
Abstract
Increasing population, demand for food, and under reduced arable land are the challenges that need to be addressed. Due to continuous effort to solve the food demand, maize breeding approaches have been enhanced by adopting multi-“omics” approaches. Physiological high-throughput phenotyping (phenomics) needs to be functionally analyzed at the level of genome (genomics), gene expression (transcriptomics), protein translation (proteomics), metabolomics profiling (metabolomics), and epi genetics modifications (epigenomics) for improved plant growth, performance, and composition. The rapid advances in “omic” technologies create opportunities to generate diverse data sets for maize breeding platform. Multi-omics approaches help to select precise genomic region in order to elevate the efficiency of maize production, protection, and adaptability for highly unpredictable looming environment. Advance next-generation sequencing technologies, reduced cost per data point generation, and time consumption have greatly reduced, which facilitate multi-omics approaches to get involved in high-throughput phenomics for rapid identification of trait-linked omics.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

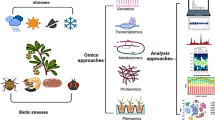
1 Introduction
Maize (Zea mays L.) is a major food and feed crop around the world. In terms of area and production, maize is the third most important food crop after rice and wheat, and India is the world’s fifth largest producer, accounting for 3% of total global production. Most people in developing countries are overly reliant on maize as a staple food due to economic necessity. It provides 50% of dietary protein for humans and can account for 70% of protein intake for people in developing countries (Deutscher 1978). In Africa and some Asian countries, nearly 90% of maize grown is for human consumption, accounting for 80–90% of total energy intake. Together with rice and wheat, it accounts for at least 30% of the food calories consumed by more than 4.5 billion people in 94 developing countries. Maize has long served as a model species in genetics, developmental biology, physiology, and, more recently, genomic research. Genetic research on Zea mays L. began with Edward East’s 1908 report on inbreeding depression and hybrid vigor, and the 1940s saw a cytogenetic breakthrough, such as transposable elements (TEs) by Barbara McClintock (Walbot 2008). The accumulated cytogenetic and genetic data, as well as the vast sequence data derived from maize genomic studies, have provided a wealth of information on the structure, function, and evolution of the maize genome. We discuss multi-omics approaches, their applications, and anticipated implementations in maize improvement to improve crop yields and biotic and abiotic stress tolerance in this book chapter.
2 Genomics
The study of genes and genomes is known as genomics, and it focuses on the structure, function, evolution, mapping, epigenomic, mutagenomic, and genome editing aspects (Muthamilarasan et al. 2019). Genomics can play an important role in elucidating genetic variation, which can improve crop breeding efficiency and lead to genetic improvement of crop species. Structural genomics includes sequence polymorphism and chromosomal organization, and it allows plant biologists to create physical and genetic maps to identify traits of interest. Functional genomics, on the other hand, provides insights into the functions of genes in relation to the regulation of the trait of interest. Epigenomics refers to the phenomenon of epigenetic changes occurring at the genomic level in the form of histone modifications, DNA, or small RNA methylations. Mutagenomics is concerned with the mutational events that orchestrate genetic modification in mutant traits. Mutagenomics and pangenomics are two recent omics approaches in crop sciences that focus on mutagenesis and the pangenome, respectively (Golicz et al. 2016; Muthamilarasan et al. 2019).
3 Structural Genomics
Structural genomics is reliant on molecular markers, which can be used to tag and map genes of interest before being used in crop breeding programs. There are different types of marker techniques. The first is non-PCR techniques such as restriction fragment length polymorphisms (RFLP). Restriction fragment length polymorphism detects DNA polymorphism by hybridizing a labeled DNA probe to a Southern blot of restriction enzyme digested DNA, resulting in a different DNA fragment profile (Agarwal et al. 2008). The second is PCR-based techniques for detecting markers such as random amplified polymorphic DNA (RAPD), amplified fragment length polymorphisms (AFLP), and single nucleotide polymorphisms (SNPs) (Williams et al. 1990; Vos et al. 1995). The RAPD markers are created by PCR-based amplification of random DNA segments with a single primer of any nucleotide sequence (Rabouam et al. 1999).
Amplification of restriction fragments from a total digest of genomic DNA is also a PCR-based technique that performs selective PCR amplification of restriction fragments (Vos et al. 1995). Single nucleotide polymorphisms are single nucleotide variations in an individual’s or organism’s genome. Sequencing of genomic PCR products derived from various individuals can be used to detect SNPs (Appleby et al. 2009). In contrast, diversity array technology (DArT) is a high-throughput technique based on microarray hybridization that involves genotyping of numerous polymorphic loci spread across the genome (Jaccoud et al. 2001). With the advent of NGS, it became possible to identify and use SNPs.
Quantitative trait loci (QTL) mapping and genome-wide association studies are two approaches used to understand and study the multiple traits in crops (GWAS). Quantitative trait loci mapping is a statistical method for connecting two types of data, namely, complex phenotypes and genotypes. Molecular markers such as SNPs and AFLPs are commonly used to map QTLs, which can then be correlated with observed phenotypic data (Kearsey 1998; Challa and Neelapu 2018). GWAS, on the other hand, has the potential to identify variants associated with traits. Based on SNPs in the sequence data, genome-wide association studies may also identify correlations between genetic variants/phenotypes in any organism’s population (Challa and Neelapu 2018). GWAS identified 48 QTLs associated with maize crop yield under heat and water stress (Millet et al. 2016).
Furthermore, numerous SNPs associated with drought-responsive TFs have been identified using maize crop GWAS (Shikha et al. 2017). Furthermore, structural variants (SVs) play an important role in the genetic control of agronomically important traits in crops. Breeders can now improve hybrid breeding by combining marker-assisted selection (MAS) with genotyping by sequencing (GBS) to improve crop quality and yield (He et al. 2014). Multiparent mapping, specifically multiparent advanced generation intercrosses (MAGIC) and nested association mapping (NAM) in model plants and crops (Yu et al. 2008; Kover et al. 2009), has revealed the vast amount of phenotypic diversity that can be achieved through experimental studies.
The MAGIC population is excellent for breeding improvement. Analyses of the relationships between genotypes and phenotypes can identify QTLs, which can then be validated using functional genomics approaches.
4 Functional Genomics and Muta-Genomics
Functional genomics will eventually make use of the vast resources and information provided by structural genomics. Hieter and Boguski (1997) define functional genomics as the development of global experimental approaches to assess gene function. Numerous biotechnological tools have been developed to identify and isolate genes of interest, clone and characterize those genes, and generate overexpression or knockout lines for functional transgenic studies (Muthamilarasan et al. 2019). Prior to genome sequencing methods, identifying candidate genes required time-consuming procedures such as suppression subtractive hybridization (SSH), expressed sequence tag (EST), and cDNA-AFLP-sequencing. As a result of the introduction of NGS, the tediousness of these approaches has decreased (Muthamilarasan et al. 2019).
The availability of crop genome sequencing has led to the identification of genes involved in disease resistance, stress resistance, and yield determination. Furthermore, using genome editing tools such as the clustered regularly interspaced short palindromic repeats (CRISPR/Cas9 system) and transcription activator-like effector nuclease (TALEN) and authentic genome engineering has been proposed to improve crops (Rinaldo and Ayliffe 2015). Genome editing tools that do not require the insertion of foreign DNA could potentially increase yield in genetically modified crops by introducing pest and disease resistance. A bread wheat mildew resistance locus o (TaMlo) mutant was created using TALEN and CRISPR/Cas9 technologies (Wang et al. 2014). Similarly, the same technique was used to create a SlMlo mutant in a tomato crop (Nekrasov et al. 2017). Numerous important crops, including soybean, rice, maize, and sorghum, have already had their genomes edited using the CRISPR/Cas9 system (Jiang et al. 2013; Lawrenson et al. 2015; Li et al. 2015; Svitashev et al. 2015). Several mutants related to crop growth, development, and stress tolerance in rice, maize, wheat, and barley have been identified using comparative genomics (Talukdar and Sinjushin 2015). TILLING has also been used to detect mutations in rice (Suzuki et al. 2008), maize (Till et al. 2004), wheat (Dong et al. 2009), barley (Caldwell et al. 2004), tomato (Minoia et al. 2010), and soybean (Cooper et al. 2008). Mutagenomics has enabled the investigation of gene function by silencing and interrupting candidate genes using reverse genetic approaches. Specific reverse genetic techniques used to screen/induce crop mutations include RNA interference (RNAi) and (VIGS). When mutant alleles are not available, reverse genetic techniques can be used to knockdown or silence the phenotype of a gene, allowing for gene function analysis (Talukdar and Sinjushin 2015). Furthermore, reverse genetic approaches such as RNAi and gene silencing technologies have been used to screen for mutations in maize (Dwivedi et al. 2008; Tomlekova 2010). As a result, both functional genomics and mutagenomics have been shown to be beneficial in terms of crop growth, yield, and stress resistance.
5 Epigenomics
The term epigenetics refers to heritable changes that are not caused by changes in the DNA sequence. These epigenetic changes are caused by DNA methylation and histone posttranslational modification (PTM) (Strahl and Allis 2000; Novik et al. 2002). The combination of epigenetics and genomics is known as epigenomics, and it has emerged as a new omics technique to better understand genetic regulation and its role in cellular growth and stress responses (Callinan and Feinberg 2006). In contrast to genomics, epigenomics can be influenced by environmental factors such as abiotic and biotic stress. Nonetheless, genome-wide studies could be conducted to investigate these epigenetic events at any developmental stage or to assess abnormalities caused by plant disease (Muthamilarasan et al. 2019). This method was found to be useful in one epigenomic study for identifying histone modifications associated with photosynthesis in maize (Offermann et al. 2006).
6 Pangenomics
The pangenome concept refers to a species’ entire genomic makeup, which can be divided into core and dispensable genes. The core gene sets are shared by all individuals, whereas the dispensable gene sets (also known as accessory genes) are individual-specific and/or present in some but not all individuals (Tettelin et al. 2005). Advances in sequencing technology and analysis tools have enabled the sequencing of multiple crop species accessions (Golicz et al. 2016). A wave of pangenomic studies in maize (Hirsch et al. 2014) has revealed that dispensable genes play important roles in crop diversity and quality improvement.
7 Transcriptomics
Transcriptomics is concerned with the transcriptome, which is the complete set of RNA transcripts produced by an organism’s genome in a cell or tissue (Raza et al. 2021). Transcriptome profiling is a dynamic technique that has emerged as a promising technique for analyzing gene expression in response to various stimuli over time (Duque et al. 2013; El-Metwally et al. 2014). This strategy enables the researcher to observe the differential expression of genes in vitro in order to comprehend the first layer function of a specific gene. Initially, transcriptome dynamics were studied using traditional profiling techniques such as cDNAs-AFLP, differential display-PCR (DD-PCR), and SSH, but the resolution was low (Nataraja et al. 2017). Following the introduction of robust techniques, RNA expression profiling using microarrays, digital gene expression profiling, NGS, RNAseq, and SAGE became possible (Kawahara et al. 2012; De Cremer et al. 2013; Duque et al. 2013). Furthermore, RNA-seq studies in maize have been conducted to identify drought stress-responsive genes (Kakumanu et al. 2012). Another method for understanding differential expression profiles in response to stress in different crop species is comparative transcriptomics. In response to heat stress, comparative transcriptomic analysis identified 16 common genes in rice, wheat, and maize compared to those in switch grass (Ding et al. 2013; Li et al. 2013).
To generate multiple transcripts in response to abiotic stress conditions, an alternative splicing (AS) transcriptomics approach was launched (Laloum et al. 2018). In response to heat and drought stress, this method has been used in crops such as rice, maize, and sorghum (Zhang et al. 2015). As a result, AS transcriptomic analyses revealed the importance of splicing factors in controlling abiotic stress responses in crops. All of these transcriptomic techniques, taken together, have the potential to play a critical role in the regulation of gene expression, resulting in crop species improvement.
8 Proteomics
Proteomics is a technique that involves profiling total expressed protein in an organism and is classified into two types.
There are four distinct parts: sequence, structural, functional, and expression proteomics (Mosa et al. 2017; Aizat and Hassan 2018). The amino acid sequence is determined by proteomics. Sequences that are typically identified in a sequential manner using high liquid chromatography with high performance (HPLC; Twyman 2013).
Structural proteomics is concerned with the structure of proteins. Comprehend their ostensible functions Structural proteomics can help be analysed using a variety of methods, including computer based modelling, as well as experimental methods such as nuclear crystallisation, electron microscopy, magnetic resonance (NMR), and protein crystal X-ray diffraction (Sali et al. 2003; Woolfson 2018).
Protein extraction and separation advances have contributed to the rapid advancement of plant proteomic research at both the sample and genome-wide scales (Nakagami et al. 2012). Traditional proteomics techniques include exchange chromatography (IEC), size exclusion chromatography (SEC), and affinity chromatography.
However, western blotting and enzyme-linked immunosorbent assay (ELISA) could be used to analyze specific proteins.
Later, more advanced techniques such as SDS-PAGE, two-dimensional gel electrophoresis (2-DE), and two-dimensional differential gel electrophoresis (2D-DIGE) were developed and used for protein separation using gel-based techniques. Simultaneously, protein microarrays/chips for detection of small amounts of protein sample have been developed for rapid protein expression analysis.
SDS-PAGE and two-dimensional gel electrophoresis are required to identify proteins and measure quantitative protein content parameters, respectively (Eldakak et al. 2013).
The identified proteins are now used to determine the molecular mass of peptides using mass spectrometry (MS), ion trap-mass spectrometry (IT-MS), or liquid chromatography (LC; Fournier et al. 2007). MALDI-TOF, electrospray ionization (ESI) and collision-induced dissociation (CID) have also been used to determine the molecular weights of proteins (Tanaka et al. 1988; McLuckey and Stephenson Jr. 1998; Baggerman et al. 2005).
9 Metabolomics
Transcriptomics, proteomics, and metabolomics also offer opportunities to decipher and understand the molecular basis of stress tolerance. The use of proteomics and metabolomics-based metabolite markers can serve as an efficient selection tool as a substitute for phenotype-based selection. This review covers the molecular mechanisms for salinity stress tolerance, recent progress in mapping and introgressing major gene/QTL (genomics), transcriptomics, proteomics, and metabolomics in major cereals, namely, rice, wheat, and maize (Kumar et al. 2022).
Breeding for drought-tolerant crops depends on omics-based approach enabling accelerated maize breeding for biotic and abiotic stress tolerance trait in crop breeding program. Increased nutrient uptake leads to increased growth as well as yield. Plant parts play a major role in nutrient uptake; majorly, nitrogen usage efficiency is mostly dependent on root traits. Maize root traits were well studied under nitrogen stress conditions. Several secondary metabolites and amino acids play important role in root biomass growth. Several transcriptomic-based experiments under nitrogen-deficient conditions increased maize root biomass production with the help of phosphatidylcholine and phosphatidyl glycerol metabolites (Chowdhury et al. 2022).
All together integrated omics is an efficient approach to enable the stress tolerance. Breeding for drought-tolerant crops depends on omics-based approach enabling accelerated maize breeding for biotic and abiotic stress tolerance trait in crop breeding program. Advanced changes in proteins and metabolites during different environmental conditions and biotic and abiotic stress conditions affect the physiological process and growth. Frequent timely alteration of proteins and metabolites in maize plants will improve growth in hybrids. Heterosis in hybrids shows that metabolites alter physiological changes for increased hybrid vigor (Li et al. 2020).
Both biotic and abiotic stresses along with timely growth and physiological and biochemical occur in maize during developmental stages. Various proteins and metabolites released vary during different developmental changes and the diverse proteins and metabolites captured during different developmental stages. Everything is interrelated. The altered genes will produce altered proteins, which combine to form altered metabolites. Different developmental stages and different genes encode for different proteins as well as metabolites. Methods used GC-LC chromatography, confocal microscopy, high-performance liquid chromatography along with ion trap tandem mass spectrometry, HPLC liquid chromatography, and genotype to phenotype prediction using genomics are not always possible for traits. The end product in the cellular regulatory processes might be a combination of gene to gene interactions, and modification leads to physiological changes. More than 200,000 metabolites including primary and secondary metabolites were identified in plants. Primary metabolites are involved in necessary plant growth and developmental activities, and the secondary metabolites are derived from primary metabolites and are involved in plant defense mechanism and biotic and abiotic stresses. Primary metabolites include carbohydrates, lipids, proteins, vitamins, and amino acids, whereas secondary metabolites are alkaloids, phenolics, sterols, steroids, lignins, and essential oils.
Maize grains have highest polyphenol content and can be well studied in metabolomics and phytochemicals. Polyphenols are known for its anticancer properties and antioxidant properties. Modifications in the metabolites are the major outcome of phenotypic outcome. Localization of nutritional phytochemicals in plant tissues is a significant information for metabolomics. Maize grains have different phytochemical substances like anthocyanins in aleurone layer and 56 other compounds including oxylipins 13-trihydroxy-octadecenoicacid and 9, 12, 13-trihydroxy-trans-10-octadecenoic acid. The genes involved in the synthesis of these substances might be expressed only in certain tissues. Combination of different methods allows more information about variable metabolites (Razgonova et al. 2022). Genetic regulatory mechanism of phenotypic traits can be well understood with metabolome studies.
In maize, the grain is the major commercial source, and improving the kernel quality not always depends on genomics but also in different metabolites. Metabolites from parental lines and hybrids were assessed, and they have different metabolites, which were clustered differently in PCA and clustering. ANOVA of the metabolites showed that 163 metabolites were exhibited significant difference between hybrids and parents. Utilizing metabolites from hybrids and parental lines heterosis was also studied, which indicates two third of all metabolites displayed 36% positive over dominance for hybrids compared to parental lines (Xu et al. 2021). Seven metabolic markers were also identified associated with multiple traits.
Maize leaf base and tip samples were subjected for metabolome analysis across diverse population of inbreed lines, and this shows that their metabolite differences are due to their tissues but not due to occurrence rate. Large number of metabolite transcripts contain housekeeping genes involved in primary metabolism as well as important cellular functions. Complex genetic architecture of the maize seedling leaves metabolome was studied using metabolome GWAS (mGWAS) and identified significant SNPs associated for the important metabolome on chromosome. Maize has different metabolite accumulation like phenylpropanoid, benzoxazinoid and flavonoids. Metabolomic difference were observed between tissue types and subpopulations clearly (Zhou et al. 2019). Metabolomics can be used as a tool for defining biosynthetic pathways and other maize physiological questions.
Metabotyping of maize hybrids under early sowing conditions could determine the metabolites responsible for chilling tolerance at vegetative stage. There are different methods for metabolomic profiling. A specialized method is named reversed-phase liquid chromatography (LC)–mass spectrometry (MS). Maize ear, late cob, leaf, stem, and tassel metabolites were studied. Specialized spectral metadata including structural characterization of candidate substrate-product pair (CSPP) network identified several new phenyl propanoids in all organs, and other metabolic classes are organ specific.
Oligolignols are abundant in LS-MS profile of stem, hydroxyl fatty acids are found in late cob and leaf extracts, and benzoxazinoids are mostly present in tassels, auxin-related compounds in late cob and tassels. Interplay of glycosylations and acylations leads to mixed glycosides present in single type of tissue. The characterized compound and varied compounds are involved in metabolite discovery and systems biology research. The spectral meta data is available in a database (DynLib spectral database, https://bioit3.irc.ugent.be/dynlib/) (Desmet et al. 2021).
Abiotic stress causes major yield loss in maize breeding. Major abiotic stresses involved are drought, heat, salt, and cold stresses. Prolonged stress conditions lead to retarded growth, biomass, and yield. Leaf metabolome of B73 inbred plants grown under long-term nonlethal drought, heat, and salt stress conditions shows that leaf metabolites are affected strongly when compared with controlled plants. Multiple amino acids like serine, threonine, tryptophan, histidine, glutamate, lysine, tyrosine, and ornitine accumulated during salt stress conditions. Several secondary metabolites like quinic acid and pipecolic acid and two unidentified phenolic compounds were also accumulated. Both salt stress and heat stress show accumulation of raffinose and its precursor galactinol. But sugar alcohol lactitol was accumulated more, and citrate and trans-3-caffeoyl quinic acid were depleted under heat stress.
In case of drought stress, hexoses were accumulated along with raffinose. In stressed leaves, multiple raffinose biosynthesis genes were upregulated and are involved in dehydration tolerance along with oxidation prevention. All together it’s shown that biosynthesis of raffinose series sugars is the major protective mechanism for all abiotic stresses including cold tolerance. Another important amino acid is a proline, which is familiar as a protective osmolyte and antioxidant during stress conditions. Proline is derived from arginine and ornithine where stress leads to upregulation genes involved in arginine and ornithin pathways. During stress, GABA accumulation is a common response, which affects tricorboxylic acid intermediates that leads to shift in carbon and nitrogen metabolism in stressed leaves (Joshi et al. 2021).
Metabolites associated with maize chilling tolerance during vegetative stage (eight leaf visible leaf stage) were identified using untargeted metabolomic approach of 30 diverse maize hybrids. Marker metabolite correlation with aerial biomass of mature plants was not affected by early sowing. Due to early sowing, the leaf metabolites in field were affected, and the metabolites involve both primary and specialized metabolism. For leaf metabolites, the balance between sugars and organic acids has higher carbohydrates (sucrose, fructose, starch, and glucose) and lower organic acids (malate, succinate) in early sowing than normal sowings. Tryptophan, shikimic acid, and quinic acid are in high contents during early sowings. Raffinose, a stress metabolite accumulation, is less in early sown hybrids (Lamari et al. 2018). Early sown hybrids showed negative correlation between aerial biomass and raffinose.
Heat stress in maize is a major constrain for maize grain development. In order to understand the maize heat tolerance mechanism, a heat-tolerant hybrid ZD309 derived from female H39_1 and male M189 were tested in heat stress environments by transcriptomic and metabolomic approaches (Liu et al. 2022). Under heat stress, growth of hybrid and its parents was deteriorated by 6 days of heat treatment compared to plants in control conditions. Plant hormone signal transduction, cystine and methionine metabolism, and alpha linolenic acid metabolism play major roles in maize heat tolerance. The genes involved in these mechanism can be utilized in maize breeding for heat tolerance.
Maize response to aphid feeding is revealed by transcriptomic and metabolomic assays. Sucking pests like corn leaf aphid directly damage plants by sucking phloem nutrients and transmit plant viruses. B73 plant leaves were infected with aphids and observed two different responses with both transcriptional and metabolic changes. Increased jasmonic acid levels increase the accumulation of benzoxazinoids. It was observed that there was a predominant effect of salicylic acid regulation and altered gene expression for prolonged induction of oxylipins (Tzin et al. 2015).
10 Conclusion
Omics analysis has been critical in identifying genetic processes, growth, development, and stress tolerance in maize. In crop science, several omics approaches, including genomics, transcriptomics, proteomics, and metabolomics, have used high-throughput techniques to interpret functional analysis, molecular mechanisms of genes, and gene networks.
Furthermore, combining GWAS with metabolomics, transcriptomics, and proteomics has shown to be a promising tool for elucidating biochemical processes and abiotic stress tolerance in some model crops including maize. The studies demonstrated how combining several omics approaches could be advantageous for identifying potential candidate genes and their pathways. The integration of some omics approaches in crop sciences has become possible thanks to advances in high-throughput technologies and computational tools.
The panomics platform, which includes integrated multi-omics such as genomics, epigenomics, transcriptomics, proteomics, proteomics, and metabolomics, would make it easier to build models to predict agronomically important traits in order to improve crops through precision breeding. Importantly, combining systems biology and complex omics datasets has improved our understanding of molecular regulator networks for crop improvement. G–P–E interactions in crops have been discovered through research. Following that, through the “genotype to phenotype” concept, integration of functional genomics with trancriptomics, proteomics, and metabolomics may result in apparent crop quality phenotypic traits under certain stresses.
References
Agarwal M, Shrivastava N, Padh H (2008) Advances in molecular marker techniques and their applications in plant sciences. Plant Cell Rep 27:617–631. https://doi.org/10.1007/s00299-008-0507-z
Aizat WM, Hassan M (2018) Proteomics in systems biology. In: Aizat W, Goh HH, Baharum S (eds) Omics applications for systems biology, Advances in experimental medicine and biology. Springer, Cham, pp 31–49. https://doi.org/10.1007/978-3-319-98758-3_3
Appleby N, Edwards D, Batley J (2009) New technologies for ultrahigh throughput genotyping in plants. In: Gustafson JP, Langridge P, Somers DJ, Totowa NJ (eds) Methods in molecular biology, plant genomics. Humana Press, New York, NY, pp 19–39. https://doi.org/10.1007/978-1-59745-427-8_2
Baggerman G, Vierstraete E, De Loof A, Schoofs L (2005) Gel-based versus gel-free proteomics: a review. Comb Chem High Throughput Screen 8:669–677. https://doi.org/10.2174/138620705774962490
Caldwell DG, McCallum N, Shaw P, Muehlbauer GJ, Marshall DF, Waugh R (2004) A structured mutant population for forward and reverse genetics in barley (Hordeum vulgare L.). Plant J 40:143–150. https://doi.org/10.1111/j.1365-313X.2004.02190.x
Callinan PA, Feinberg AP (2006) The emerging science of epigenomics. Hum Mol Genet 15(Suppl_1):R95–R101. https://doi.org/10.1093/hmg/ddl095
Challa S, Neelapu NR (2018) Genome-wide association studies (GWAS) for abiotic stress tolerance in plants. In: Wani SH (ed) Biochemical, physiological and molecular avenues for combating abiotic stress tolerance in plants. Academic, Cham, pp 135–150. https://doi.org/10.1016/B978-0-12-813066-7.00009-7
Chowdhury NB, Schroeder WL, Sarkar D, Amiour N, Quilleré I, Hirel B, Maranas CD, Saha R (2022) Dissecting the metabolic reprogramming of maize root under nitrogen-deficient stress conditions. J Exp Bot 73(1):275–291. https://doi.org/10.1093/jxb/erab435
Cooper JL, Till BJ, Laport RG, Darlow MC, Kleffner JM, Jamai A, El-Mellouki T, Liu S, Ritchie R, Nielsen N, Bilyeu KD (2008) TILLING to detect induced mutations in soybean. BMC Plant Biol 8(1):1–10. https://doi.org/10.1186/1471-2229-8-9
De Cremer K, Mathys J, Vos C, Froenicke L, Michelmore RW, Cammue BPA et al (2013) RNAseq-based transcriptome analysis of Lactuca sativa infected by the fungal necrotroph Botrytis cinerea. Plant Cell Environ 36:1992–2007. https://doi.org/10.1111/pce.12106
Desmet S, Saeys Y, Verstaen K, Dauwe R, Kim H, Niculaes C, Fukushima A, Goeminne G, Vanholme R, Ralph J, Boerjan W (2021) Maize specialized metabolome networks reveal organ-preferential mixed glycosides. Comput Struct Biotechnol J 1(19):1127–1144
Deutscher D (1978) The current status of breeding for protein quality in corn. Adv Exp Med Biol 105:281–300
Ding X, Li X, Xiong L (2013) Insight into differential responses of upland and paddy rice to drought stress by comparative expression profiling analysis. Int J Mol Sci 14:5214–5238. https://doi.org/10.3390/ijms14035214
Dong C, Dalton‐Morgan J, Vincent K, Sharp P (2009) A modified TILLING method for wheat breeding. The Plant Genome 2(1). https://doi.org/10.3835/plantgenome2008.10.0012
Duque AS, Almeida AM, da Silva AB, da Silva JM, Farinha AP, Santos D et al (2013) Chapter 3: Abiotic stress responses in plants: unraveling the complexity of genes and networks to survive. In: Vahdati K, Leslie C (eds) Abiotic stress: plant responses and applications in agriculture. INTECH Open, Rijeka, pp 49–102. https://doi.org/10.5772/45842
Dwivedi S, Perotti E, Ortiz R (2008) Towards molecular breeding of reproductive traits in cereal crops. Plant Biotechnol J 6:529–559. https://doi.org/10.1111/j.1467-7652.2008.00343.x
Eldakak M, Milad SI, Nawar AI, Rohila JS (2013) Proteomics: a biotechnology tool for crop improvement. Front Plant Sci 4:35. https://doi.org/10.3389/fpls.2013.00035
El-Metwally S, Ouda OM, Helmy M (2014) Next generation sequencing technologies and challenges in sequence assembly, 1st edn. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-0715-1
Fournier ML, Gilmore JM, Martin-Brown SA, Washburn MP (2007) Multidimensional separations-based shotgun proteomics. Chem Rev 107:3654–3686. https://doi.org/10.1021/cr068279a
Golicz AA, Batley J, Edwards D (2016) Towards plant pangenomics. Plant Biotechnol J 14:1099–1105. https://doi.org/10.1111/pbi.12499
He J, Zhao X, Laroche A, Lu ZX, Liu H, Li Z (2014) Genotyping- by- sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front Plant Sci 5:484. https://doi.org/10.3389/fpls.2014.00484
Hieter P, Boguski M (1997) Functional genomics: it’s all how you read it. Science (New York, NY) 278:601–602. https://doi.org/10.1126/science.278.5338.601
Hirsch CN, Foerster JM, Johnson JM, Sekhon RS, Muttoni G, Vaillancourt B et al (2014) Insights into the maize pan-genome and pantranscriptome. Plant Cell 26:121–135. https://doi.org/10.1105/tpc.113.119982
Jaccoud D, Peng K, Feinstein D, Kilian A (2001) Diversity arrays: a solid state technology for sequence information independent genotyping. Nucleic Acids Res 29:25. https://doi.org/10.1093/nar/29.4.e25
Jiang W, Zhou H, Bi H, Fromm M, Yang B (2013) Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res 41:e188. https://doi.org/10.1093/nar/gkt780
Joshi J, Hasnain G, Logue T, Lynch M, Wu S, Guan J-C, Alseekh S, Fernie AR, Hanson AD, McCarty DR (2021) A core metabolome response of maize leaves subjected to long-duration abiotic stresses. Meta 11:797
Kakumanu A, Ambavaram MR, Klumas C, Krishnan A, Batlang U, Myers E et al (2012) Effects of drought on gene expression in maize reproductive and leaf meristem tissue revealed by RNA-seq. Plant Physiol 160:846–867. https://doi.org/10.1104/pp.112.200444
Kawahara Y, Oono Y, Kanamori H, Matsumoto T, Itoh T, Minami E (2012) Simultaneous RNA-Seq analysis of a mixed transcriptome of rice and blast fungus interaction. PLoS One 7:e49423. https://doi.org/10.1371/journal.pone.0049423
Kearsey MJ (1998) The principles of QTL analysis (a minimal mathematics approach). J Exp Bot 49:1619–1623
Kover PX, Valdar W, Trakalo J, Scarcelli N, Ehrenreich IM, Purugganan MD et al (2009) A multiparent advanced generation inter-cross to fine map quantitative traits in Arabidopsis thaliana. PLoS Genet 5:e1000551. https://doi.org/10.1371/journal.pgen.1000551
Kumar P, Choudhary M, Halder T et al (2022) Salinity stress tolerance and omics approaches: revisiting the progress and achievements in major cereal crops. Heredity 128:497. https://doi.org/10.1038/s41437-022-00516-2
Laloum T, Martín G, Duque P (2018) Alternative splicing control of abiotic stress responses. Trends Plant Sci 23:140–150. https://doi.org/10.1016/j.tplants.2017.09.019
Lamari N, Zhendre V, Urrutia M, Bernillon S, Maucourt M, Deborde C, Prodhomme D, Jacob D, Ballias P, Rolin D, Sellier H (2018) Metabotyping of 30 maize hybrids under early-sowing conditions reveals potential marker-metabolites for breeding. Metabolomics 14(10):1–5
Lawrenson T, Shorinola O, Stacey N, Li C, Ostergaard L, Patron N et al (2015) Induction of targeted, heritable mutations in barley and Brassica oleracea using RNA-guided Cas9 nuclease. Genome Biol 16:258. https://doi.org/10.1186/s13059-015-0826-7
Li YF, Wang YI, Tang Y, Kakani VG, Mahalingam R (2013) Transcriptome analysis of heat stress response in switch grass (Panicum virgatum L.). BMC Plant Biol 13:153. https://doi.org/10.1186/1471-2229-13-153
Li Z, Liu ZB, Xing A, Moon BP, Koellhoffer JP, Huang L et al (2015) Cas9-guide RNA directed genome editing in soybean. Plant Physiol 169:960–970. https://doi.org/10.1104/pp.15.00783
Li Z, Zhu A, Song Q, Chen HY, Harmon FG, Chen ZJ (2020) Temporal regulation of the metabolome and proteome in photosynthetic and photorespiratory pathways contributes to maize heterosis. Plant Cell 32(12):3706–3722. https://doi.org/10.1105/tpc.20.00320
Liu J, Zhang L, Huang L, Yang T, Ma J, Yu T, Zhu W, Zhang Z, Tang J (2022) Uncovering the gene regulatory network of maize hybrid ZD309 under heat stress by transcriptomic and metabolomic analysis. Plan Theory 11(5):677
McLuckey SA, Stephenson JL Jr (1998) Ion/ion chemistry of high-mass multiply charged ions. Mass Spectrom Rev 17:369–407. https://doi.org/10.1002/(SICI)1098-2787(1998)17:6<369::AID-MAS1>3.0.CO;2-J
Millet EJ, Welcker C, Kruijer W, Negro S, Coupel-Ledru A, Nicolas SD et al (2016) Genome-wide analysis of yield in Europe: allelic effects vary with drought and heat scenarios. Plant Physiol 172:749–764. https://doi.org/10.1104/pp.16.00621
Minoia S, Petrozza A, D’Onofrio O, Piron F, Mosca G, Sozio G, Cellini F, Bendahmane A, Carriero F (2010) A new mutant genetic resource for tomato crop improvement by TILLING technology. BMC Res Notes 3(1):1–8. https://doi.org/10.1186/1756-0500-3-69
Mosa KA, Ismail A, Helmy M (2017) Omics and system biology approaches in plant stress research. In: Mosa KA, Ismail A, Helmy M (eds) Plant stress tolerance: an integrated omics approach. Springer, Cham, pp 21–34. https://doi.org/10.1007/978-3-319-59379-1_2
Muthamilarasan M, Singh NK, Prasad M (2019) Multi-omics approaches for strategic improvement of stress tolerance in underutilized crop species: a climate change perspective. Adv Genet 103:1–38. https://doi.org/10.1016/bs.adgen.2019.01.001
Nakagami H, Sugiyama N, Ishihama Y, Shirasu K (2012) Shotguns in the front line: phosphoproteomics in plants. Plant Cell Physiol 53:118–124. https://doi.org/10.1093/pcp/pcr148
Nataraja KN, Madhura BG, Parvathi SM (2017) Omics: modern tools for precise understanding of drought adaptation in plants. In: Zargar SM, Rai V (eds) Plant OMICS and crop breeding. Apple Academic Press, Palm Bay, FL, pp 289–320
Nekrasov V, Wang C, Win J, Lanz C, Weigel D, Kamoun S (2017) Rapid generation of a transgene-free powdery mildew resistant tomato by genome deletion. Sci Rep 7:482. https://doi.org/10.1038/s41598-017-00578-x
Novik KL, Nimmrich I, Genc B, Maier S, Piepenbrock C, Olek A et al (2002) Epigenomics: genome-wide study of methylation phenomena. Curr Issues Mol Biol 4:111–128
Offermann S, Danker T, Dreymuller D, Kalamajka R, Topsch S, Weyand K et al (2006) Illumination is necessary and sufficient to induce histone acetylation independent of transcriptional activity at the C4-specific phosphoenolpyruvate carboxylase promoter in maize. Plant Physiol 141:1078–1088. https://doi.org/10.1104/pp.106.080457
Rabouam C, Comes AM, Bretagnolle V, Humbert JF, Periquet G, Bigot Y (1999) Features of DNA fragments obtained by random amplified polymorphic DNA (RAPD) assays. Mol Ecol 8:493–503. https://doi.org/10.1046/j.1365-294X.1999.00605.x
Raza A, Tabassum J, Kudapa H, Varshney RK (2021) Can omics deliver temperature resilient ready to-grow crops? Crit Rev Biotechnol 2021:1–24. https://doi.org/10.1080/07388551.2021.1898332
Razgonova M, Zinchenko Y, Pikula K, Tekutyeva L, Son O, Zakharenko A et al (2022) Spatial distribution of polyphenolic compounds in corn grains (Zea mays L. var. Pioneer) studied by laser confocal microscopy and high-resolution mass spectrometry. Plants 11(5):630
Rinaldo AR, Ayliffe M (2015) Gene targeting and editing in crop plants: a new era of precision opportunities. Mol Breed 35:40. https://doi.org/10.1007/s11032-015-0210-z
Sali A, Glaeser R, Earnest T, Baumeister W (2003) From words to literature in structural proteomics. Nature 422:216–225. https://doi.org/10.1038/nature01513
Shikha M, Kanika A, Rao AR, Mallikarjuna MG, Gupta HS, Nepolean T (2017) Genomic selection for drought tolerance using genome-wide SNPs in maize. Front Plant Sci 8:550. https://doi.org/10.3389/fpls.2017.00550
Strahl B, Allis C (2000) The language of covalent histone modifications. Nature 403:41–45. https://doi.org/10.1038/47412
Suzuki T, Eiguchi M, Kumamaru T, Satoh H, Matsusaka H, Moriguchi K, Nagato Y, Kurata N (2008) MNU-induced mutant pools and high performance TILLING enable finding of any gene mutation in rice. Mol Genet Genomics 279(3):213–223. https://doi.org/10.1007/s00438-007-0293-2
Svitashev S, Young JK, Schwartz C, Gao H, Falco SC, Cigan AM (2015) Targeted mutagenesis, precise gene editing, and site-specific gene insertion in maize using Cas9 and guide RNA. Plant Physiol 169:931–945. https://doi.org/10.1104/pp.15.00793
Talukdar D, Sinjushin A (2015) Cytogenomics and mutagenomics in plant functional biology and breeding. In: Barh D, Khan M, Davies E (eds) Plant omics: the omics of plant science. Springer, New Delhi, pp 113–156. https://doi.org/10.1007/978-81-322-2172-2_5
Tanaka K, Waki H, Ido Y, Akita S, Yoshida Y, Yoshida T (1988) Protein and polymer analyses up to m/z 100000 by laser ionization timeof- flight mass spectrometry. Rapid Commun Mass Spectrom 2:151–153. https://doi.org/10.1002/rcm.1290020802
Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL et al (2005) Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial ‘pan-genome’. Proc Natl Acad Sci U S A 102:13950–13955. https://doi.org/10.1073/pnas.0506758102
Till BJ, Reynolds SH, Weil C, Springer N, Burtner C, Young K et al (2004) Discovery of induced point mutations in maize genes by TILLING. BMC Plant Biol 4:12. https://doi.org/10.1186/1471-2229-4-12
Tomlekova NB (2010) Induced mutagenesis for crop improvement in Bulgaria. Plant Mutat Rep 2:4–27
Twyman RM (2013) Principles of proteomics. Garland Science Press, Abingdon
Tzin V, Fernandez-Pozo N, Richter A, Schmelz EA, Schoettner M, Schäfer M, Ahern KR, Meihls LN, Kaur H, Huffaker A, Mori N (2015) Dynamic maize responses to aphid feeding are revealed by a time series of transcriptomic and metabolomic assays. Plant Physiol 169(3):1727–1743
Vos P, Hogers R, Bleeker M, Reijans M, Lee T, Hornes M et al (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414. https://doi.org/10.1093/nar/23.21.4407
Walbot V (2008) Maize genome in motion. Genome Biol 9(4):303
Wang Y, Cheng X, Shan Q, Zhang Y, Liu J, Gao C et al (2014) Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol 32:947–951. https://doi.org/10.1038/nbt.2969
Williams JGK, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV (1990) DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res 18:6531–6535. https://doi.org/10.1093/nar/18.22.6531
Woolfson M (2018) The development of structural x-ray crystallography. Phys Scr 93:1–32. https://doi.org/10.1088/1402-4896/aa9c30
Xu Y, Ma Y, Wang X, Li C, Zhang X, Li P et al (2021) Kernel metabolites depict the diversity of relationship between maize hybrids and their parental lines. Crop J 9(1):181–191
Yu J, Holland JB, McMullen MD, Buckler ES (2008) Genetic design and statistical power of nested association mapping in maize. Genetics 178:539–551. https://doi.org/10.1534/genetics.107.074245
Zhang C, Yang H, Yang H (2015) Evolutionary character of alternative splicing in plants. Bioinf Biol Insights 9:47–52. https://doi.org/10.4137/BBI.S33716
Zhou S, Kremling KA, Bandillo N, Richter A, Zhang YK, Ahern KR, Artyukhin AB, Hui JX, Younkin GC, Schroeder FC, Buckler ES (2019) Metabolome-scale genome-wide association studies reveal chemical diversity and genetic control of maize specialized metabolites. Plant Cell 31(5):937–955
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Narkhede, G.W., Kiranmayee, K.N.S.U. (2023). Maize Improvement Using Recent Omics Approaches. In: Wani, S.H., Dar, Z.A., Singh, G.P. (eds) Maize Improvement. Springer, Cham. https://doi.org/10.1007/978-3-031-21640-4_13
Download citation
DOI: https://doi.org/10.1007/978-3-031-21640-4_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-21639-8
Online ISBN: 978-3-031-21640-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)