
Abstract
Squamous epithelium is the origin of solid tumor oral squamous cell carcinoma (OSCC). Every year, nearly 400,000 OSCC patients are added to the cancer database. Presently, chemo-radiotherapy is the main important adjuvant treatment for OSCC; nevertheless, clinical resistance (drug resistance) to chemotherapy still leads to a poor prognosis of OSCC patients. Identification of potential genes and drugs might be a significant lead for the analyses of OSCC research. We aimed to identify molecular signatures for the diagnosis of OSCC patients. Statistical methods (ANOVA, limma and SAM) were used to identify differentially expressed genes (DEGs) from two datasets GSE111585 and GSE115119. Considering the cutoff values less than 0.05 and |log FC| greater than 1, we obtained 27 up-regulated and 25 down-regulated common DEGs. Protein-protein interaction (PPI) network determined hub genes (AR, ETS1, MET, PDGFB and VAV3) using STRING database. Other biomolecules: Reporter transcription factors (HIF1A, MYC, FOXP3, E2F4, WT1, PURA, ZEB1 and USF2), microRNAs (hsa-miR-589-3p, hsa-miR-155-5p and hsa-miR-301b-3p) associated with hub-genes were determined. For the hub genes, we also performed GO and KEGG enrichment analysis. We constructed gene-drug interaction using the DGIdb database and identify targeted drugs (IMATINIB, BROMOCRIPTINE, NIFEDIPINE, PIRETANIDE and TRIAMTERENE) for OSCC.
Our study suggested that the biomarkers (hub-genes, TFs and miRNAs) and the discovered drugs might be a therapeutic target of OSCC.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- Oral squamous cell carcinomas (OSCC)
- Differentially expressed genes (DEGs)
- Hub-genes
- Transcription factors (TFs)
- miRNA
- Drug discovery
1 Introduction
Oral squamous carcinoma (OSCC) is an alarming public health issue that originated from head and neck malignant tumors [1]. Among the oral malignancies its about 90% cases are detected as OSCC [2]. Recently, the number of oral cancer cases increased in Asia and Europe. In Bangladesh, lip and oral vesicle cancers are 2nd most (11.9%) loading cancer for males and 3rd most (6.5%) loading cancer for females [3]. In India, every 12 people are affected by OSCC from 100,000 people [4]. The average survival of oral cancer is more than five years. Beginning phase (I and II) oral malignant growth might be reparable by medical procedure or radiation treatment alone yet progressed tumors (stage III and IV) are the most part treated by a medical procedure followed by radiation treatment [5]. Patients who are reached cutting edge stage have possible change of survive more than 30 months, even those who are early accomplish total clinical abatement [6]. A basic figure is the absence of prognostic improvement is that a critical extent of cancer called asymptomatic injuries which are not analyzed or treated when they arrive at a high level stage. Early revelation of cancer is the best viable means to decrease death from this disease [7]. Also, most patients are prescribed to have radiotherapy and chemotherapy after medical procedure to diminish the chance of repeat. But clinical protection from chemotherapy actually prompts a helpless forecast of OSCC patients. In this analysis, two sets of data were examined to identify a molecular signature for the treatment of OSCC patients. More importantly, our study identified significant hub-genes which might be responsible for clinical resistance of OSCC and suggested candidate drugs might be a therapeutic target of OSCC.
2 Materials and methods
2.1 Conceptual Framework
To achieve the objective of this research, a logical framework was prepared through the organized steps and the methods were used in the analysis shown in Fig. 1.

(Source Created by the authors)
Schematic diagram of this study
2.2 Data Description
To identify the biomarkers of OSCC GSE111585 and GSE115119 data sets were downloaded from the NCBI with platforms GPL14715 and GPL16955, respectively. GSE111585 dataset containing six samples of SCC9 cells and 42152 genes which were partitioned into normal group and drug resistance group [8]. Dataset GSE115119 has four samples of CAL-27 cells and 42152 genes which were partitioned into normal group and drug-resistant group.
In this study, additionally we used the list of oncogenes and tumor-suppressor genes which gave a better validation of identified DEGs [9, 10].
2.3 Differentially Expressed Gene Identification Methods
In this analysis we used three different statistical methods to identify differentially expressed genes.
-
(1)
Analysis Of Variance (ANOVA) is a parametric method used to identify differentially expressed genes [11].
-
(2)
Linear Models for Microarray Data (Limma) is a popular method based on Bayesian approach to identifying differentially expressed genes of data arising from microarray experiments [12].
-
(3)
Significance Analysis of Microarrays (SAM) is an efficient technique used for analyzing microarray gene expression data. It was established in 2001 by Virginia Tusher et. al. [13]
2.4 Functional Annotation and Pathway Analysis
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) used for finding biological information of biomarker genes. GO-terms are coordinated into three autonomous controlled ontologies: Biological process, Molecular function and Cellular component. KEGG is used for pathway analysis. We used WEB-based gene set analysis toolkit (WebGestalt) for the GO and KEGG enrichment analysis [14].
2.5 Protein-Protein Interaction (PPI) Network
We constructed a PPI network using the STRING that contains multiple proteins information. Then conceptualize the PPI networks using Cytoscape [15]. We selected a confidence score ≥ 0.4 and maximum interactors = 0 for cutoff criteria.
2.6 Reporter Transcription Factors (TFs) and miRNAs
We identified TFs DEGs interactions from HTRIdb [16]. TFs were targeted by the hub genes. For targeting miRNA prediction, we used miRBD to identify a potential miRNA-mRNA interaction network [17]. Then miRNA-mRNA regulatory network was constructed by Cytoscape.
2.7 Survival Analysis of Significance Biomarkers
We completed the prognostic analysis of hub-DEGs using SurvExpress [18]. In Kaplan-Meier plots, we considered log-rank P-value < 0.05 for checking the significant survival output.
2.8 Identification of Candidate Drugs
DGIdb database used to identify desired candidate drugs [19]. In this study, highest degrees of drug-genes network were considered to find candidate drugs for OSCC.
3 Results
3.1 Identification of Differentially Expressed Genes (DEGs)
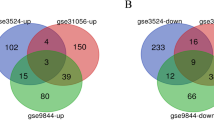
The methods ANOVA, limma and SAM identified differentially expressed genes (DEGs). The result is shown in the following Table 1. There are 2935 and 2946 common DE genes identified by the three methods for GSE111585 and GSE115119 datasets, respectively (see Fig. 2 A and B). We identified 1380 genes were up-regulated and 1555 genes were down-regulated for cisplatin-safe OSCC from GSE111585 dataset compared with parental OSCC, as like as 2226 genes were up-regulated and 720 genes were down-regulated for cisplatin-safe OSCC from GSE115119 dataset compared with parental OSCC through cluster analysis using cutoff value (|log FC| > 1 and p-value <0.05.
The up-regulated genes of those two datasets were compared with the oncogenes and down-regulated genes were compared with the genes for finding DE genes that might be responsible for tumor. Venn diagram showed that common genes of up or down-regulated from the both datasets with oncogenes or tumor-suppressor genes (TSgenes) used to obtain DEGs in cisplatin-safe OSCC cells. Diagram showed that 27 up-regulated DEGs were intersected between up-regulated genes and the list of oncogenes (see Fig. 3A) and 25 down-regulated DEGs were common between down-regulated genes and the list of TS genes (see Fig. 3B).
3.2 Protein-Protein Interaction (PPI) Network for OSCC
To identify central hub protein from differentially expressed genes (DEGs), we construct a PPI network using STRING database and Cytoscape. Figure 4 shows the PPI network for 27 common up regulated DEGs (see Fig. 3A), and hub genes are identified using hub score that has the strong association among other genes. According to high degrees and between closeness, five genes (AR, ETS1, MET, PDGFB and VAV3) are identified.
3.3 GO and KEGG Pathway Analysis for Hub DEGs
According hub-DEGs, we performed GO and KEGG pathway analysis shown in Fig. 5. The hub-DEGs are mainly involved in positive regulation of endothelial cell migration in BP category (see Fig. 5A), basal plasma membrane in the CC category (see Fig. 5B), and superoxide − generating NADPH oxidase activator activity in MF category (see Fig. 5C). KEGG enrichment analysis showed that these hub-DEGs are involved with renal cell carcinoma, Human T − cell leukemia virus 1 infection and so on (see Fig. 5D).

(Source Created by the authors)
Common DEGs and volcano plot of common DEGs. A & C for dataset GEO111585 and B & D for dataset GEO115119.
3.4 Reporter Transcription Factors (TFs) and miRNAs for OSCC
We analyzed significant transcriptional regulatory components for OSCC. We detected TFs are HIF1A, MYC, FOXP3, E2F4, WT1, PURA, ZEB1 and USF2 that are closely targeted as hub DEGs (see Table 2). These hub DEGs are also closely integrated with tumor-related miRNAs. Cytoscape was used to build a miRNA-mRNA network with setting cutoff >70 shown in Fig. 6. Among these miRNAs, some of the miRNAs are connected with multiple genes. These miRNA’s are hsa-miR-377-3p, hsa-miR-499a-5p, hsa-miR-34c-5p, hsa-miR-326, hsa-miR-34a-5p, hsa-miR-449b-5p, hsa-miR-449a, hsa-miR-589-3p which are used for next prognostic biomarker analysis.

(Source Created by the authors)
Common DEGs with oncogenes and TSgenes.

(Source Created by the authors)
PPI interaction network of hub DEGs and hub DEGs are highlighted from PPI network.
3.5 Overall Survival Significance Biomarkers for OSCC
We also performed an overall survival analysis of hub DEGs, SurvExpress is used to estimate the overall survival probabilities of the hub-DEGs. Figure 7 represents overall survival analysis of hub-DEGs which is statistically significant.
3.6 Candidate Drugs Identification Through Drug Repositioning for OSCC
We also obtain novel drugs for hub-DEGs and TFs and also constructed a drug-genes network shown in Fig. 8. Which represents the drugs with has the highest degrees (IMATINIB, BROMOCRIPTINE, NIFEDIPINE, PIRETANIDE and TRIAMTERENE), and connected with multiple genes.
4 Discussion
OSCC is a significant general medical problem with restricted treatment procedures and once again look; foundational drug opposition has disturbed the present circumstance. In this study, significant bioinformatics instruments are utilized to observe 27 up-managed DEGs that are normal with Oncogenes and 25 down-directed DEGs which are normal with TS-genes (see Fig. 3). Cross-over DEGs are utilized to build a PPI organization (see Fig. 4). PPI network recognized as AR, ETS1, MET, PDGFB and VAV3 are the center qualities those are likewise measurably huge and statistically significant (see Fig. 7). AR is related with oral disease [20].
PDGFB is associated with pancreatic cancer risk [21]. MET is associated with the progression of OSCC [22]. ETS1 is also closely related to OSCC and consider as a potential biomarker of OSCC [23]. VAV3 have been identified as important molecules in tumor genesis, tumor growth and cell migration [22].After finding the core genes, GO and KEGG examination of hub genes were performed. In GO examination, BP positively associated with “positive regulation of endothelial cell migration”, “cellular response to antibiotic”, and so on (see Fig. 5A). CC enrichment showed that hub genes were importantly associated with “basal plasma membrane”, basal part of cell”, and so on (see Fig. 5B). MF analysis enriched in “superoxide − generating NADPH oxidase activator activity”, “phosphatidylinositol − 4, and so on (see Fig. 5C).

(Source Created by the authors)
The GO (BP, CC and MF) and KEGG pathway enrichment analysis of hub-DEGs
KEGG examination described that these hub genes were associated with “Renal cell carcinoma”, “Human T − cell leukemia virus 1 infection”, and so on (see Fig. 5D). The hub DEGs targeted as reporter TFs are HIF1A, MYC, FOXP3, E2F4, WT1, PURA, ZEB1 and USF2 (see Table 2).These genes promote tumor growth, development of cancer, and are strongly associated with OSCC.
Where HIF1A TFs as an oncogene engaged in OSCC multiplication and cisplatin-safe and may act as a novel remedial objectives for OSCC treatment [24]. It also promotes human coronavirus SARS-CoV-2 replication [25]. MYC is liable for regulated tumor genesis in OSCC [26]. MYC likewise prompts to the advancement of breast cancer. Foxp3 overexpression in cancer cells predicts unfortunate endurance in oral squamous cell carcinoma [27]. E2F4 is a statistical significance gene for OSCC [28]. WT1 can be used as epigenetic oral squamous cell carcinoma biomarkers [29]. Zeb1 is responsible for oral cancer cell invasion and its suppression by resveratrol [30]. USF2 is working as a risk factor for tumor genesis [31]. Hub-DEGs are also associated with miRNAs and are also involved in the cancer cell (see Fig. 6).

(Source Created by the authors).
miRNA–mRNA network identification for hub-DEGs. Highlighted circles predicted the potential miRNAs that can regulate multiple hub-DEGs and miRNAs
hsa-miR-495-3p can be used as a biomarker for the detection of OSCC [32]. Hsa-miR-155-5pis a potential oncogenic in oral squamous cell carcinoma [33]. Hsa-miR-454-3p has been involved in head and neck squamous cell carcinoma invasion [34]. Finally, we detected candidate drugs associated with hub-DEGs such as IMATINIB, BROMOCRIPTINE, NIFEDIPINE, PIRETANIDE and TRIAMTERENE which are mostly connected with multiple genes that have also highest degrees among the other drugs (see Fig. 8). IMATINIB is an oral cancer drug that specifically represses a few protein tyrosine kinases related with human malignancy. IMATINIB is an approved drug for oral cancer [35]. BROMOCRIPTINE is used in the supported medication of metastatic breast cancer and prostate cancer-related hyperprolactinemia [36]. NIFEDIPINE is also an indicator for carcinoma associated fibroblasts (CAFs) in OSCC [37]. PIRETANIDE has a significant effect on oral cancer [38]. TRIAMTERENE is a Photosensitizing antihypertensive drug to lip cancer in non-Hispanic males [39].

(Source Created by the authors)
Overall survival analysis of hub-DEGs for OSCC.

(Source Created by the authors)
Drug-gene interaction network for OSCC. Potential drug with multiple connected hub-DEGs and TFs highlighted from the drug-gene network.
5 Conclusion
The survival rate of OSCC patient is very low for its drug resistance. We analyzed cisplatin resistance of OSCC using bioinformatics tools and found hub-genes (AR, ETS1, MET, PDGFB and VAV3), TFs (HIF1A, MYC, FOXP3, E2F4, WT1, PURA, ZEB1 and USF2) and miRNA (hsa-miR-377-3p, hsa-miR-499a-5p, hsa-miR-34c-5p, hsa-miR-326, hsa-miR-34a-5p, hsa-miR-449b-5p, hsa-miR-449a and hsa-miR-589-3p) which may be responsible for cisplatin resistance of OSCC. Furthermore, we discovered candidate drugs (IMATINIB, BROMOCRIPTINE, NIFEDIPINE, PIRETANIDE and TRIAMTERENE) which might be a therapeutic target of OSCC. These molecular signatures are also needed further validation in wet lab.
References
Siegel, R.L., Miller, K.D., Jemal, A.: Cancer Statistics. CA: Cancer J. Clin. 70, 7–30 (2020)
Neville, B.W., Day, T.A.: Oral cancer and precancerous lesions. CA: Cancer J. Clin. 52, 195–215 (2002)
Hussain, S.M.A.: Comprehensive update on cancer scenario of Bangladesh. South Asian J. Cancer. 2, 279–284 (2013)
Petersen, P.E.: Strengthening the prevention of oral cancer: the WHO perspective. Community Dentistry and Oral Epidemiology 397–3999 (2005). Wiley Online Library
Olsen, K.D.: Head and neck cancer: a multidisciplinary approach. Mayo Clin. Proc. 1308 (1999). Elsevier Limited
Hill, B.T., Price, L.A.: Lack of survival advantage in patients with advanced squamous cell carcinomas of the oral cavity receiving neoadjuvant chemotherapy prior to local therapy, despite achieving an initial high clinical complete remission rate. Am. J. Clin. Oncol. 17, 1–5 (1994)
Li, Y., et al.: Salivary transcriptome diagnostics for oral cancer detection. Clin. Cancer Res. 10, 8442–8450 (2004)
Lin, Z., et al.: Chemotherapy-induced long non-coding RNA 1 promotes metastasis and chemo-resistance of TSCC via the Wnt/β-catenin signaling pathway. Mol. Ther. 26, 1494–1508 (2018)
Liu, Y., Sun, J., Zhao, M.: ONGene: a literature-based database for human oncogenes. J. Genet Genomics. 44, 119–121 (2017)
Datta, N., Chakraborty, S., Basu, M., Ghosh, M.K.: Tumor suppressors having oncogenic functions: the double agents. Cells. 10, 46 (2021)
Park, T., et al.: Statistical tests for identifying differentially expressed genes in time-course microarray experiments. Bioinformatics 19, 694–703 (2003)
Law, W., Chen, Y., Shi, W., Smyth, G.K.: Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014)
Tusher, V.G., Tibshirani, R., Chu, G.: Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. 98(9), 5116–5121 (2001). https://doi.org/10.1073/pnas.091062498
Liao, Y., Wang, J., Jaehnig, E.J., Shi, Z., Zhang, B.: WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 47, W199–W205 (2019)
Saito, R., et al.: A travel guide to Cytoscape plugins. Nat. Methods 9, 1069 (2012)
Bovolenta, L.A., Acencio, M.L., Lemke, N.: HTRIdb: an open-access database for experimentally verified human transcriptional regulation interactions. BMC Genom. 13, 405 (2012)
Wong, N., Wang, X.: miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 43, D146–D152 (2015)
Aguirre-Gamboa, R., et al.: SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PloS One 8, e74250 (2013)
Wagner, A.H., et al.: DGIdb 2.0: mining clinically relevant drug–gene interactions. Nucleic Acids Res. 44, D1036–D1044 (2016)
Tomasovic-Loncaric, C., et al.: Androgen receptor as a biomarker of oral squamous cell carcinoma progression risk. Anticancer Res. 39, 4285–4289 (2019)
Duan, B., et al.: Genetic variants in the platelet-derived growth factor subunit B gene associated with pancreatic cancer risk. Int. J. Cancer 142, 1322–1331 (2018)
Trenkle, T., Hakim, S.G., Jacobsen, H.-C., Sieg, P.: Differential gene expression of the proto-oncogene VAV3 and the transcript variant VAV3. 1 in oral squamous cell carcinoma. Anticancer Res. 35, 2593–2600 (2015)
Arora, S., Ali, J., Ahuja, A., Khar, R.K., Baboota, S.: Floating drug delivery systems: a review. AAPS PharmSciTech 6, E372–E390 (2005)
Wang, F., et al.: LncRNA PVT1 enhances proliferation and cisplatin resistance via regulating miR-194-5p/HIF1a axis in oral squamous cell carcinoma. OncoTargets Ther. 13, 243 (2020)
Codo, A.C., et al.: Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1α/glycolysis-dependent axis. Cell Metab. 32, 437–446 (2020)
Chen, X., et al.: C-MYC and BCL-2 mediate YAP-regulated tumorigenesis in OSCC. Onco. Impact J. 9(1), 668 (2018)
Song, J.-J., et al.: Foxp3 overexpression in tumor cells predicts poor survival in oral squamous cell carcinoma. BMC Cancer. 16, 1–7 (2016)
Diniz, M.G., Silva, J.C., Souza, F.T.A., Pereira, N.B., Gomes, C.C., Gomez, R.S.: Association between cell cycle gene transcription and tumor size in oral squamous cell carcinoma. Tumor Biol. 36(12), 9717–9722 (2015). https://doi.org/10.1007/s13277-015-3735-1
Ribeiro, I.P., et al.: WT1, MSH6, GATA5 and PAX5 as epigenetic oral squamous cell carcinoma biomarkers-a short report. Cell. Oncol. 39, 573–582 (2016)
Kim, J.Y., Cho, K.H., Jeong, B.Y., Park, C.G., Lee, H.Y.: Zeb1 for RCP-induced oral cancer cell invasion and its suppression by resveratrol. Exp. Mol. Med. 52, 1152–63 (2020)
Ganguly, N., Parihar, S.P.: Human papillomavirus E6 and E7 oncoproteins as risk factors for tumorigenesis. J. Biosci. 34(1), 113–123 (2009). https://doi.org/10.1007/s12038-009-0013-7
Shoucair, I.: Extracellular microRNAs as biomarkers for the detection of nasopharyngeal carcinoma [PhD Thesis]. University of British Columbia (2021)
Han, H., et al.: TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 46, D380–D386 (2018)
Jimenez, L., Jayakar, S.K., Ow, T.J., Segall, J.E.: Mechanisms of invasion in head and neck cancer. Arch. Path. Lab. Med. 139, 1334–48 (2015)
Verweij, J., et al.: Imatinib mesylate (STI-571 Glivec®, GleevecTM) is an active agent for gastrointestinal stromal tumours, but does not yield responses in other soft-tissue sarcomas that are unselected for a molecular target: results from an EORTC Soft Tissue and Bone Sarcoma Group phase II study. Eur. J. Cancer 39, 2006–2011 (2003)
Kulman, G., Lissoni, P., Rovelli, F., Roselli, M.G., Brivio, F., Sequeri, P.: Evidence of pineal endocrine hypofunction in autistic children. Neuro Endocrinol. Lett. 21, 31–4 (2000)
A new indicator for CAFs in oral squamous cell carcinoma
Chohan, I.S.: Effects of piretanide on plasma fibrinolytic activity, platelet aggregation and platelet factor-4 release in man. J. Biosci. 10, 243–249 (1986)
Salama, A.: Squamous cell carcinoma of the lip. Lip Cancer 17–23 (2014). Springer
Acknowledgements
The authors thank the Professor Dr. Md. Nurul Haque Mollah, Bioinformatics Lab. (Dry), Dept. of Statistics, University of Rajshahi, Bangladesh and the reviewers of this work.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this paper

Cite this paper
Sarker, B., Rahaman, M.M., Khan, S., Sinha, J.D., Barman, S. (2022). Bioinformatics Analysis of Oral Squamous Cell Carcinomas and Their Interaction to Identify Molecular Signatures. In: Sk, A.A., Turki, T., Ghosh, T.K., Joardar, S., Barman, S. (eds) Artificial Intelligence. ISAI 2022. Communications in Computer and Information Science, vol 1695. Springer, Cham. https://doi.org/10.1007/978-3-031-22485-0_15
Download citation
DOI: https://doi.org/10.1007/978-3-031-22485-0_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-22484-3
Online ISBN: 978-3-031-22485-0
eBook Packages: Computer ScienceComputer Science (R0)