
Abstract
In recent years, mRNA has become an appealing platform for the development of therapeutic agents both for the prevention and treatment of cancer. Efficient delivery of mRNA into target cells is crucial for fully harnessing its therapeutic potential. However, mRNA possesses structural limitations, including its net negative charge and hydrophilicity, that impede its efficient cellular uptake. Likewise, mRNA is characterized by an intrinsic fragility, resulting in it being a highly instable molecule. Lipid nanoparticles (LNPs) have been successfully used for protecting and delivering mRNA encoding for various therapeutic proteins. This chapter is intended to give a comprehensive overview of the current approaches for mRNA synthesis and LNPs manufacturing. We provide an in-depth analysis of how mRNA technology is revolutionizing the area of cancer immunotherapy, critically reviewing the major fields of application of nanoformulated-mRNA medications and addressing the advantages and drawbacks of each one. Finally, we offer a wide landscape of future possibilities and remaining issues of current mRNA-based therapies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
Messenger RNA (mRNA) is a subtype of RNA containing the genetic information necessary to produce a specific protein. The synthesis of mRNA occurs in the cellular nucleus through a process called transcription whereby genomic DNA serves as a template (Bentley 2014; Cramer 2019). Mature mRNA that has been subjected to splicing and post-transcriptional modifications is then exported to the cytosolic compartment where its translation begins upon binding with the ribosomal subunits (Cramer 2019).
Since its discovery in 1960, different attempts have been made to exploit mRNA as a therapeutic platform for the development of gene-based therapies (Persano et al. 2017; Stadler et al. 2017; Thran et al. 2017; Guevara 2019a; Kong et al. 2019; Rybakova et al. 2019; Wei et al. 2020). mRNA-mediated transfection represents an appealing alternative to more conventional plasmid DNA (pDNA)-based strategies for inducing exogenous gene expression. This is because mRNA technology offers relevant advantages including superior transfection efficiency, especially in non-dividing and hard to transfect cells, without the risks of insertional mutagenesis (Leonhardt et al. 2014; Guevara 2019b). Indeed, unlike pDNA, mRNA does not need to enter the nucleus of the target cells to exert its function; it is sufficient that it reaches the cytosolic compartment for translation to occurs (Leonhardt et al. 2014; Andreev et al. 2016). Once mRNA has completed its function, it is rapidly degraded thus promoting only a transient expression of the protein of interest, which is convenient for safer and efficient therapeutic approaches (Huch and Nissan 2014).
However, mRNA’s application as a therapeutic molecule was limited until the second half of the last decade due to the several limitations inherent to its high fragility, poor ability to enter cells because of its net negative charge, and high immunogenicity (Karikó et al. 2008; Guevara 2019b; Bidram et al. 2021). Although the intrinsic adjuvanticity of mRNA, ascribable to the interaction with innate immune receptors, has been exploited to enhance the efficacy of mRNA-based vaccines (Kranz et al. 2016), extensive efforts have been dedicated to reducing the immunogenicity and improving the stability of mRNA molecules by incorporating chemically modified nucleotides and regulatory elements (Karikó et al. 2008; Anderson et al. 2010; Nance and Meier 2021).
Despite modified nucleotides were demonstrated to minimize the susceptibility to degradation by ribonucleases and improve the translatability of “naked” mRNA, effective delivery remained the principal obstacle for ensuring adequate production of exogenous proteins upon systemic or local mRNA administration (Pardi et al. 2018). Recent advances in non-viral mRNA delivery technologies have broadened the application of the mRNA technology in preclinical and clinical settings (Persano et al. 2017; Stadler et al. 2017; Thran et al. 2017; Guevara 2019a; Kong et al. 2019; Rybakova et al. 2019; Wei et al. 2020). A wide range of nanosized platforms has been investigated as mRNA delivery systems, such as polymeric, peptide-based, and lipid-based nanoparticles (McKinlay et al. 2017; Lou et al. 2019; Qiu et al. 2019; Kaczmarek et al. 2016; Sago et al. 2018; Veiga et al. 2018). Among these non-viral vehicles, lipid-based formulations represent the most advanced platform that has been successfully employed for in vivo mRNA delivery (Guevara et al. 2020; Pilkington et al. 2021; Schoenmaker et al. 2021).
The ongoing COVID-19 pandemic has further accelerated the development of mRNA technology, with two mRNA-based vaccines, BNT162b2 (BioNTech) and mRNA-1273 (Moderna), granted with the first historic authorization for clinical use by the Food and Drug Administration (FDA) and European Medicines Agency (EMA), while another mRNA vaccine, CVnCoV (CureVac), is currently in phase 3 clinical trials (Risma et al. 2021; Uddin and Roni 2021). This success has drawn the interest of several pharmaceutical companies and research groups in acquiring the necessary capabilities to set up the manufacturing of nanoformulated mRNA-based therapies not only for cancer immunotherapy but also for other purposes (Martin and Lowery 2020; Dolgin 2021a, b). Currently, dozens of mRNA-based therapeutics are in preclinical and clinical phases of development, with promising outcomes in diverse types of cancers (Table 1).
In this chapter, we summarize the current methods utilized for mRNA preparation and synthesis, and the progress that has been made to increase the structural stability and translation efficiency of synthetic mRNA. We describe the preparation of lipid-based nanoparticles by standard nanoprecipitation technique for mRNA encapsulation, and the major formulation parameters that can affect its ability to induce transgene expression. Finally, we critically evaluate the different forms of RNA-based therapies that have been proposed.
2 Structure, Synthesis, and Purification of in Vitro Transcribed (IVT) mRNA
2.1 Structural Organization of IVT-mRNA
Synthetic mRNAs can be classified mainly in two types, the non-replicating (or non-amplifying) and the virus-derived self-amplifying RNA (saRNA) (or replicon).
The minimum structure of conventional non-replicating in vitro transcribed (IVT) mRNA consists of all those elements present in mature eukaryotic mRNA, including an open reading frame (ORF) region that encodes the desired protein, 5′- and 3′-untranslated regions (UTRs), and 5′ cap and 3′ poly(A) tail (Fig. 1) (Chaudhary et al. 2021).

Schematic illustration of the structural organization of non-replicating mRNA (a) and self-amplifying RNA platforms (b). Non-replicating mRNA is composed of a cap structure (m7GpppN, where N can be any nucleotide), the 5′-UTR, an open ORF encoding a gene of interest, the 3′UTR, and a tail of 30–120 adenosine residues (poly(A) tail) (a). Self-amplifying RNA derives from an alphavirus genome and includes a 5′cap, nonstructural genes (nsP1–4), 26S subgenomic promoter (open arrow), the ORF encoding the desired protein, the 3′-UTR, and a poly(A) (b)
In contrast, saRNA derives from the genome backbone of an alphavirus, like the venezuelan equine encephalitis (VEE) virus, in which structural genes have been replaced by a transgene encoding the therapeutic protein of interest, whereas the genes responsible for RNA replication are maintained to preserve the auto-replicative capacity of the virus (Fig. 1) (Blakney et al. 2021). SaRNA is advantageous compared to conventional IVT-mRNA as it retains all the benefits of mRNA technology, such as rapid synthesis, transient activity, and suitability for customization, but the therapeutic effect can be achieved with a lower dose of RNA thanks to its self-replicative capacity (Vogel et al. 2018).
In the following sections, we will describe the role of the different regulatory elements in IVT-mRNA, how its sequence organization and composition can be optimized to maximize its therapeutic performances, and provide an overview of the preparation of synthetic mRNA by in vitro transcription.
2.1.1 5′ and 3′ Untranslated Regions (UTRs)
The UTR regions are non-coding elements located at the 5′ and 3′ ends of a mature mRNA (Leppek et al. 2018; Mignone et al. 2002). UTR sequences play a critical role in multiple processes conducting to mRNA translation into protein, including transport of mRNA from the nucleus to the cytosol, the assembly of the translation machinery, and mRNA decay (Leppek et al. 2018; Rabani et al. 2017; Mignone et al. 2002; Schuster and Hsieh 2019). Secondary structures (hairpins) in the UTRs are the major determinants of its regulatory function (Mignone et al. 2002). In UTRs of mRNA encoding proteins poorly expressed under normal conditions, hairpin structures are particularly abundant and form stable interactions with an average free energy of less than −50 kcal/mol (Mignone et al. 2002; Babendure et al. 2006). Secondary structures positioned in the proximity of the cap structure are more effective at inhibiting translation initiation; indeed, free energy of −30 kcal/mol is sufficient to impede the access of the 43S preinitiation complex, composed of the small ribosomal subunit (40S) bound by the initiation factors eIF1, eIF1A, eIF3, and the eIF2-Met-tRNAiMet-GTP ternary complex (eIF2-TC) (Mignone et al. 2002; Babendure et al. 2006; Pestova et al. 2007). On the other hand, secondary structures situated closer to the starting codon require a free energy higher than −50 kcal/mol to be able to inhibit translation (Mignone et al. 2002; Babendure et al. 2006). The preinitiation complex scans along the 5′-UTR until it encounters the AUG start codon; afterward, the larger 60S subunit joins the 40S to form an 80S initiation complex, and protein synthesis begins. This mechanism is known as cap-dependent translation and describes the initiation of mRNA translation in most organisms (Hinnebusch and Lorsch 2012). Some viral RNAs use a cap-independent mechanism for initiating translation, which involves an internal ribosomal entry site (IRES) able to attract ribosomal subunits independently of the cap structure (Martinez-Salas et al. 2018; Zhao et al. 2020).
The UTR sequences employed in therapeutic IVT-mRNA are retrieved from specific databases (i.e., http://utrdb.ba.itb.cnr.it/) or from coding sequences (CDS) available in sequence banks (e.g., https://www.ncbi.nlm.nih.gov/nucleotide/). Those from human α- and β-globin probably represent the most studied and characterized UTRs (Babendure et al. 2006).
Length is a critical parameter for the 5′-UTR, and an optimized sequence should not exceed 70 nt and could include the Kozak consensus sequence (GCC-(A/G)-CCAUGG) immediately upstream of the translation start codon (AUG) to enhance translation from the correct initiation codon (Asrani et al. 2018). Importantly, the start codon AUG within 5′ UTR is excluded to prevent alternative translation initiation and mutation of the amino acid sequence. In addition, secondary structure elements in the 5′ UTR region of the mRNA should be minimized to reduce the energy barrier for the scanning ribosome to reach the start codon.
The 3′-UTR has a great effect on mRNA’s stability; indeed, its optimization results in increased mRNA half-life (Holtkamp et al. 2006; Wang et al. 1999). Accordingly, the duration of mRNA expression can be regulated by varying its composition. The introduction of AU-rich elements in the 3′-UTR causes mRNA destabilization, leading to rapid mRNA decay thus shortening protein expression, while mRNAs with enriched CG-content in the 3′-UTR sequence exhibit increased stability and translation efficiency. Likewise, the proper combination of 5′- and 3′-UTR sequences can enhance the translation efficiency (Ferizi et al. 2016).
2.1.2 The Function of the Cap Structure
The introduction of a 5′ end cap is a conserved post-transcriptional modification of eukaryotic mRNAs (Ramanathan et al. 2016). mRNA molecules are capped with a 7-methylguanosine (m7G) connected by a 5′-to-5′ triphosphate bridge to the first nucleotide to form a cap 0 structure (m7GpppN). Cap 0 regulates the translation of mRNA by preventing its degradation and facilitating the assembly of the translation machinery (Ramanathan et al. 2016). In mammals, the first transcribed nucleotide is methylated in the 2′ ribose position to form a cap 1 structure (m7GpppN2′Om) and, in approximately 50% of transcripts, also the second transcribed nucleotide is 2′ O-methylated in the 2′ ribose position to form cap 2 (m7GpppN2′OmN2′Om) (Ramanathan et al. 2016).
While cap 1 structure is ubiquitously expressed in humans, the expression of cap 2 is restricted to specific tissue types, such as in striated muscles and at lower levels in brain, testes, lung, liver, and skin tissues. The function of cap structures remains largely unknown, but they are known to be involved in modulating nuclear export, splicing, turnover, translation efficiency, and decapping of mRNAs (Galloway and Cowling 2019). Cap 1 is important for self/non-self-discrimination, by preventing the recognition by interferon (IFN)-induced proteins with tetratricopeptide repeats (IFITs) or pattern recognition receptors (PRRs) (Galloway and Cowling 2019). IFIT proteins recognize non-methylated cap structures, like cap 0, and mRNA molecules with 5′-triphosphate or 5′-monophosphate ends. IFIT-1 sequesters non-methylated mRNA from the translational machinery by competing with EIF4E proteins for the binding to the cap structure (Galloway and Cowling 2019).
The importance of cap structures in preventing mRNA recognition by the innate immune system has been highlighted by the observation that cytoplasmic viruses often possess cap 1 structures and that the deletion of the viral methyltransferase responsible for the conversion of cap 0 into cap 1 resulted in viral attenuation (Bouvet et al. 2010). Retinoic acid-inducible gene 1(RIG-I) and melanoma differentiation-associated protein 5 (MDA5) are responsible for the cytoplasmic recognition of double-stranded RNA (dsRNA) with 5′ppp and cap 0 ends, and their activation induce the expression of type I IFNs (IFN-I) (IFN-α and IFN-β) and other pro-inflammatory cytokines (Ramanathan et al. 2016; Galloway and Cowling 2019). Cap 1 modification abrogates both RIG-I and MDA5 recognition of dsRNA, preventing innate immune activation. Therefore, methylation of the first transcribed nucleotide is thought to be a molecular signature that discriminates self and non-self mRNA.
Several synthetic cap analogs have been developed for the capping of IVT-mRNA (Jemielity et al. 2010; Tang et al. 2019). The anti-reverse cap analog (ARCA) is widely used for the preparation of synthetic capped mRNA. ARCA possess a cap 0 structure with a 3′-O-methyl group on the sugar adjacent to the m7G (m7,3′-OGpppG), which prevents it from incorporating in the incorrect orientation (Tang et al. 2019; Warminski et al. 2017). The variety of cap structures has been recently expanded following the observations that up to 30% of caps in animals and viral mRNAs are also methylated at the first encoded nucleotide adjacent to the 7-mG cap to obtain N6-methyladenosine (m6A) or N6,2′-O-dimethyladenosine (m6Am). In addition, multiple methylations also occur in the 5′ G cap (e.g., m2,2,7GpppN) in viral RNAs and a subset of RNAP II-transcribed cellular RNAs (Warminski et al. 2017).
2.1.3 Role of the Poly(A) Tail
The poly(A) tail is a structure characteristic of mature mRNAs that plays a significant role in mRNA translation (Weill et al. 2012). It has been shown that a gradual increase in the poly(A) tail length of IVT-mRNA to 120 bases leads to increased translation, whereas shortening of the poly(A) sequence results in faster mRNA decay (patent WO 2017/059902 Al). A further increase in the poly(A) tail size beyond 120 residues does not enhance the translation efficiency.
The poly(A) tail is bound with high affinity by the poly(A) RNA binding proteins (PABPs), which interact with eIF4G and eIF4B to promote the circularization of the mRNA molecule and ribosomal recruitment to form a polyribosome complex (Weill et al. 2012). A sufficiently long poly(A) tail is necessary to ensure the circularization of mRNA via binding of PABPs to the poly(A) tail and the cap. The minimal length of poly(A) tail required for mRNA’s stability has been determined to be 30 nt, which corresponds to the reported 25–30 nt footprint for a single PABP (Lima et al. 2017).
2.1.4 Modified Nucleotides
mRNA suffers from several limitations which impeded its use for a long time. In particular, limited stability and high immunogenicity were the most relevant issues limiting the therapeutic application of IVT-mRNA.
Chemically modified nucleotides are known to be present at low abundance in non-synthetic mRNAs (McCown et al. 2020). IVT-mRNAs incorporating modified nucleotides, commonly uridine, are termed modified mRNAs (modRNAs), while unmodified mRNAs (unmodRNA) do not contain chemically modified nucleotides. The most frequent naturally modified nucleotides are pseudouridine (Ψ), 5-methylcytidine (m5C), N6-methyladenosine (m6A), 5-methyluridine (m5U), and 2-thiouridine (S2U). The presence of modified nucleotides in the mRNA prevents its recognition as a foreign molecule by endosomal sensors, such as Toll-like receptors (TLRs) 3, TLR7, and TLR8 or cytoplasmic sensors, such as RIG-I and MDA5, responsible for the induction of type I IFNs, typically associated with antiviral responses (Karikó et al. 2005; Nelson et al. 2020). The activation of type I IFN signaling pathways causes the suppression of mRNA translation and its degradation, and it can even induce host cell death via apoptosis (Nelson et al. 2020; Palchetti et al. 2015).
It has been reported that the incorporation of modified bases in the mRNA sequence reduces innate immune activation, thus improving its translation and activity (Karikó et al. 2005, 2008). The replacement of uridine with Ψ is the predominant modification employed in the preparation of synthetic mRNA. Its incorporation into mRNA has shown to increase the resistance to RNase degradation and to limit TLR activation, with a consequent improvement of its translatability and transfection efficiency both in vitro and in vivo (Anderson et al. 2011; Svitkin 2017; Roy 2021).
Nowadays, many types of modified bases have been developed, including m5C, N1-methylpseudouridine (m1Ψ), and Ψ, and the impact of these modified nucleotides on mRNA’s activity can be sequence-dependent and cell-type-dependent. In this regard, particularly interesting has been a study in which transfection of THP-1 macrophages with m5C/Ψ-modified mRNA encoding firefly luciferase (Fluc mRNA) resulted in a higher translation rate compared to unmodified Fluc mRNA, while the incorporation of m5C/Ψ modified nucleotide into mRNA encoding enhanced green fluorescence protein (eGFP mRNA) caused a decrease in protein production (Li et al. 2016). The authors also showed that transfection with m5C/Ψ-modified mRNA generated a significantly higher expression of Fluc in THP-1 cells than in hepatocellular carcinoma Hep 3B cells.
2.1.5 Codon Optimization of the ORF
Codon optimization relies on the degeneracy of the genetic code, according to which different codons code for the same single amino acid (Mauro and Chappell 2014). The optimization of the ORF sequence is intended to replace codons with low levels of charged tRNAs with codons recognized by abundant tRNAs, so that the exogenous mRNA can be translated with higher efficiency without causing modifications to the amino acid sequence (Mauro and Chappell 2014). Using luciferase and erythropoietin coding mRNAs as model, it has been demonstrated that codon usage optimization of the ORF improves the translation rate and consequently the activity of IVT-mRNA (Thess et al. 2015). The authors found that unmodRNA incorporating codons rich in guanosine and cytosine induced higher systemic levels of erythropoietin and stronger physiological effects compared to Ψ-modified mRNA. However, in some cases high translation rate of mRNA is not desired since some proteins require a slower translation to correctly fold into biologically active forms, and the inclusion in the ORF of codons with low frequency ensures the generation of protein products of higher quality (Brule and Grayhack 2017).
In conclusion, specific codon optimization strategies should be applied depending on the type of protein encoding by the ORF sequence to improve mRNA translation rate and concomitantly ensure optimal protein expression levels.
2.2 Synthesis of IVT-mRNA
IVT-mRNA compared to more traditional gene therapy platforms, such as viral vectors and pDNA, presents the advantage that its production requires simple procedures that can be easily engineered to the required scale. Besides, once the manufacturing process is established, in principle it can be applied for any RNA sequence with essentially no size limitations. mRNA is synthesized in a cell-free system by in vitro transcription (Henderson et al. 2021), which does not foresee the use of hosts like bacteria, yeast, or mammalian cells, thus preventing the associated quality and safety concerns in the production (Fig. 2). Additionally, this host-free system allows to avoid complicated downstream purification procedures, consenting a rapid scale-up and cost-effective manufacturing. In vitro transcription utilizes a linearized pDNA or a PCR product containing a bacteriophage promoter (i.e., T7, T3 or SP6) as template for a bacteriophage DNA-dependent RNA polymerase, which recognizes the promoter within the DNA template and catalyzes the de novo synthesis of mRNA in the presence of ribonucleoside triphosphates (rNTPs) (Henderson et al. 2021). The mRNA sequence can be tailored to meet specific needs, concerning stability and translation efficiency, which can be modulated or enhanced by including further cis-acting elements such as 5′-cap structure and signal peptide (SP) or GSG linker, internal ribosome entry site (IRES) and 2A peptide sequences in multicistronic mRNAs (Mignone et al. 2002; Chng et al. 2015). Furthermore, the sequence of IVT-mRNAs can be optimized by changing the codon composition, and if needed, modified nucleotides can be inserted to improve its translatability and stability (Li et al. 2016; Thess et al. 2015). In vitro transcription has been commonly used for synthesizing both non-replicating mRNA and saRNA (Henderson et al. 2021; McKay et al. 2020).

Schematic illustration of the mRNA production process. The manufacturing of mRNA involves the preparation of a DNA template by PCR or pDNA linearization followed by a cell-free enzymatic in vitro transcription reaction. After synthesis, the mRNA is purified, concentrated, and diafiltered
The first step of in vitro RNA synthesis consists in the design of the DNA template containing a protein-encoding open reading frame (ORF) flanked by regulatory sequences (Henderson et al. 2021). Both pDNA and PCR products can be employed as a template for transcription. In a minimum composition, a pDNA must contain typical elements, such as a promoter sequence, an ORF sequence, 5′/3′-UTR sequences, a poly(A) tail, unique restriction endonuclease sites, a bacterial origin of replication (ori), and an antibiotic resistance gene (Avci-Adali et al. 2014). The ori and selectable marker in the vector backbone allow replication and selection of the plasmid in bacteria, potentially facilitating the establishment of a pDNA template bank.
In the case pDNA is utilized as template, purification, normally by chloroform/phenol extraction followed by ethanol precipitation (Dowhan 2012), and DNA linearization by digestion with a restriction enzyme producing blunt or 5′-overhang ends upstream the promoter sequence, are required steps (Henderson et al. 2021). PCR products for in vitro transcription are generated using a primer containing the desired bacteriophage promoter sequence (Henderson et al. 2021).
The RNA polymerase initiates transcription of the DNA template in the presence of natural rNTPs or with chemically modified rNTPs (5-methoxy-UTP, pseudo-UTP, etc.) to produce several copies of RNA molecules. The DNA template can be removed from the mRNA preparation by treatment with DNase (Henderson et al. 2021).
Functional RNA molecules require a cap structure at the 5′ end and a poly(A) tail at the 3′ end. Regarding the capping of the IVT-mRNA, it can be achieved by two different approaches, enzymatic capping, using vaccinia virus-derived enzymes, or a co-transcriptional method (Henderson et al. 2021; Muttach et al. 2017).
The enzymatic method utilizes a 2′-O-methyltransferase, which consents the generation of a cap 1 structure with a potential capping yield of 100%. However, the enzymatic method has exhibited some drawbacks that have recently limited its use, including a high variation in the capping efficiency, the requirement of an unstable temperature-labile cofactor (S-Adenosylmethionine), and high scale-up costs. For all these reasons, co-transcriptional methods are currently preferred for mRNA capping (Henderson et al. 2021).
In the co-transcriptional method, cap analogs are incorporated directly at the 5′-end of the IVT-mRNA by RNA polymerases, and erroneous internal incorporation of cap analogs during mRNA polymerization cannot occur since cap analogs lack a free 5′-triphosphate. The cap structure m7GpppG represents the most largely employed cap analog (Muttach et al. 2017; Kocmik et al. 2018), but many other alternative cap analogs have also demonstrated good compatibility with commonly employed T7 bacteriophage RNA polymerase. A major limitation of this first generation co-transcriptional capping is that cap analogs compete with GTP as initiator nucleotide and, therefore, the method exhibits variable capping efficiency and reproducibility (Muttach et al. 2017; Kocmik et al. 2018). Additionally, the use of this first generation of cap analogs as nucleotide initiators can cause cap incorporation in reverse directions, due to the presence of a 3′-OH group on m7G; thus, up to one half of the mRNA contains the cap in the wrong orientation and it is not translatable (Muttach et al. 2017; Kocmik et al. 2018).
This problem was solved by developing a second generation of cap structures named anti-reverse cap analogs (ARCA) with a methylated or deoxygenated 3′-OH group at the N7-methylguanosine ribose (m7,3′-OGpppG or m7,3′-dGpppG) (Kocmik et al. 2018). This prevents elongation at the “wrong” 3′-OH, avoiding the incorporation of ARCA analogs in the reverse orientation. Nevertheless, ARCA capping has the disadvantage that only cap 0 mRNAs can be prepared, and the cap structure is characterized by the presence of an unnatural 3′-O-methyl group that could be promptly recognized by innate immune receptors. The introduction of the ARCA cap analog is ensured by conducting the transcription reaction using an excess of ARCA over GTP (ARCA:GTP ratio of 4:1) (Kocmik et al. 2018; Liu et al. 2019). However, even if working at optimized conditions, the capping efficiency rarely exceeds 80%, which means that at least 20% of IVT-mRNA possesses uncapped 5′-triphosphate ends, thus requiring additional purification steps. Due to the strict preference of bacteriophage RNA polymerases for G or A, depending on the promotor, artificial mRNAs starting with a U or C at the 5′-end cannot be prepared using in vitro transcription (Henderson et al. 2021).
Trilink BioTechnologies, a US biotech company, has recently developed CleanCap® mRNA, a third generation of co-transcriptional capping technology (Henderson et al. 2021). CleanCap® results in the incorporation of N6-methyladenosine methylated cap (m6A or m6Am) at the 5′-end. This method offers important technical advantages compared to previously proposed cap analogs, since it displays superior efficiency and reproducibility (94–99% of complete capping), yields a natural unmodified cap structure (cap 1) with reduced immunogenicity, possesses a greater cost-effectiveness than enzymatic capping, and is easily scalable for large-scale manufacturing (Henderson et al. 2021).
The addition of a poly(A) tail to the synthetic mRNA can be performed either post-transcriptionally using a poly(A) polymerase or co-transcriptionally by including a poly-A tail in the DNA template. While a poly(A) tail with a maximum length of 80 nt can be incorporated into the pDNA, since longer poly(A) sequences can give stability problems due to recombination events that may occur in the host bacteria, by using the poly(A) polymerase no inherent length limitation of poly(A) tail synthesis has been found (Chaudhary et al. 2021). However, the enzymatic incorporation of poly(A) does not consent a high reproducibility, and consequently, the poly(A) products display a larger and more variable size distribution compared to those obtained using a co-transcriptional technique. A solution to address this stability issue is to include a short linker sequence in the poly(A) tail (Trepotec et al. 2019). In addition, enzymatic polyadenylation is less affordable and inadequate for scaled-up manufacturing.
Thanks to the enormous progress in the field, 5′-capped and 3′-poly-adenylated mRNAs can be easily produced in “one-pot” reaction, achieving cap and poly(A) tail additions concomitantly during the in vitro transcription of synthetic mRNA (Henderson et al. 2021).
2.3 Purification of Synthetic mRNA
IVT-mRNA purification is required to ensure the removal of contaminants that may affect the therapeutic performance of the mRNA molecules. This includes the residual DNA template, unincorporated rNTPs and cap analogs, enzymes employed in the reaction, truncated mRNA products, and double-stranded mRNA. The method chosen for purification depends on the length and abundance of the IVT-mRNA, the type of impurities (nucleic acids and/or proteins), and the type of downstream application.
Standard purification methods include lithium chloride (LiCl) precipitation, alcohol-based precipitation (i.e., ethanol precipitation), and techniques based on silica membranes (i.e., spin columns) (Henderson et al. 2021; Walker and Lorsch 2013). LiCl precipitation is employed to remove the majority of the unincorporated rNTPs and enzymes used for the synthesis of IVT-mRNA. Instead, ethanol precipitation can ensure the complete removal of free rNTPs, salts, and proteins (Walker and Lorsch 2013; Rio et al. 2010). Silica membranes selectively bind nucleic acids, allowing the elimination of salts, free rNTPs, and proteins (Baronti et al. 2018). Such membranes will retain the intact DNA template, and its removal requires pre-treatment with DNase after in vitro transcription.
None of these techniques are effective in the removal of truncated RNA and dsRNA impurities generated from abortive initiation of in vitro mRNA synthesis. Previous studies have identified dsRNA and truncated RNA as the contaminants that mostly affect IVT-mRNA’s translation efficiency, as they trigger innate immunity through their recognition by RNA sensors. To date, the most common method employed to eliminate nucleic acid contaminants from IVT-mRNAs is reversed-phase high-performance liquid chromatography (HPLC) (Karikó et al. 2011). However, this procedure requires extremely toxic solvents and is not suitable for the large-scale production of mRNA. Moreover, reversed-phase HPLC is less efficient at purifying very large molecules of mRNA.
Alternative methods have been proposed for the purification of synthetic mRNA, including ion-exchange chromatography, oligo(dT) affinity chromatography, and other separation techniques that rely on differences in size, such as size exclusion chromatography (SEC) and cross-flow filtration (CFF) (Baiersdörfer et al. 2019).
Recently, a simple, fast, and cost-effective way to eliminate dsRNA contaminants from IVT-mRNA has been reported (Baiersdörfer et al. 2019). This method is based on the selective binding of dsRNA to cellulose in an ethanol-containing buffer. The authors showed that at least 90% of the dsRNA impurities can be removed with a good recovery rate (>65%), independently from the length and nucleoside composition of the IVT-mRNA. The different purification methods can also be combined to improve the purity of IVT-mRNA.
3 Lipid Nanoparticles for Therapeutic mRNA Delivery
The inability of naked mRNA to cross the outer membranes of cells thus to reach the cytoplasmic compartment, together with its lack of stability under physiological conditions, represent the major barriers impeding the complete exploitation of mRNA-based therapies (Guevara et al. 2020).
Various strategies have been proposed for enabling efficient delivery of mRNA into target cells, including chemical modification of mRNA (Zangi et al. 2013), ionic complexation with cationic polymers, physical methods (i.e., electroporation) (Van Tendeloo et al. 2001), and viral vectors (Segel et al. 2021), but so far, lipid nanoparticles (LNPs) have demonstrated the most encouraging results (Persano et al. 2017; Stadler et al. 2017; Thran et al. 2017; Guevara 2019a; Kong et al. 2019; Rybakova et al. 2019; Wei et al. 2020; Kranz et al. 2016). The intense research efforts dedicated toward the development of LNP as mRNA delivery systems recently culminated in the approval of two mRNA-based vaccines for clinical use (Dolgin 2021a, b), and the development of a growing number of clinical trials currently ongoing (Table 1).
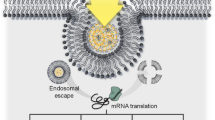
LNPs have shown to efficiently protect mRNA from hydrolysis by RNases and at the same time allow endosomal escape so to achieve mRNA delivery into the cytosol of specific cells (Sago et al. 2018; Cheng et al. 2020). Once in the cytosol, it can be sequestered by the translation machinery for initiating protein synthesis.
Compared to the most popular viral vectors, LNPs are less immunogenic, they can carry larger genetic material payloads and are easier to manufacture. Therefore, even if usually LNPs exhibit lower transfection efficiencies than viral vectors, they are becoming the preferred tool for mRNA transfection.
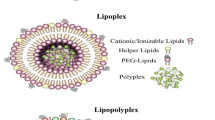
LNPs are self-assembled nanostructures with a size of approximately 100 nm, consisting of different lipid components that can be grouped in three major types: ionizable or permanently cationic lipids, helper lipids, and stealth lipids (i.e., PEGylated lipids) (Fig. 3) (Guevara 2019b; Guevara et al. 2020). Ionizable cationic lipids are usually preferred over permanently charged cationic lipids, as they exhibit higher biocompatibility and efficiency. A great number of screening studies, testing a variety of ionizable lipids composed of different combinations of hydrophilic head groups and non-polar lipid tails, have allowed to increase the number of available lipids (Billingsley et al. 2020; Miao et al. 2020; Guimaraes et al. 2019; Carrasco et al. 2021). These studies showed that the performance of ionizable lipids is controlled by the chemical and structural characteristics of both the head group and lipid tail region. The main feature of ionizable lipids is their ability to respond to an acidic pH, which is usually defined by the pKa value. A single or a mixture of ionizable lipids determines the overall pKa of the LNPs that needs to be around 6.5, as established using the anionic fluorescent dye 2-(p-toluidino)-6-naphthalenesulfonic acid (TNS) binding assay (Guevara et al. 2020; Carrasco et al. 2021). At neutral pH, these lipids are in a zwitterionic form or do not possess any charged functional groups, and only upon internalization in the endosomal compartment of the cell, where the pH is near 5, the head group of the lipids is protonated and assumes a net cationic charge that promotes its interaction with the anionic endogenous endosomal phospholipids. Ionizable lipids with a conical shape are more desirable since this conformation is incompatible with a lipid bilayer organization, thus favoring the destabilization of the endosomal membrane and allowing the mRNA payload to be released into the cytosol (Carrasco et al. 2021). Regarding the lipid tail portion, a series of features like length of the hydrophobic tail, level of unsaturation, and presence of branches have been found to affect dramatically the transfection capability of LNPs (Carrasco et al. 2021).

Schematic of mRNA LNPs. LNPs are composed of four components, such as ionizable lipid (e.g., ALC-0315, SM-102), helper lipid (e.g., DSPC, DOPE), cholesterol, and PEGylated lipid (e.g., PEG-DMG, ALC-0159). The molecules of mRNA within the LNP are confined into aqueous regions
Other components, like helper lipids, are included in the formulation to enhance the stability and delivery efficiency of LNPs, whereas PEGylated lipids are essential to reduce the opsonization of LNPs by serum proteins, which drive their undesired high accumulation in off-target organs (e.g., liver), and rapid clearance from bloodstream (Guevara et al. 2020; Patel et al. 2020). As recently demonstrated, the interaction of LNPs with serum proteins is dictated by mechanisms that are more complicated than it was initially thought and that do not rely only on the net charge of LNPs but involve other factors that still need to be determined and that deserve further attention (Miao et al. 2020).
The relative abundance of ionizable lipid, helper lipid, and stealth lipid critically determines the efficacy of LNPs and therefore needs to be opportunely optimized depending on the application and according to the administration route that is intended to be used (Guevara et al. 2020; Hassett et al. 2019; Ryals et al. 2020; Ndeupen et al. 2021). The number of possible options in the design of LNPs has been further increased due to the recent evidence that points out that different ionizable lipids can synergize in boosting mRNA transfection (Miao et al. 2020). Moreover, even if this has not been proven yet, it is highly plausible that the same synergism might be observed between helper lipids that are considered chemically and functionally equivalent.
In addition to the lipid composition, other features such as size and surface charge are known to have an enormous impact on the behavior of LNPs in vivo, and hence, it is pivotal to appropriately tune these parameters to achieve the desired therapeutic outcomes (Cheng et al. 2020; Ryals et al. 2020; Ndeupen et al. 2021; Nakamura et al. 2020; Hassett et al. 2021). The most common helper lipids that have been tested are 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and cholesterol. DSPC is a phosphatidylcholine with saturated hydrophobic tails and a cylindrical shape that allows DSPC molecules to organize in a lamellar phase, which stabilizes the structure of LNPs (Guevara et al. 2020; Hou et al. 2021). DOPE is a phosphoethanolamine with a cis-unsaturated double bond in the two oleyl fatty acid chains. It exhibits a conical shape and adopts an inverted hexagonal H(II) phase at acidic pH, which destabilizes endosomal membranes and facilitates the endosomal escape of LNPs (Guevara et al. 2020).
Nowadays, solvent injection, also known as nanoprecipitation (or antisolvent precipitation), is the most common technique used for the preparation of mRNA-loaded LNPs (Guevara et al. 2020; Hou 2021). In the last years, the implementation of microfluidic devices for LNP manufacturing has improved the reproducibility and scalability of the method. In these systems, the mixing of an organic solution, containing a mixture of lipids dissolved in an organic solvent (i.e., ethanol), with an aqueous solution, in which the mRNA molecules are dissolved, is realized in microchannels molded on a chip. The channels are designed in a way that the liquids are forced to flow in two separate microchannels and then come into contact at the crossing of channels, thus promoting the self-assembly of lipids into LNPs at the interfacial layer where the lipids are exposed to an environment with increased polarity (Fig. 4) (Guevara et al. 2020).

Schematic of mRNA-loaded LNPs preparation by a microfluidic system with a SHM configuration. One volume of ethanol solution containing a mixture of lipids and three volumes of mRNA dissolved in an acidic aqueous solution are pumped separately into two distinct inlets of the SHM chip by a syringe pump at a flow rate of 2–20 ml/minute. The SHM design favors a rapid mixing of the ethanol and aqueous phases with consequent increases of the net polarity of the lipid solution. Above a certain threshold of polarity, the lipids precipitate as form of LNPs with a size usually ranging from 70 to 100 nm. The ionic interactions taking place between the negative charges of the phosphate groups (P) in mRNA molecules and the positive charges of the amino groups (N) in cationic/ionizable lipids are the force that drives the encapsulation of the mRNA into LNPs
Microfluidic chips can be prepared with different mixing patterns, such as standard T- junction, microfluidic hydrodynamic flow focusing (HFF), microfluidic micromixer (MM), and staggered herringbone micromixer (SHM) (Riewe et al. 2020). The widely utilized NanoAssemblr™ platform employs a Y-shaped architecture incorporating SHM pattern. The channel configuration together with the flow rate are important parameters affecting dramatically the physicochemical characteristics of LNPs (Guevara et al. 2020; Riewe et al. 2020; Roces et al. 2020). LNPs with a smaller size and narrow size distribution are typically produced with a higher flow rate (Roces et al. 2020).
4 mRNA-Based Cancer Immunotherapy: Antitumor Vaccines
Cancer immunotherapy represents the major area of research where mRNA has found application as therapeutic agent. It has been demonstrated that the mRNA technology can be utilized to create different immunotherapeutic products, such as vaccines, monoclonal antibodies, chimeric antigen receptor (CAR) cells, and immunomodulatory proteins (Persano et al. 2017; Stadler et al. 2017; Thran et al. 2017; Guevara 2019a; Kong et al. 2019; Rybakova et al. 2019; Wei et al. 2020; Lai et al. 2018).
Cancer vaccines are the most advanced application of mRNA, with both prophylactic and therapeutic potentials and relying on the capability of mRNA to simultaneously deliver genetic information and act as immunoadjuvant by interacting with innate immune receptors (Persano et al. 2017; Stadler et al. 2017; Kranz et al. 2016; Guimaraes et al. 2019). The immunomodulatory properties of mRNA can be particularly relevant for the development of antitumor vaccines that require overcoming of immune tolerance, a characteristic of many malignancies (Kranz 2016). On the other hand, upon recognition of the RNA by innate immune receptors, IFN-I triggers the expression of interferon-stimulated genes (ISGs), such as IFN-inducible double-stranded RNA-activated protein kinase (PKR), and 2′,5′-oligoadenylate synthetase (OAS), with consequent decreased translation and increased decay of mRNA (De Beuckelaer et al. 2016; Yang and Shah 2020). This could be extremely deleterious for other applications of mRNA, including chimeric antigen receptor (CAR) cell therapy and gene therapy, for which maximization of the expression might be essential for ensuring a therapeutic effect. In these cases, the use of modified nucleosides, such as pseudouridine, N1-methylpseudouridine, and 2-thiouridine, is highly desirable in order to increase the translation efficiency of synthetic mRNAs (Karikó et al. 2008; Svitkin et al. 2017).
Vaccines can be designed to target tumor-associated antigens (TAAs) (overexpressed antigens, tissue differentiation antigens, and tumor germline antigens) or tumor-specific antigens (TSAs) (oncoviral antigens and neoantigens), thus promoting an antitumor response that specifically attacks and destroys cancer cells and achieves a prolonged response and prevention of relapse due to the generation of an immunological memory (Kranz et al. 2016; Hollingsworth and Jansen 2019).
Recently, several studies have pointed out the importance of neoantigens as targets for immunotherapy (Sahin et al. 2017; Cafri et al. 2020; Blass and Ott 2021). The considerable progresses made in sequencing technologies and bioinformatic tools have permitted to reveal that neoantigen-specific CD8+ and CD4+ T cells are detectable in most tumors, independently if they have a viral etiology (Hollingsworth and Jansen 2019; Sahin et al. 2017; Cafri et al. 2020; Blass and Ott 2021). Neoantigens are the consequence of somatic mutations that occur in malignant cells during cancer progression due to the high genomic instability typical of tumors. This class of TSAs is particularly appealing because, unlike TAAs, they are detectable only in cancer cells, therefore not subjected to central tolerance, and are characterized by a high immunogenicity, high affinity toward the MHC, and individual specificity (Hollingsworth and Jansen 2019; Blass and Ott 2021).
The first step in neoantigen identification is the comparison of whole exome sequencing (WES) or mRNA sequencing (mRNA-Seq) data from tumor and normal tissues obtained using high-throughput sequencing techniques (i.e., next-generation sequencing (NGS)) (Blass and Ott 2021; Esprit et al. 2020). Then, the data from the sequencing analysis are analyzed with bioinformatic tools to predict whether the identified mutations can generate tumor neoantigens (Esprit et al. 2020). Most of these bioinformatic softwares are based on HLA binding affinity and tend to ignore other important factors that have an impact on the antigen presentation process, such as the C-terminal cleavage by proteasome, efficiency of transporter associated with antigen processing (TAP)-mediated transport of peptides, expression abundance of neoantigens, tumor heterogeneity, heterogeneity and clonality of neoantigens, and loss of heterozygosity of HLA.
A vaccination platform capable of targeting multiple patient-specific antigens is highly desirable for developing personalized neoantigen vaccines with enhanced immunogenicity. In this regard, synthetic mRNA offers the possibility to easily incorporate multiple neoantigens in a single molecule that can be manufactured with a cost-effective and scalable approach.
Preliminary studies on mRNA-based neoantigen vaccines were conducted using mRNA-electroporated dendritic cells (DCs) (Wilgenhof et al. 2013). However, previous studies have reported that several factors may intervene in limiting the efficacy of DC vaccines including optimal maturation, subset of cells employed, antigen-loading efficiency, and the ability of DCs to migrate to vaccine-draining lymph nodes (Santos and Butterfield 2018). Therefore, currently most of the groups working in the area of mRNA vaccines are switching from DC platforms toward strategies that involve the delivery of neoantigen mRNAs into APCs directly in vivo, thus avoiding DC isolation and manipulation ex vivo.
The first proof of concept that in vivo delivery of tumor antigen-encoding mRNA into APCs is an achievable path for successful antitumor vaccination was reported only few years ago. For the first time, it was shown that DCs can be passively targeted in vivo upon systemic administration of mRNA-carrying lipoplexes displaying a negative net charge (Kranz et al. 2016). Vaccination with mRNA lipoplexes encoding tumor-specific or tumor-associated antigens stimulated strong type I IFN-dependent effector and memory T cell responses resulting in tumor rejection and protection from tumor rechallenge.
In situ vaccination is an alternative form of vaccine effective at eliciting antigen-specific T cell responses. This is achieved using a cytotoxic agent, alone or in combination with an immunoadjuvant, able to induce immunogenic cell death (ICD), which not only directly kills tumor cells, but also promotes the release of tumor antigens and molecular signals that promote the activation of immune cells that are able to reach even distant cancer cells.
The use of mRNA-based therapeutics has also been proposed to induce ICD in tumor cells. For this scope, it has been utilized a mRNA encoding proapoptotic proteins, caspase or PUMA, and including in the 3′-UTR microRNA (miRNA) target sites to minimize the expression of the proteins in healthy hepatocytes, thus preventing side effects due to off-target expression (Jain et al. 2018).
4.1 mRNA-Loaded LNP-Mediated Monoclonal Antibody Delivery
Monoclonal antibodies are emerging as one of the most promising classes of cancer immunotherapy, so much so that several antibodies have received approval for clinical use for the treatment of several forms of cancers (Boyiadzis and Foon 2018; Mullard 2021). Antibodies have been designed to target specific proteins expressed on tumor cells and immune cells or released into the tumor microenvironment. The use of monoclonal antibodies for targeting immune checkpoints and inhibiting their functions (immune checkpoint inhibitors, ICIs) represents one of the most investigated areas of application.
Several types of ICIs have been or are currently under investigation, and many of them have received clinical approval for the treatment of different tumors (Gravbrot et al. 2019). Despite the encouraging outcomes from preclinical and clinical studies, and the fact that many patients have already benefited from the use of monoclonal antibodies, there are still concerns regarding this type of therapeutic agents that limit their wider application in the clinic (de Miguel and Calvo 2020; Palmieri and Carlino 2018; Chames et al. 2009; Hernandez et al. 2018). Major concerns are mainly related to the complex and expensive procedures required for their manufacturing and purification (Chames et al. 2009; Hernandez et al. 2018). Therapeutic antibodies are typically full-size immunoglobulins (Ig), mostly of the IgG type, which require a wide variety of post-translational modifications, including glycosylation, disulfide bond formation, and many other modifications that cannot be introduced synthetically (Jank et al. 2019; Yang and Li 2020; Lu et al. 2020). For this reason, their preparation is commonly realized in mammalian cell lines (Lu et al. 2020; Dangi et al. 2018). Then, a purification step is required to have an injectable antibody therapeutic free from any potential harmful contaminants. Given that these modifications can directly affect the functionality of monoclonal antibodies, it is of essential importance to implement analytical assays that can ensure the quality of the product. All these aspects contribute to the elevated costs of antibody-based treatments and make these therapies poorly affordable. To achieve the synthesis of functional antibodies in procaryotic expression systems, like E. coli, that can enable faster and cheaper production, different types of antibody fragments, such as single-chain variable fragments (scFv), heavy-chain-only VH (VHH) domains, and nanobodies, have been developed (Fig. 5) (Jank et al. 2019). These are smaller than conventional antibodies and lack glycosylation. Another advantage of antibody fragments is that the small size can improve their penetration ability into tissues that are not reachable by conventional full-size antibodies, which is advantageous for many therapeutic applications. The antibody fragment technology has been also used to generate bispecific antibodies, and chimeric antigen receptors (CARs) usually consisting of scFv linked to intracellular signaling molecules, capable of triggering T cell effector activities (Hernandez et al. 2018; Jank e al. 2019; Yang and Li 2020). Yet, antibody fragments are cleared from circulation much more rapidly than conventional full-size antibodies, and since they do not have the Fc domain, fragments are unable to elicit Fc-mediated cytotoxicity (Jank et al. 2019; Yang and Li 2020).

Illustration of the different types of antibodies: monoclonal antibody, camelid heavy-chain antibody (HCAb), antigen-binding fragment (Fab), single-chain fragment variable (scFv), nanobody, bispecific T cell engagers (BiTE), and bispecific killer cells engagers (BiKE)
The use of mRNA-loaded LNPs has recently emerged as a promising approach to overcome current limitations of monoclonal antibodies by consenting the production of a specific antibody directly in the body of the patient, circumventing the complicated and expensive purification steps and thereby avoiding the batch-to-batch variation that can be found when using antibodies (Van Hoecke and Roose 2019). Since IVT-mRNA contains all the instructions for appropriate folding and assembly, and for post-translational modifications, the antibodies generated from exogenous mRNA are perfectly functional. A potential limitation of this strategy is that only antibodies with natural modifications can be produced, and it cannot be employed for delivering antibodies conjugated to synthetic molecules, such as polyethylene glycol (PEG), which can extend the blood circulation half-life of antibody fragments. However, recent studies demonstrated that mRNA-mediated delivery can enhance the serum half-life of both full-size and fragment antibodies which can improve the therapeutic efficacy of these treatments (Rybakova et al. 2019; Tiwari et al. 2018).
The first proof of the feasibility of mRNA-mediated delivery of therapeutic antibodies was reported in 2017 (Pardi et al. 2017). In that study, passive vaccination was achieved by systemic administration of LNPs carrying a modRNA encoding both light and heavy chains of an anti-HIV-1 neutralizing antibody. Similarly, the mRNA technology can be also exploited to obtain in vivo production of bispecific antibodies with two binding domains directed against a tumor antigen and CD3 marker, able to redirect and activate the antitumoral action of circulating T cells (Stadler et al. 2017). A single dose of 3–5 µg of formulated mRNA was sufficient to induce a rapid synthesis of bispecific antibodies and triggered complete eradication of advanced tumors. To obtain a comparable outcome with a recombinant bispecific antibody, it was necessary to give a dose three times higher than that of formulated mRNA. While most of the reported studies have achieved passive immunization through intravenous administration of mRNA-loaded nanoparticles, very recently it has been shown that intramuscular injection of formulated saRNA encoding an anti-Zika virus neutralizing human antibody (ZIKV-117) also induced high antibody titers and protected mice from Zika infection (Erasmus et al. 2018).
Taken together, these studies clearly show the potential of mRNA-loaded delivery systems for in vivo production of therapeutic antibodies, offering in this way a series of advantages compared recombinant antibodies, including reduced costs and prolonged serum half-life, thereby making antibody-based treatments more effective and accessible to a larger portion of patients.
4.2 mRNA-Loaded LNPs for CAR Immune Cell Engineering
CAR cell therapy is considered the most advanced modality of personalized immunotherapy in which immune cells, like T cells or NK cells, are isolated from a patient or a donor, genetically engineered ex vivo and ultimately infused into the patient (Sterner and Sterner 2021). The efficacy of adoptive T cell therapy has been demonstrated by numerous clinical trials showing remarkable outcomes in relapsed or refractory hematologic cancers. These clinical successes have led to the approval of CAR T cell products for children with acute lymphoblastic leukemia and adults with large B cell lymphoma by two of the major regulatory agencies, the FDA and the EMA (Sterner and Sterner 2021; Lin et al. 2021).
Despite these encouraging premises, CAR cell therapies suffer from considerable limitations concerning toxicity, primarily cytokine release syndrome (CRS) and neurologic adverse effects, and the high costs and complex procedures involved in the manufacturing of CAR cell-based treatments (Sterner and Sterner 2021; Lin et al. 2021). Interleukin 6 (IL-6) seems to be the major responsible for CRS since elevated levels of IL-6 have been observed in these patients and in murine models of the disease (Kishimoto 2021). While CRS may be alleviated through the administration of tocilizumab, an anti-IL-6 receptor antibody, costs, and manufacturing challenges may be addressed with the advent of mRNA technology.
Viral transduction and electroporation are the most common techniques for CAR introduction into T cells (Van Hoecke and Roose 2019). However, both strategies present limitations. Viral vectors are associated with limited genetic cargo, safety concerns, and high costs, whereas electroporation can result in reduced viability, aberrant gene expression profile, and relative low transgene expression in the surviving transfected cells (Van Hoecke and Roose 2019). Therefore, alternative strategies for CAR delivery are highly desirable.
In recent years, ionizable lipid nanoparticles encapsulating mRNA encoding CAR have been extensively tested in preclinical studies for their ability to transfect immune effector cells either in vivo or ex vivo. A large screening study has allowed to identify seven distinct formulations capable of enhanced mRNA transfection of Jurkat T cells over lipofectamine (Billingsley et al. 2020). The best performing LNP formulation of these was tested with primary human T cells, displaying a CAR transfection efficiency equivalent to those observed with electroporation, but with a significantly inferior cytotoxicity. The potent killing activity of CAR T cells generated by transfection with mRNA LNPs was proven in a coculture assay with acute lymphoblastic leukemia cells.
Lately, in vivo targeting and transfection of lymphocytes have emerged as a fascinating viable route for simple and cost-effective generation of CAR T cells directly in the body of the patient. On this regard, an injectable LNP formulation functionalized on the surface with anti-CD3 antibody was developed for active targeting and mRNA transfection of circulating T cells, to induce transient expression of CAR or TCR recognizing disease-relevant targets (Smith et al. 2017).
5 Future Prospective and Conclusions
IVT-mRNA has the unprecedented potential to address major challenges of current immunotherapies and offers the basis for the development of innovative cancer therapies. The enormous progress in LNP formulations along with a better understanding of mRNA translation regulation has allowed the development of numerous mRNA-based treatments successfully tested in preclinical settings and currently under investigation in clinical trials.
Although considerable strides have been made in the design and manufacturing of LNP formulated mRNA-based therapies, to leverage the full potential of mRNA technology it is still needed to improve the transfection and targeting efficiency of LNPs and to increase the translatability of mRNA molecules by the engineering of the RNA sequence.
As discussed throughout this chapter, for its adequate activity, eukaryotic mRNA requires five structural elements, the cap structure, poly(A) tail, protein-coding sequence, and 5′ and 3′ UTRs. These elements are pivotal in regulating translation initiation, translation termination, stability, decapping, and post-transcriptional modifications of mRNA. Thus, sequence optimization of IVT-mRNA can maximize the expression of the therapeutic protein in vivo.
In addition, alternative forms of RNA such as saRNA and most recently circular RNA (circRNA) have been proposed to enhance mRNA properties (Holdt et al. 2018; Wesselhoeft et al. 2019). circRNA is particularly appealing since, unlike linear RNA, it has no 5′ cap structure and poly(A) tail, and IRES sequences are harnessed to ensure maximum protein synthesis. Given that circRNA lacks free 5′ end cap and a 3′ poly(A) tail, this kind of RNA resists to exonuclease digestion and, therefore, has a longer half-life than conventional linear mRNA. Thus, the use of circRNA may further revolutionize the mRNA field in the coming years.
From the formulation standpoint, despite the encouraging results from passive targeting approaches by modulating physicochemical properties of LNPs, like charge and size, the ability of LNP to reach certain sites in the body or specific cell populations within organs with not relevant or absent off-target accumulation needs to be significantly improved. Indeed, it is clear from studies performed in small animal models that current LNP formulations generally suffer from low specificity, with the tendency to be sequestered by the reticuloendothelial system (RES) of the liver and spleen, or accumulate in the first draining organs (e.g., lungs) after intravenous administration. In addition, if compared to their viral counterpart, LNP-based non-viral vectors usually exhibit much lower transfection efficiencies.
The transfection efficiency of LNPs could be potentially improved, for example, by the rational design of ionizable lipids with optimized head groups and hydrophobic tails so to increase their ability to promote endosomal escape upon internalization by target cells. The incorporation of well-defined helper lipids into the formulation can also play a crucial role in enhancing the overall transfection efficiency of LNPs. Furthermore, hybrid delivery systems including, for instance, pH-responsive polymers (e.g., β-amino ester), or molecules that are known to enhance mRNA delivery by altering the endocytic pathway, can further enhance the endosomal escape of mRNA.
Selectivity of LNP formulated mRNAs can be improved by modulating the structure of the single lipids included in the formulation and the overall lipid composition of LNPs. For instance, modification of the alkyl length of ionizable lipids leads selective accumulation of mRNA-loaded LNPs in the liver or spleen (Fenton et al. 2018). In another study, the impact of cholesterol derivates on cell selectivity of LNPs was investigated. The results of this study demonstrated that the tropism of LNPs in liver endothelial cells, Kupffer cells, and hepatocytes strictly depends on cholesterol structures (Patel et al. 2020).
Finally, biodegradability and immunogenicity are important aspects that need to be considered throughout the design of novel lipid components. Indeed, biodegradability can promote fast elimination of the LNP components, thus minimizing any potential toxicity effect. mRNA-based medicines can either benefit from the intrinsic immunogenicity of lipids, especially in the case of anti-cancer vaccines, or this immunogenicity can be detrimental, by altering mRNA translation and/or causing undesirable adverse effects.
In summary, mRNA platforms are suitable for the treatment of a wide variety of pathologies since they allow the development of any protein-based therapy. However, despite the great therapeutic potential confirmed in a number of clinical trials with diverse applications, mRNA-loaded LNPs could still benefit from further studies aimed at improving the selectivity, transfection efficiency, and toxicity of LNP formulations and increase mRNA stability and translatability. All this together can ensure the development of next generation of mRNA-based therapies with superior therapeutic properties.
References
Anderson BR, Muramatsu H, Jha BK et al (2011) Nucleoside modifications in RNA limit activation of 2′-5′-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res 39:9329–9338
Anderson BR, Muramatsu H, Nallagatla SR et al (2010) Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res 38:5884–5892
Andreev DE, Terenin IM, Dmitriev SE et al (2016) Pros and cons of pDNA and mRNA transfection to study mRNA translation in mammalian cells. Gene 578:1–6
Asrani KH, Cheng L, Cheng CJ et al (2018) Arginase I mRNA therapy—a novel approach to rescue arginase 1 enzyme deficiency. RNA Biol 15:914–922
Avci-Adali M, Behring A, Steinle H et al (2014) In vitro synthesis of modified mRNA for induction of protein expression in human cells. J vis Exp 93:e51943
Babendure JR, Babendure JL, Ding JH et al (2006) Control of mammalian translation by mRNA structure near caps. RNA 12:851–861
Baiersdörfer M, Boros G, Muramatsu H et al (2019) A facile method for the removal of dsRNA contaminant from in vitro-transcribed mRNA. Mol Ther Nucleic Acids 15:26–35
Baronti L, Karlsson H, Marušič M et al (2018) A guide to large-scale RNA sample preparation. Anal Bioanal Chem 410:3239–3252
Bentley DL (2014) Coupling mRNA processing with transcription in time and space. Nat Rev Genet 15:163–175
Bidram M, Zhao Y, Shebardina NG et al (2021) mRNA-based cancer vaccines: a therapeutic strategy for the treatment of melanoma patients. Vaccines (Basel) 9:1060
Billingsley MM, Singh N, Ravikumar P et al (2020) Ionizable lipid nanoparticle-mediated mRNA delivery for human CAR T cell engineering. Nano Lett 20:1578–1589
Blakney AK, Ip S, Geall AJ (2021) An update on self-amplifying mRNA vaccine development. Vaccines (Basel) 9:97
Blass E, Ott PA (2021) Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol 18:215–229
Bouvet M, Debarnot C, Imbert I et al (2010) In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog 6:e1000863
Boyiadzis M, Foon KA (2018) Approved monoclonal antibodies for cancer therapy. Expert Opin Biol Ther 8:1151–1158
Brule CE, Grayhack EJ (2017) Synonymous codons: choose Wisely for expression. Trends Genet 33(4):283–297
Cafri G, Gartner JJ, Zaks T et al (2020) mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J Clin Invest 130:5976–5988
Carrasco MJ, Alishetty S, Alameh MG et al (2021) Ionization and structural properties of mRNA lipid nanoparticles influence expression in intramuscular and intravascular administration. Commun Biol 4:956
Chames P, Van Regenmortel M, Weiss E et al (2009) Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol 157:220–233
Chaudhary N, Weissman D, Whitehead KA (2021) mRNA vaccines for infectious diseases: principles, delivery and clinical translation. Nat Rev Drug Discov 20:817–838
Cheng Q, Wei T, Farbiak L et al (2020) Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat Nanotechnol 15:313–320
Chng J, Wang T, Nian R et al (2015) Cleavage efficient 2A peptides for high level monoclonal antibody expression in CHO cells. Mabs 7:403–412
Cramer P (2019) Organization and regulation of gene transcription. Nature 573:45–54
Dangi AK, Sinha R, Dwivedi S et al (2018) Cell line techniques and gene editing tools for antibody production: a review. Front Pharmacol 9:630
De Beuckelaer A, Pollard C, Van Lint S et al (2016) Type I interferons interfere with the capacity of mRNA Lipoplex vaccines to elicit cytolytic T cell responses. Mol Ther 24:2012–2020
de Miguel M, Calvo E (2020) Clinical challenges of immune checkpoint inhibitors. Cancer Cell 38:326–333
Dolgin E (2021a) mRNA flu shots move into trials. Nat Rev Drug Discov 20:801–803
Dolgin E (2021b) The tangled history of mRNA vaccines. Nature 597:318–324
Dowhan DH (2012) Purification and concentration of nucleic acids. Curr Protoc Essent Lab Tech 6:5.2.1–5.2.21
Erasmus JH, Khandhar AP, Guderian J et al (2018) A nanostructured lipid carrier for delivery of a replicating viral RNA provides single, low-dose protection against Zika. Mol Ther 26:2507–2522
Esprit A, de Mey W, Bahadur Shahi R et al (2020) Neo-Antigen mRNA Vaccines. Vaccines (Basel) 8:776
Fenton OS, Kauffman KJ, McClellan RL et al (2018) Customizable lipid nanoparticle materials for the delivery of siRNAs and mRNAs. Angew Chem Int Ed Engl 57:13582–13586
Ferizi M, Aneja MK, Balmayor ER et al (2016) Human cellular CYBA UTR sequences increase mRNA translation without affecting the half-life of recombinant RNA transcripts. Sci Rep 6:39149
Galloway A, Cowling VH (2019) mRNA cap regulation in mammalian cell function and fate. Biochim Biophys Acta Gene Regul Mech 1862:270–279
Gravbrot N, Gilbert-Gard K, Mehta P et al (2019) Therapeutic monoclonal antibodies targeting immune checkpoints for the treatment of solid tumors. Antibodies (Basel) 4:51
Guevara ML, Jilesen Z, Stojdl D et al (2019a) Codelivery of mRNA with α-galactosylceramide using a new lipopolyplex formulation induces a strong antitumor response upon intravenous administration. ACS Omega 4:13015–13026
Guevara ML, Persano S, Persano F (2019b) Lipid-based vectors for therapeutic mRNA-based anti-cancer vaccines. Curr Pharm Des 25:1443–1454
Guevara ML, Persano F, Persano S (2020) Advances in lipid nanoparticles for mRNA-based cancer immunotherapy. Front Chem 8:589959
Guimaraes PPG, Zhang R, Spektor R et al (2019) Ionizable lipid nanoparticles encapsulating barcoded mRNA for accelerated in vivo delivery screening. J Control Release 316:404–417
Hassett KJ, Benenato KE, Jacquinet E et al (2019) Optimization of lipid nanoparticles for intramuscular administration of mRNA vaccines. Mol Ther Nucleic Acids 15:1–11
Hassett KJ, Higgins J, Woods A et al (2021) Impact of lipid nanoparticle size on mRNA vaccine immunogenicity. J Control Release 335:237–246
Henderson JM, Ujita A, Hill E et al (2021) Cap 1 messenger RNA synthesis with co-transcriptional CleanCap® Analog by in vitro transcription. Curr Protoc 1:e39
Hernandez I, Bott SW, Patel AS et al (2018) Pricing of monoclonal antibody therapies: higher if used for cancer? Am J Manag Care 24:109–112
Hinnebusch AG, Lorsch JR (2012) The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb Perspect Biol 4:a011544
Holdt LM, Kohlmaier A, Teupser D (2018) Circular RNAs as therapeutic agents and targets. Front Physiol 9:1262
Hollingsworth RE, Jansen K (2019) Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 4:7
Holtkamp S, Kreiter S, Selmi A et al (2006) Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 108:4009–4017
Hou X, Zaks T, Langer R et al (2021) Lipid nanoparticles for mRNA delivery. Nat Rev Mater 10:1–17
Huch S, Nissan T (2014) Interrelations between translation and general mRNA degradation in yeast. Wiley Interdiscip Rev RNA 5:747–763
Jain R, Frederick JP, Huang EY et al (2018) MicroRNAs enable mRNA therapeutics to selectively program cancer cells to self-destruct. Nucleic Acid Ther 28:285–296
Jank L, Pinto-Espinoza C, Duan Y et al (2019) Current approaches and future perspectives for nanobodies in stroke diagnostic and therapy. Antibodies (Basel) 8:5
Jemielity J, Kowalska J, Rydzika AM et al (2010) Synthetic mRNA cap analogs with a modified triphosphate bridge—synthesis, applications and prospects. New J Chem 34:829–844
Kaczmarek JC, Patel AK, Kauffman KJ et al (2016) Polymer-lipid nanoparticles for systemic delivery of mRNA to the lungs. Angew Chem Int Ed Engl 55:13808–13812
Karikó K, Buckstein M, Ni H et al (2005) Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23:165–175
Karikó K, Muramatsu H, Welsh FA et al (2008) Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther 16:1833–1840
Karikó K, Muramatsu H, Ludwig J et al (2011) Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res 39:e142
Kishimoto T (2021) IL-6: from arthritis to CAR-T-cell therapy and COVID-19. Int Immunol 33:515–519
Kocmik I, Piecyk K, Rudzinska M et al (2018) Modified ARCA analogs providing enhanced translational properties of capped mRNAs. Cell Cycle 17:1624–1636
Kong N, Tao W, Ling X et al (2019) Synthetic mRNA nanoparticle-mediated restoration of p53 tumor suppressor sensitizes p53-deficient cancers to mTOR inhibition. Sci Transl Med 11:eaaw1565
Kranz LM, Diken M, Haas H et al (2016) Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534:396–401
Lai I, Swaminathan S, Baylot V et al (2018) Lipid nanoparticles that deliver IL-12 messenger RNA suppress tumorigenesis in MYC oncogene-driven hepatocellular carcinoma. J Immunother Cancer 6:125
Leppek K, Das R, Barna M (2018) Functional 5’ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Biol 19:673
Leonhardt C, Schwake G, Stögbauer TR et al (2014) Single-cell mRNA transfection studies: delivery, kinetics and statistics by numbers. Nanomedicine 10:679–688
Li B, Luo X, Dong Y (2016) Effects of chemically modified messenger RNA on protein expression. Bioconjug Chem 27:849–853
Lima SA, Chipman LB, Nicholson AL et al (2017) Short poly(A) tails are a conserved feature of highly expressed genes. Nat Struct Mol Biol 24:1057–1063
Lin H, Cheng J, Mu W et al (2021) Advances in universal CAR-T cell therapy. Front Immunol 12:744823
Liu Y, Chin JM, Choo EL et al (2019) Messenger RNA translation enhancement by immune evasion proteins: a comparative study between EKB (vaccinia virus) and NS1 (influenza A virus). Sci Rep 9:11972
Lou B, De Koker S, Lau CYJ et al (2019) mRNA polyplexes with post-conjugated GALA peptides efficiently target, Transfect, and activate antigen presenting cells. Bioconjug Chem 30:461–475
Lu RM, Hwang YC, Liu IJ et al (2020) Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci 27:1
Martin C, Lowery D (2020) mRNA vaccines: intellectual property landscape. Nat Rev Drug Discov 19:578
Martinez-Salas E, Francisco-Velilla R, Fernandez-Chamorro J et al (2018) Insights into structural and mechanistic features of viral IRES elements. Front Microbiol 8:2629
Mauro VP, Chappell SA (2014) A critical analysis of codon optimization in human therapeutics. Trends Mol Med 20:604–613
McCown PJ, Ruszkowska A, Kunkler CN et al (2020) Naturally occurring modified ribonucleosides. Wiley Interdiscip Rev RNA 11:e1595
McKinlay CJ, Vargas JR, Blake TR et al (2017) Charge-altering releasable transporters (CARTs) for the delivery and release of mRNA in living animals. Proc Natl Acad Sci USA 114:E448–E456
McKay PF, Hu K, Blakney AK et al (2020) Self-amplifying RNA SARS-CoV-2 lipid nanoparticle vaccine candidate induces high neutralizing antibody titers in mice. Nat Commun 11:3523
Miao L, Lin J, Huang Y et al (2020) Synergistic lipid compositions for albumin receptor mediated delivery of mRNA to the liver. Nat Commun 11:2424
Mignone F, Gissi C, Liuni S et al (2002) Untranslated regions of mRNAs. Genome Biol 3:REVIEWS0004
Mullard A (2021) FDA approves 100th monoclonal antibody product. Nat Rev Drug Discov 20(7):491–495
Muttach F, Muthmann N, Rentmeister A (2017) Synthetic mRNA capping. Beilstein J Org Chem 13:2819–2832
Nakamura T, Kawai M, Sato Y et al (2020) The effect of size and charge of lipid nanoparticles prepared by microfluidic mixing on their lymph node transitivity and distribution. Mol Pharm 17:944–953
Nance KD, Meier JL (2021) Modifications in an emergency: the role of N1-methylpseudouridine in COVID-19 vaccines. ACS Cent Sci 7:748–756
Ndeupen S, Qin Z, Jacobsen S et al (2021) The mRNA-LNP platform’s lipid nanoparticle component used in preclinical vaccine studies is highly inflammatory. bioRxiv [Preprint] 23:2021.03.04.430128
Nelson J, Sorensen EW, Mintri S et al (2020) Impact of mRNA chemistry and manufacturing process on innate immune activation. Sci Adv 6:eaaz6893
Palchetti S, Starace D, De Cesaris P et al (2015) Transfected poly(I:C) activates different dsRNA receptors, leading to apoptosis or immunoadjuvant response in androgen-independent prostate cancer cells. J Biol Chem 290:5470–5483
Palmieri DJ, Carlino MS (2018) Immune checkpoint inhibitor toxicity. Curr Oncol Rep 20(9):72
Pardi N, Hogan MJ, Naradikian MS et al (2018) Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. J Exp Med 215:1571–1588
Pardi N, Hogan MJ, Pelc RS et al (2017) Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 543:248–251
Patel S, Ashwanikumar N, Robinson E et al (2020) Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA. Nat Commun 11:983
Persano S, Guevara ML, Li Z et al (2017) Lipopolyplex potentiates anti-tumor immunity of mRNA-based vaccination. Biomaterials 125:81–89
Pilkington EH, Suys EJA, Trevaskis NL et al (2021) From influenza to COVID-19: Lipid nanoparticle mRNA vaccines at the frontiers of infectious diseases. Acta Biomater 131:16–40
Pestova TV, Lorsch JR, Hellen CUT (2007) Translational control in biology and medicine. In: Mathews MB, Sonenberg N, Hershey JWB (Eds) Cold Spring Harbor Laboratory Press; Cold Spring Harbor, pp 87–128
Qiu Y, Man RCH, Liao Q et al (2019) Effective mRNA pulmonary delivery by dry powder formulation of PEGylated synthetic KL4 peptide. J Control Release 314:102–115
Rabani M, Pieper L, Chew GL et al (2017) A massively parallel reporter assay of 3’ UTR sequences identifies in vivo rules for mRNA degradation. Mol Cell 68:1083-1094.e5
Ramanathan A, Robb GB, Chan SH (2016) mRNA capping: biological functions and applications. Nucleic Acids Res 44:7511–7526
Riewe J, Erfle P, Melzig S et al (2020) Antisolvent precipitation of lipid nanoparticles in microfluidic systems—a comparative study. Int J Pharm 579:119167
Rio DC, Ares M Jr, Hannon GJ et al (2010) Ethanol precipitation of RNA and the use of carriers. Cold Spring Harb Protoc 2010:pdb.prot5440
Risma KA, Edwards KM, Hummell DS et al (2021) Potential mechanisms of anaphylaxis to COVID-19 mRNA vaccines. J Allergy Clin Immunol 147:2075-2082.e2
Roces CB, Lou G, Jain N et al (2020) Manufacturing considerations for the development of lipid nanoparticles using microfluidics. Pharmaceutics 12:1095
Roy B (2021) Effects of mRNA modifications on translation: an overview. Methods Mol Biol 2298:327–356
Ryals RC, Patel S, Acosta C et al (2020) The effects of PEGylation on LNP based mRNA delivery to the eye. PLoS ONE 15:e0241006
Rybakova Y, Kowalski PS, Huang Y et al (2019) mRNA delivery for therapeutic anti-HER2 antibody expression in vivo. Mol Ther 27:1415–1423
Sago CD, Lokugamage MP, Paunovska K et al (2018) High-throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing. Proc Natl Acad Sci USA 115:E11427
Sahin U, Derhovanessian E, Miller M et al (2017) Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547:222–226
Santos PM, Butterfield LH (2018) Dendritic cell-based cancer vaccines. J Immunol 200:443–449
Schoenmaker L, Witzigmann D, Kulkarni JA et al (2021) mRNA-lipid nanoparticle COVID-19 vaccines: structure and stability. Int J Pharm 601:120586
Schuster SL, Hsieh AC (2019) The untranslated regions of mRNAs in cancer. Trends Cancer 5:245–262
Segel M, Lash B, Song J et al (2021) Mammalian retrovirus-like protein PEG10 packages its own mRNA and can be pseudotyped for mRNA delivery. Science 373:882–889
Smith TT, Stephan SB, Moffett HF et al (2017) In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nanotechnol 12:813–820
Stadler CR, Bähr-Mahmud H, Celik L et al (2017) Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat Med 23:1241
Sterner RC, Sterner RM (2021) CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J 11:69
Svitkin YV, Cheng YM, Chakraborty T et al (2017) N1-methyl-pseudouridine in mRNA enhances translation through eIF2α-dependent and independent mechanisms by increasing ribosome density. Nucleic Acids Res 45:6023–6036
Tang X, Zhang S, Fu R et al (2019) Therapeutic prospects of mRNA-based gene therapy for glioblastoma. Front Oncol 9:1208
Thess A, Grund S, Mui BL et al (2015) Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol Ther 23:1456–1464
Thran M, Mukherjee J, Pönisch M et al (2017) mRNA mediates passive vaccination against infectious agents, toxins, and tumors. EMBO Mol Med 9:1434–1447
Tiwari PM, Vanover D, Lindsay KE et al (2018) Engineered mRNA-expressed antibodies prevent respiratory syncytial virus infection. Nat Commun 9:3999
Trepotec Z, Geiger J, Plank C et al (2019) Segmented poly(A) tails significantly reduce recombination of plasmid DNA without affecting mRNA translation efficiency or half-life. RNA 25:507–518
Uddin MN, Roni MA (2021) Challenges of storage and stability of mRNA-based COVID-19 vaccines. Vaccines (Basel) 9:1033
Van Hoecke L, Roose K (2019) How mRNA therapeutics are entering the monoclonal antibody field. J Transl Med 17:54
Van Tendeloo VF, Ponsaerts P, Lardon F et al (2001) Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood 98:49–56
Veiga N, Goldsmith M, Granot Y et al (2018) Specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat Commun 9:4493
Vogel AB, Lambert L, Kinnear E et al (2018) Self-amplifying RNA vaccines give equivalent protection against influenza to mRNA vaccines but at much lower doses. Mol Ther 26:446–455
Walker SE, Lorsch J (2013) RNA purification–precipitation methods. Methods Enzymol 530:337–343
Wang Z, Day N, Trifillis P et al (1999) An mRNA stability complex functions with poly(A)-binding protein to stabilize mRNA in vitro. Mol Cell Biol 19:4552–4560
Warminski M, Sikorski PJ, Kowalska J et al (2017) Applications of phosphate modification and labeling to study (m)RNA caps. Top Curr Chem (cham) 375:16
Wei T, Cheng Q, Min YL et al (2020) Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat Commun 11:3232
Weill L, Belloc E, Bava FA et al (2012) Translational control by changes in poly(A) tail length: recycling mRNAs. Nat Struct Mol Biol 19:577–585
Wesselhoeft RA, Kowalski PS, Parker-Hale FC et al (2019) RNA circularization diminishes immunogenicity and can extend translation duration in vivo. Mol Cell 74:508-520.e4
Wilgenhof S, Van Nuffel AMT, Benteyn D et al (2013) A phase IB study on intravenous synthetic mRNA electroporated dendritic cell immunotherapy in pretreated advanced melanoma patients. Ann Oncol 24:2686–2693
Yang E, Li MMH (2020) All about the RNA: interferon-stimulated genes that interfere with viral RNA processes. Front Immunol 11:605024
Yang EY, Shah K (2020) Nanobodies: next generation of cancer diagnostics and therapeutics. Front Oncol 10:1182
Zangi L, Lui KO, von Gise A et al (2013) Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat Biotechnol 31:898–907
Zhao J, Li Y, Wang C et al (2020) IRESbase: a comprehensive database of experimentally validated internal ribosome entry sites. Genomics Proteomics Bioinform 18:129–139
Acknowledgements
The authors gratefully acknowledge partial financial support from the Horizon 2020 Marie Skłodowska-Curie Actions (H2020-MSCA) program (Grant No. 843838 to S.P.).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Guevara, M.L., Persano, F., Persano, S. (2022). Lipid Nanoparticles to Harness the Therapeutic Potential of mRNA for Cancer Treatment. In: Jurga, S., Barciszewski, J. (eds) Messenger RNA Therapeutics. RNA Technologies, vol 13. Springer, Cham. https://doi.org/10.1007/978-3-031-08415-7_14
Download citation
DOI: https://doi.org/10.1007/978-3-031-08415-7_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-08414-0
Online ISBN: 978-3-031-08415-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)