hepatitis
Autoimmune hepatitis (AIH) is characterized by the natural history of successive bursts of varying intensity causing inflammatory destruction of hepatocytes. The actual trigger of the inflammation is unknown. The disease is not considered curable, but the established treatment is effective in the great majority of cases[146]. For patients progressing to decompensated cirrhosis liver transplantation remains the therapy of choice.
Studies of the microbiome in AIH have revealed a consistently increased abundance of Veillonella, Streptococcus, Lactobacilli, Lachnospiraceae, Bacteroides, Roseburia, Ruminococcacae, and Klebsiella. In contrast, the depletion of Bifidobacteria and Clostridiales has been reported [147]. The presence of a sufficient abundance of Bifidobacteria could also increase the chances of disease remission after therapy [148].
The mechanisms by which these bacteria might affect the pathogenesis of AIH are unknown. So far, there is no established cause and effect relationship. One hypothesis suggests that microbiota changes could lead to lower metabolic production of SCFA, increased intestinal permeability resulting in innate immunity (by RIP3) activation of liver macrophages. [149,150,151]. Moreover, the spectrum of bile acids and their metabolites has also been implicated in the regulation of T cells balancing Th17 and Treg response. Some proof-of-concept studies in animal models have confirmed these proposed mechanisms. Improvement in AIH by dietary fiber, probiotics (including Bifidobacteria and Lactobacilli), or butyrate supplementation via increase in Treg/T17 ratio, expression of tight junction proteins, decreased LPS translocation/TLR activation, and a decreased E. coli protein in the liver were displayed [150, 152, 153]. Moreover, FMT attenuated liver injury, bacterial translocation, and improved the imbalance between helper and regulatory splenic T cells [154].
6.6 Primary Biliary Cholangitis
Primary biliary cholangitis (PBC) is characterized by inflammatory destruction of the ducts transporting bile from the liver to the digestive tract. The actual trigger of the inflammation is still unknown with autoimmunity likely involved in its pathogenesis due to the frequent presence of anti-mitochondrial or specific anti-nuclear autoantibodies (anti-gp210 or anti-sp100) and the presence of lymphocytic infiltrate in the proximity of the bile ducts [155]. Without treatment, more than 50% of cases progress to cirrhosis and end-stage liver disease. Since 1987 [156], a naturally occurring secondary bile acid ursodeoxycholic acid (UDCA) has been successfully used for treatment. However, approximately 20–40% of cases do not respond to UDCA therapy [157, 158]. Second-line add-on therapy with obeticholic acid or fibrates has been used in these patients [159].
Several studies of microbiome, mainly in the Asian population, have reported an increased abundance of several species: Haemophilus, Veillonella, Clostridiales, Lactobacilli, Streptococci, Pseudomonas, Klebsiella, Bifidobacterium, and an unknown genus from the Enterobacteriaceae family [144]. In contrast, several species have been reported reduced such as Bacteroidetes, Sutterella, Oscillospira, and Faecalibacterium. The common ground for these changes is not completely understood but it could be associated with the decreased metabolic output of the butyric acid. In addition, the decrease in Faecalibacterium was associated with non-response to UDCA. As stated above, Bacteroidetes and Faecalibacterium prausnitzi are known butyrate-producing bacteria and a sufficient butyrate concentration in the gut is indispensable for healthy mucin production ensuring a normal function of the intestinal barrier [139].
In PBC, the autoantigen of anti-mitochondrial antibodies displays structural similarities with the human E2 component of the mitochondrial pyruvate dehydrogenase complex (PDC-E2). Since PDC-E2 is also a commonly occurring enzyme among the various bacterial species, exposition to this antigen through the disrupted intestinal barrier in a genetically susceptible individual may explain the origin of autoimmunity in PBC [160].
6.7 Primary Sclerosing Cholangitis
Primary sclerosing cholangitis (PSC) is characterized by the progressive fibrosing damage of the intrahepatic and extrahepatic bile ducts leading to impairment of bile flow and eventually biliary cirrhosis.
Natural history is marked by variable progression rate toward end-stage liver disease and less frequently to cholangiocarcinoma [155]. The trigger of the inflammation is not known, but autoimmunity is suspected due to its association with inflammatory bowel disease in 60–80% of cases, and frequent detection of autoantibodies against the cytoplasm of neutrophils.
Currently, there is no established treatment with liver transplantation remaining the only curative option. UDCA therapy is recommended by some authorities for its proven effect in lowering the markers of cholestasis and improving the quality of life. The studies have explored the mechanisms linking the genetic predisposition with the immune system and the microbiome. The microbial composition can influence the balance of the immune system directly when microbes or their fragments cross the dysfunctional intestinal barrier. Moreover, products of bacterial metabolism also influence the host immune system, as some are being absorbed into the bloodstream.
Current understanding of the pathogenesis of PSC highlights the central role of the microbiome in the maintenance of chronic inflammation by shifting the mucosal homeostasis toward intestinal barrier dysfunction, activation of several lineages of the immune system, and homing of gut-tropic lymphocytes in the liver endothelium [161]. Studies have reported compositional changes of the gut microbiome in PSC compared with healthy controls and newer studies are emerging with data on the functional differences.
Microbiome studies in patients with PSC have revealed consistent enrichment in various taxa including Clostridiales, Streptococcus salivarius, Veillonella dispar, Ruminococcus gnavus, Bacteroides fragilis, Enterobacteriaceae, Lactobacilli, Blautia, Enterococcus, Rothia. A shotgun metagenomic sequencing of the fecal microbiome also showed a markedly reduced gene richness compared to healthy controls [144]. Authors have concluded that Veillonella species were more prevalent, with decreased abundance after UDCA therapy; however, the mechanisms by which Veillonella is more abundant and how it may affect the natural history of the disease have not yet been deciphered [144].
Interestingly, patients with associated inflammatory bowel disease have a distinct profile of the microbiome compared to patients with pure PSC or healthy controls [162, 163]. A recent study of the fungal microbiota in PSC patients reported increased diversity with increased abundance of Exophiala genus and Sordariomycetes class and a decrease in Sacharomycetacae [164]. Bile microbiota has also been studied in PSC patients, but the results are not consistently different from healthy controls [144].
In contrast, oral microbiome changes copied those of the fecal microbiota displaying an increased abundance of Streptococcus salivarius, Veillonella parvula, Actinomyces, and Bifidobacterium in PSC patients compared to healthy controls [165].
Functional studies have revealed lowered content of the butyrate, a different metabolite content, and a decreased total bile acid pool with a lower conversion from primary to secondary bile acids [166]. Increased concentration of secondary bile acids in the liver bile has been linked to inflammation, cholestasis, gallstone formation, and carcinogenesis, as well as to modulation of FXR or TGR5 receptors on the natural killer cells, liver or intestinal macrophages, or intestinal dendritic cells. Bile acids acting on both receptors modulate the immune response against inflammation by suppressing the nuclear factor NF-kB signaling pathways and modulating the balance between the Treg and Th17 cells in the gut-liver axis.
Data from the studies with vancomycin have suggested that the observed increased conversion of primary to secondary bile acids can be reversed. Treatment with vancomycin resulted in depletion of the Gram-positive Firmicutes including the Clostridium species, which are known for their dehydroxylation activity. Indeed, vancomycin decreased fecal secondary bile acids and their postprandial plasma concentration [167, 168]. This concept has been clinically tested in small trials of patients with refractory PSC receiving oral vancomycin demonstrating a positive effect while larger randomized trials are warranted [169, 170]. Other antibiotics or fecal microbial transplantation [171] have so far reported less promising results in comparison with vancomycin alone [161, 172].
6.8 Liver Cirrhosis, Acute Decompensation and Acute-On Chronic Liver Failure
Cirrhosis is the final stage of the sufficiently long-lasting chronic liver diseases of various etiologies, characterized by an increased collagen deposition, distorted architecture and, gradually decreasing volume of hepatocytes despite their intensive regeneration [70, 173]. Being the increasing cause of morbidity and mortality globally, the prevalence of cirrhosis in Slovakia is highest in the world and, liver-related mortality is the number-one cause of death in 25–45 years old [7, 174].
Recently, dysbiosis has been proposed as the key factor associated with the transition from a pre-cirrhotic stage of CLD to cirrhosis, and from compensated to decompensated cirrhosis with the time-to-decompensation of 10 years, time-to-ACLF 2 years, and time-to-death 2 months [80, 86, 175]. Gut microbiome signatures could thus become the biomarkers discriminating asymptomatic-stage cirrhosis in population for prevention, for prediction of decompensation in diagnosed yet stable/asymptomatic cirrhosis, as well as for prediction of prognosis after decompensation.
After first defining the metagenomic signature for non-invasive detection of advanced fibrosis (pre-cirrhotic stage of CLD), Loomba et al. have also detected a 19–microbes-containing signature, distinguishing cirrhosis—with an unprecedented area under receiver operating characteristic (AUROC) of 0.91 and validated it against various geographical regions and degrees of fibrosis [32, 176]. Moreover, with the 7% of adults (not aware of any liver disease) having fibrosis, this direction of research is absolutely crucial for a modern 3PM hepatology for several other reasons [177]: First, cirrhosis fulfills all the WHO criteria for screening except one—the widely available, affordable, and patient-acceptable diagnostic marker of pre-clinical stage of disease, malleable by the recall policy (median time-to decompensation 10 years); second, current two sets of non-invasive diagnostic modalities (serological and imaging) are either not universally available, or not affordable; third, to collect a stool sample and store/transport it for examination is conceivable in a mass context akin to a colorectal cancer screening; and, fourth, the only impediment (cost) is falling exponentially over the last 15 years. Our SIRIUS Microbiome Study has been designed to detect fibrosis in community and to try to find a link with a region-relevant microbiome signature (NCT05486767).
In a more advanced stage of cirrhosis, i.e., after a decompensating event has materialized (decompensated cirrhosis, median time-to-ACLF 2 years), microbiome and gut microbial metabolome can serve as a predictive biomarkers and therapeutic targets [63, 83].
Cirrhosis is among diseases with the most profound dysbiosis (as compared to AUROCs in obesity, colorectal cancer, inflammatory bowel diseases, type 2 diabetes mellitus) [178].
Dysbiosis in cirrhosis is characterized by a reduction in Bacteroidetes, Lachnospiraceae, Ruminococcaceae, and Clostridium incertae sedis XIV; and increase in Proteobacteria, Fusobacteria, Clostridium cluster XI, Streptococcaceae, Streptecoccus spp., Veilonella spp., Enterobacteriaceae, Enterococcaceae, Lactobacillaceae, Alcaligenaceae, etc., which were derived from a stool samples, mucosal biopsies, salivary samples, etc. [14, 23, 59, 71, 80, 179,180,181].
Based on deviations of cirrhotic microbiome, the cirrhosis dysbiosis ratio (CDR) was developed by Bajaj et al. and has become the prime example of how to utilize a complex microbiome output in a user-friendly way to personalize risk of patients with cirrhosis and to predict outcome—especially hepatic encephalopathy, and rehospitalizations; however, CDR requires wider external validation. Taxa selected in abovementioned studies are being scrutinized as signature predictors of a response to therapy or outcome, with the European Microb-PREDICT being the eponymous example-endeavor in the field, whose results are expected shortly. An absolute 3PM match is the sum of a microbiome-based tools to (1) predict the risk of ACLF; (2) predict therapeutic benefit and personalize it; 3) monitor effect of therapy, with the cautionary note concerning the effect size and drug treatment confounders [80].
7 Microbiome as a Therapeutic Target in Cirrhosis and AD/ACLF
Of the three next-generation therapeutic domains defined by Schnabl and described below, most are being scrutinized against liver syndromes at the right side of CLD spectrum—i.e., in cirrhosis, AD, and ACLF. Diets have been shown to modulate microbiome and outcome in cirrhotics differently if of Turkish (vegetables, fermented milk) and American (typical Western diet) type [57]. Probiotics clearly need a more precision and personalization in cirrhosis but have already shown a potential in encephalopathy and hospitalization-rate [182, 183] . The so-called next-generation probiotics are being awaited by the community with much hope. Of quite a few antibiotics studied in cirrhosis, rifaximin has received the most focused attention with a clear effect in hepatic encephalopathy but with, as of now, contradictory performances in other indications [80]. Fecal microbial transplantation has been shown to be safe and improve encephalopathy, rehospitalizations, and ACLF, with a proven safety even over the long-term follow-up [114, 184,185,186]. Less odious/more acceptable capsule formulations of FMT have been introduced by a Bajaj’s group and at least six more studies, registered in ClinicalTrials.gov are underway [187].
8 Hepatocellular Carcinoma and Microbiome
Ever since the landmark antibiotic study by Schwabe’s group confirmed the role of microbiome in the evolution of liver cancer (HCC, hepatocellular carcinoma), the field has been the focus of much interest in hepatology [188, 189]). The microbiome-HCC pathogenetic pathways have been summarized recently and are the next-step evolution of the same mechanisms which lead to CLDs and cirrhosis [190,191,192]. At present, however, rough-level microbiome analyses based on diversity and phyla have not found a differences between cirrhosis with- and without HCC; and, although the deeper-level granularity analyses revealed small differences, predicting HCC in cirrhosis based on gut microbiome analysis remains an unmet need [193,194,195,196].
One promising direction of investigation in the predictive arm of 3PM is the intratumoral (and liver parenchymal) microbiome analysis, but the field is in its statu nascendi and more studies are needed [197, 198]. As for the other areas of 3PM, a microbiome-targeted prevention (primary and secondary) of HCC is scrutinized widely via all the above-mentioned pathways but, the real-life output is still to be awaited from more than forty NCT-registered studies with rifaximin, nine with norfloxacin, and one with probiotic; no HCC preventive studies with FXR agonists are underway as of 2021. As stated above, an absolutely new 3PM direction touching indirectly the HCC arena is the prediction of a microbiome-dependent effectivity of the new anti-tumor therapies such as PD-1-based immunotherapy [199,200,201].
9 Predictive Potential of Microbiome Analysis in Liver Diseases
Currently, there is ample evidence to suggest a predictive potential of a microbiome analysis in many liver diseases. Akin to a FOBT for colorectal cancer screening, microbiome signature can pick up cirrhosis in a general population with AUROC of 0.91. The above-mentioned associations of CLDs with microbiome / metabolome signatures notwithstanding, predicting early stage CLDs is as of now an unmet need, however. In a more advanced stages of CLD, such as in compensated cirrhosis, microbiome analysis can predict deterioration (decompensation) and, in decompensated cirrhosis, it can predict outcome.
10 Microbiome as a Target for Preventive and Personalized Medicine in Liver Diseases
Naturally, the most efficient primary prevention in this regard in hepatology is to aim at a healthy microbiome. As to ascertain healthy microbiome by daily living is often beyond the reach of the common people of the Western and westernized world, 3PM-aware medicine can step in with the region- and/or person-appropriate selection of measures from the three domains of Bugs as drugs, Drug the bug, and Drugs from bugs. The spectrum of modalities is wide, spanning from the dietary intervention to FMT and engineered phage therapy for cytolysin-positive Enterococcus faecalis. Of course, ideal prevention is as far left in this spectrum as possible but, thanks to the research done it is now clear that to claim diet and probiotics effective, it must be personalized and precise. There is accumulating evidence to suggest that the personalized diet designed according to the microbiome pattern of an individual, as well as the effectivity of probiotics predicted according to the microbiome of the recipient are the directions to be taken. As hepatology—for its inherent tight junction of liver with gut - is the area of research contributing vastly to the microbiome movement, we can expect a real-life prediction, prevention, and personalization shortly.
11 Technological Challenges of Microbiome Analysis
Once the diversity of the microbial world is catalogued, it will make astronomy look like a pitiful science—(Julian Davies)
As to MS being the biggest data challenge ever (bigger than astrophysics), a teaspoon of a stool contains the data filling the memory of a ton of DVD’s [16]. Conceiving of the handling these data with respect to an evolution of patient’s microbiome over time or its comparisons between individuals, diseases, populations and of modeling the outcomes, a hundreds-to-thousands of years of computing time would be necessary. Not to speak of a microbiome metabolomics combined with a foodomics—the next steps of MS and 3PM. With the groundbreaking accrual of the NIH’s Human Microbiome Project (HMP) 4.5 trillion bases freely available for analysis, MS got the necessary first impetus [10]. Then the QIIME produced an unprecedented half a million of catalogued sequences for a reasonable computing time and money (QIIME—Quantitative Insight into Microbial Ecology, pronounced chime) [202, 203]. This fascinating translation of teradata to the point on a graph—the distance metric of an evolutionary history - was described by Lozupone of Knight’s then Colorado lab under the name UniFrac [56, 204] (Fig. 1).

The Healthy Microbiome Map. This schematic drawing has been derived from the landmark US National Institute of Health’s Human Microbiome Project (HMP). By subsequently applying of certain reductive tools to the big data provided by HMP, it was possible to get output which was understandable to a non-experts, even to a lay public: Each dot represents the microbiome of one person from one body site. It can be seen that in these meticulously selected healthy people, dots tend to gravitate together to form the “continents” of the healthy microbiome map
These endeavors have enabled opening the current chapter of MS which gave birth to such projects as The American Gut, The Microsetta Initiative, The Earth Microbiome Project, The FoodOmics, The Microb-Predict and our SIRIUS Microbiome Project (NCT05486767) [205, 206]. However, to really understand a microbiome means to understand its function rather than the composition. And, if a dynamic mapping of a microbiome with a GPS navigation can be considered a reality around the corner, understanding and leveraging the host–microbiome interactions is as of yet an unmet need. The metabolomic pathway of microbiome → protein sequence → protein structure → protein function → molecular interactions → therapies, is just being scrutinized against the computing capacity of today’s machines and community grids [20]. Over the last decade, the size of a dataset of a sequenced proteins has grown exponentially (from <10M to 175M by UniProt.org), and the size of the database of protein structures started to move (from 60K to 160K, by PDB) [207, 208]. However, even before the technology will have allowed us to leverage microbiome in our real-life 3PM clinical practice by the user-friendly gadgets, we could and should take the pains to understand more of the predictive power of this “dark matter” of our patient’s personal universes [209, 210]. Because, it is quite safe to assume that most of our patients suffer chronic dysbiosis. And we can provide them with a general advice with a subsequent more and more personalized stewardship based on a lifestyle analysis and possibly repeated sequencing of a gut microbiome - as eponymously exemplified by Larry Smarr [211,212,213]. Subsequently, real-life MS will translate to the lifelong endeavor of monitoring, understanding and manipulating microbiome for a better health—which is the 3PM at its best.
12 Visions and Perspectives of Microbiome Analysis in 3PM
From the teleological perspective, MS and 3PM are the perfect match. The reason for 3PM to act is the technological singularity we live on the brink of: Not much more than a decade-long revolution brought about by MS has provided a new meaning to the Heidegger‘s Question Concerning Technology, as well as to the Technological Singularity theory. Pausing over the meaning of a personalized medicine’s person, one recalls the famous “When I think of it I know but when you ask me I do not know.” If sobering before, talking the Person in the era of MS has become the experience outright humbling: Some thirty trillion human-person cells and 20,000 genes make but a 43% and 1% of the individual, respectively [47]. What are the remaining 39–100 trillion cells and 99% genes of the “human” body? Yes. They are the the human microbiota and microbiome, respectively. However, it is a holobiome’s feature other than a mere quantity that makes it the prime ally for the next-generation 3PM. After the last major hurdles of price and computing time will have been overcame shortly a lay, user-friendly output will become one of the most if not The Most personal and predictive tools for an unprecedently dynamic and targeted disease prevention and therapy ever. Because, at a variance with the human genome, microbiome is malleable.
What Elon Musk said to world leaders at their summit about the artificial intelligence (AI) in politics, Rob Kight conveyed to the medical community about AI in MS: “Do not think of it as of a science fiction. Think of it as of a science fact.” Imagine it is morning. You have just flushed your smart toilet and now you are looking at your face in a smart mirror. The mirror is mass-spectrometering your exhaled oral microbiome metabolic output and displays the result in a lay language and pictures. A smart toilet has already sequenced your microbiome’s terrabites and sent the result to your smart-phone app called by Rob Knight the microbiome GPS which will help you by QR to choose the right yogurt [10] (Fig. 2).

The healthy microbiome map as a template against which new samples can be scrutinized. This schematic drawing has been derived from the landmark US National Institute of Health’s Human Microbiome Project (HMP). Here you can see the example of a result from a stool sample (fecal microbiome) of a hypothetical healthy individual: the dot fits inside its respective healthy area (this time the “stool continent”). This reassuring result is clearly understandable to a layperson and it can be used for predictive and preventive purposes
If your “GPS”- “You are here“ position has moved away from the healthy area of the map, you are informed about the ensuing health risks and advised what to do to compensate for it (e.g., “Exercise 20 minutes more,” or “Try probiotic Lactobacillus,“ or “Consult your gastroenterologist for FMT”) (Fig. 3).

The microbiome “GPS.” This schematic drawing has been derived from the landmark US National Institute of Health’s Human Microbiome Project (HMP). The healthy microbiome map serves as a template against which samples from individuals and patients can be scrutinized. Here is the example of result from stool sample (fecal microbiome) of a hypothetical patient with liver cirrhosis: the (red) dot is located outside the healthy area. This particular dislocation of fecal microbiome is typical for cirrhosis and is called "oralization." As it is potentially malleable, the so-called microbiome GPS can drive patient back to the healthy area (“continent”) by specific measures (“What To Do To Get Back Here?”) from lifestyle changes through pre–pro–post-biotics to fecal microbial transplantation. Microbiome GPS for various diseases provides the opportunity to be leveraged by predictive–preventive–personalized medicine
The current price is prohibitive but, as a DNA sequencing is a million-times cheaper than 15 years ago, this technology is thought to be around the corner (genome.gov/ sequencing costs). Naturally, expected revolution in microbiome-based prediction, prevention, and personalization in medicine will inevitably concern all its areas, hepatology included [57, 60, 80, 101, 175]. After all, it has been in the realm of hepatology where Schnabl et al. proposed the next-generation approach summing the otherwise difficult-to-grasp plethora of microbiome-directed therapeutics (such as fermented foods/prebiotics, probiotics, synbiotics, postbiotics, and parabiotics) to three domains, mentioned above: “Bug as Drug,” “Drug the Bug,” and “Drugs from Bugs”; these next-generation microbiome-directed approaches have specific precision and 3PM as the leading principles [101, 214,215,216,217,218].
13 Conclusions and Recommendations
Considering the modest cumulative effect of past microbiome-based therapies in liver diseases, the next-generation approach is being launched, based on the cornerstones of prediction, prevention, and personalization as well as very specific precision, in all the three newly delineated therapeutic domains (Drugs for Bugs, Bugs as Drugs, and Drugs from Bugs). Moreover, shifts from rough-level analysis of microbiome composition to an ultimate granularity of strains are expected and, most importantly, focus on metabolic aspects will prevail. Based on a pre-emptive analysis of a functional potential of a donor stool, ideal FMT donors will be determined for particular liver diseases together with a more acceptable FMT delivery modalities.
According to the accumulated knowledge to date by the scientific research in the field of human microbiome we can undoubtedly assume that the predictive potential, potential for prevention and potential for personalization in liver diseases is simply enormous. 3PM/PPPM must essentially get ready to use this potential for the patients as well as for those who want to avoid a health deterioration. We suggest, from the point of view of liver diseases to:
-
start education activities of the population in order to increase the knowledge about liver diseases in relation to microbiome, lifestyle, and healthy diet;
-
start preventive and predictive monitoring of the population willing to implement particular suggestions for supporting their health;
-
include the knowledge on microbiome health into the routine processes of healthcare education in the specific context of liver diseases (prehabilitation, ERAS protocols, pain chronification prediction, suboptimal health monitoring as mentioned and discussed in the other chapters of this publication);
-
extend the potential of laboratory diagnostics in order to be able to provide the patient with concrete information on his/her microbiome—patterns, of dysbiosis, nutritional status, fitness status, immunity/autoimmunity status, inflammation markers monitoring, and other related factors.
Abbreviations
- 3PM:
-
Preventive, predictive, and personalized medicine
- ACLF:
-
Acute-on-chronic liver failure
- AD:
-
Acute decompensation of cirrhosis
- AH:
-
Alcoholic hepatitis
- AIH:
-
Autoimmune hepatitis
- ALD:
-
Alcohol-associated liver disease
- AUD:
-
Alcohol use disorder
- DAMPs:
-
Damage-associated molecular patterns
- ERAS:
-
Early recovery after surgery
- FFAR:
-
Free fatty acid receptor
- FGF:
-
Fibroblast growth factor
- FMT:
-
Fecal microbial transplantation
- FOBT:
-
Fecal occult blood test
- FXR:
-
Farnesoid-X-receptor
- GPR:
-
G-protein-coupled receptor
- GPS:
-
Global positioning system
- LPS:
-
Lipopolysaccharide
- MALT:
-
Mucosa-associated lymphatic tissue (in Gut referred to as GALT)
- MAMPs:
-
Microbe-associated molecular patterns
- MS:
-
Microbiome science
- NAFLD:
-
Non-alcoholic fatty liver disease
- PAMPs:
-
Pathogen-associated molecular patterns
- PBC:
-
Primary biliary cholangitis
- PPPM:
-
The predictive, preventive, and personalized medicine
- PRR:
-
Pattern recognition receptor superfamily, including membrane and cytosolic receptors such as TLR, RLR, NOD, CLR
- PSC:
-
Primary sclerosing cholangitis
- RIP:
-
Receptor-interacting protein kinase family
- SCFA:
-
Short-chain fatty acids
- SIBO:
-
Small intestinal bacterial overgrowth
- TGR5:
-
Takeda G-protein-coupled bile acid receptor
References
National Research Council (US) Committee on Metagenomics: Challenges and Functional Applications (2007) The new science of metagenomics: revealing the secrets of our microbial planet. In: The new science of metagenomics: revealing the secrets of our microbial planet. National Academies Press, Washington, DC, pp 1–158. https://doi.org/10.17226/11902
Evogeneao: The Tree of Life. http://www.evogeneao.com/en. Accessed 1 Sep 2022
Falkowski PG, Fenchel T, Delong EF (2008) The microbial engines that drive Earth’s biogeochemical cycles. Science 320:1034–1039. https://doi.org/10.1126/SCIENCE.1153213
Lynch SV, Pedersen O (2016) The human intestinal microbiome in health and disease. N Engl J Med 375:2369–2379. https://doi.org/10.1056/NEJMRA1600266
Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, Sonnenburg JL (2016) Diet-induced extinctions in the gut microbiota compound over generations. Nature 529:212–215. https://doi.org/10.1038/NATURE16504
Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M et al (2012) Human gut microbiome viewed across age and geography. Nature 486:222–227. https://doi.org/10.1038/NATURE11053
Sepanlou SG, Safiri S, Bisignano C, Ikuta KS, Merat S, Saberifiroozi M et al (2020) The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol 5:245–266. https://doi.org/10.1016/S2468-1253(19)30349-8
Zhu Q, Mai U, Pfeiffer W, Janssen S, Asnicar F, Sanders JG et al (2019) Phylogenomics of 10,575 genomes reveals evolutionary proximity between domains Bacteria and Archaea. Nat Commun 10:5477. https://doi.org/10.1038/S41467-019-13443-4
Woese CR, Fox GE (1977) Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci U S A 74:5088–5090. https://doi.org/10.1073/PNAS.74.11.5088
Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI (2007) The human microbiome project. Nature 449:7164. https://doi.org/10.1038/nature06244
de Vos WM, Tilg H, van Hul M, Cani PD (2022) Gut microbiome and health: mechanistic insights. Gut 71:1020–1032. https://doi.org/10.1136/GUTJNL-2021-326789
Fülling C, Dinan TG, Cryan JF (2019) Gut microbe to brain signaling: what happens in Vagus…. Neuron 101:998–1002. https://doi.org/10.1016/J.NEURON.2019.02.008
Albillos A, de Gottardi A, Rescigno M (2020) The gut-liver axis in liver disease: pathophysiological basis for therapy. J Hepatol 72:558–577. https://doi.org/10.1016/J.JHEP.2019.10.003
Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L et al (2014) Alterations of the human gut microbiome in liver cirrhosis. Nature 513:59–64. https://doi.org/10.1038/NATURE13568
Rinninella E, Raoul P, Cintoni M, Franceschi F, Miggiano GAD, Gasbarrini A et al (2019) What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms 7:14. https://doi.org/10.3390/MICROORGANISMS7010014
Allaband C, McDonald D, Vázquez-Baeza Y, Minich JJ, Tripathi A, Brenner DA et al (2019) Microbiome 101: studying, analyzing, and interpreting gut microbiome data for clinicians. Clin Gastroenterol Hepatol 17:218–230. https://doi.org/10.1016/J.CGH.2018.09.017
Belzer C, de Vos WM (2012) Microbes inside—from diversity to function: the case of Akkermansia. ISME J 6:1449–1458. https://doi.org/10.1038/ISMEJ.2012.6
Thursby E, Juge N (2017) Introduction to the human gut microbiota. Biochem J 474:1823–1836. https://doi.org/10.1042/BCJ20160510
Korpela K, de Vos WM (2018) Early life colonization of the human gut: microbes matter everywhere. Curr Opin Microbiol 44:70–78. https://doi.org/10.1016/J.MIB.2018.06.003
Muller EEL, Faust K, Widder S, Herold M, Martínez Arbas S, Wilmes P (2018) Using metabolic networks to resolve ecological properties of microbiomes. Curr Opin Syst Biol 8:73–80. https://doi.org/10.1016/J.COISB.2017.12.004
Knights D, et al. (2014) Rethinking “enterotypes”. Cell Host Microbe. 8;16(4):433–7. https://doi.org/10.1016/j.chom.2014.09.013.
Arumugam M, Raes J, Pelletier E, Paslier D, Yamada T, Mende DR et al (2011) Enterotypes of the human gut microbiome. Nature 473:174–180. https://doi.org/10.1038/NATURE09944
Bajaj JS, Heuman DM, Hylemon PB, Sanyal AJ, White MB, Monteith P et al (2014) Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol 60:940–947. https://doi.org/10.1016/J.JHEP.2013.12.019
Hsu CL, Duan Y, Fouts DE, Schnabl B (2021) Intestinal virome and therapeutic potential of bacteriophages in liver disease. J Hepatol 75:1465–1475. https://doi.org/10.1016/J.JHEP.2021.08.003
Rinella ME (2015) Nonalcoholic fatty liver disease: a systematic review. JAMA 313:2263–2273. https://doi.org/10.1001/JAMA.2015.5370
Kundu P, Blacher E, Elinav E, Pettersson S (2017) Our gut microbiome: the evolving inner self. Cell 171:1481–1493. https://doi.org/10.1016/J.CELL.2017.11.024
Ni JJ, Xu Q, Yan SS, Han BX, Zhang H, Wei XT et al (2022) Gut microbiota and psychiatric disorders: a two-sample Mendelian Randomization Study. Front Microbiol 12:737197. https://doi.org/10.3389/FMICB.2021.737197
Earle KA, Billings G, Sigal M, Lichtman JS, Hansson GC, Elias JE et al (2015) Quantitative imaging of gut microbiota spatial organization. Cell Host Microbe 18:478–488. https://doi.org/10.1016/J.CHOM.2015.09.002
Bäckhed F, Fraser CM, Ringel Y, Sanders ME, Sartor RB, Sherman PM et al (2012) Defining a healthy human gut microbiome: current concepts, future directions, and clinical applications. Cell Host Microbe 12:611–622. https://doi.org/10.1016/J.CHOM.2012.10.012
Cho I, Blaser MJ (2012) The human microbiome: at the interface of health and disease. Nat Rev Genet 13:260–270. https://doi.org/10.1038/NRG3182
Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS (2014) Bile acids and the gut microbiome. Curr Opin Gastroenterol 30:332–338. https://doi.org/10.1097/MOG.0000000000000057
Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A et al (2017) Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab 25:1054–1062.e5. https://doi.org/10.1016/J.CMET.2017.04.001
Cryan JF, O’riordan KJ, Cowan CSM, Sandhu KV, Bastiaanssen TFS, Boehme M et al (2019) The microbiota-gut-brain axis. Physiol Rev 99:1877–2013. https://doi.org/10.1152/PHYSREV.00018.2018
Chambers ES, Preston T, Frost G, Morrison DJ (2018) Role of gut microbiota-generated short-chain fatty acids in metabolic and cardiovascular health. Curr Nutr Rep 7:198–206. https://doi.org/10.1007/S13668-018-0248-8
Muccioli GG, Naslain D, Bäckhed F, Reigstad CS, Lambert DM, Delzenne NM et al (2010) The endocannabinoid system links gut microbiota to adipogenesis. Mol Syst Biol 6:392. https://doi.org/10.1038/MSB.2010.46
Dong F, Hao F, Murray IA, Smith PB, Koo I, Tindall AM et al (2020) Intestinal microbiota-derived tryptophan metabolites are predictive of Ah receptor activity. Gut Microb 12:1–24. https://doi.org/10.1080/19490976.2020.1788899
Knauf C, Abot A, Wemelle E, Cani PD (2020) Targeting the enteric nervous system to treat metabolic disorders? “Enterosynes” as therapeutic gut factors. Neuroendocrinology 110:139–146. https://doi.org/10.1159/000500602
Zhang X, Han Y, Huang W, Jin M, Gao Z (2021) The influence of the gut microbiota on the bioavailability of oral drugs. Acta Pharm Sin B 11:1789–1812. https://doi.org/10.1016/J.APSB.2020.09.013
Kumar K, Jaiswal SK, Dhoke GV, Srivastava GN, Sharma AK, Sharma VK (2018) Mechanistic and structural insight into promiscuity based metabolism of cardiac drug digoxin by gut microbial enzyme. J Cell Biochem 119:5287–5296. https://doi.org/10.1002/JCB.26638
Bertani B, Ruiz N (2018) Function and biogenesis of lipopolysaccharides. EcoSal Plus 8. https://doi.org/10.1128/ECOSALPLUS.ESP-0001-2018
Peng Z, Cheng S, Kou Y, Wang Z, Jin R, Hu H et al (2020) The gut microbiome is associated with clinical response to anti-PD-1/PD-L1 immunotherapy in gastrointestinal cancer. Cancer Immunol Res 8:1251–1261. https://doi.org/10.1158/2326-6066.CIR-19-1014
Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI (2009) The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 1:6ra14. https://doi.org/10.1126/SCITRANSLMED.3000322
Nagpal R, Wang S, Solberg Woods LC, Seshie O, Chung ST, Shively CA et al (2018) Comparative microbiome signatures and short-chain fatty acids in mouse, rat, non-human primate, and human feces. Front Microbiol 9:2897. https://doi.org/10.3389/FMICB.2018.02897
Kronsten VT, Tranah TH, Pariante C, Shawcross DL (2022) Gut-derived systemic inflammation as a driver of depression in chronic liver disease. J Hepatol 76:665–680. https://doi.org/10.1016/J.JHEP.2021.11.008
Kolodziejczyk AA, Federici S, Zmora N, Mohapatra G, Dori-Bachash M, Hornstein S et al (2020) Acute liver failure is regulated by MYC- and microbiome-dependent programs. Nat Med 26:1899–1911. https://doi.org/10.1038/S41591-020-1102-2
Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM et al (1979) Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021(371):595–602. https://doi.org/10.1126/science.abf3363
Rowan-Nash AD, Korry BJ, Mylonakis E, Belenky P (2019) Cross-domain and viral interactions in the microbiome. Microbiol Mol Biol Rev 83:e00044. https://doi.org/10.1128/MMBR.00044-18
Hartmann P, Lang S, Zeng S, Duan Y, Zhang X, Wang Y et al (2021) Dynamic changes of the fungal microbiome in alcohol use disorder. Front Physiol 12:699253. https://doi.org/10.3389/FPHYS.2021.699253
Barlow JT, Leite G, Romano AE, Sedighi R, Chang C, Celly S et al (2021) Quantitative sequencing clarifies the role of disruptor taxa, oral microbiota, and strict anaerobes in the human small-intestine microbiome. Microbiome 9:214. https://doi.org/10.1186/S40168-021-01162-2
Augustyn M, Grys I, Kukla M (2019) Small intestinal bacterial overgrowth and nonalcoholic fatty liver disease. Clin Exp Hepatol 5:1–10. https://doi.org/10.5114/CEH.2019.83151
Acharya C, Sahingur SE, Bajaj JS (2017) Microbiota, cirrhosis, and the emerging oral-gut-liver axis. JCI Insight 2:e94416. https://doi.org/10.1172/JCI.INSIGHT.94416
Horvath A, Rainer F, Bashir M, Leber B, Schmerboeck B, Klymiuk I et al (2019) Biomarkers for oralization during long-term proton pump inhibitor therapy predict survival in cirrhosis. Sci Rep 9:12000. https://doi.org/10.1038/S41598-019-48352-5
Swidsinski A, Loening-Baucke V, Verstraelen H, Osowska S, Doerffel Y (2008) Biostructure of fecal microbiota in healthy subjects and patients with chronic idiopathic diarrhea. Gastroenterology 135:568. https://doi.org/10.1053/J.GASTRO.2008.04.017
Tamboli CP, Neut C, Desreumaux P, Calambel JF (2004) Dysbiosis as a prerequisite for IBD. Gut 53:1057
Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R (2012) Diversity, stability and resilience of the human gut microbiota. Nature 489(489):220–230. https://doi.org/10.1038/nature11550
Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT et al (2012) Structure, function and diversity of the healthy human microbiome. Nature 486:207–214. https://doi.org/10.1038/NATURE11234
Bajaj JS, Idilman R, Mabudian L, Hood M, Fagan A, Turan D et al (2018) Diet affects gut microbiota and modulates hospitalization risk differentially in an international cirrhosis cohort. Hepatology 68:234–247. https://doi.org/10.1002/HEP.29791
Volta U, Bonazzi C, Bianchi FB, Baldoni AM, Zoli M, Pisi E (1987) IgA antibodies to dietary antigens in liver cirrhosis. Ric Clin Lab 17:235–242. https://doi.org/10.1007/BF02912537
Schnabl B, Brenner DA (2014) Interactions between the intestinal microbiome and liver diseases. Gastroenterology 146:1513–1524. https://doi.org/10.1053/J.GASTRO.2014.01.020
Wiest R, Albillos A, Trauner M, Bajaj JS, Jalan R (2017) Targeting the gut-liver axis in liver disease. J Hepatol 67:1084–1103. https://doi.org/10.1016/J.JHEP.2017.05.007
Spadoni I, Zagato E, Bertocchi A, Paolinelli R, Hot E, di Sabatino A et al (2015) A gut-vascular barrier controls the systemic dissemination of bacteria. Science 350:830–834. https://doi.org/10.1126/SCIENCE.AAD0135
Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M et al (2016) A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 167:1339–1353.e21. https://doi.org/10.1016/J.CELL.2016.10.043
Clària J, Stauber RE, Coenraad MJ, Moreau R, Jalan R, Pavesi M et al (2016) Systemic inflammation in decompensated cirrhosis: Characterization and role in acute-on-chronic liver failure. Hepatology 64:1249–1264. https://doi.org/10.1002/HEP.28740
Almeida JI, Tenreiro MF, Martinez-Santamaria L, Guerrero-Aspizua S, Gisbert JP, Alves PM et al (2022) Hallmarks of the human intestinal microbiome on liver maturation and function. J Hepatol 76:694–725. https://doi.org/10.1016/J.JHEP.2021.10.015
Donaldson GP, Lee SM, Mazmanian SK (2016) Gut biogeography of the bacterial microbiota. Nat Rev Microbiol 14:20–32. https://doi.org/10.1038/NRMICRO3552
Fukui H (2015) Gut-liver axis in liver cirrhosis: How to manage leaky gut and endotoxemia. World J Hepatol 7:425–442. https://doi.org/10.4254/WJH.V7.I3.425
Kinashi Y, Hase K (2021) Partners in leaky gut syndrome: intestinal dysbiosis and autoimmunity. Front Immunol 12:673708. https://doi.org/10.3389/FIMMU.2021.673708
Muñoz L, Borrero MJ, Úbeda M, Conde E, del Campo R, Rodríguez-Serrano M et al (2019) Intestinal immune dysregulation driven by dysbiosis promotes barrier disruption and bacterial translocation in rats with cirrhosis. Hepatology 70:925–938. https://doi.org/10.1002/HEP.30349
Acharya C, Bajaj JS (2021) chronic liver diseases and the microbiome-translating our knowledge of gut microbiota to management of chronic liver disease. Gastroenterology 160:556–572. https://doi.org/10.1053/J.GASTRO.2020.10.056
Trebicka J, Macnaughtan J, Schnabl B, Shawcross DL, Bajaj JS (2021) The microbiota in cirrhosis and its role in hepatic decompensation. J Hepatol 75(Suppl 1):S67–S81. https://doi.org/10.1016/J.JHEP.2020.11.013
Ponziani FR, Zocco MA, Cerrito L, Gasbarrini A, Pompili M (2018) Bacterial translocation in patients with liver cirrhosis: physiology, clinical consequences, and practical implications. Expert Rev Gastroenterol Hepatol 12:641–656. https://doi.org/10.1080/17474124.2018.1481747
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D et al (2007) Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56:1761–1772. https://doi.org/10.2337/DB06-1491
Moon AM, Singal AG, Tapper EB (2020) Contemporary epidemiology of chronic liver disease and cirrhosis. Clin Gastroenterol Hepatol 18:2650. https://doi.org/10.1016/J.CGH.2019.07.060
Asrani SK, Devarbhavi H, Eaton J, Kamath PS (2019) Burden of liver diseases in the world. J Hepatol 70:151–171. https://doi.org/10.1016/J.JHEP.2018.09.014
Asrani SK, Kouznetsova M, Ogola G, Taylor T, Masica A, Pope B et al (2018) Increasing health care burden of chronic liver disease compared with other chronic diseases, 2004-2013. Gastroenterology 155:719–729.e4. https://doi.org/10.1053/J.GASTRO.2018.05.032
Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM et al (2015) Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 148:547–555. https://doi.org/10.1053/J.GASTRO.2014.11.039
Pimpin L, Cortez-Pinto H, Negro F, Corbould E, Lazarus JV, Webber L et al (2018) Burden of liver disease in Europe: epidemiology and analysis of risk factors to identify prevention policies. J Hepatol 69:718–735. https://doi.org/10.1016/J.JHEP.2018.05.011
Moreau R, Jalan R, Gines P, Pavesi M, Angeli P, Cordoba J et al (2013) Acute-on-chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology 144:1426. https://doi.org/10.1053/J.GASTRO.2013.02.042
Sarin SK, Choudhury A (2016) Acute-on-chronic liver failure: terminology, mechanisms and management. Nat Rev Gastroenterol Hepatol 13:131–149. https://doi.org/10.1038/NRGASTRO.2015.219
Trebicka J, Bork P, Krag A, Arumugam M (2021) Utilizing the gut microbiome in decompensated cirrhosis and acute-on-chronic liver failure. Nat Rev Gastroenterol Hepatol 18:167–180. https://doi.org/10.1038/S41575-020-00376-3
Bajaj JS, Khoruts A (2020) Microbiota changes and intestinal microbiota transplantation in liver diseases and cirrhosis. J Hepatol 72:1003–1027. https://doi.org/10.1016/J.JHEP.2020.01.017
Acharya C, Bajaj JS (2019) Altered microbiome in patients with cirrhosis and complications. Clin Gastroenterol Hepatol 17:307–321. https://doi.org/10.1016/J.CGH.2018.08.008
Bajaj JS, Reddy KR, O’Leary JG, Vargas HE, Lai JC, Kamath PS et al (2020) Serum levels of metabolites produced by intestinal microbes and lipid moieties independently associated with acute-on-chronic liver failure and death in patients with cirrhosis. Gastroenterology 159:1715–1730.e12. https://doi.org/10.1053/J.GASTRO.2020.07.019
Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B et al (2018) The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol 15:397–411. https://doi.org/10.1038/S41575-018-0011-Z
Fairfield B, Schnabl B (2021) Gut dysbiosis as a driver in alcohol-induced liver injury. JHEP Rep 3:100220. https://doi.org/10.1016/J.JHEPR.2020.100220
Arroyo V, Moreau R, Jalan R (2020) Acute-on-chronic liver failure. N Engl J Med 382:2137–2145. https://doi.org/10.1056/NEJMRA1914900
Han S, Yang Z, Zhang T, Ma J, Chandler K, Liangpunsakul S (2021) Epidemiology of alcohol-associated liver disease. Clin Liver Dis 25:483–492. https://doi.org/10.1016/J.CLD.2021.03.009
Lee BP, Vittinghoff E, Dodge JL, Cullaro G, Terrault NA (2019) National trends and long-term outcomes of liver transplant for alcohol-associated liver disease in the United States. JAMA Intern Med 179:340–348. https://doi.org/10.1001/JAMAINTERNMED.2018.6536
Skladany L, Molcan P, Vnencakova J, Vrbova P, Kukla M, Laffers L et al (2021) Frailty in nonalcoholic fatty liver cirrhosis: a comparison with alcoholic cirrhosis, risk patterns, and impact on prognosis. Can J Gastroenterol Hepatol 2021:5576531
Skladany L, Koller T, Adamcova Selcanova S, Vnencakova J, Jancekova D, Durajova V et al (2021) Challenging management of severe chronic disorders in acute pandemic situation: chronic liver disease under COVID-19 pandemic as the proof-of-principle model to orchestrate the measures in 3PM context. EPMA J 12:1–14. https://doi.org/10.1007/s13167-021-00231-8
Singal AK, Bataller R, Ahn J, Kamath PS, Shah VH (2018) ACG clinical guideline: alcoholic liver disease. Am J Gastroenterol 113:175–194. https://doi.org/10.1038/AJG.2017.469
Dasarathy J, McCullough AJ, Dasarathy S (2017) Sarcopenia in alcoholic liver disease: clinical and molecular advances. Alcohol Clin Exp Res 41:1419–1431. https://doi.org/10.1111/acer.13425
Schnabl B (2016) Liver capsule: mechanisms of alcoholic hepatitis. Hepatology 64:276. https://doi.org/10.1002/HEP.28488
Szabo G, Thursz M, Shah VH (2022) Therapeutic advances in alcohol-associated hepatitis. J Hepatol 76:1279–1290. https://doi.org/10.1016/J.JHEP.2022.03.025
Habash NW, Sehrawat TS, Shah VH, Cao S (2022) Epigenetics of alcohol-related liver diseases. JHEP Rep 4:100466. https://doi.org/10.1016/j.jhepr.2022.100466
Mutlu EA, Gillevet PM, Rangwala H, Sikaroodi M, Naqvi A, Engen PA et al (2012) Colonic microbiome is altered in alcoholism. Am J Physiol Gastrointest Liver Physiol 302:G966. https://doi.org/10.1152/AJPGI.00380.2011
Kim SS, Eun JW, Cho HJ, Song DS, Kim CW, Kim YS et al (2021) Microbiome as a potential diagnostic and predictive biomarker in severe alcoholic hepatitis. Aliment Pharmacol Ther 53:540–551. https://doi.org/10.1111/APT.16200
German MN, Musto J, Lucey MR (2021) Novel treatments for alcoholic hepatitis. Curr Opin Gastroenterol 37:179–186. https://doi.org/10.1097/MOG.0000000000000725
European Association for the Study of Liver (2012) EASL clinical practical guidelines: management of alcoholic liver disease. J Hepatol 57:399–420. https://doi.org/10.1016/j.jhep.2018.03.018
Llopis M, Cassard AM, Wrzosek L, Boschat L, Bruneau A, Ferrere G et al (2016) Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut 65:830–839. https://doi.org/10.1136/GUTJNL-2015-310585
Lang S, Schnabl B (2020) Microbiota and fatty liver disease-the known, the unknown, and the future. Cell Host Microbe 28:233–244. https://doi.org/10.1016/J.CHOM.2020.07.007
Duan Y, Llorente C, Lang S, Brandl K, Chu H, Jiang L et al (2019) Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 575:505–511. https://doi.org/10.1038/s41586-019-1742-x
Smirnova E, Puri P, Muthiah MD, Daitya K, Brown R, Chalasani N et al (2020) Fecal microbiome distinguishes alcohol consumption from alcoholic hepatitis but does not discriminate disease severity. Hepatology 72:271–286. https://doi.org/10.1002/HEP.31178
Sarin SK, Pande A, Schnabl B (2019) Microbiome as a therapeutic target in alcohol-related liver disease. J Hepatol 70:260–272. https://doi.org/10.1016/J.JHEP.2018.10.019
Ferrere G, Wrzosek L, Cailleux F, Turpin W, Puchois V, Spatz M et al (2017) Fecal microbiota manipulation prevents dysbiosis and alcohol-induced liver injury in mice. J Hepatol 66:806–815. https://doi.org/10.1016/J.JHEP.2016.11.008
Philips CA, Pande A, Shasthry SM, Jamwal KD, Khillan V, Chandel SS et al (2017) Healthy donor fecal microbiota transplantation in steroid-ineligible severe alcoholic hepatitis: a pilot study. Clin Gastroenterol Hepatol 15:600–602. https://doi.org/10.1016/J.CGH.2016.10.029
DeFilipp Z, Bloom PP, Torres Soto M, Mansour MK, Sater MRA, Huntley MH et al (2019) Drug-resistant E. coli bacteremia transmitted by fecal microbiota transplant. N Engl J Med 381:2043–2050. https://doi.org/10.1056/NEJMoa1910437
Safety Alert Regarding Use of Fecal Microbiota for Transplantation and Additional Safety Protections Pertaining to Monkeypox Virus, FDA. https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/safety-alert-regarding-use-fecal-microbiota-transplantation-and-additional-safety-protections-0. Accessed 1 Sep 2022
Grander C, Adolph TE, Wieser V, Lowe P, Wrzosek L, Gyongyosi B et al (2018) Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut 67:892–902. https://doi.org/10.1136/gutjnl-2016-313432
Jiang L, Lang S, Duan Y, Zhang X, Gao B, Chopyk J et al (2020) Intestinal virome in patients with alcoholic hepatitis. Hepatology 72:2182–2196. https://doi.org/10.1002/hep.31459
Yang AM, Inamine T, Hochrath K, Chen P, Wang L, Llorente C et al (2017) Intestinal fungi contribute to development of alcoholic liver disease. J Clin Invest 127:2829–2841. https://doi.org/10.1172/JCI90562
Chu H, Duan Y, Lang S, Jiang L, Wang Y, Llorente C et al (2020) The Candida albicans exotoxin Candidalysin promotes alcohol-associated liver disease. J Hepatol 72:391. https://doi.org/10.1016/j.jhep.2019.09.029
Lang S, Duan Y, Liu J, Torralba MG, Kuelbs C, Ventura-Cots M et al (2020) Intestinal fungal dysbiosis and systemic immune response to fungi in patients with alcoholic hepatitis. Hepatology 71:522–538. https://doi.org/10.1002/HEP.30832
Bajaj JS, Kassam Z, Fagan A, Gavis EA, Liu E, Cox IJ et al (2017) Fecal microbiota transplant from a rational stool donor improves hepatic encephalopathy: a randomized clinical trial. Hepatology 66:1727–1738. https://doi.org/10.1002/HEP.29306
Eslam M, Sanyal AJ, George J, Sanyal A, Neuschwander-Tetri B, Tiribelli C et al (2020) MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 158:1999–2014.e1. https://doi.org/10.1053/J.GASTRO.2019.11.312
Byrne CD, Targher G (2015) NAFLD: a multisystem disease. J Hepatol 62:S47–S64. https://doi.org/10.1016/J.JHEP.2014.12.012
Loomba R, Adams LA (2019) The 20% rule of NASH progression: the natural history of advanced fibrosis and cirrhosis caused by NASH. Hepatology 70:1885–1888. https://doi.org/10.1002/HEP.30946
Caldwell S, Argo C (2010) The natural history of non-alcoholic fatty liver disease. Dig Dis 28:162–168. https://doi.org/10.1159/000282081
Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F et al (2016) The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 63:764–775. https://doi.org/10.1002/HEP.28356
Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD et al (2013) Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 57:601–609. https://doi.org/10.1002/HEP.26093
Bäckhed F, Ding H, Wang T, Hooper LV, Gou YK, Nagy A et al (2004) The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A 101:15718–15723. https://doi.org/10.1073/PNAS.0407076101
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444:1027–1031. https://doi.org/10.1038/NATURE05414
Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA et al (2011) Linking long-term dietary patterns with gut microbial enterotypes. Science 334:105–108. https://doi.org/10.1126/SCIENCE.1208344
Buzzetti E, Pinzani M, Tsochatzis EA (2016) The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 65:1038–1048. https://doi.org/10.1016/J.METABOL.2015.12.012
Mehal WZ (2013) The Gordian Knot of dysbiosis, obesity and NAFLD. Nat Rev Gastroenterol Hepatol 10:637–644. https://doi.org/10.1038/NRGASTRO.2013.146
Aron-Wisnewsky J, Vigliotti C, Witjes J, Le P, Holleboom AG, Verheij J et al (2020) Gut microbiota and human NAFLD: disentangling microbial signatures from metabolic disorders. Nat Rev Gastroenterol Hepatol 17:279–297. https://doi.org/10.1038/s41575-020-0269-9
Lee G, You HJ, Bajaj JS, Joo SK, Yu J, Park S et al (2020) Distinct signatures of gut microbiome and metabolites associated with significant fibrosis in non-obese NAFLD. Nat Commun 11:1–13. https://doi.org/10.1038/s41467-020-18754-5
Caesar R, Reigstad CS, Bäckhed HK, Reinhardt C, Ketonen M, Lundén GÖ et al (2012) Gut-derived lipopolysaccharide augments adipose macrophage accumulation but is not essential for impaired glucose or insulin tolerance in mice. Gut 61:1701–1707. https://doi.org/10.1136/GUTJNL-2011-301689
Wree A, McGeough MD, Peña CA, Schlattjan M, Li H, Inzaugarat ME et al (2014) NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J Mol Med (Berl) 92:1069–1082. https://doi.org/10.1007/S00109-014-1170-1
Mouzaki M, Wang AY, Bandsma R, Comelli EM, Arendt BM, Zhang L et al (2016) Bile acids and dysbiosis in non-alcoholic fatty liver disease. PloS One 11:e0151829. https://doi.org/10.1371/JOURNAL.PONE.0151829
Zhang C-X, Hu J, Hu K-W, Zhang C, Wang L, Xu J-M (2011) Noninvasive analysis of portal pressure by contrast-enhanced sonography in patients with cirrhosis. J Ultrasound Med 30:205–211
Jiao N, Baker SS, Chapa-Rodriguez A, Liu W, Nugent CA, Tsompana M et al (2018) Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 67:1881. https://doi.org/10.1136/GUTJNL-2017-314307
Schwenger KJP, Bolzon CM, Li C, Allard JP (2019) Non-alcoholic fatty liver disease and obesity: the role of the gut bacteria. Eur J Nutr 58:1771–1784. https://doi.org/10.1007/S00394-018-1844-5
Schwiertz A, Taras D, Schäfer K, Beijer S, Bos NA, Donus C et al (2010) Microbiota and SCFA in lean and overweight healthy subjects. Obesity 18:190–195. https://doi.org/10.1038/OBY.2009.167
Zeevi D, Korem T, Zmora N, Israeli D, Rothschild D, Weinberger A et al (2015) Personalized nutrition by prediction of glycemic responses. Cell 163:1079–1094. https://doi.org/10.1016/J.CELL.2015.11.001
Cuevas-Sierra A, Milagro FI, Guruceaga E, Cuervo M, Goni L, García-Granero M et al (2022) A weight-loss model based on baseline microbiota and genetic scores for selection of dietary treatments in overweight and obese population. Clin Nutr 41:1712. https://doi.org/10.1016/J.CLNU.2022.06.008
Qiu X, Wu Q, Li W, Tang K, Zhang J (2022) Effects of Lactobacillus supplementation on glycemic and lipid indices in overweight or obese adults: a systematic review and meta-analysis. Clin Nutr 41:1787–1797. https://doi.org/10.1016/J.CLNU.2022.06.030
Sharpton SR, Maraj B, Harding-Theobald E, Vittinghoff E, Terrault NA (2019) Gut microbiome-targeted therapies in nonalcoholic fatty liver disease: a systematic review, meta-analysis, and meta-regression. Am J Clin Nutr 110:139–149. https://doi.org/10.1093/AJCN/NQZ042
Hu W, Gao W, Liu Z, Fang Z, Wang H, Zhao J et al (2022) Specific strains of Faecalibacterium prausnitzii Ameliorate nonalcoholic fatty liver disease in mice in association with gut microbiota regulation. Nutrients 14:2945. https://doi.org/10.3390/NU14142945
Craven L, Rahman A, Nair Parvathy S, Beaton M, Silverman J, Qumosani K et al (2020) Allogenic fecal microbiota transplantation in patients with nonalcoholic fatty liver disease improves abnormal small intestinal permeability: a randomized control trial. Am J Gastroenterol 115:1055–1065. https://doi.org/10.14309/AJG.0000000000000661
Witjes JJ, Smits LP, Pekmez CT, Prodan A, Meijnikman AS, Troelstra MA et al (2020) Donor fecal microbiota transplantation alters gut microbiota and metabolites in obese individuals with steatohepatitis. Hepatol Commun 4:1578–1590. https://doi.org/10.1002/HEP4.1601
Manns MP, Lohse AW, Vergani D (2015) Autoimmune hepatitis—update 2015. J Hepatol 62:S100–S111. https://doi.org/10.1016/j.jhep.2015.03.005
Boonstra K, Beuers U, Ponsioen CY (2012) Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: a systematic review. J Hepatol 56:1181–1188. https://doi.org/10.1016/j.jhep.2011.10.025
Zheng Y, Ran Y, Zhang H, Wang B, Zhou L (2021) The microbiome in autoimmune liver diseases: metagenomic and metabolomic changes. Front Physiol 12:715852. https://doi.org/10.3389/FPHYS.2021.715852
Li B, Selmi C, Tang R, Gershwin ME, Ma X (2018) The microbiome and autoimmunity: a paradigm from the gut-liver axis. Cell Mol Immunol 15:595–609. https://doi.org/10.1038/CMI.2018.7
Volk M, Reau N (2021) Diagnosis and management of autoimmune hepatitis in adults and children: a patient-friendly summary of the 2019 AASLD guidelines. Clin Liver Dis 17:85–89. https://doi.org/10.1002/CLD.1080
Wei Y, Li Y, Yan L, Sun C, Miao Q, Wang Q et al (2020) Alterations of gut microbiome in autoimmune hepatitis. Gut 69:569–577. https://doi.org/10.1136/GUTJNL-2018-317836
Liwinski T, Casar C, Ruehlemann MC, Bang C, Sebode M, Hohenester S et al (2020) A disease-specific decline of the relative abundance of Bifidobacterium in patients with autoimmune hepatitis. Aliment Pharmacol Ther 51:1417–1428. https://doi.org/10.1111/APT.15754
Wu JL, Zou JY, de Hu E, Chen DZ, Chen L, Lu F et al (2017) Sodium butyrate ameliorates S100/FCA-induced autoimmune hepatitis through regulation of intestinal tight junction and toll-like receptor 4 signaling pathway. Immunol Lett 190:169–176. https://doi.org/10.1016/J.IMLET.2017.08.005
Zhang H, Liu M, Liu X, Zhong W, Li Y, Ran Y et al (2020) Bifidobacterium animalis ssp. Lactis 420 mitigates autoimmune hepatitis through regulating intestinal barrier and liver immune cells. Front Immunol 11:569104. https://doi.org/10.3389/fimmu.2020.569104
Zhang H, Liu M, Zhong W, Zheng Y, Li Y, Guo L et al (2021) Leaky gut driven by dysbiosis augments activation and accumulation of liver macrophages via RIP3 signaling pathway in autoimmune hepatitis. Front Immunol 12:624360. https://doi.org/10.3389/FIMMU.2021.624360
Liu Q, Tian H, Kang Y, Tian Y, Li L, Kang X et al (2021) Probiotics alleviate autoimmune hepatitis in mice through modulation of gut microbiota and intestinal permeability. J Nutr Biochem 98:108863. https://doi.org/10.1016/J.JNUTBIO.2021.108863
de Hu E, Chen DZ, Wu JL, Lu F, Chen L, Zheng MH et al (2018) High fiber dietary and sodium butyrate attenuate experimental autoimmune hepatitis through regulation of immune regulatory cells and intestinal barrier. Cell Immunol 328:24–32. https://doi.org/10.1016/J.CELLIMM.2018.03.003
Liang M, Liwen Z, Jianguo S, Juan D, Fei D, Yin Z et al (2021) Fecal microbiota transplantation controls progression of experimental autoimmune hepatitis in mice by modulating the TFR/TFH immune imbalance and intestinal microbiota composition. Front Immunol 12:728723. https://doi.org/10.3389/FIMMU.2021.728723
Chapman MH, Thorburn D, Hirschfield GM, Webster GGJ, Rushbrook SM, Alexander G et al (2019) British Society of Gastroenterology and UK-PSC guidelines for the diagnosis and management of primary sclerosing cholangitis. Gut 68:1356–1378. https://doi.org/10.1136/GUTJNL-2018-317993
Poupon RE, Balkau B, Eschwege E, Poupon R (1991) A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. UDCA-PBC Study Group. N Engl J Med 324:1548–1554. https://doi.org/10.1056/NEJM199105303242204
Gazda J, Janicko M, Drazilova S, Grgurevic I, Kanizaj TF, Koller T et al (2021) External validation of UDCA response score in Slovak and Croatian patients with primary biliary cholangitis. Can J Gastroenterol Hepatol 2021:9928065. https://doi.org/10.1155/2021/9928065
Beuers U, Trauner M, Jansen P, Poupon R (2015) New paradigms in the treatment of hepatic cholestasis: from UDCA to FXR, PXR and beyond. J Hepatol 62:S25–S37. https://doi.org/10.1016/j.jhep.2015.02.023
Hirschfield GM, Chazouillères O, Cortez-Pinto H, Macedo G, de Lédinghen V, Adekunle F et al (2021) A consensus integrated care pathway for patients with primary biliary cholangitis: a guideline-based approach to clinical care of patients. Expert Rev Gastroenterol Hepatol 15:929–939. https://doi.org/10.1080/17474124.2021.1945919
Huang MX, Yang SY, Luo PY, Long J, Liu QZ, Wang J et al (2021) Gut microbiota contributes to sexual dimorphism in murine autoimmune cholangitis. J Leukoc Biol 110:1121–1130. https://doi.org/10.1002/JLB.3MA0321-037R
Shah A, MacDonald GA, Morrison M, Holtmann G (2020) Targeting the gut microbiome as a treatment for primary sclerosing cholangitis: a conceptional framework. Am J Gastroenterol 115:814–822. https://doi.org/10.14309/AJG.0000000000000604
Kummen M, Holm K, Anmarkrud JA, Nygård S, Vesterhus M, Høivik ML et al (2017) The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut 66:611–619. https://doi.org/10.1136/GUTJNL-2015-310500
Bajer L, Kverka M, Kostovcik M, Macinga P, Dvorak J, Stehlikova Z et al (2017) Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J Gastroenterol 23:4548–4558. https://doi.org/10.3748/WJG.V23.I25.4548
Lemoinne S, Kemgang A, Ben Belkacem K, Straube M, Jegou S, Corpechot C et al (2020) Fungi participate in the dysbiosis of gut microbiota in patients with primary sclerosing cholangitis. Gut 69:92–102. https://doi.org/10.1136/GUTJNL-2018-317791
Lapidot Y, Amir A, Ben-Simon S, Veitsman E, Cohen-Ezra O, Davidov Y et al (2020) Alterations of the salivary and fecal microbiome in patients with primary sclerosing cholangitis. Hepatol Int 15:191–201. https://doi.org/10.1007/S12072-020-10089-Z
Torres J, Palmela C, Brito H, Bao X, Ruiqi H, Moura-Santos P et al (2018) The gut microbiota, bile acids and their correlation in primary sclerosing cholangitis associated with inflammatory bowel disease. United Eur Gastroenterol J 6:112–122. https://doi.org/10.1177/2050640617708953
Vrieze A, Out C, Fuentes S, Jonker L, Reuling I, Kootte RS et al (2014) Impact of oral vancomycin on gut microbiota, bile acid metabolism, and insulin sensitivity. J Hepatol 60:824–831. https://doi.org/10.1016/J.JHEP.2013.11.034
Tan LZ, Reilly CR, Steward-Harrison LC, Balouch F, Muir R, Lewindon PJ (2019) Oral vancomycin induces clinical and mucosal remission of colitis in children with primary sclerosing cholangitis-ulcerative colitis. Gut 68:1533–1535. https://doi.org/10.1136/gutjnl-2018-316599
Ali AH, Damman J, Shah SB, Davies Y, Hurwitz M, Stephen M et al (2020) Open-label prospective therapeutic clinical trials: oral vancomycin in children and adults with primary sclerosing cholangitis. Scand J Gastroenterol 55:1–10. https://doi.org/10.1080/00365521.2020.1787501
Dao A, Abidian M, Lestrange A, Mattar M, Rangnekar A, Charabaty A (2019) Oral Vancomycin induces and maintains remission of ulcerative colitis in the subset of patients with associated primary sclerosing cholangitis. Inflamm Bowel Dis 25:E90–E91. https://doi.org/10.1093/IBD/IZZ027
Allegretti JR, Kassam Z, Carrellas M, Mullish BH, Marchesi JR, Pechlivanis A et al (2019) Fecal microbiota transplantation in patients with primary sclerosing cholangitis: a pilot clinical trial. Am J Gastroenterol 114:1071–1079. https://doi.org/10.14309/AJG.0000000000000115
Kummen M, Thingholm LB, Rühlemann MC, Holm K, Hansen SH, Moitinho-Silva L et al (2021) Altered gut microbial metabolism of essential nutrients in primary sclerosing cholangitis. Gastroenterology 160:1784–1798.e0. https://doi.org/10.1053/J.GASTRO.2020.12.058
Ginès P, Krag A, Abraldes JG, Solà E, Fabrellas N, Kamath PS (2021) Liver cirrhosis. Lancet 398:1359–1376. https://doi.org/10.1016/S0140-6736(21)01374-X
Health Statistics Yearbook of the Slovak Republic (2020) Národné centrum zdravotníckych infromácií. https://www.nczisk.sk/en/Publications/Health_Statistics_Yearbooks/Pages/default.aspx. Accessed 1 Sep 2022
Tilg H, Cani PD, Mayer EA (2016) Gut microbiome and liver diseases. Gut 65:2035–2044. https://doi.org/10.1136/GUTJNL-2016-312729
Oh TG, Kim SM, Caussy C, Fu T, Guo J, Bassirian S et al (2020) A universal gut-microbiome-derived signature predicts cirrhosis. Cell Metab 32:878–888.e6. https://doi.org/10.1016/J.CMET.2020.06.005
Ginès P, Graupera I, Lammert F, Angeli P, Caballeria L, Krag A et al (2016) Screening for liver fibrosis in the general population: a call for action. Lancet Gastroenterol Hepatol 1:256–260. https://doi.org/10.1016/S2468-1253(16)30081-4
Pasolli E, Truong DT, Malik F, Waldron L, Segata N (2016) Machine learning meta-analysis of large metagenomic datasets: tools and biological insights. PLoS Comput Biol 12:e1004977. https://doi.org/10.1371/JOURNAL.PCBI.1004977
Chen Y, Yang F, Lu H, Wang B, Chen Y, Lei D et al (2011) Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 54:562–572. https://doi.org/10.1002/HEP.24423
Bajaj JS, Betrapally NS, Hylemon PB, Heuman DM, Daita K, White MB et al (2015) Salivary microbiota reflects changes in gut microbiota in cirrhosis with hepatic encephalopathy. Hepatology 62:1260–1271. https://doi.org/10.1002/HEP.27819
Bajaj JS, Hylemon PB, Ridlon JM, Heuman DM, Daita K, White MB et al (2012) Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am J Physiol Gastrointest Liver Physiol 303:G675. https://doi.org/10.1152/AJPGI.00152.2012
Holte K, Krag A, Gluud LL (2012) Systematic review and meta-analysis of randomized trials on probiotics for hepatic encephalopathy. Hepatol Res 42:1008–1015. https://doi.org/10.1111/J.1872-034X.2012.01015.X
Dhiman RK, Rana B, Agrawal S, Garg A, Chopra M, Thumburu KK et al (2014) Probiotic VSL#3 reduces liver disease severity and hospitalization in patients with cirrhosis: a randomized, controlled trial. Gastroenterology 147:1327–1337.e3. https://doi.org/10.1053/J.GASTRO.2014.08.031
Bajaj JS, Fagan A, Gavis EA, Kassam Z, Sikaroodi M, Gillevet PM (2019) Long-term outcomes of fecal microbiota transplantation in patients with cirrhosis. Gastroenterology 156:1921–1923.e3. https://doi.org/10.1053/J.GASTRO.2019.01.033
Kao D, Roach B, Park H, Hotte N, Madsen K, Bain V et al (2016) Fecal microbiota transplantation in the management of hepatic encephalopathy. Hepatology 63:339–340. https://doi.org/10.1002/HEP.28121
Shasthry SM (2020) Fecal microbiota transplantation in alcohol related liver diseases. Clin Mol Hepatol 26:294–301. https://doi.org/10.3350/CMH.2020.0057
Bajaj JS, Salzman NH, Acharya C, Sterling RK, White MB, Gavis EA et al (2019) Fecal microbial transplant capsules are safe in hepatic encephalopathy: a phase 1, randomized, placebo-controlled trial. Hepatology 70:1690–1703. https://doi.org/10.1002/HEP.30690
Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I et al (2012) Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 21:504–516. https://doi.org/10.1016/J.CCR.2012.02.007
Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S et al (2013) Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499:97–101. https://doi.org/10.1038/NATURE12347
Schwabe RF, Greten TF (2020) Gut microbiome in HCC—mechanisms, diagnosis and therapy. J Hepatol 72:230–238. https://doi.org/10.1016/J.JHEP.2019.08.016
Toffanin S, Cornella H, Harrington A, Llovet JM (2012) HCC is promoted by bacterial translocation and TLR-4 signaling: a new paradigm for chemoprevention and management. Hepatology 56:1998–2000. https://doi.org/10.1002/HEP.26080
Darnaud M, Faivre J, Moniaux N (2013) Targeting gut flora to prevent progression of hepatocellular carcinoma. J Hepatol 58:385–387. https://doi.org/10.1016/J.JHEP.2012.08.019
Ponziani FR, Bhoori S, Castelli C, Putignani L, Rivoltini L, del Chierico F et al (2019) Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology 69:107–120. https://doi.org/10.1002/HEP.30036
Ren Z, Li A, Jiang J, Zhou L, Yu Z, Lu H et al (2019) Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 68:1014–1023. https://doi.org/10.1136/gutjnl-2017-315084
Liu Y, Li S, Li W, Wang P, Ding P, Li L et al (2019) RstA, a two-component response regulator, plays important roles in multiple virulence-associated processes in enterohemorrhagic Escherichia coli O157:H7. Gut Pathog 11:53
Behary J, Amorim N, Jiang XT, Raposo A, Gong L, McGovern E et al (2021) Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat Commun 12:187. https://doi.org/10.1038/S41467-020-20422-7
Chakladar J, Wong LM, Kuo SZ, Li WT, Yu MA, Chang EY et al (2020) The liver microbiome is implicated in cancer prognosis and modulated by alcohol and Hepatitis B. Cancers (Basel) 12:1–16. https://doi.org/10.3390/CANCERS12061642
Komiyama S, Yamada T, Takemura N, Kokudo N, Hase K, Kawamura YI (2021) Profiling of tumour-associated microbiota in human hepatocellular carcinoma. Sci Rep 11:10589. https://doi.org/10.1038/S41598-021-89963-1
Routy B, le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R et al (2018) Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359:91–97. https://doi.org/10.1126/SCIENCE.AAN3706
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV et al (2018) Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359:97–103. https://doi.org/10.1126/SCIENCE.AAN4236
Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML et al (2018) The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359:104–108. https://doi.org/10.1126/SCIENCE.AAO3290
Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R (2008) Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods 5:235–237. https://doi.org/10.1038/NMETH.1184
Navas-Molina JA, Peralta-Sánchez JM, González A, McMurdie PJ, Vázquez-Baeza Y, Xu Z et al (2013) Advancing our understanding of the human microbiome using QIIME. Methods Enzymol 531:371–444. https://doi.org/10.1016/B978-0-12-407863-5.00019-8
Lozupone CA, Hamady M, Kelley ST, Knight R (2007) Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 73:1576–1585. https://doi.org/10.1128/AEM.01996-06
Thompson SK, Hoye TR (2017) Organic chemistry: molecular structure assignment simplified. Nature 547:410–411. https://doi.org/10.1038/547410A
Microb-Predict - MICROB-PREDICT. https://microb-predict.eu/. Accessed 1 Sep 2022
Gligorijević V, Renfrew PD, Kosciolek T, Leman JK, Berenberg D, Vatanen T et al (2021) Structure-based protein function prediction using graph convolutional networks. Nat Commun 12:1–14. https://doi.org/10.1038/s41467-021-23303-9
Greener JG, Kandathil SM, Jones DT (2019) Deep learning extends de novo protein modelling coverage of genomes using iteratively predicted structural constraints. Nat Commun 10:1–13. https://doi.org/10.1038/s41467-019-11994-0
Loos RJF (2012) Genetic determinants of common obesity and their value in prediction. Best Pract Res Clin Endocrinol Metab 26:211–226. https://doi.org/10.1016/J.BEEM.2011.11.003
Knights D, Parfrey LW, Zaneveld J, Lozupone C, Knight R (2011) Human-associated microbial signatures: examining their predictive value. Cell Host Microbe 10:292–296. https://doi.org/10.1016/J.CHOM.2011.09.003
Why Professor Larry Smarr freezes his own faeces - BBC News. https://www.bbc.com/news/av/health-23621357. Accessed 1 Sep 2022
Huang S, Haiminen N, Carrieri A-P, Hu R, Jiang L, Parida L et al (2020) Human skin, oral, and gut microbiomes predict chronological age. mSystems 5:e00630. https://doi.org/10.1128/MSYSTEMS.00630-19
Oh J, Conlan S, Polley EC, Segre JA, Kong HH (2012) Shifts in human skin and nares microbiota of healthy children and adults. Genome Med 4:1–11. https://doi.org/10.1186/GM378/FIGURES/4
Koutnikova H, Genser B, Monteiro-Sepulveda M, Faurie JM, Rizkalla S, Schrezenmeir J et al (2019) Impact of bacterial probiotics on obesity, diabetes and non-alcoholic fatty liver disease related variables: a systematic review and meta-analysis of randomised controlled trials. BMJ Open 9:e017995. https://doi.org/10.1136/BMJOPEN-2017-017995
Sáez-Lara MJ, Robles-Sanchez C, Ruiz-Ojeda FJ, Plaza-Diaz J, Gil A (2016) Effects of probiotics and synbiotics on obesity, insulin resistance syndrome, type 2 diabetes and non-alcoholic fatty liver disease: a review of human clinical trials. Int J Mol Sci 17:928. https://doi.org/10.3390/IJMS17060928
Sharpton SR, Schnabl B, Knight R, Loomba R (2021) Current concepts, opportunities, and challenges of gut microbiome-based personalized medicine in nonalcoholic fatty liver disease. Cell Metab 33:21–32. https://doi.org/10.1016/J.CMET.2020.11.010
Acharya C, Bajaj JS (2020) Transmitting diet-related microbial benefit through fecal microbiota transplant in NASH: can microbiota cut through the fat? Hepatol Commun 4:1559–1561. https://doi.org/10.1002/HEP4.1596
Long-Smith C, O’Riordan KJ, Clarke G, Stanton C, Dinan TG, Cryan JF (2020) Microbiota-gut-brain axis: new therapeutic opportunities. Annu Rev Pharmacol Toxicol 60:477–502. https://doi.org/10.1146/ANNUREV-PHARMTOX-010919-023628
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Skladany, L., Koller, T., Kukla, M., Soltys, K. (2023). Gut Microbiome and Liver Diseases from the Perspective of 3PM: The Predictive, Preventive, and Personalized Medicine. In: Podbielska, H., Kapalla, M. (eds) Predictive, Preventive, and Personalised Medicine: From Bench to Bedside. Advances in Predictive, Preventive and Personalised Medicine, vol 17. Springer, Cham. https://doi.org/10.1007/978-3-031-34884-6_9
Download citation
DOI: https://doi.org/10.1007/978-3-031-34884-6_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-34883-9
Online ISBN: 978-3-031-34884-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)