
Abstract
The most frequent special histological type of breast cancer is represented by invasive lobular carcinoma (ILC), which makes up about 15% of all invasive breast carcinomas. The molecular signature of ILC is the dysregulation of E-cadherin due to CDH1 abnormalities. Although CDH1 germline mutations are very uncommon in women with early-onset and/or familial ILC, they are the most common detrimental non-BRCA mutations and are thought to be the origin of a significant fraction of lobular breast cancer. Since the morphology and immunophenotype of hereditary and non-hereditary ILCs are nearly identical, no specific histopathological findings can be used to distinguish between the two. High-throughput sequencing studies revealed that ILCs represent a separate entity at the genomic level. This chapter addresses the very important topic of ILC morpho-molecular characteristics in the setting of germline and/or somatic CDH1 abnormalities.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

1 Introduction
Invasive lobular carcinoma (ILC) is the most common special type of breast cancer and accounts for ~15% of invasive breast carcinomas [1]. Dysregulation of E-cadherin due to CDH1 aberrations is considered the molecular hallmark of ILC [2, 3]. Although the frequency of CDH1 germline mutations is very low (~1%) in women with early-onset or familial ILC, these mutations represent the most frequent deleterious non-BRCA mutations, and they are considered founder genetic events in a substantial proportion of lobular breast cancer [4,5,6]. No specific histopathological features can help discriminate between hereditary and non-hereditary ILCs because their morphology and immunophenotype are substantially identical [7]. However, ILCs display peculiar clinic-pathologic characteristics as compared to other breast cancer histotypes [1]. Moreover, high-throughput sequencing analyses showed that ILCs also represent a distinct entity at the genomic level [2, 3, 8, 9]. This chapter provides a comprehensive overview of the morpho-molecular characteristics of ILC in the context of germline and/or somatic CDH1 aberrations.
2 Pathology of Lobular Breast Cancer
Individuals with ILC typically have a diagnosis at an older age and come to the physician’s attention with larger tumors than patients with invasive breast cancer (IBC) of no special type [10]. Hereditary ILC is often bilateral and multicentric, appearing as ill-defined palpable mass(es) or widespread breast nodularities [11]. Classic ILC is composed of non-to-poorly cohesive small, roundish, monomorphic neoplastic elements, with uniform nuclei, inconspicuous nucleoli, and infrequent mitotic figures interspersed into a variably dense fibrous stroma arranged in loose or linear growth pattern. ILC exhibits a targetoid concentric distribution around ducts and lobules and is usually associated with little host reaction [1, 12,13,14,15,16].
It is possible to identify different ILC variants, including solid, alveolar, trabecular, tubule-lobular, signet ring cell, pleomorphic, and histiocytoid which differ from classical ILC in their morphologic characteristics and behavior (Fig. 11.1).

Invasive lobular carcinoma, histiocytoid variant. These tumors are morphologically characterized by sheets/cords of cells with abundant granular cytoplasm and variably eccentric nuclei. Among the possible differential diagnoses of histiocytoid lobular carcinoma, it is worth mentioning some non-neoplastic conditions, such as reactive histiocytic infiltrates and fat necrosis. Hematoxylin and eosin, original magnification 100×; inset 400×. Note. Personal archive
The traditional ILC and other ILC variants are occasionally mixed [13]. The discohesive tumor cells that make up the solid variant of ILC grow in solid nests and may exhibit pleomorphism or enhanced mitotic activity. The tumor cells of alveolar ILC are grouped in distinct clusters or aggregates of 20 cells or more, which are divided by thin fibrous septa. Tumor cells develop in bands thicker than two cells in the trabecular ILC. The tubule-lobular type of ILC has a hybrid tubular and lobular appearance. The growth pattern of pleomorphic ILC is identical to that of classic ILC, but the tumor cells exhibit increased cytological atypia and pleomorphism as well as a higher rate of mitosis [1, 12,13,14,15,16].
Classic ILC are of low or intermediate histological grade and the majority are characterized by the positivity of hormone receptors and lack of HER2 expression; however, HER2-positive and/or triple-negative (estrogen and progesterone receptor-negative and HER-2 negative) phenotypes have been reported, particularly in ILC variants [1, 12,13,14,15,16,17,18]. Consistently, more than 80% of ILCs fall into the category of luminal molecular subtypes according to gene expression profile studies [3, 19]. Her-2-enriched and basal-like lobular tumors are rare, usually of non-classic variant, and associated with a worse prognosis [20]. Similar to invasive ductal carcinoma (IDC), tumor staging, and nodal status are important prognostic factors also in patients with ILC. Moreover, a high Ki67 proliferation index was found to be associated with a high risk of early and late recurrence [19, 20].
In addition to traditional prognostic and predictive factors, other actional biomarkers, such as tumor-infiltrating lymphocytes (TILs) and PD-L1 expression, have been recently included in the pathological characterization of IBC. PD-L1 expression in ILC has been observed both on lymphocyte and tumor cells. Overall, the level of TILs and PD-L1 reported in ILCs are lower than those observed in IDC and with different patterns, suggesting that ILC may be associated with a distinct immune microenvironment [21,22,23,24].
As mentioned above, most ILCs are currently classified as HER-2 negative. According to the American Society of Clinical Oncology (ASCO) and College of American Pathologists (CAP), the HER2 test positivity is defined by protein overexpression (score 3+) at immunohistochemistry (IHC) and/or HER2 score 2+ with gene amplification at in situ hybridization (ISH), while score 2+/ISH negative, score 1+ and score 0 were considered negative [25]. However, the introduction of novel anti-HER2 antibody-drug conjugates requires an in-depth categorization of this “HER2-negative” group, distinguishing tumors with no HER2 expression by IHC (or in less than 10% of tumor cells; score 0) from those with low HER2 expression (HER2-low IBC) showing immunohistochemistry HER2 score 1+ or 2+/ISH- [26,27,28]. Considering ILC, fewer cases have been observed among HER2-low IBC compared to HER2-zero tumors [29, 30].
Non-invasive lobular neoplasia, including lobular carcinoma in situ (LCIS) and atypical lobular hyperplasia (ALH), are frequently seen in combination with ILC [31,32,33,34]. ALH and LCIS are considered risk indicators and non-obligate precursors of invasive breast cancer [35, 36]. The neoplastic cells of ALH/LCIS morphologically resemble those of ILC distending the acini with the maintenance of the lobular architecture. Moreover, akin to the invasive counterpart, these types of non-invasive lobular neoplasia lack E-cadherin expression, confirming the early oncogenicity of CDH1 alterations in hereditary and non-hereditary lobular breast cancer [35,36,37] (Fig. 11.2).

Histological features of lobular carcinoma in situ (LCIS). (a) Monomorphic proliferation of polygonal discohesive cells with clear cytoplasm that distend the acini with the maintenance of the lobular architecture. (b) Non-invasive lesion with lobular phenotype, showing eccentric large pleomorphic nuclei, conspicuous nucleoli and large eosinophilic granular cytoplasm, consistent pleomorphic lobular carcinoma in situ. Hematoxylin and eosin, original magnification 200×. Adapted from: Guerini-Rocco and Fusco. Premalignant and preinvasive lesions of the breast. In: Breast Cancer: Innovations in Research and Management. Veronesi U, Goldhirsh A, Veronesi P, et al., editors. Springer International Publishing; 2017. p. 103–20 [33]
3 CDH1 Aberrations: The Hallmark of Lobular Breast Cancer
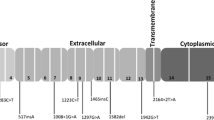
The CDH1 gene (16q22.1) encodes for the E-cadherin protein, which is responsible for cell adhesion and suppresses cell motility and invasion [38, 39]. The rationale for the use of E-cadherin as a biomarker in ILC is related to its very biology. This protein has an extracellular domain responsible for cell-to-cell adhesion via homodimerization with other E-cadherin molecules on adjacent cells [40]. The intracellular domain interacts with the actin cytoskeleton indirectly, through a complex formed by several mediators such as α-, β-, and p120-catenins. Therefore, the presence and functionality of E-cadherin are crucial not only in maintaining cell-to-cell adhesion but through the interaction with these mediators, in different intracellular pathways [40, 41] (Fig. 11.3).

Molecular events mediated by loss of E-cadherin in hereditary lobular carcinoma. E-cadherin is a 120 kDa glycoprotein encoded by the CDH1 gene, located on chromosome 16q22.1, and belongs to the classical Cadherin subgroup. It has an extracellular domain formed by five extracellular ~100 amino acid residue motifs, termed extracellular cadherin repeats. The calcium binding sites are located in the pockets between the repeats. This extracellular domain is mainly responsible for cell-to-cell adhesion via homodimerization with other E-cadherin molecules present on adjacent cells. E-cadherin has a single transmembrane domain that links the extracellular domain with the smaller intracellular domain. The intracellular domain interacts with the actin cytoskeleton indirectly, through a complex formed by several mediators such as α-, β-, and p120-catenins. Therefore, the presence and functionality of E-cadherin are crucial not only in maintaining cell-to-cell adhesion but through the interaction with these mediators, which plays also a role in a variety of intracellular pathways
The loss of E-cadherin functionality caused by CDH1 mutations results in the facilitation of epithelial-to-mesenchymal transition and tumorigenesis [42]. This molecular aberration is directly reflected by the non-to-poorly cohesive morphological appearance of lobular carcinoma cells and by the loss of immunohistochemical expression of E-cadherin and cytoplasmic expression of p120-catenins [43]. However, up to 15% of ILC may show E-cadherin expression and abnormal E-cadherin immunoreactivity has been seen in other breast cancer subtypes, including total absence or diminished membrane staining, and punctate or cytoplasmic expression [44, 45] (Fig. 11.4).

Spectrum of E-cadherin immunoreactivity in breast cancer. Representative micrographs of (a) lobular carcinoma showing loss of E-cadherin immunohistochemical expression (dashed arrow) and adjacent normal terminal duct-lobular units with strong membranous E-cadherin staining (full arrow); invasive breast cancers of no special type showing partial loss (b) and strong (c) membranous immunoreactivity for E-cadherin. E-cadherin immunohistochemistry, original magnification 200×. Adapted from: Corso G, Figueiredo J, De Angelis SP, et al. E-cadherin deregulation in breast cancer. J Cell Mol Med 2020;24:5930–6 [50]
In the TCGA series, CDH1 genomic aberrations have been detected in nearly 12% of all breast cancers including truncating, missense and splice-site mutations, copy number, and structural variants. Somatic CDH1 mutations have been reported in 50–80% of lobular breast cancer [2, 3, 6] (Fig. 11.5). These mutations mostly co-occur with heterozygous loss of 16q and they are frequently associated with downregulation of CDH1 transcript and protein levels [46]. Interestingly, the complete loss of CDH1 expression alone is not sufficient for invasive carcinoma development, as demonstrated in transgenic animal models. Indeed, other genetic alterations, such as Smad4 and p53, are required to promote invasiveness and metastasis [47,48,49]. Besides alterations affecting the CDH1 gene, epigenetic modifications and upregulation of transcriptional inhibitors have also been described as mechanisms of E-cadherin inactivation [50]. An important and frequent epigenetic modification is hypermethylation of the CDH1 promoter. This alteration has been studied in hereditary and non-hereditary lobular breast cancers, which suggests epigenetic silencing as an alternative CDH1 downregulation mechanism. CDH1 DNA hypermethylation has been demonstrated to be inversely proportional to E-cadherin levels in tumor cells [51].

Distribution of CDH1 mutations in breast cancer. (a) Oncoprint visualization of the CDH1 mutations across different histological subtypes of breast cancer. (b) Lollipop plot presenting frequencies and types of CDH1 mutations. TGCA Combined Study (3835 samples) from https://www.cbioportal.org/, accessed 20th July 2022)
Interestingly, it has been observed that CDH1 promoter hypermethylation is associated with reduced HR expression, increased disease progression, a higher metastatic rate, and a more aggressive clinical course overall. It is more frequent in patients presenting with sentinel lymph node metastases at diagnosis and is correlated with disease progression to distant metastases [52, 53]. This has led to the proposal of CDH1 hypermethylation as a prognostic biomarker to predict poorer outcomes [54]. Another mechanism of E-cadherin inactivation is represented by the overexpression of its transcriptional inhibitors, namely Snail, SLUG, zinc finger E-box-binding (ZEB1 and 2), and TWIST transcription factors [55]. Among these molecules, the one with the highest affinity for the CDH1 promoter is Snail, which acts by recruiting the mSin3A/Histone Deacetylase1 and 2 (HDAC1/2). Subsequent deacetylation of histones H3 and H4 results in silencing of the gene, thus effectively inhibiting E-cadherin synthesis [56, 57]. ZEB1 and ZEB2 behave similarly to Snail in suppressing CDH1 transcription, but their mechanisms of action appear to be independent. Thus, it has been hypothesized that at least two transcriptional downregulation complexes of E-cadherin do exist, but whether they participate in tumorigenesis within the same cell remains to be established [58]. High levels of ZEB1 have been found in aggressive BCs and associated with advanced-stage and lymph node metastases. Therefore, ZEB1 has been proposed as an additional prognostic biomarker in breast cancers, in particular in lobular breast cancer [41, 50, 59,60,61].
E-cadherin and many RTKs tend to co-localize at the basolateral portion of the cell membrane. In particular, the complex formed by the E-cadherin intracellular domain and EGFR has been extensively studied to be involved in adhesion-dependent bidirectional crosstalk. On one hand, cell-to-cell adhesion via E-cadherin inhibits the EGFR signaling pathway, including downstream mediators such as MAPK/ERK with downregulation of cell cycle progression and cellular proliferation [62]. Conversely, it has been demonstrated that cell adhesion transiently activates the EGFR/MAPK signaling cascade, and has a role in tissue growth [63]. Moreover, the upregulation of several RTKs pathways is known to inhibit E-cadherin-dependent cell-to-cell adhesion and promote epithelial-to-mesenchymal transition (EMT), suggesting that E-cadherin plays a role in tumorigenesis even when not directly affected by inactivating mutations [64]. E-cadherin is also known to form a complex with β-catenin. The E-cadherin/ β-catenin complex is crucial in maintaining not only cell-to-cell adhesion but also tissue’s architectural homeostasis. Beta-catenin is well known for being a central component of the WNT signal transduction pathway. It has been demonstrated that when catenin is bound by E-cadherin, the result is the promotion of tissue stasis by inhibition of cell proliferation and architectural stabilization. The disruption of the cadherin-catenin complex causes an increase of cytoplasmic un-bound β-catenin. This alters the WNT signaling pathway shifting the balance toward cell growth and proliferation. This effect has been demonstrated to be unrelated to E-cadherin adhesive properties and to be entirely dependent on its β-catenin binding region. In addition, β-catenin has an inhibitory effect on PTEN, a well-known tumor suppressor gene, further promoting uncontrolled cell proliferation [65, 66]. Another signaling pathway influenced by the interaction between E-cadherin and catenins at the cell membrane is that of the Rho GTPases. The Rho GTPases are a family of proteins involved in the interaction of E-cadherin with the cytoskeleton, a process influenced also by p120-catenin. They promote and regulate the organization of the cytoskeletal network during the formation of adherens junctions. The two Rho GTPase subfamilies most known for being influenced by E-cadherin are Rac and Rho. In normal conditions, E-cadherin activates Rac1 and inhibits Rho through the interaction of p120, increasing cell adhesion and cellular structural stability. Loss of E-cadherin causes an increase in unbound p120, which in turn creates an inversion of this balance. This not only promotes loss of cell-to-cell adhesion by disruption of the adherens junctions but also enhances cellular motility and migration due to rearrangement of the cytoskeletal network. Therefore, the Rho GTPase family has an important role in the process of EMT mediated by E-cadherin loss [67, 68]. Moreover, increased levels of p120 upregulate the NF-kB pathway, which contributes to tumorigenesis by promoting inflammation, cell proliferation, and apoptosis escape [69]. During EMT, when cells have detached from their tissue of origin they start to migrate within the extracellular matrix. E-cadherin loss has been demonstrated to enhance cellular motility in this new environment by upregulation of secretion and activity of metalloproteinases (MMP) [70]. These molecules play a role in matrix digestion and remodeling and, when their activity is increased, tumor cell migration is facilitated. In addition, MMPs have been shown to inactivate E-cadherin by cleavage of its extracellular domain, further demonstrating the close interplay of these two effectors in tumor spread [71]. Besides the loss of cell-to-cell adhesion and EMT, E-cadherin loss also increases the resistance of cells to apoptotic stimuli. This effect is mediated by the inverse relationship between E-cadherin expression and the Notch pathway. Reduction in E-cadherin levels is correlated with upregulation of this pathway, leading to an increase in intracellular levels of Bcl-2. The Bcl-2 family of proteins is known to be involved in the regulation of programmed cell death. Specifically, they have an anti-apoptotic role, thus their upregulation following E-cadherin loss promotes tumor resistance to apoptotic stimuli and improves the survival of neoplastic cells [72]. The interplay between E-cadherin and a plethora of intracellular signaling pathways demonstrates how the role of this molecule in tumorigenesis goes well beyond the loss of cell-to-cell adhesion. This also highlighted the need for detailed characterization and reporting of CDH1 variants identified, especially at the germline level.
4 The Genomic Landscape of Lobular Breast Cancer
During the last decades, broad genomic profiling with high-throughput next-generation sequencing technologies has shown that breast cancers are highly heterogeneous at the molecular level harboring few recurrent genomic aberrations and potentially actionable drivers [2, 4, 5, 73,74,75,76,77]. Overall, PIK3CA and TP53 are the most frequently mutated genes with different mutation rates based on breast cancer subtype. Nearly 40% of estrogen receptor-positive/luminal breast cancer harbor somatic driver mutations in the PIK3CA gene. TP53 mutations can be detected in 20–30% of luminal tumors but nearly 85% of basal-like/triple-negative breast cancers. Indeed, these triple-negative tumors show also high genomic instability and DNA repair gene aberrations, including BRCA1/2 alterations [75, 77]. ILC represents a special breast cancer type also at the genomic level. As mentioned above, ILC is characterized by a higher rate of CDH1 mutations as compared to IDC (63% versus 2% in the TCGA study). Other recurrently mutated genes (reported rate > 2%) in ILC included: PIK3CA, TBX3, RUNX1, FOXA1, ERBB2, ERBB3, PTEN, MAP3K1, AKT1, ARID1A, and TP53. Besides CDH1 heterozygous deletion (16q loss) detected in more than 90% of the cases, other recurrent copy number variations involve gain of CCND1, FGFR1, and MYC genes. Although amplification of the HER2 gene is not frequently seen in ILC, somatic mutations of ERBB2 have been reported in 2%–15% of cases [2,3,4,5,6, 8, 9, 78]. Overall, as compared to estrogen receptor-positive luminal breast cancer, invasive lobular carcinoma is enriched for CDH1 mutations and loss, mutation of TBX3 and FOXA1, mutation, and loss of PTEN with activation of AKT pathway but low mutation rate of GATA-3 [3] (Fig. 11.6).

Recurrent genomic alterations in CDH1-mutated invasive lobular carcinoma. Oncoprint visualization of the most frequently mutated genes in lobular breast carcinomas harboring somatic CDH1 mutations. TGCA Firehose Legacy series (99 samples) from https://www.cbioportal.org/, accessed 20th July 2022
Triple-negative (hormone receptors-negative and HER2-negative) ILC is a rare disease accounting for nearly 1% of triple-negative breast cancers and it has a poor prognosis. Although no significant differences in gene mutation frequency have been found compared to hormone receptor-positive/her2-negative cases, enrichment for alterations in ErbB and androgen receptor signaling pathways were observed in triple-negative ILC. Moreover, these tumors show a genomic profile distinct from triple-negative IDCs, including higher frequencies of CDH1, ERBB2, PI3KCA, and FOXA1 mutations [8, 79, 80].
Considering primary and metastatic ILC, similar repertoires of genomic alterations have been described. However, in the metastatic setting higher frequencies of TP53, ESR1, NF1, and ERRB2 alterations have been reported. Indeed, these genomic alterations may represent mechanisms of endocrine therapy resistance. Moreover, a higher tumor mutational burden has been observed in metastatic ILC as compared to primary tumors [81].
5 Conclusion
Lobular breast cancers display peculiar characteristics including morphologic, phenotypic, and transcriptomic features, genomic aberrations, immune microenvironment composition, and clinical behavior. Given the rarity of and maybe low awareness about hereditary CDH1-related ILC, few studies have been specifically focused on this entity and, so far, similar characteristics have been reported. Dedicated investigations are warranted to elucidate the molecular profiles of ILC that arise in women harboring CDH1 germline mutations. Indeed, there are numerous questions to be uncovered in the molecular mechanisms driving tumorigenesis and disease progression. A focused characterization of the molecular profile of hereditary CDH1-related ILC may enhance our understanding of these tumors and ultimately might aid in establishing effective prevention, screening, and tailored treatment strategies for women carrying CDH1 germline mutations.
References
Breast Tumours (2019) WHO Classification of Tumours. International Agency for Research on Cancer
Pereira B, Chin SF, Rueda OM et al (2016) The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun 7:11479
Ciriello G, Gatza ML, Beck AH et al (2015) Comprehensive molecular portraits of invasive lobular breast cancer. Cell 163:506–519
Grote I, Bartels S, Christgen H et al (2022) ERBB2 mutation is associated with sustained tumor cell proliferation after short-term preoperative endocrine therapy in early lobular breast cancer. Mod Pathol
WHO Classification of Tumours Editorial Board (2019) WHO classification of breast tumours: WHO classification of tumours, vol 2. World Health Organization
Ross JS, Wang K, Sheehan CE et al (2013) Relapsed classic E-cadherin (CDH1)-mutated invasive lobular breast cancer shows a high frequency of HER2 (ERBB2) gene mutations. Clin Cancer Res 19:2668–2676
Corso G, Montagna G, Figueiredo J et al (2020) Hereditary gastric and breast cancer syndromes related to CDH1 germline mutation: a multidisciplinary clinical review. Cancers (Basel) 12
Desmedt C, Zoppoli G, Gundem G et al (2016) Genomic characterization of primary invasive lobular breast cancer. J Clin Oncol 34:1872–1881
Guiu S, Wolfer A, Jacot W et al (2014) Invasive lobular breast cancer and its variants: how special are they for systemic therapy decisions? Crit Rev Oncol Hematol 92:235–257
Chen Z, Yang J, Li S et al (2017) Invasive lobular carcinoma of the breast: a special histological type compared with invasive ductal carcinoma. PLoS One 12:e0182397
McCart Reed AE, Kutasovic JR, Lakhani SR et al (2015) Invasive lobular carcinoma of the breast: morphology, biomarkers and 'omics. Breast Cancer Res 17:12
Pagni F, Guerini-Rocco E, Schultheis AM et al (2019) Targeting immune-related biological processes in solid tumors: we do need biomarkers. Int J Mol Sci 20
Arias-Stella JA, Alvarado-Cabrero I, Pareja F (2018) Special types of invasive breast carcinoma. Practical Atlas of breast pathology. Springer, pp 263–292
Walker RA, Hanby A, Pinder SE et al (2012) Current issues in diagnostic breast pathology. J Clin Pathol 65:771–785
De Schepper M, Vincent-Salomon A, Christgen M et al (2022) Results of a worldwide survey on the currently used histopathological diagnostic criteria for invasive lobular breast cancer. Mod Pathol
Christgen M, Cserni G, Floris G et al (2021) Lobular breast cancer: histomorphology and different concepts of a special spectrum of tumors. Cancers (Basel) 13
Fusco N, Sajjadi E, Venetis K et al (2022) Low-risk triple-negative breast cancers: clinico-pathological and molecular features. Crit Rev Oncol Hematol 103643
Venetis K, Crimini E, Sajjadi E, et al. HER2 low, ultra-low, and novel complementary biomarkers: expanding the spectrum of HER2 positivity in breast cancer. Front Mol Biosci 2022:fmolb.2022.834651
Conforti F, Pala L, Pagan E et al (2019) Endocrine-responsive lobular carcinoma of the breast: features associated with risk of late distant recurrence. Breast Cancer Res 21:153
Iorfida M, Maiorano E, Orvieto E et al (2012) Invasive lobular breast cancer: subtypes and outcome. Breast Cancer Res Treat 133:713–723
Thompson ED, Taube JM, Asch-Kendrick RJ et al (2017) PD-L1 expression and the immune microenvironment in primary invasive lobular carcinomas of the breast. Mod Pathol 30:1551–1560
Dill EA, Gru AA, Atkins KA et al (2017) PD-L1 expression and intratumoral heterogeneity across breast cancer subtypes and stages: an assessment of 245 primary and 40 metastatic tumors. Am J Surg Pathol 41:334–342
Desmedt C, Salgado R, Fornili M et al (2018) Immune infiltration in invasive lobular breast cancer. J Natl Cancer Inst 110:768–776
Sobral-Leite M, Van de Vijver K, Michaut M et al (2018) Assessment of PD-L1 expression across breast cancer molecular subtypes, in relation to mutation rate, BRCA1-like status, tumor-infiltrating immune cells and survival. Onco Targets Ther 7:e1509820
Wolff AC, Hammond MEH, Allison KH et al (2018) Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J Clin Oncol 36:2105–2122
Tarantino P, Hamilton E, Tolaney SM et al (2020) HER2-low breast cancer: pathological and clinical landscape. J Clin Oncol 38:1951–1962
Venetis K, Crimini E, Sajjadi E et al (2022) HER2 low, ultra-low, and novel complementary biomarkers: expanding the spectrum of HER2 positivity in breast cancer. Front Mol Biosci 9:834651
Sajjadi E, Venetis K, Ivanova M et al (2022) Improving HER2 testing reproducibility in HER2-low breast cancer. Cancer Drug Resist 5 (Accepted)
Mutai R, Barkan T, Moore A et al (2021) Prognostic impact of HER2-low expression in hormone receptor positive early breast cancer. Breast 60:62–69
Tarantino P, Jin Q, Tayob N et al (2022) Prognostic and biologic significance of ERBB2-low expression in early-stage breast cancer. JAMA Oncol
Lopez-Garcia MA, Geyer FC, Lacroix-Triki M et al (2010) Breast cancer precursors revisited: molecular features and progression pathways. Histopathology 57:171–192
Sciarra A, Lopez G, Corti C et al (2019) Columnar cell lesion and apocrine hyperplasia of the breast: is there a common origin? The role of prolactin-induced protein. Appl Immunohistochem Mol Morphol 27:508–514
Guerini-Rocco E, Fusco N (2017) Premalignant and preinvasive lesions of the breast. In: Veronesi U, Goldhirsh A, Veronesi P et al (eds) Breast cancer: innovations in research and management. Springer International Publishing, pp 103–120
Sciarra A, Lopez G, Corti C, et al (2017) Columnar cell lesion and apocrine hyperplasia of the breast: is there a common origin? The role of prolactin-induced protein. Appl Immunohistochem Mol Morphol
Sakr RA, Schizas M, Carniello JV et al (2016) Targeted capture massively parallel sequencing analysis of LCIS and invasive lobular cancer: repertoire of somatic genetic alterations and clonal relationships. Mol Oncol 10:360–370
Lee JY, Schizas M, Geyer FC et al (2019) Lobular carcinomas in situ display intralesion genetic heterogeneity and clonal evolution in the progression to invasive lobular carcinoma. Clin Cancer Res 25:674–686
Girardi A, Magnoni F, Vicini E et al (2022) CDH1 germline mutations in families with hereditary lobular breast cancer. Eur J Cancer Prev 31:274–278
Massari G, Magnoni F, Favia G et al (2021) Frequency of CDH1 germline mutations in non-gastric cancers. Cancers (Basel) 13
Paço A, Leitão-Castro J, Freitas R (2021) Epigenetic regulation of CDH1 is altered after HOXB7-silencing in MDA-MB-468 triple-negative breast cancer cells. Genes (Basel) 12
Halbleib JM, Nelson WJ (2006) Cadherins in development: cell adhesion, sorting, and tissue morphogenesis. Genes Dev 20:3199–3214
Paredes J, Figueiredo J, Albergaria A et al (2012) Epithelial E- and P-cadherins: role and clinical significance in cancer. Biochim Biophys Acta 1826:297–311
Corso G (2022) Pleiotropic cancer manifestations of germline CDH1 mutations: risks and management. J Surg Oncol 125:1326–1331
Rätze MAK, Koorman T, Sijnesael T et al (2022) Loss of E-cadherin leads to Id2-dependent inhibition of cell cycle progression in metastatic lobular breast cancer. Oncogene 41:2932–2944
Arps DP, Healy P, Zhao L et al (2013) Invasive ductal carcinoma with lobular features: a comparison study to invasive ductal and invasive lobular carcinomas of the breast. Breast Cancer Res Treat 138:719–726
Christgen M, Kandt LD, Antonopoulos W et al (2022) Inter-observer agreement for the histological diagnosis of invasive lobular breast carcinoma. J Pathol Clin Res 8:191–205
Cerami E, Gao J, Dogrusoz U et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401–404
Luo W, Fedda F, Lynch P et al (2018) CDH1 gene and hereditary diffuse gastric cancer syndrome: molecular and histological alterations and implications for diagnosis and treatment. Front Pharmacol 9:1421
Park JW, Jang SH, Park DM et al (2014) Cooperativity of E-cadherin and Smad4 loss to promote diffuse-type gastric adenocarcinoma and metastasis. Mol Cancer Res 12:1088–1099
Park JW, Kim M-S, Voon DC et al (2018) Multi-omics analysis identifies pathways and genes involved in diffuse-type gastric carcinogenesis induced by E-cadherin, p53, and Smad4 loss in mice. Mol Carcinog 57:947–954
Corso G, Figueiredo J, De Angelis SP et al (2020) E-cadherin deregulation in breast cancer. J Cell Mol Med 24:5930–5936
Shargh SA, Sakizli M, Khalaj V et al (2014) Downregulation of E-cadherin expression in breast cancer by promoter hypermethylation and its relation with progression and prognosis of tumor. Med Oncol 31:250
Shinozaki M, Hoon DS, Giuliano AE et al (2005) Distinct hypermethylation profile of primary breast cancer is associated with sentinel lymph node metastasis. Clin Cancer Res 11:2156–2162
Sebova K, Zmetakova I, Bella V et al (2011) RASSF1A and CDH1 hypermethylation as potential epimarkers in breast cancer. Cancer Biomark 10:13–26
Liu J, Sun X, Qin S et al (2016) CDH1 promoter methylation correlates with decreased gene expression and poor prognosis in patients with breast cancer. Oncol Lett 11:2635–2643
Schmalhofer O, Brabletz S, Brabletz T (2009) E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev 28:151–166
Bolós V, Peinado H, Pérez-Moreno MA et al (2003) The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with snail and E47 repressors. J Cell Sci 116:499–511
Peinado H, Ballestar E, Esteller M et al (2004) Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol 24:306–319
Comijn J, Berx G, Vermassen P et al (2001) The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell 7:1267–1278
Figueiredo J, Melo S, Carneiro P et al (2019) Clinical spectrum and pleiotropic nature of CDH1 germline mutations. J Med Genet 56:199–208
Li M, Rao X, Cui Y et al (2021) The keratin 17/YAP/IL6 axis contributes to E-cadherin loss and aggressiveness of diffuse gastric cancer. Oncogene
Xiang S, Liu YM, Chen X et al (2015) ZEB1 expression is correlated with tumor metastasis and reduced prognosis of breast carcinoma in Asian patients. Cancer Investig 33:225–231
Qian X, Karpova T, Sheppard AM et al (2004) E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J 23:1739–1748
Pece S, Gutkind JS (2000) Signaling from E-cadherins to the MAPK pathway by the recruitment and activation of epidermal growth factor receptors upon cell-cell contact formation. J Biol Chem 275:41227–41233
Thiery JP (2002) Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2:442–454
Gottardi CJ, Wong E, Gumbiner BM (2001) E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion-independent manner. J Cell Biol 153:1049–1060
Nelson WJ, Nusse R (2004) Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303:1483–1487
Deplazes J, Fuchs M, Rauser S et al (2009) Rac1 and Rho contribute to the migratory and invasive phenotype associated with somatic E-cadherin mutation. Hum Mol Genet 18:3632–3644
Bruner HC, Derksen PWB (2018) Loss of E-cadherin-dependent cell-cell adhesion and the development and progression of cancer, vol 10. Cold Spring Harb Perspect Biol
Vlahopoulos SA, Cen O, Hengen N et al (2015) Dynamic aberrant NF-κB spurs tumorigenesis: a new model encompassing the microenvironment. Cytokine Growth Factor Rev 26:389–403
Munshi HG, Ghosh S, Mukhopadhyay S et al (2002) Proteinase suppression by E-cadherin-mediated cell-cell attachment in premalignant oral keratinocytes. J Biol Chem 277:38159–38167
Davies G, Jiang WG, Mason MD (2001) Matrilysin mediates extracellular cleavage of E-cadherin from prostate cancer cells: a key mechanism in hepatocyte growth factor/scatter factor-induced cell-cell dissociation and in vitro invasion. Clin Cancer Res 7:3289–3297
Ferreira AC, Suriano G, Mendes N et al (2012) E-cadherin impairment increases cell survival through notch-dependent upregulation of Bcl-2. Hum Mol Genet 21:334–343
Christinat A, Pagani O (2013) Practical aspects of genetic counseling in breast cancer: lights and shadows. Breast 22:375–382
Stephens PJ, Tarpey PS, Davies H et al (2012) The landscape of cancer genes and mutational processes in breast cancer. Nature 486:400–404
The Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490:61–70
Curtis C, Shah SP, Chin SF et al (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486:346–352
Ellis MJ, Perou CM (2013) The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Discov 3:27–34
Corso G, Veronesi P, Sacchini V et al (2018) Prognosis and outcome in CDH1-mutant lobular breast cancer. Eur J Cancer Prev 3:237–238
Bareche Y, Venet D, Ignatiadis M et al (2018) Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Ann Oncol 29:895–902
Conforti F, Pala L, Pagan E et al (2021) Biological and clinical features of triple negative invasive lobular carcinomas of the breast. Clinical outcome and actionable molecular alterations. Breast 59:94–101
Pareja F, Ferrando L, Lee SSK et al (2020) The genomic landscape of metastatic histologic special types of invasive breast cancer. NPJ Breast Cancer 6:53
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Porta, F.M., Blanco, M.C., Ivanova, M., Fusco, N., Guerini-Rocco, E. (2023). Pathology and Somatic Alterations in Hereditary Lobular Breast Cancers. In: Corso, G., Veronesi, P., Roviello, F. (eds) Hereditary Gastric and Breast Cancer Syndrome. Springer, Cham. https://doi.org/10.1007/978-3-031-21317-5_11
Download citation
DOI: https://doi.org/10.1007/978-3-031-21317-5_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-21316-8
Online ISBN: 978-3-031-21317-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)