
Abstract
2021 marked the 25th anniversary of the discovery of the HFE gene, a major genetic breakthrough which revolutionised the understanding and diagnosis of hereditary haemochromatosis (HH). The original description of the condition ‘bronze diabetes’ is now rare with earlier presentations being the norm due to increased awareness and easier diagnosis. Importantly, a diagnosis of HH can be made quite readily now in primary care via HFE genotyping. Liver biopsy is reserved for fibrosis staging although non-invasive methods such as transient elastography are becoming complementary. Cirrhosis impacts on survival, and certain manifestations, such as arthritis, do not respond well to phlebotomy; the latter is a source of long-term morbidity in many patients, hence the need to diagnose the condition early. Asymptomatic HFE positive individuals may not necessarily benefit from immediate therapeutic venesection, e.g. pre-menopausal females with normal ferritin, elderly patients and compound heterozygotes. Importantly, the National Blood Service will consider treated HH individuals for maintenance phlebotomy, which provides benefit to other patients. Clinicians require a low index of suspicion for diagnosis and should implement cascade screening once a case is identified. The identification of HFE spawned a new era of molecular genetic research and a number of rarer inherited forms of iron overload have since been recognised; advanced genetic testing is now more readily available for clinicians and novel molecular therapies are in development.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
FormalPara Key Learning Points-
Understanding the pathophysiology of haemochromatosis
-
Pathway for diagnosis
-
Investigation and when/how to treat
-
Non-HFE syndromes
A 55-year-old moderately obese woman presents with a history of worsening arthralgia. Her serum ferritin level is raised at 650 μg/L with normal CRP, haemoglobin and liver function tests. What would you do next?
-
(a)
Recommend venesection
-
(b)
Arrange HFE genotyping
-
(c)
Check the transferrin saturation
-
(d)
Arrange a liver ultrasound
The patient is diagnosed with HH (C282Y homozygous) following further investigation. Which of the following apply?
-
(a)
The patient could be observed initially
-
(b)
Therapeutic venesection should be commenced
-
(c)
First degree relatives require only a ferritin check
-
(d)
First degree relatives should have HFE genotyping
Background
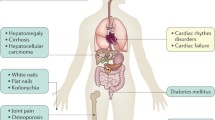
Hereditary Haemochromatosis (HH) is an autosomal recessive disorder characterised by organ damage due to dietary iron accumulation; the condition may also be referred to as HFE-related or type 1 Haemochromatosis. The presentation occurs earlier, and with greater severity, in men due to the lack of specific iron losses that occur via menstruation in women. Typically, HH manifests above the age of 40 years. Iron is mainly deposited in the liver and in the synovial tissue of joints but eventually in the pancreas, skin, heart, the gonadotrophin-secreting cells of the anterior pituitary and rarely the adrenal and parathyroid glands. It is not clear why iron accumulates preferentially in certain extra-hepatic tissues. Disease manifestations thereby include hepatic fibrosis, arthropathy, diabetes mellitus due to lack of insulin, pigmentation, cardiomyopathy and impotence. Fatigue is a common early symptom and, together with arthralgia, affect quality of life in patients with HH. A serum ferritin >1000 μg/L at diagnosis equates to a five-fold relative mortality risk. Hepatic cirrhosis is associated with significantly reduced survival and a 100-fold increased risk of hepatocellular carcinoma (HCC), the commonest cause of death in HH [1]. The high rate of HCC is due to a combination of cirrhosis and the carcinogenic properties of iron, and the risk remains despite iron removal. The discovery of the HFE gene in 1996 provided a significant breakthrough for the diagnosis of HH [2]. Until then, genetic testing relied on HLA linkage following the identification of this association in the 1970s. Homozygosity for the C282Y mutation on chromosome 6p accounts for 90% of cases of HH; thus a specific tool was immediately available for non-invasive diagnosis, screening and prevalence estimation. The advent of HFE has enabled characterisation of the natural history and expression of HH, and a greater understanding of the hepatic siderosis associated with chronic liver disease.
Iron Pathophysiology
In normal individuals, gastrointestinal iron absorption is homeostatically regulated according to body iron status. An important concept is that there is no physiological mechanism in humans to excrete excess iron when overload occurs. In HH, the gene defect results in an inability to appropriately reduce iron absorption such that body iron accumulation ensues (Fig. 12.1). When following the natural history of the condition, the initial laboratory finding is that of a raised plasma transferrin saturation as iron absorbed from the intestine loads on to the transferrin carrier protein at an increased rate compared with normal individuals. Subsequently, as tissue iron loading occurs the serum ferritin starts to rise proportionately. Total body iron, which is 4 g in a normal adult, typically rises above 10 g in a patient with HH. In C282Y homozygotes serum ferritin concentrations >1000 μg/L are associated with the development of liver fibrosis.

Iron pathophysiology in HH. Under normal circumstances iron absorption is regulated to match insensible losses with total adult body iron around 4 g. In haemochromatosis the iron storage compartment greatly increases due to unopposed intestinal uptake as normal feedback mechanisms are disrupted. Note the size of the transferrin iron pool, approximately 0.1% of total body iron, and that transferrin saturates early during the natural history of haemochromatosis
Disease Expression
After the discovery of HFE it was quickly recognised that around 0.5% of white people are homozygous for the C282Y mutation and thereby genetically predisposed to developed HH [3]. However, only a proportion of these individuals have evident symptoms or signs. Many will be presymptomatic with biochemical iron loading or have little or no evidence of iron loading, particularly pre-menopausal females (Fig. 12.2). When taking a history from a patient with HH it is prudent to ask about previous iron gains (oral iron supplementation, amenorrhoea, early menopause) and iron losses (blood donation, gut pathology, menorrhagia). As a rule, symptoms tend not to occur until the serum ferritin level is elevated. Although significant hepatic fibrosis is unlikely with serum ferritin values <1000 μg/L, joint disease can certainly occur and may be irreversible. The ‘penetrance’ of C282Y homozygosity is generally described as ‘low’ but varies according to the definition and study population; for example, evidence of pathology may be seen in a third of males but biopsy-proven cirrhosis occurs in only 1% overall. A recent UK biobank study, which included nearly 3000 homozygotes, described significant a disease burden in HH [4]. Despite the evident disease burden and potential preventability, population screening is not currently recommended as effective pilot studies remain lacking. Environmental factors which modify iron loading and hence expression of the disease include excess alcohol, iron-rich diet and blood donation. Other factors such as obesity may influence hepatic fibrosis per se. Genetic modifiers are important determinants of disease expression in C282Y homozygotes. Genome-wide association studies in HH have identified the transferrin gene and TMPRSS6 gene as direct modifiers of iron overload. In addition, variants which contribute to chronic liver disease progression have been associated with development of cirrhosis in homozygotes [5].

The iceberg of pathology in C282Y homozygotes ranging from genetic predisposition only to significant organ damage including cirrhosis, arthropathy and diabetes. Various factors determine progression as listed. Venesection prior to the onset of organ damage ensures normal survival and prior to the onset of symptoms prevents morbidity associated with the disorder
Diagnosis
HH can be diagnosed in most cases by non-invasive means. A compatible genotype combined with biochemical evidence of iron loading is sufficient. The combination of elevated serum ferritin and transferrin saturation (>50% in males and 45% in females) is highly suggestive of the condition—it is advisable to repeat these on a fasting sample in the first instance. Hyperferritinaemia with a normal transferrin saturation is typically due to excess alcohol and/or in the context of non-alcoholic fatty liver and the metabolic syndrome. As well as the common homozygous genotype C282Y/C282Y, which accounts for the majority of cases, the compound heterozygous form C282Y/H63D accounts for 5–10% of cases and is associated with mild iron burden. Of note, EASL guidelines recognise C282Y homozygosity as a diagnostic genotype for HH, whereas other genotypes such as C282Y/H63D and H63D/H63D require exclusion of additional causes of hyperferritinaemia [6]. Liver biopsy is reserved for those individuals without a recognisable genotype or in those where there is a risk of significant liver fibrosis. The latter is important to identify as surveillance for HCC is required in those with incipient or established cirrhosis. Histologically, iron is deposited initially in peri-portal hepatocytes with later spill over into bile duct epithelium and Kupffer cells (Fig. 12.3).

Biopsy and imaging findings in severe HH. The top left panel shows a low power haematoxylin and eosin stain demonstrating parenchymal nodules surrounded by fibrous tissue (cirrhosis). On high power (top right) brown pigment is noted within hepatocytes. With Perls’ reagent (bottom left) the pigment stains blue and is confirmed as iron. Magnetic resonance imaging (MRI) demonstrates a low signal intensity liver compared with muscle on T2-weighted imaging (bottom right)
In terms of non-invasive exclusion of significant fibrosis, in homozygotes where serum aminotransferase values are normal, hepatomegaly is absent and the serum ferritin is below 1000 μg/L there is negligible risk [7]. In addition, a serum hyaluronic acid level >46.5 ng/mL is associated with 100% sensitivity and specificity for cirrhosis. Transient elastography has been shown to reduce the requirement for biopsy in at risk patients and can accurately classify severe fibrosis in around 60% of homozygotes with ferritin >1000 μg/L and raised transaminases [8]. Cirrhosis is less common at presentation over recent decades due to greater clinical awareness and access to HFE gene testing. Magnetic resonance imaging (MRI), specifically T2-weighted sequences, can be used to demonstrate hepatic iron deposition non-invasively and specific software may be used for quantification. This is not required in HH as serum ferritin and quantitative phlebotomy are reliable indicators of the degree of iron overload. However, the technique may be useful when HFE analysis is negative but iron overload is suspected biochemically. MRI can also be used to quantify levels of steatosis and fibrosis when characterising patients with unexplained significant hyperferritinaemia.
It is important to screen first degree relatives of C282Y homozygotes as there will be a significant pick up of pre-morbid disease and indeed those already with unrecognised symptoms due to HH—the so-called cascade screening approach. Children should not be tested until adulthood, as iron is required for growth, although spousal testing for C282Y heterozygosity (10–15% risk in Caucasian populations) may obviate this need.
Treatment
Regular removal of blood, typically approximately 500 mL every week, remains a proven and effective method to clear excess iron. As a rule this will drop the serum ferritin by around 50 μg/L per visit. It is advisable to venesect until the serum ferritin falls below 50 μg/L and, from a practical perspective, to then maintain a level between 20 and 100 μg/L long term. Some advocate maintaining the transferrin saturation below 50% at all times based on a recent French study which was retrospective in nature [9, 10]. Some patients do not tolerate venesection well and therefore may be restricted to a half unit or fortnightly removal instead. It is not necessary to check the serum ferritin at every visit when undergoing therapeutic venesection, particularly in the early stages when the ferritin level is high and can fluctuate. The haemoglobin level should be maintained above 12 g/dL for men and 11 g/dL for women—if the haemoglobin drops below these values a reduction in venesection frequency would be indicated. The frequency of maintenance phlebotomy depends on age, gender and the degree of initial iron accumulation although a typical interval is 3 months. Patients in the maintenance phase may be able to enrol as blood donors, certainly in the UK. Since October 2012, NHS Blood and Transplant have allowed stable patients with HH, who are otherwise eligible, to donate (up to every 6 weeks if necessary) and since 2018 they have been coded specifically to ensure regular phlebotomy whatever their blood group. This means for the majority of patients in the maintenance phase there is an option to give blood rather than have it discarded through a hospital or community venesection service. Younger homozygotes in the very early stages of iron accumulation should be encouraged to become donors from the start. HH blood donors will require separate measurement of serum ferritin which can be annual once established. Patients should be encouraged to join a patient society such as Haemochromatosis UK which also provides venesection booklets for recording of phlebotomies and laboratory values; the booklets are useful for the patient’s clinician to see how they are progressing.
Venesection treatment prior to onset of cirrhosis or diabetes ensures normal survival, and has been associated with regression of hepatic fibrosis [11]. Interestingly, longitudinal studies have shown that rates of iron accumulation are variable and progressive iron loading does not always occur particularly in females [12]. This begs the question of whether all homozygotes require immediate treatment; asymptomatic pre-menopausal females with normal ferritin and well elderly patients could be observed. In addition, compound heterozygotes should have lifestyle advice offered and venesection only if there is convincing evidence of iron overload, e.g. via MRI or liver biopsy. Of note, HCC can occur in non-cirrhotic patients and despite iron depletion. HH is a relatively uncommon indication for liver transplantation, usually in the context of HCC, and outcomes are comparable with other forms of chronic liver disease.
Some patients do not tolerate venesection at all, although this is quite rare—typical reasons would be anaemia, poor veins or needle phobia. There is some evidence that proton pump inhibitors reduce iron absorption and the need for venesection during the maintenance phase. The once daily oral iron chelator deferasirox has shown reasonable efficacy for therapeutic iron depletion at a dose of 10 mg/kg in a phase 1/2 trial [13]. Novel therapies which interfere with iron homeostasis at a molecular level are in development.
Non-HFE Haemochromatosis
Since the discovery of the HFE gene, several other gene defects have been associated with primary iron overload (Table 12.1). Apart from the distinct phenotype associated with classical ferroportin disease, these other types resemble HFE-related disease though more severe. A number of private mutations in the HFE gene itself have also been identified which in some patients, often in conjunction with C282Y heterozygosity, explain the observed iron overload from a genetic perspective.
Juvenile haemochromatosis (JH) was first described in the late 1970s, is severe and seen typically under the age of 30 affecting both sexes equally. Inheritance is recessive and hypogonadism and cardiomyopathy are usually evident. Heart failure may indeed be life-threatening but salvageable with aggressive iron-chelation therapy. Mutations in the HJV gene on chromosome 1 account for the majority of JH (type 2A), with homozygosity for G320V accounting for half of cases. JH is rarely associated with HAMP gene mutations on chromosome 19 (type 2B). This gene encodes an antimicrobial peptide known as ‘hepcidin’ which is principally synthesised in hepatocytes and acts as an iron regulatory hormone within the body. An intermediate severity form of haemochromatosis is seen with homozygosity for transferrin receptor 2 (TfR2) mutations (type 3), though this can sometimes explain JH [14].
A specific form of iron overload is associated with mutations in the ferroportin (SLC40A1) gene also known as type 4 haemochromatosis. The ferroportin protein controls iron export from a number of cell types including enterocytes and macrophages where it has a role in iron entry from the gut and in iron recycling, respectively. Mutations in SLC40A1 occur similarly in non-Caucasians unlike HFE. The classical disorder is characterised by a raised ferritin with normal or low transferrin saturation and a tendency towards anaemia following venesection. Iron loading occurs predominantly within the reticulo-endothelial system and splenic iron uptake may be observed on MRI. At a microscopic level the distribution of iron in the liver is different to HH with Kupffer cell iron deposition occurring early. The clinical significance of iron loading and the benefit of venesection remain unclear. The differential diagnosis includes hereditary hyperferritinaemia with or without cataracts which requires sequencing of the ferritin light chain (FTL) gene [15].
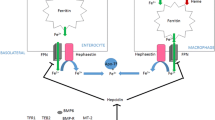
Discovery of additional iron regulatory genes has considerably advanced our understanding of the molecular control of iron homeostasis. Until then, it was known that C282Y abrogates the binding of beta2-microglobulin to HFE thus preventing cell surface expression and interaction with transferrin receptors. This provided a ready explanation for how HFE might interfere with iron entry into cells but did not explain how HFE influences whole body iron control. The hepcidin peptide acts a negative regulator of iron absorption. In the iron deficient state, hepcidin synthesis is reduced in order to stimulate gastrointestinal uptake but in the setting of secondary iron overload, hepcidin expression is increased to suppress iron absorption. Recessive mutations in HFE, HJV and TfR2 all cause paradoxical suppression of hepcidin synthesis with subsequent iron overload (Fig. 12.4). Ferroportin is directly inhibited by hepcidin, preventing release of iron from enterocytes and macrophages [16]. Thus when hepcidin levels are low, in the context of haemochromatosis, ferroportin is readily expressed and releases iron into the circulation from the gut and from macrophages. As hepcidin appears central to the molecular control of iron balance, modulating its activity may represent a future viable therapy for disorders of iron loading. For example, interfering RNAs targeting TMPRSS6 (the gene product of which inhibits hepcidin production by hepatocytes) have been shown to increase hepcidin expression and ameliorate iron overload in mouse models of haemochromatosis.

This schema illustrates how hepcidin synthesis is coordinated by a number of haemochromatosis-related genes in the liver. Hepcidin exerts its downstream effect on ferroportin which is located on the cell surface of macrophages and enterocytes and which is internalised following hepcidin binding. In haemochromatosis hepcidin synthesis is reduced which allows ferroportin to export iron freely into the circulation
Conclusion
The discovery of HFE 25 years ago provided a simple tool for diagnosis and improved our understanding of the natural history of what is the commonest autosomal genetic disorder in Caucasians. HH is entirely preventable but as yet population screening has not been advocated. Therefore, a low index of suspicion for HH in both primary and secondary care is needed for timely diagnosis and further reduction of the morbidity and mortality historically linked with this disorder. Cascade screening for HFE homozygosity is an important consideration after a diagnosis of HH. Although venesection is the mainstay for iron depletion, patients with HH can on the whole donate blood safely and enhance the donor pool for the benefit of others. For those intolerant of venesection alternative therapies are emerging including oral iron chelation and ultimately molecular correction of iron homeostasis. Finally, we are now in an era where patients with unexplained iron overload can be diagnosed via an exome or indeed whole genome panel approach.
Answers
-
1.
(c) The initial step is to check the transferrin saturation (preferably fasting). If this is raised, then an HFE gene test should be performed. A raised ferritin with normal transferrin saturation might relate to obesity/insulin resistance for example.
-
2.
(b) and (d) Given the patient has a raised ferritin and is symptomatic, therapeutic venesection should be initiated. First degree relatives require HFE genotyping in order to identify homozygotes. Serum ferritin is reasonable to check at the same time but gene testing is the priority.
References
Niederau C, Fischer R, Sonnenberg A, et al. Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N Engl J Med. 1985;313:1256–62.
Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399–408.
Merryweather-Clarke AT, Pointon JJ, Shearman JD, Robson KJ. Global prevalence of putative haemochromatosis mutations. J Med Genet. 1997;34:275–8.
Pilling LC, Tamosauskaite J, Jones G, et al. Common conditions associated with hereditary haemochromatosis genetic variants: cohort study in UK Biobank. BMJ. 2019;364:k5222.
Buch S, Sharma A, Ryan E, et al. Variants in PCSK7, PNPLA3 and TM6SF2 are risk factors for the development of cirrhosis in hereditary haemochromatosis. Aliment Pharmacol Ther. 2021;53(7):830–43.
EASL Clinical Practice Guidelines on haemochromatosis. European association for the study of the liver. J Hepatol. 2022;77:479–502.
Guyader D, Jazquelinet C, Moirand R, et al. Noninvasive prediction of fibrosis in C282Y homozygous hemochromatosis. Gastroenterology. 1998;115:929–36.
Legros L, Bardou-Jacquet E, Latournerie M, et al. Non-invasive assessment of liver fibrosis in C282Y homoyzgous HFE hemochromatosis. Liver Int. 2015;35:1731–8.
Bardo-Jacquet E, Laine F, Guggenbuhi P, et al. Worse outcomes of patients with HFE hemochromatosis with persistent increases in transferrin saturation during maintenance therapy. Clin Gastroenterol Hepatol. 2017;15:1620–7.
Fitzsimons EJ, Cullis JO, Thomas DW, et al. Diagnosis and therapy of genetic haemochromatosis (review and 2017 update). Br J Haematol. 2018;181:293–303.
Falize L, Guillygomarc'h A, Perrin M, et al. Reversibility of hepatic fibrosis in treated genetic hemochromatosis: a study of 36 cases. Hepatology. 2006;44:472–7.
Gurrin LC, Osborne NJ, Constantine CC, et al. The natural history of serum iron indices for HFE C282Y homozygosity associated with hereditary hemochromatosis. Gastroenterology. 2008;135:1945–52.
Phatak P, Brissot P, Wurster M, et al. A phase 1/2, dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary hemochromatosis. Hepatology. 2010;52:1671–779.
Griffiths WJH, Besser M, Bowden DJ, Kelly DA. Juvenile haemochromatosis. Lancet Child Adolesc Health. 2021;5:524–30.
Bhuva M, Sen S, Elsey T, et al. Sequence analysis of exon 1 of the ferritin light chain (FTL) gene can reveal the rare disorder ‘hereditary hyperferritinaemia without cataracts’. Br J Haematol. 2019;184:1037–40.
Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–3.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Griffiths, W.J.H. (2022). Hereditary Haemochromatosis. In: Cross, T. (eds) Liver Disease in Clinical Practice. In Clinical Practice. Springer, Cham. https://doi.org/10.1007/978-3-031-10012-3_12
Download citation
DOI: https://doi.org/10.1007/978-3-031-10012-3_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-10011-6
Online ISBN: 978-3-031-10012-3
eBook Packages: MedicineMedicine (R0)