
Summary
Decaprenylphosphoryl-β-d-ribose 2′-epimerase 1(DprE1) is a new and competent target that could be exploited for drug discovery to tackle the problem of drug-resistant tuberculosis (TB). It is a flavoprotein that essentially contributes to mycobacterial cell wall biosynthesis. The enzyme is involved in the synthesis of Araf molecules, which are the building blocks in the synthesis of lipoarabinomannans and arabinogalactans. Benzothiazinones were the first molecules to be reported as DprE1 inhibitors. Since then, a number of new and novel compounds have been reported as DprE1 inhibitors. These inhibitors exhibit either covalent or non-covalent binding to the enzyme. Four DprE1 inhibitors, namely BTZ-043, Macozinone, OPC-167832, and TBA-7371, are currently in clinical trials. This chapter attempts to discuss DprE1 as a potential druggable target and its inhibitors for the discovery of anti-TB agents.
Graphical Abstract

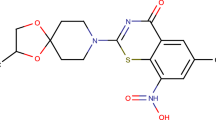
DprE1 inhibitors in clinical trials
The biggest disease today is not leprosy or tuberculosis, but rather the feeling of being unwanted.
Mother Teresa
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
Target identification serves as the commencing step of any drug discovery program. Medicinal chemists worldwide have been actively involved in unraveling novel tuberculosis (TB) targets and their promising inhibitors. Such a target should possess three essential characteristics:
-
(i)
significance for the growth and persistence of bacteria;
-
(ii)
selectivity for the bacteria over the host; and
-
(iii)
drug approachability, i.e., the absence of structural barriers in the bacteria which would block the approach of the drug to the target.
Numerous first-line and second-line anti-TB drugs (Fig. 1) have been used clinically to treat TB for a long time, but the real fact is that the exact targets for many of them have not yet been recognized [1]. There are several targets identified to date for the inhibition of the active, replicating, and dormant forms of TB. Figure 2 depicts various targets and some promising TB inhibitors [2]. To date, a number of novel techniques, strategies, and programs have been undertaken as part of the TB drug discovery process. The overall picture is such that new drug discovery for the management of TB has become a daunting task for the researchers as the biggest culprit is Mycobacterium tuberculosis (M. tb) bacterium itself which exists in replicating as well as in dormant forms [3,4,5].

Chemical structures of first (1–4) and second (5–19)-line anti-TB drugs

It is needed that novel anti-TB agents, either individually or in combination, have a shorter duration of treatment and manage drug resistance (DR) cases effectively with minimum or no toxicity. Recently, the decaprenylphosphoryl-β-d-ribose 2′-epimerase 1 (DprE1) enzyme possessing all the desirable requirements has emerged as a prospective novel target for the discovery of new anti-TB drugs [6]. DprE1, a flavoprotein present in the periplasm of the M. tb cell wall, is indispensable for cell wall synthesis. The significance of DprE1 as a potential druggable target for the discovery of anti-TB agents has been thoroughly discussed in the following sections of this chapter.
2 Special Features of the Enzyme Decaprenylphosphoryl-β-d-Ribose 2′-Epimerase 1 Which Make It a Target
To combat DR in mycobacterial strains, it is an urgent need to develop a drug that would be able to decrease the duration of treatment and kill the mycobacteria both in replicating and dormant forms completely. This is possible only when a valid target, which is essential for the growth of bacteria and its survival, is identified for the drug. Though many targets have been recognized and validated and a number of new anti-TB agents with potential anti-TB activity, the biggest threat of DR-TB still keeps on challenging the medicinal chemists’ fraternity [2].
DprE1 has evolved as a new competent target that could be exploited for anti-TB drug discovery to tackle the problem of DR. It was postulated a few decades ago that blocking the biosynthetic process of mycobacterial cell wall would be the best way to win the battle against TB. DprE1 is a vital enzyme in the M. tb cell wall synthesis. In fact, inhibition of DprE1 causes cessation of generation of DPA required for the formation of Araf residues. Reduced levels of Araf residues hamper the supply of these essential building blocks of the mycobacterial cell wall, i.e., AG and LAM, which subsequently affect the biosynthesis of the M. tb cell wall. Thus, it clearly demonstrates that inhibiting DprE1 could hamper the growth as well as the survival of M. tb. Moreover, DprE1 is an ideal target as it is present only in mycobacteria and not in humans, which certainly underlines its importance as an anti-TB drug target for designing, developing, and discovering novel anti-TB agents. These special features of the DprE1 enzyme significantly make it a valuable drug target that could be utilized effectively for the discovery of novel anti-TB agents with enhanced biological potential and minimum toxicity [1, 7, 8].
2.1 Location and Role
Considering its function to provide overall strength to the cell and protect it from virulence and pathogenicity, the cell wall is the most important component of a bacterial cell [9]. Therefore, cell wall biosynthesis has been considered the most promising target for most drugs, including antibiotics. Biosynthesis of the cell wall in M. tb consists of a number of processes that are ideal drug targets for discovering anti-TB drugs [10]. Several anti-TB agents, such as isoniazid (1) and ethambutol (3) of the first-line category, along with other second-line agents, actually interfere in the cell wall biosynthesis during different stages (Fig. 1) [11, 12]. With this strategy in mind to block the biosynthesis of the cell wall, various novel anti-TB targets, as shown in Fig. 2, have been recognized, which could be exploited further to develop novel anti-TB drugs.
The composition of the M. tb cell wall is quite complex as it is built of two unique complexes known as peptidoglycan-arabinogalactan-mycolic acid (PAM) complex or mAGP complex (mycolyl-arabinogalactan–peptidoglycan) and lipoarabinomannan (LAM) [13]. PAM complex is mainly composed of three layers:
-
(i)
a highly impermeable lining of mycolic acid;
-
(ii)
arabinogalactan polysaccharide (AG); and
-
(iii)
peptidoglycan (PG), posing from outer side to inner side of the cell.
PG is covalently bound to AG through a phosphodiester linkage that gets attached to the mycolic acid, forming the PAM complex [14]. The second element, LAM, is a non-covalently bound lipopolysaccharide comprising d-arabinofuranose (Araf) and mannopyranosyl residues. Both the components (PAM and LAM) are a prerequisite to maintaining cell wall integrity and impart a crucial role in the M. tb virulence and pathogenesis [12, 15, 16].
Synthesis of Araf residues, essential building blocks of AG and LAM, is a crucial biosynthetic step. Biosynthesis of AG and LAM involves the addition of Araf residues to the galactan and mannan domains, respectively, catalyzed by a specific enzyme known as arabinosyltransferase. The arabinosyltransferase enzyme uses the sugar decaprenylphosphoryl-β-d-arabinose (DPA) generated by the epimerization of decaprenylphosphoryl-β-d-ribose (DPR). DPA is the only source for the Araf residues in M. tb. Without DPA, it is difficult for the bacteria to survive in latent as well as virulent forms as the cell wall synthesis would be ceased completely [12, 15, 17].
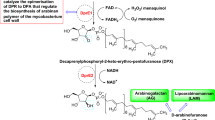
A heterodimeric enzyme, i.e., DprE, consists of two enzymes–DprE1 and DprE2–the key proteins involved in the biosynthesis of DPA. DprE1 is a FAD-dependent enzyme that converts DPR to decaprenylphosphoryl-2-keto-β-d-erythro-pentofuranose (DPX) by oxidation, and DPX is then further reduced to DPA in the presence of decaprenylphosphoryl-D-2-keto-erythro-pentose reductase (DprE2). DprE1 is required for the growth and survival of M. tb. Hence, blockade of DPA synthesis by inhibition of the DprE1 enzyme would be a key strategy to stop the biosynthesis of the M. tb cell wall [6, 18].
2.2 Mechanism of Action
Biosynthesis of DPA involves oxidation of DPR to DPX in the first step and reduction of DPX to DPA in the second step (Fig. 3). Oxidation of DPR is catalyzed by a flavoenzyme, DprE1, using flavin adenine dinucleotide (FAD) as an oxidant which gets reduced to FADH2. Now, to start a new cycle of oxidation of DPR, FADH2 has to be re-oxidized to its oxidizing form, i.e., FAD. Despite being an oxidase enzyme, DprE1 showed comparatively low reactivity with oxygen. Actually, DprE1 uses a natural membrane-embedded electron acceptor, menaquinone, present in M. tb to re-oxidize FADH2 to FAD. In the second step, the reduction of intermediate DPX to DPA is catalyzed by DprE2 in the presence of cofactor NADH. Considering the above fact, DprE1 could be considered an oxidoreductase enzyme rather than a true oxidase. Therefore, the DprE1, DprE2, or DprE1-DprE2 complex could be exploited as potential TB targets to design and develop small molecule therapeutics [10, 19, 20].

2.3 Crystal Structure
The discovery of the crystal structure of DprE1 shed more light on the identification of the active sites of the enzyme and the possible mechanism of action of its inhibitors which proved beneficial for the medicinal chemists to design and develop novel anti-TB drugs with improved clinical potential. There have been several reports wherein crystal structures of DprE1 from M. smegmatis and M. tb co-crystallized with or without covalent/non-covalent inhibitors [7]. Neres et al. [19] and Batt et al. [20] were the first groups to report the crystal structure of the DprE1 enzyme in the year 2012. Thereafter, approximately 35 crystal structures of DprE1 have been reported to date, which are available in the protein data bank (PDB).Footnote 1 Piton et al. [7] and Chikhale et al. [2] have enlisted 23 of the available structures of DprE1 systematically according to their date of release, PDB IDs, resolution, and source of Mycobacterium species [2, 7]. A summary of the remaining structures of DprE1 from 2018 onwards has been presented in Table 1.
The crystal structure of DprE1 contains several active sites for binding inhibitors. The enzyme DprE1 (PDB code 4P8L; Fig. 4) consists of two active binding domains, similar to the other oxidoreductases such as vanillyl alcohol oxidase. These two active binding domains include:

© 2017 Elsevier
Structure of DprE1 (PDB code 4P8L). A deep blue color represents the FAD-binding domain, whereas light blue color depicts the substrate-binding domain. Substrate-binding pocket is highlighted on the lime green surface. Orange and hot pink depict the two disordered loops [7]. Reprinted with permission from Ref. [7] Copyright
-
(i)
a FAD-binding domain with residues 7–196, 413–461; and
-
(ii)
a substrate-binding domain with residues 197–412 (Fig. 4).
The cofactor FAD is located deep inside the FAD-binding domain of the enzyme, with the isoalloxazine ring of FAD lying at the interface of the substrate-binding domain. Two disordered loops are present above the substrate-binding domain, confirmed by the electron density map obtained in all of the crystal structures of DprE1 [7]. Interestingly, these two loops, disordered loop I with amino acid residues 269–303 and disordered loop II with amino acid residues 316–330, might be interacting with the cell membrane, with certain proteins involved in the biosynthesis of DPA, a substrate for DprE1 or with DPR [20]. These two disordered loops actually keep the substrate-binding domain wide open, facilitating the accommodation of the substrate in the domain. Thus, these loops could be considered as the entrance gate for the substrate approaching the substrate-binding domain [7]. Recent developments of various DprE1 inhibitors have been discussed in the following sections of this chapter.
3 Insight into DprE1 Inhibitors
Previously in the year 2018, we have published from our lab an extensive review on DprE1 and its inhibitors as an anti-TB target [2]. Some important developments in discovering DprE1 inhibitors as potential anti-TB drugs have been discussed here.
The discovery of the co-crystal structure of DprE1 bound to benzothiazinone (BTZ) offered an important insight into the mechanistic view of DprE1 inhibitors that helped develop newer anti-TB agents. DprE1 inhibitors can be categorized or differentiated based on their binding interactions, viz. covalent or non-covalent binding to the enzyme. Here, DprE1 inhibitors are classified as covalent and non-covalent binding and miscellaneous inhibitors.
3.1 Covalent Binding Inhibitors
3.1.1 Benzothiazinones as the Leading Inhibitors of DprE1
Benzothiazinones (BTZs) evolved as a new class of anti-TB agents in 2009. They offered hope for the design and development of newer and effective drug candidates for the treatment of DR-TB [24]. BTZ scaffold demonstrated sub-micro molar MIC value against M. tb. BTZs have been proved to inhibit the DprE1 enzyme covalently. In a successful attempt to improvise the pharmacological properties of BTZs, their piperazine-containing analogs (PBTZ) were synthesized.
In the biological systems, the nitro functional group gets reduced primarily to the corresponding nitroso group, then to the hydroxylamine, and finally to the amine [25]. It is hypothesized that benzothiazinone-nitrosoarene derivatives bind with Cys387 residue of the enzyme via interaction with the thiol group of the residue, thereby forming a covalent adduct and, thus, behaving suicide inhibitors [26]. Later in 2013, Tiwari et al. [27] proposed another plausible mechanism behind the formation of the nitroso derivative. They proposed that the redox reaction responsible for converting the nitro into the nitroso derivative is facilitated by the thiol group of cysteine residue present in the enzyme. It was also hypothesized that this conversion does not require FADH2 as the catalyst. Further, the cine addition of thiolate would initiate the redox reaction that generates the nitroso derivative.
For the first time, BTZs were reported as nitro benzothiazinones, the active molecules that could inhibit DprE1 from Mollmann lab, Germany; Makarov lab, Moscow; and Cole lab at the Global Health Institute, Ecole Polytechnique, Switzerland [16, 28]. BTZ043 (29) was found to be the most potent compound, exhibiting antimycobacterial activity with a minimum inhibitory concentration (MIC) value of 1 ng/mL, whereas MIC values for Isoniazid and ethambutol (EMB) were found in the range of 0.02–0.2 mg/mL and 1–5 mg/mL respectively. In order to determine the site of action of BTZ, radiolabeled studies were performed. These results demonstrated that BTZ targeted the biosynthesis of arabinogalactan, which in turn is an important component in the covalent linking of mycolic acid and peptidoglycan layer, thus inhibiting the biosynthesis of the M. tb cell wall.

Gao et al. [29] attempted to establish the structure-activity relationship (SAR) of BTZs. In order to do so, they prepared a series of N-alkyl and heterocycle substituted BTZ compounds. These derivatives showed potent inhibition against the M. tb strains. It was noted that increasing the bulk or substituting bulky groups in the BTZ motif amplified the inhibitory activity. Also, the trifluoro substituent played a pivotal role in deciding the activity of the piperazine or piperidine analogs. Some compounds with spiro-piperidine moiety exhibited activity comparable to that of BTZ043. This indicated that the presence of sulfur in the azaspiro ring increased the activity. These compounds also demonstrated good bioavailability. Further, an attempt was made to substitute the oxygen of the BTZ043 carbonyl group with a sulfur atom, generating SKLB-TB1001 (30). Compound (30) showed promising in vitro activity and good ADMET properties, and it was found to be efficient in treating acute infection in a mouse model. Its MIC value was found to be 0.02 μg/ml. Subsequent work resulted in the design and synthesis of substituted 2-piperazine-benzothiazinone (PBTZ) (31) derivatives with enhanced lipophilicity [30]. SAR studies for this series indicated that the presence of hydrophilic groups such as secondary or tertiary amines, alcohols, etc., on the N-4 piperazine ring led to compounds with diminished or complete loss of activity.

Benzothiazinones BTZ043 (29) and PBTZ0169 (31) were explored substantially due to their imposing DprE1 inhibition. To enhance the activity and pharmacokinetic parameters, Peng et al. [31] synthesized 1,3-benzothiazin-4-one derivatives with 4-carbonylpiperazine (32). MIC value of this most active compound was found to be 0.0131 μM.

BTZ-SO (33) and BTZ-SO2 (34) were prepared to study the effect of oxidation of sulfur on the biological activity of BTZ [32]. BTZ-SO was found to produce impressive antimycobacterial activity, whereas BTZ-SO2 failed to exhibit anti-TB activity.

3.1.2 Benzothiazole Containing DprE1 Inhibitors
Langde el at. [33] discovered some new molecules containing benzothiazole, possessing antimycobacterial activity by high throughput screening of database from AstraZeneca Pharmaceuticals drug library, containing more than 100,000 compounds. Initial searching gave benzothiazole N-oxide as the lead molecule (8-BTO) (35). The most potent compound was found to possess a MIC value of 1 μg/mL. It was further taken up to optimize and develop a new series of derivatives. Benzothiazole oxide and benzothiazole were found to show good potency, but this potency came along with the side effect of mutagenicity. It was noted that substitution of the nitro group rendered the compound inactive. Furthermore, the authors tried to sterically hinder the nitro group so as to prevent the generation of reactive intermediate derivatives responsible for mutagenic properties. The IC50 value for 8-BTO (35) was found to be 0.026 μM.

3.1.3 Triazole Scaffold Containing DprE1 Inhibitors
Stanley et al. [34] performed a cell-based HTS assay and reported various novel inhibitors. In this study, screening of the literature reported 20,000 antibacterial agents against M. tb. Additionally, a dataset of 341,808 compounds was also screened against M. tb using a 7H12 medium. 1-(4-(tert.Butyl)benzyl)-3-nitro-1H-1,2,4-triazole (36) came out as an initial hit showing an IC90 value of 0.5 μM. It was observed that compounds having nitro groups showed good activity, whereas compounds devoid of the nitro group exhibited reduced activity. Also, compounds with nitro substitution were bound covalently with the enzyme. These observations suggest the importance of the nitro group, which in turn gets reduced to some reactive species to provide interaction to the target.

Karabanovich et al. [35] prepared various 3,5-dinitrophenyl-1,2,4-triazole containing compounds exhibiting magnificent and selective antimycobacterial activity. Among the 23 compounds, it was found that S-substituted 4-alkyl-5-(3,5-dinitrophenyl)-4H-1,2,4-triazole-3-thiols (37) and their 3-nitro-5-(trifluoromethyl)phenyl (38) analogs exhibited the highest in vitro activity against M. tb H37Rv.

Ali et al. [36] synthesized seventeen novel 1,2,3-triazole derivatives (39) using ‘click chemistry’ methodology and evaluated them in vitro for their inhibitory activity against M. tb H37Rv strain. Among the synthesized derivatives, six compounds were found to have significant activity with MIC values ranging from 3.12 to 0.78 µM with nil or negligible cytotoxicity against mouse bone marrow-derived macrophages. These six compounds possessed MIC values lesser than 6.25 µg/ mL along with a high affinity for the active site of DprE1.
3.1.4 Quinoxalines as DprE1 Inhibitors
Magnet et al. [37], while performing an experiment of screening a collection of kinase inhibitors containing 12,000 compounds against M. tb, reported the quinoxaline scaffold active against M. tb. Three compounds were obtained as initial hits having activity lesser than 10 μM. During the studies, it was observed that these compounds were non-mutagenic and non-toxic. All three hits contained quinoxaline as a basic scaffold (40) and were reported as specific inhibitors of DprE1. The mechanism of action or binding mode of these molecules is essentially the same as BTZs.

3.1.5 Nitrobenzamide-Based DprE1 Inhibitors
Nitrobenzamide derivatives depict another example of the application of high throughput screening (HTS) in drug discovery. Christophe et al. [38] performed screening of a collection of 56,984 molecules for checking drug-likeness by applying Lipinski’s rule of five, and then the short-listed molecules were screened for their anti-TB activity at a single dose concentration. Four hundred eighty-six molecules from the selected molecules were checked using the serial dilution method. About 8% of these molecules showed MIC value comparable to isoniazid. Cluster analysis indicated that 69 compounds had a similar structure as isoniazid, and out of these, 24 compounds had benzamide as the common structural feature (41). These derivatives were further exploited to produce a series of compounds with improved activity [39]. SAR study was also undertaken to optimize the benzamide derivatives. It was noted that nitro groups at positions 3 and 5 were essential for the potency of the compounds as reduction of nitro to the corresponding hydroxylamine derivatives with no activity. An enhancement in the activity profile was observed when the amide nitrogen was substituted with benzyloxy or phenoxymethyl moieties. Cyclic benzamides demonstrated MIC values as low as 80 nM, but they lacked potency during the intracellular assay. Compound (42) was screened for antimycobacterial activity on replicating as well as non-replicating strains. Unfortunately, the results demonstrated that it was effective only in the replicating cultures and was inactive against the non-replicating strains. These results concluded that compound (42) demonstrated activity by interacting with the Cys387 residue of the DprE1.

Furthermore, two more benzamide derivatives, CT325 (43) and CT319 (44), were synthesized by taking BTZ structure into consideration, which was found to bind to the active site of the DprE1 enzyme [18]. Both of them showed good inhibitory activity with specificity towards DprE1. It was seen that compound (43) interacted covalently with the enzyme acting as an irreversible inhibitor, whereas compound (44) formed a non-covalent bond with the enzyme.

3.2 Non-covalent Inhibitors
3.2.1 Benzothiazinone Containing DprE1 Inhibitors
Earlier, benzothiazinones were known to interact only covalently with the enzyme. But the scenario changed when Makarov et al. [40] tried to replace the nitro group of the BTZ with a pyrrole ring. These efforts resulted in the discovery of active pyrrole-BTZ compounds (45 and 46) having a MIC value of 0.16 μg/mL. The IC50 values were as low as < 8 μM with appreciable ADMET and in vivopharmacokinetic parameters. Unfortunately, they failed to impress the animal models. Molecular docking studies revealed that pyrrole-BTZs bind to the same cavity of DprE1 as BTZ, with pyrrole rings located close to Cys387 residue. Surprisingly, any covalent interaction with the enzyme was absent, indicating that Pyr-BTZ compounds act as non-covalent inhibitors.

3.2.2 Benzothiazole Based DprE1 Inhibitors
Benzothiazoles have been considered a boon for designing and developing DprE1 inhibitors. Wang et al. [41] performed cell-based phenotype screening and reported a small molecule TCA1 (47) as a DprE1 inhibitor. The Discovery of TCA1 (47) was serendipitous, as it was obtained during the screening of a collection of 70,000 molecules for their inhibitory activity against replicating and non-replicating M. tb strains. Compound (47) showed promising activity both in vitro and in vivo. Initially, it was observed that the compound (47) exhibited antimycobacterial action by downregulation of persistent genes and cell wall inhibition via interfering with mycolic acid synthesis. The discovery of TCA1 (47) offered a pathway for further developing DprE1 inhibitors by serving as a template molecule. Liu et al. [21] performed synthesis, molecular docking, and pharmacological evaluation along with SAR studies to optimize the benzothiazoles as DprE1 inhibitors. Three positions were modified, i.e., thiophene moiety, benzothiazole core, and carbamate group. It was observed that these structural changes were indeed beneficial for inhibitory activity, but they also caused CYP2C9 inhibition. Subsequently, to develop DprE1 inhibitors with no CYC2C9 inhibition, the authors carried out further studies using compound TCA007 (48) as a template molecule.

In another study conducted by Chikhale et al. [42], a series of benzothiazolyl pyrimidine carboxamides was reported. This series provided information that compounds having para-substituents on the phenyl ring of the compound demonstrate favorable activity though the unsubstituted compound (49) offered the best results in terms of MIC value of 0.08 and MBC of 7.7 μM.

Fauzia et al. [43] reported anti-TB activity of benzothiazole and 1,2,3-triazole based bis-heterocycles against M. tb. Three compounds (50–52) showed good activity among the series. Furthermore, a molecular docking study was conducted to investigate the binding interactions between the compounds and DprE1. It was found that compound (51) possessed potent inhibitory properties owing to hydrogen bonding and hydrophobic interactions. It was seen that compound (51) interacted with Tyr60, Gly117, Ala375, Ser378, Asn385, Lys418, and Trp437 residues via H-bond.

3.2.3 Imidazopyridine Based DprE1 Inhibitors
Gawad et al. [44] reported 6-(4-nitrophenoxy)-2-substituted-1H-imidazo[4,5-b]pyridine derivatives to explore the potential of 1H-imidazo[4,5-b]pyridine nucleus. In this study, the nitro group was intentionally substituted at the sixth position because of its proven binding with Cys387 residue of the DprE1 enzyme. Some of the derivatives have shown good anti-TB activity. The most potent compounds were found to have MIC values ranging from 0.5 to 0.8 μM. Interestingly, docking studies of these compounds yielded excellent docking scores. Binding interactions shown by these compounds were similar to that of the lead molecule TCA1 (47). Earlier it was reported that the nitro group got reduced and interacted with Cys387, but no such interaction was seen here. Information obtained from the docking studies and the in vitro studies indicated that further structural modifications could help develop better compounds as DprE1 inhibitors.

3.2.4 Quinoxalines as DprE1 Inhibitors
Neres et al. [45] performed phenotype screening of a collection of 266 compounds against M. tb. This resulted in the discovery of novel quinoxaline derivatives with promising bactericidal activity. The lead compound (54) was highly effective against M. tb, having an IC50 value of 6.1 μM. SAR studies disclosed that the absence of the 6-trifluoromethyl group rendered the compounds inactive, and its presence at the para-position of the C3 benzyl amine moiety affected the DprE1 inhibition significantly. Substituents like methoxyl and halogens on the phenyl ring of the benzylamino group yielded compounds with moderate activity. All the derivatives were active against both replicating and non-replicating strains of M. tb.

3.2.5 Thiadiazole Containing Inhibitors
Batt et al. [46] simultaneously utilized phenotype screening and target-based drug design strategy to report a series of novel DprE1 inhibitors. This study was performed using a library of 177 compounds with known M. tb inhibitory activity. These compounds were then tested for their enzymatic assay for DprE1 selectivity. Compound (55) was found to be the most potent derivative with an IC50 value of 0.054 μM, and it demonstrated the greatest binding affinity (Kd of 0.25 μM) for the enzyme.

3.2.6 Azaindoles as DprE1 Inhibitors
Earlier various imidazopyridine-based compounds were reported as antimycobacterials, but these possessed very mild activity [44, 47, 48]. The imidazopyridine scaffold (56) was morphed into 1,4-azaindoles (57 and 58) to explore and enhance the activity. Shirude et al. [49, 50] attempted to improve 1,4-azaindoles wherein they synthesized 23 compounds. The authors claimed that this novel class of inhibitors exhibited cellular activity via non-covalent inhibition of DprE1. The most potent derivatives exhibited MIC in the range of 0.39–0.78 μM. These derivatives were found to be better than the already reported DprE1 inhibitors. Yet they exhibited a couple of pitfalls, like inhibiting the PDE6 protein complex, which plays an essential role in the proper functioning of the human eyes and has shown not-so-good pharmacokinetic properties. In continuation, to optimize the lead (58), Shirude et al. [51] further reported other 27 compounds to overcome the pitfalls of the earlier compounds. These newer derivatives offered an optimal pharmacokinetic profile. Moreover, these were devoid of any inhibition of PDE6. Further, SAR was developed for the azaindole series (Fig. 5). It was noted that three essential structural features were necessary for the activity. The core 1,4-azaindole with a substituent on the sixth position was the minimum requirement for the activity. The amide chain was a requirement for optimal potency and binding affinity. It also influenced the physicochemical parameters. Small substituents were necessary for cellular potency. The hydrophobic pocket of the enzyme gets filled with an aromatic core at the N-1 position during its binding with the enzyme; thus, this aromatic core was found to be one of the requirements. One of the most potent compounds, TBA-7371 (60), from the azaindole series is under clinical trials.

SAR and SPR of 1,4-azaindoles. Adapted with permission from Ref. [51]

3.2.7 Benzimidazole as DprE1 Inhibitors
Manjunatha et al. [52] used a scaffold morphing approach to modify the already known DprE1 inhibitors. They undertook azaindole TBA7371 (60) core as the template molecule and performed scaffold morphing to report benzimidazole derivatives (61 and 62). They demonstrated potent DprE1 inhibition with improved aqueous solubility and increased plasma function.
The benzimidazole derivative (61) and TBA7371 (60) were docked with the M. tb DprE1 enzyme (PDB ID 4KW5 binding site). One of the plausible binding modes for compound (61) from unconstrained docking is shown in Fig. 6a. It was seen that carbonyl oxygen was involved in H-bonding with Ser228, whereas amidic NH was bound with FAD carbonyl oxygen. The benzimidazole core generated CH−π contacts with Trp230 and Tyr314 residues. The molecule was also found to have hydrophobic interactions with various amino acid residues. The binding mode of the overlaid azaindoles showed a similar binding mode for the amide group (Fig. 6b).

a Binding mode of compound 61 in the DprE1 active site. b Docked poses of 60 (magenta) and 61 (green). Reprinted with permission from Ref. [52] Copyright© 2019 American Chemical Society
3.2.8 Pyrazolopyridine Based DprE1 Inhibitors
Panda et al. [53] performed a whole-cell screening assay against M. tb strains and reported a new series of pyrazolopyridones as the active scaffold against M. tb. In order to determine the selectivity of the compounds over DprE1, an overexpression assay was performed wherein gene Rv3790 was overexpressed for MIC study. These compounds showed higher MIC values than the earlier reported compounds. These derivatives were found to be interacting non-covalently with DprE1 and were effective against both replicating and non-replicating strains. The IC50 value for compound (63) was reported to be 0.04 μM.

3.2.9 Aminoquinolone Scaffold Containing DprE1 Inhibitors
Naik et al. [54] reported 4-aminoquinoline piperidine amides as novel DprE1 inhibitors based on the whole-cell assay. AstraZeneca corporate collections of approximately 320,000 compounds were screened to yield a compound as the lead molecule (64). It was found to be reasonably active against DprE1. Based on the lead molecule (64), various other 4-aminoquinoline piperidine amides were prepared and evaluated for pharmacological activity. Various studies, such as mass spectrometry and enzyme kinetic studies, concluded that these derivatives exhibited non-covalent and reversible inhibition of the enzyme. Analogs were found to possess excellent cidal properties in vitro against both replicating and non-replicating M. tb strains.

3.2.10 Hydantoins as DprE1 Inhibitors
A target-based HTS study conducted by GlaxoSmithKline (GSK) led to the emergence and identification of a novel hydantoin-based hit motif as a DprE1 inhibitor. This report offered a totally different scaffold from the other known DprE1 inhibitors. In 2018, Rogacki et al. [55] explored the report results and optimized the hits while developing a SAR for the series. Compound (65) was taken as the starting point for hit-to-lead optimization as it had exhibited good DprE1 enzyme inhibitory activity (pIC50 = 7.0). Additionally, it was characterized by good solubility, lack of cytotoxicity, and acceptable lipophilicity.

The authors carried out SAR studies to explore and optimize the hit (65) and divided the structure into five sub-structures, i.e., acetyl linker, substituents at C-5 of the hydantoin ring, phenyl ring at C-5, hydantoin ring substituents on the nitrogen (N-3) of the hydantoin ring, and the hydantoin moiety itself. The acetyl linker was seen as a potential liability because aromatic ketones are reactive groups, which may lead to enhanced metabolic instability of the compound. An attempt was made to modulate the linker by altering its length, removal of carbonyl group, or its substitution with known bioisosteric groups. It was observed that while most of the modulations resulted in the loss of activity, consistent low toxicity for the compounds throughout the series was worth noticing. Both methylations of the methylene group and bioisosteric replacement of the carbonyl moiety resulted in activity loss. These observations proved the importance of acetyl linker in the hit molecule (65).
C-5 of hydantoin had two substituents. The carbonitrile moiety has been known to react with cysteine and/or serine residues, leading to possible off-target covalent binding. Exploring this position indicated that the nitrile group was unnecessary for activity and could be exchanged for more active derivatives. Alteration of the methyl substituent resulted in either reduced or loss of activity. These results suggested that this part of the scaffold has a limited scope of modification. Modifications around position one by placing substituents on the nitrogen or exchanging it with carbon led to a decrease in enzymatic activity. Further, N-1 acyl substitution was tried, but the resulting derivatives lacked biological activity. This data suggested the importance of unsubstituted N1-nitrogen atoms for binding to the enzyme. All the changes made at N-3 resulted in activity loss, indicating a lack of scope for modification at this position.
The authors acknowledged the possibility of hydantoin scaffold giving some undesired effects like inhibition of hERG potassium channels, the fatal hydantoin syndrome, and cardiovascular risks. This served as the liability for further development, so the authors planned to replace the hydantoin core. Structurally similar rings such as succinimide, imidazolidin-2-one, imidazole, and pyrazole were considered as alternatives to the hydantoin core since they differ in aspect of one or more hydrogen bond donors/acceptors while at the same time they have the same geometry as the hydantoin motif. Unfortunately, none of the core replacements served the purpose as these derivatives were found to be inactive. These results indicated the importance of hydantoin core. The authors suggested that the hydantoin core could also interact with the protein. It was also reported that the lead compound (65) exhibited reversible binding to the enzyme.
Recently, in 2020, Balabon et al. [56] expanded the exploration of SAR for the hydantoin family through 80 new compounds. Based on the results of their earlier studies, they tried to optimize the phenyl ring wherein they replaced cyano with different substituents. But the results were disappointing for most of the substituents. The only notable exceptions were the methyl ester and the fused bicyclic analog. The most potent compounds were (66 and 67), having tetrazole and sulphonamide moieties as the substituents on the phenyl ring.

In vivo studies for the two most potent compounds (66 and 67) were carried out, and the efficacy of these derivatives was examined in a C57BL/6 J mouse model. No sign of adverse reaction was observed in any of the animals. The bioavailability of compound (67) was lower, and it exhibited significant blood exposure with a Cmax value of 6380 ng/mL and an AUC value of 31,400 h * ng/mL. Also, it depicted the greatest reduction of Log10 CFUs (0.5). Although the value is lesser than that of moxifloxacin, it still indicates the capability of hydantoin derivatives to reach the lungs of the animals after oral administration. These results indicated that this chemical family displays no appreciable cytotoxicity or cardiotoxicity (hERG), an appropriate physiological profile, and satisfactory metabolic stability. Although the results are encouraging, these compounds need further research to improve in vivo efficacy.
3.3 Miscellaneous Inhibitors
Wisely et al. [57] conducted virtual screening of a dataset consisting of 4.1 million compounds against the enzyme DrpE1. For the screening purpose, the co-crystal structure of DprE1 CT319 (PDB ID:4FDO) was taken. Initially, 500 hits were identified, out of which 41 compounds were isolated based on the structural diversity, binding affinity, and binding conformation. Compounds (68 and 69) were obtained as the most potent compounds. Molecular docking studies depicted that –NH of the amide group formed H-bond with adjacent Tyr 60 via the hydroxyl group and with the phenylalanine 320 residue through the carbonyl group. Leu317 formed a hydrogen bond with the amide of the carbonyl group. The planar orientation of the inhibitor allowed it to fit well into the active cavity.

A series consisting of some novel 11α-substituted bile acid derivatives along with N-alkyl and N-acyl derivatives of C-11 amino bile acid esters were reported as anti-TB agents by Vandana et al. [58]. Among the reported series, four compounds (70–73) showed significant activity against the M. tb H37Ra strain. Docking studies revealed that the docking scores for these compounds varied from −9.951 to −4.995, while the reference compound showed a docking score of −7.953. It was found that compound (71) was stabilized via various interactions such as van der Waals and electrostatic interactions with amino acids. Interestingly a prominent pi–pi interaction was seen between the triazole ring of compound (71) and the imidazole ring in His132 residue. Additionally, some H-bonding is also observed within the active site. The authors also predicted ADME properties of these active compounds (Table 2) using in silicotechniques. These compounds demonstrated acceptable oral bioavailability along with low susceptibility towards acid hydrolysis.

Jebiti et al. [59] performed molecular docking studies for some acyl thiourea derivatives against the DprE1 enzyme (PDB id: 4FDO). The results revealed that these thiourea derivatives interacted with the enzyme in a similar fashion as the co-crystallized ligand. The docking score for the most active compound (74) was −8.13, while the score was −6.77 for the co-crystallized ligand.

Shaikh et al. [60] reported novel triazole-based benzothiazinone derivatives as anti-TB agents. They carried out docking of the most potent compounds (75 and 76) to examine the binding interactions with the enzyme. The results from docking studies depicted that these derivatives interacted with the active site via van der Waals and electrostatic interactions. The benzothiazinone moiety and m-chloro group of the phenyl ring participated in van der Waals interactions, whereas the triazole ring was involved in interaction with Lys 418, Cys 317, and Ile386 residues. Additionally, H-bonding interactions facilitated steric and electrostatic interactions by anchoring the 3D-position of the compound within the active site. Moreover, the stability of the compound (76) in the active site is facilitated by pi-pi stacking interactions.

Chitre et al. [61] synthesized novel agents by hybridizing pyrazine and thiazolidine derivatives (77). These derivatives exhibited MIC values in the micro molar range. Molecular docking studies were reported for the novel hybrid molecule (77). The result showed that N-(4-oxo-2 substituted thiazolidin-3yl)pyrazine-2-carbohydrazide derivative fitted well in the active site and was located near the native ligand having a similar orientation. Their docking scores were in the range of −7.83 to −6.00 (native ligand: −7.953). These compounds fitted well in the active site via bonded and non-bonded interactions. They were found to be involved in electrostatic as well as van der Waals interactions with the amino acid residues. Thiazolidinone ring was found to be interacting with Lys418, Gly117, and Pro116 residues, whereas 3-ethoxy-4-hydroxyphenyl ring was involved in binding with Leu363 and Asp389 residues. The compound was further stabilized by pi-pi stacking between pyrazine ring and His132 residue.

Bhalerao et al. [62] performed the docking study of some thiazole-based compounds (PDB code:4FDO). The docking study revealed that the compounds fitted snugly into the active site by acquiring a similar orientation as the native ligand. The docking score for the test compounds ranged between −7.31 and −6.00, and it was −7.95 for the active compound. The highest docking score was −7.84 for the compound (78).

Yagao et al. [63] performed virtual screening of 6.2 million small molecules against DprE1 (PDB ID: 4FDO) using ICM 3.8.2 modeling software. Based on whether the compounds are occupying the binding pocket, many compounds were excluded. Further, the next filter used was Lipinski’s rule of five for drug-likeness. Based on these two filters, 63 compounds came out of the whole database. Further, molecular docking was carried out that resulted in the finding of the compound (79). Docking studies illustrated that the ligand-binding pocket of the enzyme is in a zig-zag shape, and the compounds also bind in a zig-zag manner in this cavity by indulging in hydrophobic interactions with the amino acid residues.

Kasa et al. [64] reported the interaction studies of M. tb protein DprE1 (PDB ID: 4P8C) with various triazole-based pyrrole-pyrimidine analogs (80) by performing molecular docking. The compound (80a) came out with the highest Mol Dock score of −157.926 and was demonstrated to interact via hydrogen bonding and pi-lone pair interactions with the enzyme. It was noted that the results from in silico studies of the active compounds supported the activity data, indicating the importance of the triazole ring for exhibiting anti-TB activity.

Yalcin et al. [65] performed molecular docking to evaluate the potential of fluoro-substituted chalcone derivatives (81–83) as DprE1 inhibitors. They synthesized fluoro and non-fluoro chalcone derivatives and evaluated their anti-proliferative and anti-TB activity. The crystal structure used for the study was PDB ID: 4P8H. The synthesized chalcone derivatives had both cis and trans isomers. Among the synthesized compounds, (82g) was found to have the best binding affinity and inhibitory constant of −8.3 kcal/mol and 0.812 mM, respectively. The binding features of the compound (82g) are shown in Fig. 7. It can be seen that hydroxyl groups present in the structure form hydrogen bonds with the residues in the active site. The other phenyl moiety is involved in π-σ interaction with the Trp 230 residue.


Molecular docking results for 82 g. a Analysis with Chimera. b Analysis with Autodock Tools. Reprinted with permission from Ref. [65] Copyright© 2018 Elsevier
Docking results of these test compounds revealed that compound (82g) generated superior binding properties than the (81g) molecule. This may indicate that trans-configuration could be crucial for enhancing the binding properties of the molecules. However, no such relation was found on the other pair of isomers.
Recently in 2020, Kumar et al. [66] used a hybrid approach to synthesize some novel pyrazole-quinoline chalcones and pyrazole-coumarin chalcones and evaluated their anti-TB activity. Docking these compounds with the DprE1 enzyme demonstrated that the active compounds showed good binding to the target, having docking scores from −7.047 to −9.353 kcal/mol. Based on the molecular docking results, the authors proposed that the anti-TB activity of these could be due to inhibition of the DprE1 enzyme. The MIC value for the most potent compound (84) was observed to be 3.125 μg/ml.

GlaxoSmithKline (GSK) conducted an HTS campaign to identify potential DprE1 inhibitors. This campaign led to the emergence of a novel series of 2-((2,3-dihydrobenzo[b][1,4]dioxin-6-yl)amino)-N-phenylpropanamides. Surprisingly, the structure of this series is unrelated to the already known inhibitors. Compound (85) represents the initial HTS hit that demonstrated DprE1 inhibition with a pIC50 value of 7.2. Whitehurst et al. [67] extracted some compound analogs (85) using similarity-based clusters of HTS hit. Further, these analogs and sub-structures of GSK compound collection were analyzed to gain early SAR information. For defining SAR, compound (85) was divided into three main components: the central alanine linker, left-hand side aminobenzodioxane, and right-side C-terminal alanine. SAR studies revealed that the compound (85) showed the maximum activity. Some derivatives were indeed found with equal activity. However, further research is required to assess the risk associated with the compound (85) and develop this new structure into a viable lead.

Hariguchi et al. [68] applied a phenotypic screening method to identify and optimize compounds with anti-TB activity containing carbostyril as the core. Carbostyril core has been chosen because it has been reported to have good ADMET properties and has been used in numerous drugs as the core moiety. As a result of these efforts, OPC-167832 (86) came into the lime light, which has potent in vitro and in vivo activities. Subsequently, the authors mapped DprE1 inhibition as the mode of action. They also reported preclinical data, including in vivoefficacy of the regimens composed of this compound.

3.4 Covalent vs. Non-covalent Binding Inhibitors
In the above three subsections, it is observed that anti-TB activity can be achieved by blocking DprE1 both covalently and non-covalently. To date, researchers have reported both covalent and non-covalent inhibitors with high potency and minimum toxicity. So, an obvious question arises, which type of inhibition is better. To answer this, we must understand the difference in their mechanism of action. Covalent inhibitors work by binding to the Cys387 residue of the enzyme. The binding is irreversible; thus, the inhibitors act as suicide substrates. Some reports also revealed that mutation in the Cys387 residue might develop resistance towards covalent inhibitors [69].
On the other hand, non-covalent inhibitors bind reversibly to the enzyme, leading to inefficient inhibition or development of resistance by incrementing the bacterial load [70]. However, compounds from both categories have found a place in clinical trials. So, we consider the future of both the types of inhibitors, covalent and non-covalent, as bright in the direction of our search for new anti-TB drugs.
4 Patented DprE1 Inhibitors
During the last decade, many compounds have been patented as DprE1 inhibitors. The very first patent on DprE1 inhibitor was obtained on the benzothiazinone derivative BTZ043 (29) [71]. Subsequently, many patent applications were filed for various benzothiazinones such as PBTZ169 (31), BTZ-SO (33), and compounds (87a and 87b) [72,73,74]. Recently patent applications for the two benzothiazinone derivatives (88, 89) have been filed [75, 76], claiming almost equal potency to PBTZ169 (31) but with a lower cLogP value. Compounds (88, 89) exhibited MIC values of 0.005 μM and 0.022 μM respectively. Another benzothiazinone derivative (90) yielded a MIC value lower than 0.000063 μg/ml against resistant M. tb strains [77].

A carbostyril derivative, OPC-167832 (86), has also been patented as a DprE1 inhibitor. The claims mentioned in the application illustrate that the compound is specifically active against mycobacteria and is orally active with no gastrointestinal disturbances [78]. Benzothiazole derivative, TCA1 (47), and the azaindole derivative, TBA-7371 (60), have also been patented as DprE1 inhibitors [79, 80]. TBA-7371 (60) has been mentioned as pathogen-specific for M. tb and M. smegmatis. Recently, another azaindole amide derivative (91) has been reported under publication [81]. Compound (91) showed a MIC value of 0.1953 μg/ml against M. tb. Another invention claimed arylamide-substituted thiophenimide (92) esters as DprE1 inhibitors. The compound (92) exhibited excellent in vivo activity with Log10CFU = 4.42 [82]. Another invention reported nitrofuran derivatives (93) as DprE1 inhibitors with a MIC value of 1 μM [83]. Yet another patented compound (94) has been claimed to have excellent activity against M. tb with a MIC value less than 0.0625 μM [84].

5 DprE1 Inhibitors in Clinical Trials
The discovery of benzothiazinones has marked the onward journey of DprE1 inhibitors in our quest for discovering novel anti-TB agents [24]. Two benzothiazinone derivatives, BTZ043 (29) and PBTZ-169 (31), are currently in phase 1 clinical trial. Macozinone (MCZ, PBTZ-169) (31), a piperazino benzothiazinone derivative, is obtained as a result of optimization of benzothiazinone lead molecule BTZ043 (29). PBTZ-169 (31) was found to have many merits over the lead BTZ-043, amongst which better pharmacodynamics, easier method of synthesis, and absence of chiral centers in its structure are some of them. The drug has additive effects with other anti-TB agents, both marketed and underdeveloped, while showing harmonious effects with bedaquiline (17) and clofazimine (95) in the preclinical stage [85]. Currently, it is in the second phase in Russia, whereas in Europe, it is in phase 1 [86, 87]. Another agent, TBA-7371 (60), is in phase 2 clinical trials [88]. TBA-7371, along with sutezolid (96), entered phase 1 clinical trials. It is developed by AstraZeneca in collaboration with TB Alliance. It is believed that TBA-7371 (60) does have the potential to be used for the treatment of resistant cases of TB because it is devoid of any pre-existing resistance or cross-resistance with other drugs [89, 90]. OPC-167832 (86) is also reported to be in phase 1 clinical trials [68].

6 Microbial Resistance and DprE1 Inhibitors
The main issue with the already existing anti-TB drugs is the development of microbial resistance. First-line agents like isoniazid, pyrazinamide, etc., are vulnerable to the development of resistance. A decade ago, when BTZs came into the light, they attracted researchers worldwide due to the new target and the sub-micromolar MIC values they exhibited. Since then, continuous research has been going on DprE1 inhibitors. But it is important to evaluate whether this target is also vulnerable to resistance and, if yes, to what extent. To meet this particular requirement, Foo et al. [69] reported DprE1-mediated BTZ’s resistance in M. tb. Results of the study revealed that the C387 residue of the enzyme served as the site of mutation, leading to the development of resistance towards BTZ. Additionally, it was proved that five mutations on the C387 residue were responsible for developing resistance. These mutations were caused by substituting different amino acids such as glycine, alanine, arginine, serine, and threonine. It was also observed that mutations with C387T, C387A, and C387S had a greater impact than C387N and C387G. The authors also claimed that the decreased potency of covalent inhibitors results from a mutation at C387 residue. While mutations at Ty38C residue resulted in resistance towards non-covalent inhibitors.
Warrier et al. [91] also studied the development of microbial resistance by overexpressing some genes, i.e., rv0560c, rv0558, and rv0559c. The authors claimed that rv0560c is a gene responsible for S-adenosyl-l-methionine-dependent methyltransferase, an enzyme that methylates the inhibitors and reduces their activity.
7 Conclusion
Since its discovery in 2009, DprE1 has been perceived as the best druggable target to combat TB [2, 92].The discovery of BTZ043 served as the starting point for researching novel covalent DprE1 inhibitors. The revelation of the mechanism of action of BTZ as covalent inhibitors was a breakthrough in the field of DprE1 inhibitors. Since then, continuous research has been carried out by researchers worldwide. Both covalent and non-covalent inhibitors have been looked out as potential anti-TB agents. Two such candidates, i.e., Macozinone and TBA-7371, are already in clinical trials. We keep our fingers crossed and hope to emerge some novel DprE1 inhibitors as efficient anti-TB agents.
Mother Teresa once said, “The biggest disease today is not leprosy or tuberculosis, but rather the feeling of being unwanted.” At present, the world needs new and effective drugs for eradicating an infectious disease like TB as it is the need of the hour to prevent and address TB patients’ sufferings, a suffering due to ineffective treatment due to development of drug resistance, poor drug compliance by the patients, the cost of treatment, and the feeling of unwanted by the society.
Core Messages
-
DprE1 has evolved as a new competent target that could be exploited for anti-TB drug discovery.
-
Covalent and non-covalent inhibitors have been looked out as potential anti-TB agents.
-
Four DprE1 inhibitors, namely BTZ-043, Macozinone, OPC-167832, and TBA-7371, are currently in clinical trials.
References
Manina G, Pasca MR, Buroni S, De Rossi E, Riccardi G (2010) Decaprenylphosphoryl-β-d-ribose 2′-epimerase from Mycobacterium tuberculosis is a magic drug target. Curr Med Chem 17(27):3099–3108
Chikhale RV, Barmade MA, Murumkar PR, Yadav MR (2018) Overview of the development of DprE1 inhibitors for combating the menace of tuberculosis. J Med Chem 61(19):8563–8593
Yuan T, Sampson NS (2018) Hit generation in TB drug discovery: from genome to granuloma. Chem Rev 118(4):1887–1916
Campanico A, Moreira R, Lopes F (2018) Drug discovery in tuberculosis. New drug targets and antimycobacterial agents. Eur J Med Chem 150:525–545
Wellington S, Hung DT (2018) The expanding diversity of Mycobacterium tuberculosis drug targets. ACS Infect Dis 4(5):696–714
Richter A, Rudolph I, Mollmann U, Voigt K, Chung CW, Singh OM, Rees M, Mendoza-Losana A, Bates R, Ballell L, Batt S (2018) Novel insight into the reaction of nitro, nitroso and hydroxylamino benzothiazinones and of benzoxacinones with Mycobacterium tuberculosis DprE1. Sci Rep 8(1):1–12
Piton J, Foo CSY, Cole ST (2017) Structural studies of Mycobacterium tuberculosis DprE1 interacting with its inhibitors. Drug Discov Today 22(3):526–533
Riccardi G, Pasca MR (2014) Trends in discovery of new drugs for tuberculosis therapy. J Antibiot 67(9):655–659
Brennan PJ (2003) Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis 83(1–3):91–97
Riccardi G, Pasca MR, Chiarelli LR, Manina G, Mattevi A, Binda C (2013) The DprE1 enzyme, one of the most vulnerable targets of Mycobacterium tuberculosis. Appl Microbiol Biotechnol 97(20):8841–8848
Rombouts Y, Brust B, Ojha AK, Maes E, Coddeville B, Elass-Rochard E, Kremer L, Guerardel Y (2012) Exposure of mycobacteria to cell wall-inhibitory drugs decreases production of arabinoglycerolipid related to mycolyl-arabinogalactan-peptidoglycan metabolism. J Bio Chem 287(14):11060–11069
Wolucka BA (2008) Biosynthesis of D-arabinose in mycobacteria—a novel bacterial pathway with implications for antimycobacterial therapy. FEBS J 275(11):2691–2711
Bhutani I, Loharch S, Gupta P, Madathil R, Parkesh R (2015) Structure, dynamics, and interaction of Mycobacterium tuberculosis (Mtb) DprE1 and DprE2 examined by molecular modeling, simulation, and electrostatic studies. PLoS ONE 10(3):e0119771
Alderwick LJ, Birch HL, Mishra AK, Eggeling L, Besra GS (2007) Structure, function and biosynthesis of the Mycobacterium tuberculosis cell wall: arabinogalactan and lipoarabinomannan assembly with a view to discovering new drug targets. Biochem Soc Trans 35(5):1325–1328
Meniche X, de Sousa-d’Auria C (2008) Partial redundancy in the synthesis of the D-arabinose incorporated in the cell wall arabinan of Corynebacterineae. Microbiology (Reading) 154(8):2315–2326
Makarov V, Manina G, Mikusova K, Möllmann U, Ryabova O, Saint-Joanis B, Dhar N, Pasca MR, Buroni S, Lucarelli AP, Milano A (2009) Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 324(5928):801–804
Mikusova K, Huang H, Yagi T, Holsters M, Vereecke D, D’Haeze W, Scherman MS, Brennan PJ, McNeil MR, Crick DC (2005) Decaprenylphosphoryl arabinofuranose, the donor of the D-arabinofuranosyl residues of mycobacterial arabinan, is formed via a two-step epimerization of decaprenylphosphoryl ribose. J Bacteriol 187(23):8020–8025
Abdel-Magid AF (2015) Decaprenylphosphoryl-β-d-ribose 2′-epimerase 1 (DprE1): a novel therapeutic target for the treatment of tuberculosis. ACS Med Chem Lett 6:373–374
Neres J, Pojer F, Molteni E, Chiarelli LR, Dhar N, Boy-Rottger S, Buroni S, Fullam E, Degiacomi G, Lucarelli AP, Read RJ, Giuseppe Z, Edmondson DE, Rossi ED, Pasca MR, McKinney JD, Dyson PJ, Riccardi G, Mattevi A, Cole ST, Binda C (2012) Structural basis for benzothiazinone-mediated killing of Mycobacterium tuberculosis. Sci Trans Med 4(150):150ra121–150ra121
Batt SM, Jabeen T, Bhowruth V, Quill L, Lund PA, Eggeling L, Alderwick LJ, Futterer K, Besra GS (2012) Structural basis of inhibition of Mycobacterium tuberculosis DprE1 by benzothiazinone inhibitors. Proc Natl Acad Sci 109(28):11354–11359
Liu R, Lyu X, Batt SM, Hsu MH, Harbut MB, Vilcheze C, Cheng B, Ajayi K, Yang B, Yang Y, Guo H (2017) Determinants of the inhibition of DprE1 and CYP2C9 by Antitubercular thiophenes. Angew Chem Int Ed Engl 56(42):13011–13015
Piton J, Vocat A, Lupien A, Foo CS, Riabova O, Makarov V, Cole ST (2018) Structure-based drug design and characterization of sulfonyl-piperazine benzothiazinone inhibitors of DprE1 from Mycobacterium tuberculosis. Antimicrob Agents Chemother 62(10):e00681-e718
Manina G, Bellinzoni M, Pasca MR, Neres J, Milano A, de Jesus Lopes Ribeiro AL, Buroni S, Skovierova H, Dianiskova P, Mikusova K, Marak J (2010) Biological and structural characterization of the Mycobacterium smegmatis nitroreductase NfnB, and its role in benzothiazinone resistance. Mol Microbiol 77(5):1172–1185
Ribeiro AL, Degiacomi G, Ewann F, Buroni S, Incandela ML, Chiarelli LR, Mori G, Kim J, Contreras-Dominguez M, Park YS, Han SJ (2011) Analogous mechanisms of resistance to benzothiazinones and dinitrobenzamides in Mycobacterium smegmatis. PLoS ONE 6(11):e26675
Spain JC (1995) Biodegradation of nitroaromatic compounds. Annu Rev Microbiol 49(1):523–555
Trefzer C, Rengifo-Gonzalez M, Hinner MJ, Schneider P, Makarov V, Cole ST, Johnsson K (2010) Benzothiazinones: prodrugs that covalently modify the decaprenylphosphoryl-β-d-ribose 2′-epimerase DprE1 of Mycobacterium tuberculosis. J Am Chem Soc 132(39):13663–13665
Tiwari R, Moraski GC, Krchnnak V, Miller PA, Colon-Martinez M, Herrero E, Oliver AG, Miller MJ (2013) Thiolates chemically induce redox activation of BTZ043 and related potent nitroaromatic anti-tuberculosis agents. J Am Chem Soc 135(9):3539–3549
Stewart CT (2010) New benzothiazinone derivatives and their use as antibacterial agents. EP2029583B1, 09 July 2010
Gao C, Ye TH, Wang NY, Zeng XX, Zhang LD, Xiong Y, You XY, Xia Y, Xu Y, Peng CT, Zuo WQ (2013) Synthesis and structure–activity relationships evaluation of benzothiazinone derivatives as potential anti-tubercular agents. Bioorg Med Chem Lett 23(17):4919–4922
Makarov V, Lechartier B, Zhang M, Neres J, van der Sar AM, Raadsen SA, Hartkoorn RC, Ryabova OB, Vocat A, Decosterd LA, Widmer N (2014) Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol Med 6(3):372–383
Peng CT, Gao C, Wang NY, You XY, Zhang LD, Zhu YX, Xv Y, Zuo WQ, Ran K, Deng HX, Lei Q (2015) Synthesis and anti-tubercular evaluation of 4-carbonyl piperazine substituted 1,3-benzothiazin-4-one derivatives. Bioorg Med Chem Lett 25(7):1373–1376
Tiwari R, Miller PA, Cho S, Franzblau SG, Miller MJ (2015) Syntheses and antituberculosis activity of 1, 3-benzothiazinone sulfoxide and sulfone derived from BTZ043. ACS Med Chem Lett 6(2):128–133
Landge S, Mullick AB, Nagalapur K, Neres J, Subbulakshmi V, Murugan K, Ghosh A, Sadler C, Fellows MD, Humnabadkar V, Mahadevaswamy J (2015) Discovery of benzothiazoles as antimycobacterial agents: synthesis, structure–activity relationships and binding studies with Mycobacterium tuberculosis decaprenylphosphoryl-β-d-ribose 2′-oxidase. Bioorg Med Chem Lett 23(24):7694–7710
Stanley SA, Grant SS, Kawate T, Iwase N, Shimizu M, Wivagg C, Silvis M, Kazyanskaya E, Aquadro J, Golas A, Fitzgerald M (2012) Identification of novel inhibitors of M. tuberculosis growth using whole cell based high-throughput screening. ACS Chem Biol 7(8):1377–1384
Karabanovich G, Dusek J, Savkova K, Pavlis O, Pavkova I, Korabecny J, Kucera T, Koccovaa Vlcckovaa H, Huszar S, Konyarikova Z, Konecna K (2019) Development of 3, 5-dinitrophenyl-containing 1, 2, 4-triazoles and their trifluoromethyl analogues as highly efficient anti-tubercular agents inhibiting decaprenylphosphoryl-β-d-ribofuranose 2′-oxidase. J Med Chem 62(17):8115–8139
Ali AA, Gogoi D, Chaliha AK, Buragohain AK, Trivedi P, Saikia PJ, Gehlot PS, Kumar A, Chaturvedi V, Sarma D (2017) Synthesis and biological evaluation of novel 1, 2, 3-triazole derivatives as anti-tubercular agents. Bioorg Med Chem Lett 27(16):3698–3703
Magnet S, Hartkoorn RC, Szekely R, Pato J, Triccas JA, Schneider P, Szantai-Kis C, Orfi L, Chambon M, Banfi D, Bueno M (2010) Leads for anti-tubercular compounds from kinase inhibitor library screens. Tuberculosis 90(6):354–360
Christophe T, Jackson M, Jeon HK, Fenistein D, Contreras-Dominguez M, Kim J, Genovesio A, Carralot JP, Ewann F, Kim EH, Lee SY (2009) High content screening identifies decaprenyl-phosphoribose 2′ epimerase as a target for intracellular antimycobacterial inhibitors. PLoS Pathog 5(10):e1000645
Brodin PR (2011) Anti-infective compounds. WO2011113606 A1, 22 Sept 2011
Makarov V, Neres J, Hartkoorn RC, Ryabova OB, Kazakova E, Šarkan M, Huszár S, Piton J, Kolly GS, Vocat A, Conroy TM (2015) The 8-pyrrole-benzothiazinones are non-covalent inhibitors of DprE1 from Mycobacterium tuberculosis. Antimicrob Agents Chemother 59(8):4446–4452
Wang F, Sambandan D, Halder R, Wang J, Batt SM, Weinrick B, Ahmad I, Yang P, Zhang Y, Kim J, Hassani M (2013) Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proc Natl Acad Sci 110(27):E2510–E2517
Chikhale R, Menghani S, Babu R, Bansode R, Bhargavi G, Karodia N, Rajasekharan MV, Paradkar A, Khedekar P (2015) Development of selective DprE1 inhibitors: design, synthesis, crystal structure and anti-tubercular activity of benzothiazolylpyrimidine-5-carboxamides. Eur J Med Chem 96:30–46
Mir F, Shafi S, Zaman MS, Kalia NP, Rajput VS, Mulakayala C, Mulakayala N, Khan IA, Alam MS (2014) Sulfur rich 2-mercaptobenzothiazole and 1, 2, 3-triazole conjugates as novel anti-tubercular agents. Eur J Med Chem 76:274–283
Gawad J, Bonde C (2018) Synthesis, biological evaluation and molecular docking studies of 6-(4-nitrophenoxy)-1H-imidazo[4,5-b]pyridine derivatives as novel anti-tubercular agents: future DprE1 inhibitors. Chem Cent J 12(1):1–11
Neres J, Hartkoorn RC, Chiarelli LR, Gadupudi R, Pasca MR, Mori G, Venturelli A, Savina S, Makarov V, Kolly GS, Molteni E (2014) 2-Carboxyquinoxalines kill Mycobacterium tuberculosis through non-covalent inhibition of DprE1. ACS Chem Biol 10:705–714
Batt SM, Cacho Izquierdo M, Castro Pichel J, Stubbs CJ, Vela-Glez Del Peral L, Pérez-Herrán E, Dhar N, Mouzon B, Rees M, Hutchinson JP, Young RJ (2015) Whole cell target engagement identifies novel inhibitors of Mycobacterium tuberculosis decaprenylphosphoryl-β-d-ribose oxidase. ACS Infect Dis 1(12):615–626
Brodin P (2010) Anti-infective compounds. WO2010003533A2, 11 Nov 2010
Brodin P (2011) Anti-infective pyrido(1,2-a)Pyrimidines. WO2011085990A1, 21 July 2011
Shirude PS, Shandil R, Sadler C, Naik M, Hosagrahara V, Hameed S, Shinde V, Bathula C, Humnabadkar V, Kumar N, Reddy J (2013) Azaindoles: non-covalent DprE1 inhibitors from scaffold morphing efforts, kill Mycobacterium tuberculosis and are efficacious in vivo. J Med Chem 56(23):9701–9708
Chatterji M, Shandil R, Manjunatha MR, Solapure S, Ramachandran V, Kumar N, Saralaya R, Panduga V, Reddy J, Prabhakar KR, Sharma S (2014) 1, 4-Azaindole, a potential drug candidate for treatment of tuberculosis. Antimicrob Agents Chemother 58(9):5325–5331
Shirude PS, Shandil RK, Manjunatha MR, Sadler C, Panda M, Panduga V, Reddy J, Saralaya R, Nanduri R, Ambady A, Ravishankar S (2014) Lead optimization of 1, 4-azaindoles as antimycobacterial agents. J Med Chem 57(13):5728–5737
Manjunatha MR, Shandil R, Panda M, Sadler C, Ambady A, Panduga V, Kumar N, Mahadevaswamy J, Sreenivasaiah M, Narayan A, Guptha S (2019) Scaffold morphing to identify novel DprE1 inhibitors with antimycobacterial activity. ACS Med Chem Lett 10(10):1480–1485
Panda M, Ramachandran S, Ramachandran V, Shirude PS, Humnabadkar V, Nagalapur K, Sharma S, Kaur P, Guptha S, Narayan A, Mahadevaswamy J (2014) Discovery of pyrazolopyridones as a novel class of non-covalent DprE1 inhibitor with potent antimycobacterial activity. J Med Chem 57(11):4761–4771
Naik M, Humnabadkar V, Tantry SJ, Panda M, Narayan A, Guptha S, Panduga V, Manjrekar P, Jena LK, Koushik K, Shanbhag G (2014) 4-aminoquinolone piperidine amides: non-covalent inhibitors of DprE1 with long residence time and potent antimycobacterial activity. J Med Chem 57(12):5419–5434
Rogacki MK, Pitta E, Balabon O, Huss S, Lopez-Roman EM, Argyrou A, Blanco-Ruano D, Cacho M, Vande Velde CM, Augustyns K, Ballell L (2018) Identification and profiling of hydantoins—a novel class of potent antimycobacterial DprE1 inhibitors. J Med Chem 61(24):11221–11249
Balabon O, Pitta E, Rogacki MK, Meiler E, Casanueva R, Guijarro L, Huss S, Lopez-Roman EM, Santos-Villarejo A, Augustyns K, Ballell L (2020) Optimization of hydantoins as potent antimycobacterial decaprenylphosphoryl-β-d-ribose oxidase (DprE1) inhibitors. J Med Chem 63(10):5367–5386
Wilsey C, Gurka J, Toth D, Franco J (2013) A large scale virtual screen of DprE1. Comput Biol Chem 47:121–125
Pore VS, Divse JM, Charolkar CR, Nawale LU, Khedkar VM, Sarkar D (2015) Design and synthesis of 11α-substituted bile acid derivatives as potential anti-tuberculosis agents. Bioorg Med Chem Lett 25(19):4185–4190
Haribabu J, Subhashree GR, Saranya S, Gomathi K, Karvembu R, Gayathri D (2015) Synthesis, crystal structure, and in vitro and in silico molecular docking of novel acyl thiourea derivatives. J Mol Struct 1094:281–291
Shaikh MH, Subhedar DD, Arkile M, Khedkar VM, Jadhav N, Sarkar D, Shingate BB (2016) Synthesis and bioactivity of novel triazole incorporated benzothiazinone derivatives as anti-tubercular and antioxidant agent. Bioorg Med Chem Lett 26(2):561–569
Chitre TS, Asgaonkar KD, Miniyar PB, Dharme AB, Arkile MA, Yeware A, Sarkar D, Khedkar VM, Jha PC (2016) Synthesis and docking studies of pyrazine–thiazolidinone hybrid scaffold targeting dormant tuberculosis. Bioorg Med Chem Lett 26(9):2224–2228
Bhalerao MB, Dhumal ST, Deshmukh AR, Nawale LU, Khedkar V, Sarkar D, Mane RA (2017) New bithiazolyl hydrazones: novel synthesis, characterization and anti-tubercular evaluation. Bioorg Med Chem Lett 27(2):288–294
Gao Y, Xie J, Tang R, Yang K, Zhang Y, Chen L, Li H (2019) Identification of a pyrimidinetrione derivative as the potent DprE1 inhibitor by structure-based virtual ligand screening. Bioorg Chem 85:168–178
Raju KS, AnkiReddy S, Sabitha G, Krishna VS, Sriram D, Reddy KB, Sagurthi SR (2019) Synthesis and biological evaluation of 1H-pyrrolo[2,3-d]pyrimidine-1,2,3-triazole derivatives as novel anti-tubercular agents. Bioorg Med Chem Lett 29(2):284–290
Yalcin G, Burmaoglu S, Yildiz I, Algul O (2018) Molecular docking studies on fluoro-substituted chalcones as potential DprE1 enzyme inhibitors. J Mol Struct 1164:50–56
Kumar G, Siva Krishna V, Sriram D, Jachak SM (2020) Pyrazole–coumarin and pyrazole–quinoline chalcones as potential antitubercular agents. Arch Pharm 353(8):e2000077
Whitehurst BC, Young RJ, Burley GA, Cacho M, Torres P, del Peral LV (2020) Identification of 2-((2,3-dihydrobenzo [b][1,4]dioxin-6-yl) amino)-N-phenylpropanamides as a novel class of potent DprE1 inhibitors. Bioorg Med Chem Lett 30(12):127192. https://doi.org/10.1016/j.bmcl.2020.127192
Hariguchi N, Chen X, Hayashi Y, Kawano Y, Fujiwara M, Matsuba M, Shimizu H, Ohba Y, Nakamura I, Kitamoto R, Shinohara T (2020) OPC-167832, a novel carbostyril derivative with potent antituberculosis activity as a DprE1 inhibitor. Antimicrob Agents Chemother 64(6):e02020-e2119. https://doi.org/10.1128/AAC.02020-19
Foo CS, Lechartier B, Kolly GS, Boy-Röttger S, Neres J, Rybniker J, Lupien A, Sala C, Piton J, Cole ST (2016) Characterization of DprE1-mediated benzothiazinone resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 60(11):6451–6459
Singh J, Petter RC, Baillie TA, Whitty A (2011) The resurgence of covalent drugs. Nat Rev Drug Discov 10:307–317
Makarov V (2011) Benzothiazinone derivatives and their use as antibacterial agents. US7863268B2, 4 Jan 2011
Makarov V (2014) 2-piperazin-1-yl-4H-1,3-benzothiazin-4-one derivatives and their use for the treatment of mammalian infections. US8796264B2, 5 Aug 2014
Milller MJ (2016) 1,3-Benzothiazinone sulfoxide and sulfone compounds. US9481683B2, 1 Nov 2016
Chao G (2018) Benzothiazine derivative, a preparation method, and uses thereof. CN108456204A, 28 Aug 2018
Chunhua Q (2020) Benzothiazinone derivatives, preparation method thereof, and application as anti-tuberculosis drugs. CN111303075A, 19 Jun 2020
Chunhua Q (2020) Benzothiazinone compound, preparation method thereof, and application as anti-tuberculosis medicine. CN111269197A, 12 Jun 2020
Florian K (2019) New anti-microbial compounds, their use for the treatment of mammalian infections and a new metabolic mechanism. EP3515920A1, 31 Jul 2019
Shimizu H (2018) Heterobicyclic compounds and their use for the treatment of tuberculsosis. US10053446B2, 21 Aug 2018
Chatterjee AK (2016) Compounds for treatment of drug resistant and persistent tuberculosis. US20160194299A1, 7 Jul 2016
Shirude PS (2015) Azaindole compounds, synthesis thereof, and methods of using the same. US9163020B2, 20 Oct 2015
Changlun A (2020) Azaindole amide compounds and preparation method and application thereof. CN111393435A, 10 Jul 2020
Haihong H (2020) 2-Arylamino-substituted thienylimide ester compound and preparation method and application thereof. CN110759889A, 7 Feb 2020
Lin D (2018) Nitrofuran antituberculous component. CN108558858A, 21 Sep 2018
Desai R (2019) Condensed azaheteroaryl compounds having antibacterial activity against tuberculosis bacteria. WO2019239382A1, 19 Dec 2019
https://www.newtbdrugs.org/pipeline/compound/macozinone-mcz-pbtz-169. Accessed on 09 Oct 2020
https://clinicaltrials.gov/ct2/show/record/NCT03334734. Accessed on 09 Oct 2020
http://im4tb.org/our-pipeline/#:~:text=PBTZ169%20(macozinone)%20%E2%80%93%20currently%20in,to%20treat%20multidrug%2Dresistant%20tuberculosis. Accessed on 09 Oct 2020
https://www.tballiance.org/portfolio/compound/tba-7371-dpre1-inhibitor. Accessed on 09 Oct 2020
https://www.tballiance.org/news/tb-alliance-moves-two-novel-tuberculosis-drugs-human-trials. Accessed on 09 Oct 2020
Degiacomi G, Belardinelli JM, Pasca MR, Rossi ED, Riccardi G, Chiarelli LR (2020) Promiscuous targets for antitubercular drug discovery: the paradigm of DprE1 and MmpL3. Appl Sci 10(2):623
Warrier T, Kapilashrami K, Argyrou A, Ioerger TR, Little D, Murphy KC, Nandakumar M, Park S, Gold B, Mi J, Zhang T (2016) N-methylation of a bactericidal compound as a resistance mechanism in Mycobacterium tuberculosis. Proc Natl Acad Sci 113(31):E4523–E4530
Murumkar PR, Sharma MK, Gupta P, Patel NM, Yadav MR (2022) Selection of suitable protein structure from Protein Data Bank: An important step in Structure based Drug Design Studies. Mini Rev Med Chem. https://doi.org/10.2174/1389557522666220512151454
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Yadav, M.R., Murumkar, P.R., Ghuge, R.B., Barot, R.R., Chauhan, M. (2023). Exploring Decaprenylphosphoryl-β-d-Ribose 2′-Epimerase 1 (DprE1): A Target for Anti-tubercular Drugs. In: Rezaei, N. (eds) Tuberculosis. Integrated Science, vol 11. Springer, Cham. https://doi.org/10.1007/978-3-031-15955-8_24
Download citation
DOI: https://doi.org/10.1007/978-3-031-15955-8_24
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-15954-1
Online ISBN: 978-3-031-15955-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)