
Abstract
The biophysical properties of authentic infectious prion protein (PrP)-based mammalian prions have long impeded determination of their detailed structures. However, considerable recent progress has been made using cryo-electron microscopy. Three near-atomic resolution structures of ex vivo prions have now been reported, one of hamster 263K scrapie and the others of wildtype and glycophosphatidylinositol (GPI)-deficient forms of the mouse RML strain. Each of these highly infectious prion fibrils have ordered cores with parallel in-register intermolecular β-stack (PIRIBS) architectures that share major structural motifs. However, the 263K fibril differs from the RML structures in the detailed conformations of those motifs and the overall shapes of the fibril cross-sections. Such motif variations likely contribute to the strain-dependent templating that underpins conformationally faithful prion propagation. In the wild-type prion structures, N-linked glycans and GPI anchors project outward from the fibril surface. The wildtype and anchorless (and severely glycan deficient) RML fibrils have similar folds, indicating that these post-translational modifications do not substantially alter the core structure of this strain. However, in the wild-type structures, the GPI anchors follow the twisting fibril axis and are likely to bind cellular membranes. This binding may contribute to the pathognomonic membrane distortions of wild-type prion diseases. Analysis of the 263K structure with molecular dynamics simulations has suggested a mechanism for the hamster-to-mouse transmission barrier. These initial high-resolution structures provide foundations for understanding prion molecular pathogenesis, but given the multitude of mammalian prion strains, much further work will be required to characterize the full range of prion structures.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Structures
- Cryo-EM
- NMR
- Glycans
- Glycophosphatidylinositol
- Parallel in-register β-sheet
- Strains
- Transmission barrier
- β-arch
1 Introduction
As early as the 1960s, researchers were perplexed by the unusual properties of the scrapie agent and proposed that they might be self-propagating states of proteins (Griffith 1967; Pattison and Jones 1967). In the ensuing decades, the infectious scrapie agent and related pathogens of the transmissible spongiform encephalopathies (TSEs) were dubbed prions (Prusiner 1982), and the protein involved became prion protein or PrP, with the infectious form often called PrPSc (Prusiner 1998). For nearly seven decades after the first proposals of protein structure-based pathogens, the 3D structures that allow prions to replicate as deadly infectious agents remained enigmatic. One of the key mysteries was how distinct strains can be propagated faithfully, passage after passage, in a single host genotype if prions carried no agent-specific nucleic acid genome. Another question was what, mechanistically, controls transmission barriers when prions are passed from one host genotype to another. In other words, why do some PrP sequence mismatches between hosts matter so much more than others? These mysteries have been difficult to explain with any clarity without detailed knowledge of prion structures. However, near-atomic cryo-EM structures of highly infectious brain-derived prions (Kraus et al. 2021a, b; Hoyt et al. 2021; Manka et al. 2021), as well as much more innocuous synthetic recombinant PrP fibrils (Gallagher-Jones et al. 2018; Glynn et al. 2020; Wang et al. 2020; Li and Jaroniec 2021), have begun to emerge. Here we review those structures and their new mechanistic implications for prion replication, strain fidelity, species barriers, and pathogenesis. We focus on overtly fibrillar forms of prions because those are the only ones for which highly resolved structures are available.
2 Development of Initial Parallel In-Register and 4-Rung β-Solenoid Models for PrPSc Fibrils
The accumulation in the literature of a variety of coarse empirical descriptors of prion fibrils allowed increasingly grounded structural models to be proposed (Groveman et al. 2014; Spagnolli et al. 2019). Ultrastructural imaging indicated that prions could be fibrillar, with properties of amyloids (Merz et al. 1981; Prusiner et al. 1983; Gabizon et al. 1987; Hope et al. 1988; Silveira et al. 2005) while other studies have described infectious units that are smaller than elongated fibrils (Silveira et al. 2005; Tzaban et al. 2002; Sajnani et al. 2012; Vanni et al. 2020; Cortez et al. 2021). Diffraction studies of ex vivo prion fibrils showed that, as is typical of amyloid fibrils, PrP polypeptide chains run perpendicular to the fibril axis with spacings of ~4.9 Å. Measurements of the intermolecular distances between specific labeled residues in synthetic recombinant PrP fibrils using electron paramagnetic resonance (EPR) (Cobb et al. 2008; Cobb et al. 2007) and solid-state NMR (Tycko et al. 2010; Helmus et al. 2011; Groveman et al. 2014; Theint et al. 2017, 2018; Shannon et al. 2019) provided strong evidence that such fibrils could assemble with parallel in-register intermolecular β-sheet or stack (PIRIBS) architectures. In PIRIBS structures, residues in one molecule are aligned along the fibril axis with the corresponding residues of adjacent molecules in the stack, that is, in-register (Fig. 3.1a). Although these types of studies established that certain PrP residues were within PIRIBS structures in synthetic PrP fibrils, they did not establish the overall folds of the polypeptides.

Parallel in-register intermolecular β-sheet/stack (PIRIBS) versus 4-rung β-solenoid (4RβS) models for prion fibrils. (a) Trimeric segment of PIRIBS-based fibril as determined by high-resolution cryo-EM of 263K prions (Kraus et al. 2021b). A single monomeric unit is highlighted in orange. In PIRIBS (and not 4RβS) architectures, each amino acid residue in one monomer is aligned with the corresponding residue in the adjacent monomers (aqua blue circles). A dashed line circumscribes a representative arch (the middle arch), by which we mean a loop that bends back on itself. We have previously referred to these motifs as β-arches, but now simply call them arches because some do not meet all of the criteria of β-arches in which sidechains within β-strands on the opposing flanks of the arch interact directly. (b) Trimeric stack assembled from a 4RβS protofilament model proposed for GPI-anchorless RML prion based on lower resolution data (Spagnolli et al. 2019). These models were each drawn using PDB coordinates as reported in (Kraus et al. 2021b; Spagnolli et al. 2019) using ChimeraX (Pettersen et al. 2021). In the case of the 4RβS illustration, the published coordinates of the monomer were used and stacked manually using in Powerpoint to depict the concept of a 4RβS protofilament without intending to accurately represent any proposed interfaces between monomers
Nonetheless, based on such initial findings, Groveman and colleagues envisioned PIRIBS-based models for infectious prion fibrils, which typically have much larger proteinase K (PK)-resistant cores and are much more infectious than the synthetic fibrils studied in the EPR and NMR studies (Groveman et al. 2014). More recently, a quite distinct 4-rung β-solenoid (4RβS) model was proposed for the GPI-anchorless RML (aRML) prion fibril based on brain-derived prion fiber diffraction patterns, low-resolution cryo-EM imaging, and H/D-exchange data (Spagnolli et al. 2019) (Fig. 3.1b). Among the key arguments for the 4RβS model were meridional diffraction signals at 9.6, 6.4, and 4.8 Å, corresponding to second-, third-, and fourth-order diffraction of a β-sheet with a 19.2 Å spacing of features along the fibril axis (Wille and Requena 2018). In the proposed 4RβS model, single PrP molecules provide four successive, distinct rungs along the axis of a protofilament (Spagnolli et al. 2019), in contrast to a single rung in PIRIBS models. In such an arrangement, the interfaces between monomers in the stack, for example, would be ~19 to 20 Å and would be consistent with a 19.2 Å diffraction. This model also postulates that two intertwined protofilaments comprise the overall fibril, whereas, in the PIRIBS models, a single PrP molecule spans the entire fibril cross-section. In any case, when these widely divergent PIRIBS and 4RβS models were proposed, there was insufficient empirical data on ex vivo prions to discriminate between these architectures.
3 Cryo-EM of Synthetic PrP Fibrils
In the last couple of years, cryo-EM combined with single particle analysis and helical reconstruction (Scheres 2020) has revealed near-atomic resolution structures for recombinant human PrP PrP94-178 (rhu94-178) (Glynn et al. 2020), human PrP23-144 (rhu23-144) (Li and Jaroniec 2021), full-length human PrP23-231 (Wang et al. 2020), mutant full-length human E196K PrP23-231 (rhu23-231 E196K) (Wang et al. 2021), and a much shorter synthetic peptide (residues 168–176) of bank vole PrP (Gallagher-Jones et al. 2018). Importantly, as is true of most of the synthetic fibrils mentioned above, the PK-resistant cores of these fibrils are much smaller than those found in bona fide tissue-derived infectious PrPSc fibrils. Such synthetic fibrils are likely to be either non-infectious or many orders of magnitude less infectious per unit protein (Li et al. 2018; Kraus et al. 2017; Groveman et al. 2017; Caughey and Kraus 2019). Nonetheless, these studies provided important initial clues to how various recombinant PrP constructs can assemble into fibrils in vitro.
Each of these synthetic PrP fibrils has a PIRIBS architecture. However, their ordered fibrillar cores are comprised of different sequences. Fibrils of the N- and C-terminally truncated rhu94-178 fibrils have two closely packed, symmetrical protofilaments (Glynn et al. 2020). The core of each protofilament contains a β-arch of residues 106–145 (Fig. 3.2). These same residues comprise the ordered core of fibrils formed from rhu23-144, but with a quite distinct conformation and a fibril cross-section comprising four identical protofilaments (Li and Jaroniec 2021). The human PrP23-144 sequence corresponds to that expressed in humans with a form of Gerstmann–Sträussler–Scheinker (GSS) syndrome linked to expression of PrP with the rare Y145Stop mutation (Ghetti et al. 2018), but whether the respective conformations of the synthetic and in vivo fibrils are similar remains to be determined.

Cross-sections of infectious brain-derived hamster 263K prion fibrils and likely non-infectious synthetic recombinant human PrP fibrils. The underlying images are taken and adapted with permission from projections of density maps derived from single-particle cryo-EM analyses of fibrils of 263K prions (Kraus et al. 2021a, b), synthetic rhuPrP94-178 (Glynn et al. 2020), and rhuPrP23-231 (Wang et al. 2020). Note that, the synthetic fibrils have two identical symmetrically arranged protofilaments, whereas the 263K fibril core is comprised of a single filament. Blue lines trace the polypeptide backbones of residues 106–145 in one of the protofilaments (top left) that, in the 263K structure, form the N arch (bottom panel, also see Fig. 3.3d). Red lines trace backbones of the respective disulfide arches and additional C-terminal strands within residues 170–227 as it occurs in synthetic PrP 23-231 fibrils (top right) and 263K prion fibrils (bottom panel)
Fibrils derived from rhuPrP23-231 have two protofilaments, with the ordered cores being formed by C-terminal residues 170–229 (Fig. 3.2). In contrast to the rhu94-178 (Glynn et al. 2020) or rhu23-144 (Li and Jaroniec 2021) fibrils, these protofilament cores feature a arch linked at the base by the natural disulfide bond formed between Cys179 and Cys214. This disulfide arch is related to disulfide arches suggested previously by multiple EPR and ssNMR studies of synthetic human and rodent PrP fibrils containing this C-terminal domain (Groveman et al. 2014; Cobb et al. 2007, 2008; Tycko et al. 2010). A disulfide arch also dominates the PIRIBS core of fibrils of the familial human prion disease-linked E196K mutant of rhu23-231 PrP (Wang et al. 2021). However, this arch has a distinct conformation, showing that the disulfide arches can differ between fibrils formed from mutant versus wild-type human PrP sequences.
4 Near-Atomic Cryo-EM Structures of Infectious Tissue-Derived Prions
As of this writing, three high-resolution cryo-EM structures of fully infectious, ex vivo prion fibrils have been reported, including those of the hamster 263K scrapie strain (Kraus et al. 2021a, b) and both wildtype (wt) and GPI-anchorless (a) forms of the mouse RML scrapie strain (Hoyt et al. 2021; Manka et al. 2021). Each of these bona fide protease-resistant PrPSc (PrPRes) preparations was shown to contain approximately 109 50% lethal doses (LD50) per mg protein. The wildtype 263K and RML prions also have GPI-anchors and abundant N-linked glycans, whereas the aRML strain is deficient in these post-translational modifications (Chesebro et al. 2005). As noted above, these ex vivo prions have much larger proteinase K-resistant cores than those of the synthetic PrP fibrils described above. Indeed, this span of ~140 to 150 residues of the highly infectious prion fibrils is also larger than those of most, if not all, other neuropathologic protein amyloids.
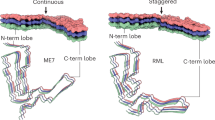
That said, and consistent with what has been seen so far with synthetic PrP amyloids, the ex vivo prion fibril structures also have PIRIBS cores (Kraus et al. 2021a, b; Hoyt et al. 2021; Manka et al. 2021) (Fig. 3.3) with single monomers comprising the entire cross-sections of these fibrils. Occasionally, laterally aligned duplexes of fibrils can be seen (Hoyt et al. 2021; Manka et al. 2021), but not regularly enough to be resolved as discrete subpopulations by single-particle cryo-EM analysis. Importantly, the aRML and wtRML fibril cross-sections are strikingly similar (compare refs (Hoyt et al. 2021; Manka et al. 2021)), but each is distinct from the 263K cross-section in overall shape as well as conformational detail (Fig. 3.3b–d).

Structures of hamster 263K and mouse aRML prion strains (Kraus et al. 2021b; Hoyt et al. 2021). (a) Lateral view of hamster 263K fibril (density map projection). (b) Enlarged cross-sectional views of fibril density maps. Presumed positions of the structurally variable and mostly unresolved N-linked glycans and GPI anchor are indicated on the 263K map. aRML is deficient in these post-translational modifications. (c) Atomic models (monomeric subunits). (d) Ribbon diagrams with structural motifs outlined in the 263K model that are analogous to, but distinct from, those in aRML. Panel a adapted with permission from (Kraus et al. 2021b)
Among the key features of the 263K (Kraus et al. 2021a, b) and RML (Hoyt et al. 2021; Manka et al. 2021) prions are 3 arches. These include two types of arch motifs that are seen, albeit with conformational variation, in synthetic fibrils, that is, those spanning ~113 to 131 and ~170 to 229 (Fig. 3.3d). As we expected from our initial modeling of a PIRIBS architecture for infectious prions (Groveman et al. 2014), the much larger cores of ex vivo prions have both of these arches at once, whereas the synthetic fibrils have only one or the other. We now refer to the more N-terminal of these arches as the “N arch.” As with the synthetic fibrils, we refer to the C-terminal arch as the “disulfide arch” (Fig. 3.3d). An additional feature of 263K and RML prions is another arch, namely the middle arch, that occurs between the N- and disulfide arches. The middle arch shares its N-terminal flank with the N arch. Another shared feature of the 263K and RML prions is a steric zipper between the extreme N-terminal residues of the core against the head of the middle arch.
Although the 263K and RML prion structures share these key structural motifs, their conformational details are substantially different between these strains (Kraus et al. 2021a, b; Hoyt et al. 2021; Manka et al. 2021). Notably, the N arches of 263K and RML fibrils have strikingly different heads or tips despite having identical glycine- and hydrophobic amino acid sequences spanning residues 113–138 (hamster numbering). Possibly, the conformational options of these head regions are influenced by the sequence differences that are immediately N- and C-terminal to the shared stretch of residues in the loop. The C-terminal half of the prion fibril cores of these strains also have marked conformational differences. For example, whereas in 263K the disulfide arch is nearly aligned with the N arch, these β-arches in the RML strains are almost perpendicular to one another, giving the cross-section a V-shape (Fig. 3.3c, d). The extreme C-terminal residues, where the GPI anchors are attached in the wild-type structures, project in opposite directions. In 263K, residues 219–227 flank the disulfide arch, whereas in the RML structures, the analogous residues flank residues 166–171. The otherwise similar aRML and wtRML structures differ in the C-terminal residues that could be assigned in the resolved map, with the ordered cores of aRML and wtRML extending to residues 230 and 225, respectively. This may be due to presence of structurally heterogeneous GPI anchors on the latter, which may compromise the resolution of the adjacent residues. Similarly, the resolved amyloid core for 263K prions (95–227) did not include the extreme C-terminal residues linked most closely to the glycolipid.
With respect to the mechanism by which these prions grow, the cross-sectional differences between the 263K and the RML prion fibrils clearly give them distinct templates on the fibril tips where the incorporation of new monomers occurs (Kraus et al. 2021a, b; Hoyt et al. 2021; Manka et al. 2021). Presumably contributing to these distinct templates is the difference in sequence between the hamster and mouse PrP sequences at 8 positions within the fibril core (e.g., see Figure S8 of (Kraus et al. 2021b)). The purely conformational, as opposed to sequence, determinants of prion strain should be clarified by analyses of strains isolated from hosts of the same genotype.
5 PrPC to PrPSc Conversion
Given the respective structures of PrPC and PrPSc that are now known, it is clear that complete refolding of the secondary and tertiary structures of PrPC is required (Fig. 3.4) (Kraus et al. 2021a, b). The steps involved, and the involvement of monomeric or oligomeric intermediates, remain unclear. Among the major conformational changes that must occur are dissociation of the PrPC’s β1-Helix 1-β2 loop from Helices 2 and 3 (Kraus et al. 2021a, b; Hoyt et al. 2021; Manka et al. 2021), a process that has been predicted and described previously as a “banana-peeling model” (Adrover et al. 2010). The α-helices also must be rearranged into extended chains, and the small intramolecular β1-β2 sheet dissociated. Contributing to the complexity of the conversion process is the polarity of the 263K and RML fibrils with opposite ends that are not equivalent (Kraus et al. 2021a, b; Hoyt et al. 2021). Notably, deviations from planarity of each monomer within the prion fibril stack mean that, for example, the hydrophobic heads of the N β-arches protrude at one end and recede at the other. This might affect the initial points of contact of the PrPSc template with incoming PrP molecules, and consequently, the sequence of events and kinetics of growth at each end. Given the in-register polypeptide alignment within the PrPSc end product, it seems likely that certain residues of an incoming monomer initiate contact with the analogous residues on the PrPSc template. Then adjacent residues might “zip” onto the polypeptide track of the template, forming periodic intermolecular β-sheets and loops along the way. Whatever the actual conversion mechanism, it will also likely be influenced crucially by interactions with anionic cofactors. Such cofactors have been shown to be important in conversion (Wong et al. 2001) and the assembly of infectious prions in vitro (Shaked et al. 2001; Deleault et al. 2003, 2005, 2007, 2010, 2012a, b; Supattapone 2020; Geoghegan et al. 2007; Wang et al. 2010; Miller et al. 2013) and are likely to electrostatically compensate for the stacking of positively charged and mutually repulsive, residues along the axis of the fibril (Kraus et al. 2021b; Groveman et al. 2014, 2015). Interactions with membranes can also affect conversion reactions (Baron et al. 2002, 2006; Baron and Caughey 2003; Rouvinski et al. 2014; Wegmann et al. 2008). In the membrane-bound context of wild-type forms of PrPC and PrPSc, their relative topologies and modes of contact should be constrained by C-terminal tethering of each to the same phospholipid bilayer (Fig. 3.4).

Hypothetical depictions of the membrane-bound hamster PrPC monomer (residues 95–231) and corresponding residues in each monomer of the 263K prion multimer. Polypeptides (aqua blue) are shown with N-linked glycans (yellow) and GPI anchors (blue) imbedded in a phospholipid membrane. The PrPC and 263K structures were drawn using PDB coordinates referenced in (James et al. 1997; Kraus et al. 2021b), respectively. In the PrPC structure, the serpentine line at the N-terminus represents residues 95–124 that are disordered in the NMR-based PrPC structure. The GPI and N-linked glycan illustrations show single representative structures that, in actuality, are heterogeneous (Rudd et al. 1999; Stahl et al. 1992). Adapted with permission from (Kraus et al. 2021b). Graphics by Austin Athman
6 Impacts of Glycans and GPI Anchors
Multiple studies have documented profound effects of GPI anchors and N-linked glycans, or lack thereof, on prion disease pathogenesis [e.g. (Chesebro et al. 2005, 2010; Sevillano et al. 2020; Bett et al. 2013; Cancellotti et al. 2010; Klingeborn et al. 2011; Wiseman et al. 2015; Race et al. 2018; Makarava et al. 2020)]. For example, in hosts expressing PrP without the GPI anchor signal sequence, PrPSc accumulates in the large extracellular amyloid plaques. Such hosts include genetically engineered transgenic mice (Chesebro et al. 2005, 2010; Klingeborn et al. 2011; Raymond et al. 2012; Rangel et al. 2013) or humans expressing anchorless PrP mutants such as Y145X 163X, Y226X, Q227X, and G131V (Ghetti et al. 2018). Genetic manipulations of the N-linked glycosylation of the host’s PrP molecules can also markedly affect prion disease phenotypes [e.g., (Sevillano et al. 2020; Cancellotti et al. 2010; Wiseman et al. 2015)]. However, these post-translational modifications do not seem to substantially alter the core structures of at least 3 murine prions (RML, ME7, and 22L) strains, as probed by infrared spectroscopy (Baron et al. 2011). This conclusion is confirmed in much greater detail by the new cryo-EM structures of the wtRML and aRML fibril cores, which, as noted above, are quite similar (Hoyt et al. 2021; Manka et al. 2021). Also, fundamental RML strain phenotypes including incubation period and neuropathological lesion profile are maintained through passages from wildtype mice into anchorless PrP mice and back again (Chesebro et al. 2010), although more subtle long-term effects on inhibitor sensitivity have been reported (Mahal et al. 2012). Still, the overall similarity of the aRML and wtRML core structures, together with their divergence from the 263K structure, are consistent with polypeptide core structures “encoding” the fundamental self-replicative properties of strains as postulated previously (Bessen et al. 1995; Bessen and Marsh 1994; Telling et al. 1996). Nonetheless, the phenotypes of those strains can be affected profoundly by the GPI anchors and glycans available in a given type of host or tissue (e.g. (Chesebro et al. 2005, 2010; Sevillano et al. 2020; Bett et al. 2013; Cancellotti et al. 2010; Klingeborn et al. 2011; Wiseman et al. 2015; Race et al. 2018; Makarava et al. 2020)).
Such phenotypic effects are likely due to different interactions of wildtype and anchorless prion fibrils with their tissue environments, as mediated by the glycans and GPI anchors on their surfaces (Fig. 3.4). With prion fibrils tethered to the membrane, glycans and bound membranes would blanket the C-terminal half of the fibril cores and restrict the access of other macromolecules to the polypeptide. Presumably, this would slow, or even preclude, easy access of proteostatic or innate immune macromolecules that might be involved in prion clearance or fragmentation. Access to PrPSc might be particularly limited within distorted membrane invaginations that are pathognomonic lesions of prion disease (Rouvinski et al. 2014; Wegmann et al. 2008; Caughey et al. 2009; Jeffrey et al. 2011, 2017; Jeffrey 2013). Among the more intriguing of those lesions are spiral twisted membrane inclusions (Jeffrey 2013; Jeffrey et al. 2017). As these spiral structures can be immunogold-stained for PrPd, it is tempting to speculate that PrPSc fibrils lie at their cores, with the spiraling GPI anchors of the fibril(s) pulling and distorting cocoon-like membranes that wrap them. Membrane attachments might also enhance prion replication by promoting fragmentation due to stresses imposed by membrane dynamics. Fragmentation is thought to be key in prion replication in vivo (Meisl et al. 2021). Also, cell-to-cell spreading might be facilitated via prion binding to membranous particles such as exosomes and tunneling nanotubes (Caughey et al. 2009; Gousset et al. 2009; Vassileff et al. 2020). Such mechanisms, as well as toxic effects of GPI-mediated membrane distortions, might help to account for more the rapid disease progression that has been observed in wild-type hosts (Chesebro et al. 2005, 2010; Klingeborn et al. 2011). Ultimately, however, like wild-type prions, anchorless prions can be highly infectious and lethal for the host (Chesebro et al. 2010).
7 Structure-Based Modeling of Transmission Barriers
When prions are transmitted between hosts of different PRNP genotypes, profound inefficiencies, that is, transmission barriers, can be observed [e.g., (Prusiner et al. 1990)]. For an infection to take hold, the incoming PrPSc must be able to convert and recruit the heterologous PrPC of the new host. Although there is considerable sequence homology between the PrP sequences of different mammalian hosts, mismatches of as little as a single residue can inhibit such heterologous conversions (e.g., (Prusiner et al. 1990; Scott et al. 1993; Goldmann et al. 1994; Kocisko et al. 1995; Priola et al. 1994; Priola and Chesebro 1995; Bossers et al. 1997; Raymond et al. 1997, 2000; Asante et al. 2015)). Modeling based on the new high-resolution 263K prion structures, together with knowledge of key mismatches controlling the hamster 263K-to-mouse transmission barrier (Scott et al. 1993; Priola et al. 2001), has suggested a plausible molecular mechanism for this barrier (Kraus et al. 2021b). Specifically, the sequence mismatch at residue 155 (hamster numbering), which is N in hamsters and Y in mice, had been shown to be particularly influential (Scott et al. 1993; Priola et al. 2001). In the 263K structure, the sidechain of N155 is in a tightly packed area, and in silico modeling suggests that attempts to incorporate a bulkier Y sidechain at this position would cause steric clashes and require adjustments in hydrogen bonding and the polypeptide backbone to form a hybrid prion structure (Kraus et al. 2021b). We suspect that these effects slow the kinetics of conversion and/or the stability of the product to an extent that greatly reduces the efficiency of infection. In contrast, several other sequence mismatches between the hamster and mouse PrP sequences are much less inhibitory, presumably due to the positions of those residues on the outside of the fibril core or in less tightly packed interior positions. Given the multitude of prion strains/conformations and the variety of PrP sequence mismatches that influence their transmission efficiencies, we assume that the mechanisms of transmission barriers will be diverse.
8 Conclusions
The availability of high-resolution 3D structures of fully infectious prions is now helping us understand how prions replicate with conformational fidelity, how they interact with their tissue environments to cause disease, and how sequence mismatches between hosts can result in transmission barriers. So far, only three such structures are available, and much more work will be needed to characterize the entire spectrum of PrP-based prion structures. Such work will provide important structural foundations for the rational design and discovery of drugs or vaccines that can block propagation, promote clearance, and/or detoxify prions in infected individuals.
References
Asante EA, Smidak M, Grimshaw A, Houghton R, Tomlinson A, Jeelani A, Jakubcova T, Hamdan S, Richard-Londt A, Linehan JM, et al. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature. 2015;522:478–81. https://doi.org/10.1038/nature14510.
Baron GS, Caughey B. Effect of glycosylphosphatidylinositol anchor-dependent and – independent prion protein association with model raft membranes on conversion to the protease-resistant Isoform. J Biol Chem. 2003;278:14883–92.
Baron GS, Wehrly K, Dorward DW, Chesebro B, Caughey B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. EMBO J. 2002;21:1031–40.
Baron GS, Magalhaes AC, Prado MA, Caughey B. Mouse-adapted scrapie infection of SN56 cells: greater efficiency with microsome-associated versus purified PrP-res. J Virol. 2006;80:2106–17.
Baron GS, Hughson AG, Raymond GJ, Offerdahl DK, Barton KA, Raymond LD, Dorward DW, Caughey B. Effect of glycans and the glycophosphatidylinositol anchor on strain dependent conformations of scrapie prion protein: improved purifications and infrared spectra. Biochemistry. 2011;50:4479–90. https://doi.org/10.1021/bi2003907.
Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol. 1994;68:7859–68.
Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT Jr, Caughey B. Nongenetic propagation of strain-specific phenotypes of scrapie prion protein. Nature. 1995;375:698–700.
Bett C, Kurt TD, Lucero M, Trejo M, Rozemuller AJ, Kong Q, Nilsson KP, Masliah E, Oldstone MB, Sigurdson CJ. Defining the conformational features of anchorless, poorly neuroinvasive prions. PLoS Pathog. 2013;9:e1003280. https://doi.org/10.1371/journal.ppat.1003280.
Bossers A, Belt PBGM, Raymond GJ, Caughey B, de Vries R, Smits MA. Scrapie susceptibility-linked polymorphisms modulate the in vitro conversion of sheep prion protein to protease-resistant forms. Proc Natl Acad Sci U S A. 1997;94:4931–6.
Cancellotti E, Bradford BM, Tuzi NL, Hickey RD, Brown D, Brown KL, Barron RM, Kisielewski D, Piccardo P, Manson JC. Glycosylation of PrPC determines timing of neuroinvasion and targeting in the brain following transmissible spongiform encephalopathy infection by a peripheral route. J Virol. 2010;84:3464–75. https://doi.org/10.1128/JVI.02374-09.
Caughey B, Kraus A. Transmissibility versus pathogenicity of self-propagating protein aggregates. Viruses. 2019;11:1044. https://doi.org/10.3390/v11111044.
Caughey B, Baron GS, Chesebro B, Jeffrey M. Getting a grip on prions: oligomers, amyloids, anchors and pathological membrane interactions. Annu Rev Biochem. 2009;78:177–204.
Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–9.
Chesebro B, Race B, Meade-White K, LaCasse R, Race R, Klingeborn M, Striebel J, Dorward D, McGovern G, Jeffrey M. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathog. 2010;6:e1000800. https://doi.org/10.1371/journal.ppat.1000800.
Cobb NJ, Sonnichsen FD, McHaourab H, Surewicz WK. Molecular architecture of human prion protein amyloid: a parallel, in-register beta-structure. Proc Natl Acad Sci U S A. 2007;104:18946–51.
Cobb NJ, Apetri AC, Surewicz WK. Prion protein amyloid formation under native-like conditions involves refolding of the C-terminal alpha-helical domain. J Biol Chem. 2008;283:34704–11. https://doi.org/10.1074/jbc.M806701200.
Cortez LM, Nemani SK, Duque Velasquez C, Sriraman A, Wang Y, Wille H, McKenzie D, Sim VL. Asymmetric-flow field-flow fractionation of prions reveals a strain-specific continuum of quaternary structures with protease resistance developing at a hydrodynamic radius of 15 nm. PLoS Pathog. 2021;17:e1009703. https://doi.org/10.1371/journal.ppat.1009703.
Deleault NR, Lucassen RW, Supattapone S. RNA molecules stimulate prion protein conversion. Nature. 2003;425:717–20.
Deleault NR, Geoghegan JC, Nishina K, Kascsak R, Williamson RA, Supattapone S. Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. J Biol Chem. 2005;280:26873–9.
Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A. 2007;104:9741–6.
Deleault NR, Kascsak R, Geoghegan JC, Supattapone S. Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry. 2010;49:3928–34. https://doi.org/10.1021/bi100370b.
Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, Supattapone S. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc Natl Acad Sci U S A. 2012a;109:8546–51. https://doi.org/10.1073/pnas.1204498109.
Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, Rees JR, Supattapone S. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci U S A. 2012b;109:E1938–46. https://doi.org/10.1073/pnas.1206999109.
Gabizon R, McKinley MP, Prusiner SB. Purified prion proteins and scrapie infectivity copartition into liposomes. Proc Natl Acad Sci U S A. 1987;84:4017–21.
Gallagher-Jones M, Glynn C, Boyer DR, Martynowycz MW, Hernandez E, Miao J, Zee CT, Novikova IV, Goldschmidt L, McFarlane HT, et al. Sub-angstrom cryo-EM structure of a prion protofibril reveals a polar clasp. Nat Struct Mol Biol. 2018;25:131–4. https://doi.org/10.1038/s41594-017-0018-0.
Geoghegan JC, Valdes PA, Orem NR, Deleault NR, Williamson RA, Harris BT, Supattapone S. Selective incorporation of polyanionic molecules into hamster prions. J Biol Chem. 2007;282:36341–53.
Ghetti B, Piccardo P, Zanusso G. Dominantly inherited prion protein cerebral amyloidoses – a modern view of Gerstmann-Straussler-Scheinker. Handb Clin Neurol. 2018;153:243–69. https://doi.org/10.1016/B978-0-444-63945-5.00014-3.
Glynn C, Sawaya MR, Ge P, Gallagher-Jones M, Short CW, Bowman R, Apostol M, Zhou ZH, Eisenberg DS, Rodriguez JA. Cryo-EM structure of a human prion fibril with a hydrophobic, protease-resistant core. Nat Struct Mol Biol. 2020;27:417–23. https://doi.org/10.1038/s41594-020-0403-y.
Goldmann W, Hunter N, Smith G, Foster J, Hope J. PrP genotype and agent effects in scrapie: change in allelic interaction with different isolates of agent in sheep, a natural host of scrapie. J Gen Virol. 1994;75(Pt 5):989–95. https://doi.org/10.1099/0022-1317-75-5-989.
Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT, Chenouard N, de Chaumont F, Martino A, Enninga J, et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol. 2009;11:328–36. https://doi.org/10.1038/ncb1841.
Griffith JS. Self-replication and scrapie. Nature. 1967;215:1043–4.
Groveman BR, Dolan MA, Taubner LM, Kraus A, Wickner RB, Caughey B. Parallel in-register intermolecular beta-sheet architectures for prion-seeded prion protein (PrP) amyloids. J Biol Chem. 2014;289:24129–42. https://doi.org/10.1074/jbc.M114.578344.
Groveman BR, Kraus A, Raymond LD, Dolan MA, Anson KJ, Dorward DW, Caughey B. Charge neutralization of the central lysine cluster in prion protein (PrP) promotes PrP(Sc)-like folding of recombinant PrP amyloids. J Biol Chem. 2015;290:1119–28. https://doi.org/10.1074/jbc.M114.619627.
Groveman BR, Raymond GJ, Campbell KJ, Race B, Raymond LD, Hughson AG, Orru CD, Kraus A, Phillips K, Caughey B. Role of the central lysine cluster and scrapie templating in the transmissibility of synthetic prion protein aggregates. PLoS Pathog. 2017;13:e1006623. https://doi.org/10.1371/journal.ppat.1006623.
Helmus JJ, Surewicz K, Apostol MI, Surewicz WK, Jaroniec CP. Intermolecular alignment in Y145Stop human prion protein amyloid fibrils probed by solid-state NMR spectroscopy. J Am Chem Soc. 2011;133:13934–7. https://doi.org/10.1021/ja206469q.
Hope J, Reekie LJD, Hunter N, Multhaup G, Beyreuther K, White H, Scott AC, Stack MJ, Dawson M, Wells GAH. Fibrils from brains of cows with new cattle disease contain scrapie-associated protein. Nature. 1988;336:390–2.
Hoyt F, Standke HG, Artikis E, Schwartz CL, Hansen B, Li K, Hughson AG, Manca M, Thomas OR, Raymond GJ, et al. Structure of anchorless RML prion reveals motif variation between strains. bioRXiv. 2021; https://doi.org/10.1101/2021.12.22.473909.
James TL, Liu H, Ulyanov NB, Farr-Jones S, Zhang H, Donne DG, Kaneko K, Groth D, Mehlhorn I, Prusiner SB, et al. Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc Natl Acad Sci U S A. 1997;94:10086–91.
Jeffrey M. Review: membrane-associated misfolded protein propagation in natural transmissible spongiform encephalopathies (TSEs), synthetic prion diseases and Alzheimer’s disease. Neuropathol Appl Neurobiol. 2013;39:196–216. https://doi.org/10.1111/nan.12004.
Jeffrey M, McGovern G, Siso S, Gonzalez L. Cellular and sub-cellular pathology of animal prion diseases: relationship between morphological changes, accumulation of abnormal prion protein and clinical disease. Acta Neuropathol. 2011;121:113–34. https://doi.org/10.1007/s00401-010-0700-3.
Jeffrey M, Gonzalez L, Simmons MM, Hunter N, Martin S, McGovern G. Altered trafficking of abnormal prion protein in atypical scrapie: prion protein accumulation in oligodendroglial inner mesaxons. Neuropathol Appl Neurobiol. 2017;43:215–26. https://doi.org/10.1111/nan.12302.
Klingeborn M, Race B, Meade-White KD, Rosenke R, Striebel JF, Chesebro B. Crucial role for prion protein membrane anchoring in the neuroinvasion and neural spread of prion infection. J Virol. 2011;85:1484–94. https://doi.org/10.1128/JVI.02167-10.
Kocisko DA, Priola SA, Raymond GJ, Chesebro B, Lansbury PT Jr, Caughey B. Species specificity in the cell-free conversion of prion protein to protease-resistant forms: a model for the scrapie species barrier. Proc Natl Acad Sci U S A. 1995;92:3923–7.
Kraus A, Raymond GJ, Race B, Campbell KJ, Hughson AG, Anson KJ, Raymond LD, Caughey B. PrP P102L and nearby lysine mutations promote spontaneous in vitro formation of transmissible prions. J Virol. 2017;91:e01276. https://doi.org/10.1128/JVI.01276-17.
Kraus A, Hoyt F, Schwartz CL, Hansen B, Hughson AG, Artikis E, Race B, Caughey B. Structure of an infectious mammalian prion. bioRxiv. 2021a; https://doi.org/10.1101/2021.02.14.431014.
Kraus A, Hoyt F, Schwartz CL, Hansen B, Artikis E, Hughson AG, Raymond GJ, Race B, Baron GS, Caughey B. High-resolution structure and strain comparison of infectious mammalian prions. Mol Cell. 2021b;81:4540–51.
Li Q, Wang F, Xiao X, Kim C, Bohon J, Kiselar J, Safar JG, Ma J, Surewicz WK. Structural attributes of mammalian prion infectivity: Insights from studies with synthetic prions. J Biol Chem. 2018;293:18494–503. https://doi.org/10.1074/jbc.RA118.005622.
Li Q, Jaroniec CP, Surewicz W. Cryo-EM structure of disease-related prion fibrils provides insights into seeding barriers. bioRXiv. 2021; https://doi.org/10.1101/2021.08.10.455830.
Mahal SP, Jablonski J, Suponitsky-Kroyter I, Oelschlegel AM, Herva ME, Oldstone M, Weissmann C. Propagation of RML prions in mice expressing PrP devoid of GPI anchor leads to formation of a novel, stable prion strain. PLoS Pathog. 2012;8:e1002746. https://doi.org/10.1371/journal.ppat.1002746.
Makarava N, Chang JC, Molesworth K, Baskakov IV. Posttranslational modifications define course of prion strain adaptation and disease phenotype. J Clin Invest. 2020;130:4382–95. https://doi.org/10.1172/JCI138677.
Manka SW, Zhang W, Wenborn A, Betts J, Joiner S, Saibil HR, Collinge J, Wadsworth JD. 2.7 Å cryo-EM structure of ex vivo RML prion fibrils. bioRxiv. 2021; https://doi.org/10.1101/2021.12.13.472424.
Meisl G, Kurt T, Condado-Morales I, Bett C, Sorce S, Nuvolone M, Michaels TCT, Heinzer D, Avar M, Cohen SIA, et al. Scaling analysis reveals the mechanism and rates of prion replication in vivo. Nat Struct Mol Biol. 2021;28:365–72. https://doi.org/10.1038/s41594-021-00565-x.
Merz PA, Somerville RA, Wisniewski HM, Iqbal K. Abnormal fibrils from scrapie-infected brain. Acta Neuropathol. 1981;54:63–74.
Miller MB, Wang DW, Wang F, Noble GP, Ma J, Woods VL Jr, Li S, Supattapone S. Cofactor molecules induce structural transformation during infectious prion formation. Structure. 2013;2061–2068:21. https://doi.org/10.1016/j.str.2013.08.025.
Pattison IH, Jones KM. The possible nature of the transmissible agent of scrapie. Vet Rec. 1967;80:2–9.
Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021;30:70–82. https://doi.org/10.1002/pro.3943.
Priola SA, Chesebro B. A single hamster amino acid blocks conversion to protease-resistant PrP in scrapie-infected mouse neuroblastoma cells. J Virol. 1995;69:7754–8.
Priola SA, Caughey B, Race RE, Chesebro B. Heterologous PrP molecules interfere with accumulation of protease-resistant PrP in scrapie-infected murine neuroblastoma cells. J Virol. 1994;68:4873–8.
Priola SA, Chabry J, Chan K. Efficient conversion of normal prion protein (PrP) by abnormal hamster PrP is determined by homology at amino acid residue 155. J Virol. 2001;75:4673–80.
Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–44.
Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–83.
Prusiner SB, McKinley MP, Bowman KA, Bendheim PE, Bolton DC, Groth DF, Glenner GG. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell. 1983;35:349–58.
Prusiner SB, Scott M, Foster D, Pan KM, Groth D, Mirenda C, Torchia M, Yang SL, Serban D, Carlson GA, et al. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell. 1990;63:673–86.
Race B, Williams K, Hughson AG, Jansen C, Parchi P, Rozemuller AJM, Chesebro B. Familial human prion diseases associated with prion protein mutations Y226X and G131V are transmissible to transgenic mice expressing human prion protein. Acta Neuropathol Commun. 2018;6:13. https://doi.org/10.1186/s40478-018-0516-2.
Rangel A, Race B, Klingeborn M, Striebel J, Chesebro B. Unusual cerebral vascular prion protein amyloid distribution in scrapie-infected transgenic mice expressing anchorless prion protein. Acta Neuropathol Commun. 2013;1:25. https://doi.org/10.1186/2051-5960-1-25.
Raymond GJ, Hope J, Kocisko DA, Priola SA, Raymond LD, Bossers A, Ironside J, Will RG, Chen SG, Petersen RB, et al. Molecular assessment of the transmissibilities of BSE and scrapie to humans. Nature. 1997;388:285–8.
Raymond GJ, Bossers A, Raymond LD, O’Rourke KI, McHolland LE, Bryant PK III, Miller MW, Williams ES, Smits M, Caughey B. Evidence of a molecular barrier limiting susceptibility of humans, cattle and sheep to chronic wasting disease. EMBO J. 2000;19:4425–30.
Raymond GJ, Race B, Hollister JR, Offerdahl DK, Moore RA, Kodali R, Raymond LD, Hughson AG, Rosenke R, Long D, et al. Isolation of novel synthetic prion strains by amplification in transgenic mice coexpressing wild-type and anchorless prion proteins. J Virol. 2012;86:11763–78. https://doi.org/10.1128/JVI.01353-12.
Rouvinski A, Karniely S, Kounin M, Moussa S, Goldberg MD, Warburg G, Lyakhovetsky R, Papy-Garcia D, Kutzsche J, Korth C, et al. Live imaging of prions reveals nascent PrPSc in cell-surface, raft-associated amyloid strings and webs. J Cell Biol. 2014;204:423–41. https://doi.org/10.1083/jcb.201308028.
Rudd PM, Endo T, Colominas C, Groth D, Wheeler SF, Harvey DJ, Wormald MR, Serban H, Prusiner SB, Kobata A, et al. Glycosylation differences between the normal and pathogenic prion protein isoforms. Proc Natl Acad Sci U S A. 1999;96:13044–9.
Sajnani G, Silva CJ, Ramos A, Pastrana MA, Onisko BC, Erickson ML, Antaki EM, Dynin I, Vazquez-Fernandez E, Sigurdson CJ, et al. PK-sensitive PrP is infectious and shares basic structural features with PK-resistant PrP. PLoS Pathog. 2012;8:e1002547. https://doi.org/10.1371/journal.ppat.1002547.
Scheres SHW. Amyloid structure determination in RELION-3.1. Acta Crystallogr D Struct Biol. 2020;76:94–101. https://doi.org/10.1107/S2059798319016577.
Scott M, Groth D, Foster D, Torchia M, Yang SL, DeArmond SJ, Prusiner SB. Propagation of prions with artificial properties in transgenic mice expressing chimeric PrP genes. Cell. 1993;73:979–88.
Sevillano AM, Aguilar-Calvo P, Kurt TD, Lawrence JA, Soldau K, Nam TH, Schumann T, Pizzo DP, Nystrom S, Choudhury B, et al. Prion protein glycans reduce intracerebral fibril formation and spongiosis in prion disease. J Clin Invest. 2020;130:1350–62. https://doi.org/10.1172/JCI131564.
Shaked GM, Meiner Z, Avraham I, Taraboulos A, Gabizon R. Reconstitution of prion infectivity from solubilized protease-resistant PrP and nonprotein components of prion rods. J Biol Chem. 2001;276:14324–8.
Shannon MD, Theint T, Mukhopadhyay D, Surewicz K, Surewicz WK, Marion D, Schanda P, Jaroniec CP. Conformational dynamics in the core of human Y145Stop prion protein amyloid probed by relaxation dispersion NMR. ChemPhysChem. 2019;20:311–7. https://doi.org/10.1002/cphc.201800779.
Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. The most infectious prion protein particles. Nature. 2005;437:257–61.
Spagnolli G, Rigoli M, Orioli S, Sevillano AM, Faccioli P, Wille H, Biasini E, Requena JR. Full atomistic model of prion structure and conversion. PLoS Pathog. 2019;15:e1007864. https://doi.org/10.1371/journal.ppat.1007864.
Stahl N, Baldwin MA, Hecker R, Pan KM, Burlingame AL, Prusiner SB. Glycosylinositol phospholipid anchors of the scrapie and cellular prion proteins contain sialic acid. Biochemistry. 1992;31:5043–53. https://doi.org/10.1021/bi00136a600.
Supattapone S. Cofactor molecules: essential partners for infectious prions. Prog Mol Biol Transl Sci. 2020;175:53–75. https://doi.org/10.1016/bs.pmbts.2020.07.009.
Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;2079-2082:274.
Theint T, Nadaud PS, Aucoin D, Helmus JJ, Pondaven SP, Surewicz K, Surewicz WK, Jaroniec CP. Species-dependent structural polymorphism of Y145Stop prion protein amyloid revealed by solid-state NMR spectroscopy. Nat Commun. 2017;8:753. https://doi.org/10.1038/s41467-017-00794-z.
Theint T, Xia Y, Nadaud PS, Mukhopadhyay D, Schwieters CD, Surewicz K, Surewicz WK, Jaroniec CP. Structural studies of amyloid fibrils by paramagnetic solid-state nuclear magnetic resonance spectroscopy. J Am Chem Soc. 2018;140:13161–6. https://doi.org/10.1021/jacs.8b06758.
Tycko R, Savtchenko R, Ostapchenko VG, Makarava N, Baskakov IV. The alpha-helical C-terminal domain of full-length recombinant PrP converts to an in-register parallel beta-sheet structure in PrP fibrils: evidence from solid state nuclear magnetic resonance. Biochemistry. 2010;49:9488–97. https://doi.org/10.1021/bi1013134.
Tzaban S, Friedlander G, Schonberger O, Horonchik L, Yedidia Y, Shaked G, Gabizon R, Taraboulos A. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry. 2002;41:12868–75.
Vanni I, Pirisinu L, Acevedo-Morantes C, Kamali-Jamil R, Rathod V, Di Bari MA, D’Agostino C, Marcon S, Esposito E, Riccardi G, et al. Isolation of infectious, non-fibrillar and oligomeric prions from a genetic prion disease. Brain. 2020;143:1512–24. https://doi.org/10.1093/brain/awaa078.
Vassileff N, Cheng L, Hill AF. Extracellular vesicles – propagators of neuropathology and sources of potential biomarkers and therapeutics for neurodegenerative diseases. J Cell Sci. 2020;133 https://doi.org/10.1242/jcs.243139.
Wang F, Yin S, Wang X, Zha L, Sy MS, Ma J. Role of the highly conserved middle region of prion protein (PrP) in PrP-lipid interaction. Biochemistry. 2010;49:8169–76. https://doi.org/10.1021/bi101146v.
Wang LQ, Zhao K, Yuan HY, Wang Q, Guan Z, Tao J, Li XN, Sun Y, Yi CW, Chen J, et al. Cryo-EM structure of an amyloid fibril formed by full-length human prion protein. Nat Struct Mol Biol. 2020;27:598–602. https://doi.org/10.1038/s41594-020-0441-5.
Wang LQ, Zhao K, Yuan HY, Li XN, Dang HB, Ma Y, Wang Q, Wang C, Sun Y, Chen J, et al. Genetic prion disease-related mutation E196K displays a novel amyloid fibril structure revealed by cryo-EM. Sci Adv. 2021;7:eabg9676. https://doi.org/10.1126/sciadv.abg9676.
Wegmann S, Miesbauer M, Winklhofer KF, Tatzelt J, Muller DJ. Observing fibrillar assemblies on scrapie-infected cells. Pflugers Arch. 2008;456:83–93.
Wille H, Requena JR. The structure of PrP(Sc) prions. Pathogens. 2018;7 https://doi.org/10.3390/pathogens7010020.
Wiseman FK, Cancellotti E, Piccardo P, Iremonger K, Boyle A, Brown D, Ironside JW, Manson JC, Diack AB. The glycosylation status of PrPC is a key factor in determining transmissible spongiform encephalopathy transmission between species. J Virol. 2015;89:4738–47. https://doi.org/10.1128/JVI.02296-14.
Wong C, Xiong L-W, Horiuchi M, Raymond LD, Wehrly K, Chesebro B, Caughey B. Sulfated glycans and elevated temperature stimulate PrPSc dependent cell-free formation of protease-resistant prion protein. EMBO J. 2001;20:377–86.
Acknowledgments
We thank Austin Athman for graphics assistance. This work was supported by the Intramural Research Program of the NIAID. AK is supported by Case Western Reserve University, the Britton Fund, and the Clifford V. Harding and Mina K. Chung Professorship in Pathology. Molecular graphics and analyses performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Caughey, B., Artikis, E., Kraus, A. (2023). PrP Prion Structures. In: Zou, WQ., Gambetti, P. (eds) Prions and Diseases. Springer, Cham. https://doi.org/10.1007/978-3-031-20565-1_3
Download citation
DOI: https://doi.org/10.1007/978-3-031-20565-1_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-20564-4
Online ISBN: 978-3-031-20565-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)