
Abstract
Strontium stannate (SrSnO3) is a perovskite, which has been extensively studied due to its potential technological applications. In this work, SrSnO3 was doped with transition metals (Cu, Fe, Ni) by the modified Pechini method and evaluated in the catalytic reduction of NO with CO. The perovskite structure was obtained as the major phase for all samples. The catalytic activity of SrSnO3 was highly improved by all the dopants, as only 10% of conversion was obtained for SrSnO3, whereas samples with 5% of Cu presented the highest conversions, reaching 100% of NO into N2 and 100% of CO into CO2 at 550 °C. Conversions of 85% of NO into N2 and 90% of CO into CO2 were obtained for Ni doped samples at 600 °C, while 62% of conversion was obtained for Fe-doped samples, for both reactions. These results indicate that Cu is the best dopant, which leads to the greatest NO conversion with a smaller amount of dopant.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

1 Introduction
Atmospheric pollution is related to the increase of concentration of harmful compounds, such as carbon monoxide (CO), nitrogen oxides (NOx), sulfur oxides (SOx), carbon dioxide (CO2), volatile organic compounds (VOC) and halogenated compounds.
Nitrogen oxides (NOx) come from natural and human sources, and they are very harmful compounds because they are toxic to human beings, contribute to acid rain, to the destruction of the ozone layer, increase the greenhouse effect and photochemical fog [1,2,3]. New processes, including new catalysts, have been developed to reduce the emission of pollutants into the atmosphere. Catalysts are used in car exhausts and in industries in order to decrease pollution caused by emission of harmful gases. For instance, NOx elimination does not take place spontaneously, but it can be done using appropriate catalysts. Most of these catalysts are based on noble metals [4], zeolites modified with transition metals [5] and oxide compounds [6].
Perovskite compounds have ABO3 stoichiometry and are interesting alternatives to replace the traditional NOx catalysts, due to their greater thermal stability, lower cost, great versatility, and excellent redox property [6]. Most of the perovskites used for NO abatement have lanthanum in the A site and cobalt, nickel, manganese, or iron in the B site [6, 7]. These catalysts have good catalytic activity but not always a good selectivity for N2 [8].
With the aim of developing new catalytic compounds with selectivity for NO abatement, perovskites containing Sn4+ in the B site and with substitution of lanthanum for alkaline earth metals have been studied by our research group. Among these compounds, SrSnO3 was well studied by our research group, after doping with different transition metals, and applied in the NO reduction by CO.
1.1 Catalytic Abatement of NO
Conversion reaction of NO into N2 and O2 is extremely slow due to the high dissociation energy of NO molecules, and it hardly occurs at room temperature [9]. The use of catalysts favour the conversion process, but due to the strong adsorption of NO molecules on the catalyst surface, the formation of undesirable intermediates as N2O and NO2 and catalytic deactivation takes place. To avoid these problems, selective catalytic reduction (SCR) is used in the decomposition of NO. In this process, NO conversion is thermodynamically favoured by the presence of reducing agents, as CO, NH3, H2, CH4, etc., which decreases the Gibbs free energy (ΔrGo), compared to the direct decomposition of NO into N2 and O2 [1, 10, 11].
The use of carbon monoxide (CO) is a promising alternative because it has smaller values of ΔrGº in a broad temperature range and CO is also a pollutant and abatement of both gases is environmentally interesting [12]. The reaction mechanism of NO reduction by CO is very complex. According to literature, in the first step, NO molecules adsorb on the catalyst surface, with dissociation on atomic oxygen and atomic nitrogen, followed by N2 formation. In the second step, CO molecules adsorb on the catalyst surface, react with atomic oxygen or lattice oxygen to form CO2. These reactions are presented in Eqs. (1) to (7) [13].


During the catalytic process, the metallic cation is active in the reaction mechanism, being oxidized by NO and reduced by CO, due to the oxygen transfer to this last molecule. Therefore, the behaviour of the metallic catalyst is directly related to the catalyst efficiency [7]. According to Zhang [14], when NO is adsorbed on the perovskite surface, it receives an electron from the metal to form the nitrosyl species (NO−), which is the first important step of the reduction reaction, Eq. (8). N–O bond in the nitrosyl species is weaker than in NO one favouring the reaction. At low temperatures, adsorbed species on the perovskite surface are dissociated to form N2 and N2O, Eqs. (9) and (10). N2O is more stable than NO at low temperatures, but it only decomposes above 300 °C, despite the weaker N–O bond. At higher temperatures, N2O decomposition is easier than NO one and it is favoured when a reducing agent (CO) is added to the reaction, Eqs. (11) and (12). Because of this mechanism, N2O has been observed as a reaction intermediate by different authors [13,14,15,16].
1.2 ASnO3 Perovskites
ASnO3 stannates have interesting physical properties, many of them associated with impurities in the crystalline lattice of structural defects. Alkaline earth stannates have interesting optical, electrical and magnetic properties, with applications as thermally stable capacitors, gas sensors, humidity sensors [17] and as photocatalysts [18]. Despite their technological importance, they are not as studied as the corresponding titanates [19].
The three ASnO3 compounds have classic perovskite structure (BaSnO3, CaSnO3 and SrSnO3). BaSnO3 has an ideal cubic structure (space group Pm3m), while SrSnO3 and CaSnO3 have orthorhombic structure (Pbnm). Despite this, the octahedral site of Sn4+ is maintained as well as their tridimensional arrangements, while different tiltings among the octahedra lead to meaningful changes in the A site (Ca, Sr and Ba) and in O2− site [19, 20]. This distortion is periodically repeated so that a long-range order is observed. Moreover, tilting among the octahedra decreases the conduction band edge with a corresponding increase of the band gap. Therefore, many changes of properties are observed when different modifiers are added to the A site of the stannates with perovskite structure [19, 21].
SrSnO3 (Fig. 1) is a n-type semiconductor with band gap value above 3.0 eV [19], a valence band (HOMO) corresponding to the non-bridging 2p orbital of O2− and a conduction band (LUMO) corresponding to the antibonding orbital resulting from the interaction of the 5 s orbital of Sn4+ and 2p orbital of O2−.

Orthorhombic structure of SrSnO3 [22]
A partial replacement of Sr2+ and/or Sn4+ cations leads to structural defects, especially when this replacement is done by a cation with different oxidation state. Therefore, different compounds are formed, but the perovskite structure is maintained. Moreover, cationic vacancies in A and B sites or anionic vacancies are also usual and lead to structural defects and to interesting physical and chemical properties [23, 24]. These structural defects are directly related to the catalytic activity as they act on the reaction mechanisms. For instance, oxygen vacancies are mostly responsible for oxygen mobility inside the crystalline lattice and the more vacancies, the greater the oxygen mobility. Ionic vacancies may favour or not the adsorption of reagents in gaseous phase, but they certainly promote the ion transportation inside the lattice [25].
1.3 Perovskites Applied in the Reduction of NO by CO
The catalytic properties of perovskites have been studied since the 1950s, for CO oxidation. In 1972, Libby showed its potential application for purification of exhaust gases. Thereafter, perovskite oxides have been recognized as valuable catalysts for NO reduction by CO, due to its high catalytic activity, selectivity, and stability [26, 27].
Some authors believe that ions in the A site are catalytic inactive and influence the thermal stability of the perovskites, while ions in the B site are catalytic active, with a relatively great distance from one another (0.4 nm), leaving a big space for small molecules to interact selectively in only one active site [28]. For this reason, the choice of the cation in the B site is of great importance for the catalysts as well as for the modification of their catalytic properties, which are determined by the nature of this cation [29].
Most works published concerning perovskites applied in the NO reduction by CO have lanthanum as lattice modifier, associated to transition metals in the B site, as displayed in Table 1.
He et al. [32] studied the influence of the preparation method and of doping on La0.8Ce0.2B0.4Mn0.6O3 (B = Cu or Ag) systems. According to the authors, after adsorption of the NO molecules on the perovskite surface, an electron is transferred from the B site to the NO molecule and the NO− molecule is formed simultaneously to the oxidation of the B cation (Cu+ ↔ Cu2+ and Mn3+ ↔ Mn4+). The presence of Cu+ and the redox reaction (Cu+ ↔ Cu2+) may benefit the NO adsorption and its reduction by CO. When the particle size decreases (40–50 nm) and the surface area increases (13.9;17.9; 5.7 e 14.2 m2 g−1), more defects and larger amounts of Cu+ ions are formed on the surface and the catalytic activity for NO and CO abatement increases.
Dai et al. [33] evaluated different perovskite type catalysts and correlated the catalytic activity to structural defects (especially oxygen vacancies) and to the redox property (especially of transition metal ions). According to the authors, the oxygen vacancies favor the O2 and the NOx activation and the change of the oxidation state of the B site promotes the redox process of the catalyst. Together, the two effects influence the oxygen mobility in the lattice and improve the catalytic performance for NO reduction by CO. The importance of the oxygen vacancies in the catalytic performance was confirmed by other authors [29, 34], who reported that oxygen vacancies in perovskites have an important role on the catalytic reduction of NO, forming adsorption sites for NO and CO and favoring the dissociation of adsorbed NO species.
In the present work, different transition metals, Fe, Ni and Cu were added into the SrSnO3 lattice to improve the redox ability of the perovskite and form oxygen vacancies. The pure and doped samples were evaluated in the SCR of NO by CO.
2 Materials and Methods
The synthesis of pure and doped (Cu, Fe e Ni) strontium stannate was done by the modified Pechini method, also known as polymeric precursor method. The procedure was carried out in two steps: the synthesis of the tin citrate followed by synthesis of the polymeric resin and calcination. Pure SrSnO3 and Fe-doped SrSnO3 was also synthesized using metallic tin, using a route with only one step.
2.1 Reagents
The precursors used in the synthesis of pure and doped strontium stannate were: citric acid (99%, Cargill), tin chloride (99.9%, J.T. Baker), tin (99.99%, Vetec), ammonium hydroxide (99.9%, Sigma), copper (II) nitrate (99.0%, Vetec), iron (III) nitrate (99.0%, Vetec), nickel (II) nitrate (99.0%, Vetec), nitric acid (65.0%, Dinamica), ethylene glycol (99.5%, Vetec).
2.2 Modified Pechini Method
2.2.1 Two Steps Route
Tin citrate was prepared by dissolution of citric acid in water heated at 70 °C, followed by addition of dihydrated tin chloride, with a stoichiometric ratio of 3 mol of citric acid to 1 mol of tin chloride. The becker was immersed in a container with ice and ammonium hydroxide was slowly added to the solution to induce the precipitation of tin citrate, under a pH value below 3. A molar ratio of 1 mol of tin chloride to 1 mol of ammonium hydroxide was used. The suspension was stored in a refrigerator and supernatant was changed to eliminate the chloride, until a negative result of the chloride test using AgNO3. Tin citrated was vacuum filtered, dried in a stove at 60 °C for 24 h, deagglomerated and stored in a refrigerator. Gravimetry was done to determine the amount of tin oxide obtained from the tin citrate.
For the synthesis of the polymeric resin, solutions of the different precursors were prepared. Strontium citrate solution was prepared by dissolution of citric acid and strontium nitrate in a small amount of water. For doped SrSnO3, solutions of copper citrate, nickel citrate and iron citrate were also prepared, similarly to the procedure described to strontium citrate. Tin citrate solution was prepared by solubilization of the previously prepared powder in a small amount of nitric acid. Then strontium citrate solution was added into the tin citrate solution under heating at 70 °C and magnetic stirring, followed by addition of the dopant citrate solution (copper citrate or nickel citrate or iron citrate), considering the desired stoichiometry (1, 5, 10 mol %). After homogenization for 15 min, ethylene glycol was added to the solution and heated up to ~90 °C to obtain the polymeric resin. A molar stoichiometric ratio of citric acid:metal of 3:1 was used, besides a mass ratio of citric acid:ethylene glycol of 60:40.
Each polymeric resin was heat treated at 300 °C for 4 h to obtain the polymeric powder precursor, which was deagglomerated and sieved at 100 mesh before milling in an attritor mill for 4 h, in ethanolic medium. After drying, the material was dried, and heat treated with a heating rate of 1 °C min−1 up to 300 °C for 7 h under oxygen atmosphere. The resulting powder was calcined from 400 to 800 °C, with a heating rate of 5 °C min−1 por 4 h in a muffle oven.
2.2.2 One Step Route
First, tin (Sn) was dissolved in an aqueous solution of nitric acid at 0.1 mol.L−1 in an ice bath, followed by addition of citric acid. The pH of the solution was adjusted to 3–4 by addition of ammonium hydroxide. The previously prepared strontium citrate solution was added to tin citrate solution under stirring at 60 °C, followed by addition of the iron citrate solution for the doped sample. After 20 min of stirring, ethylene glycol was added, and the solution was heated to 90–100 °C. The same stoichiometric ratios described before were used, as well as the same milling and heat treatment procedure.
2.3 Characterization of the Catalysts
The polymeric powder precursors of the pure and doped SrSnO3 heat treated under oxygen atmosphere were characterized by thermogravimetry (TG) and differential thermal analysis (DTA) using a thermal analyser DTG-60H from Shimadzu. In the analysis, 10 mg of sample were weighed in an alumina crucible, heated with a heating rate of 10 °C min−1 up to 1200 °C under a synthetic air atmosphere with flow of 50 mL min−1.
After calcination from 400 to 800 °C, samples were characterized by X-ray diffraction (XRD), infrared spectroscopy (IR), Raman spectroscopy, UV–visible spectroscopy (UV–vis) and specific surface area.
XRD patterns were obtained in a XRD-6000 diffractometer from Shimadzu operating at 40 kV and 40 mA with CuKα radiation. The θ–2θ XRD patterns were collected in the range of 15–80°, using a step size of 0.02° and step time of 2 s. IR spectra were obtained in an IRPrestige-21 spectrophotometer from Shimadzu, in mid infrared range, from 300 to 2000 cm−1 using KBr pellets or in the far infrared range, from 240 to 650 cm−1 using Nujol oil. UV–vis spectra were obtained in a UV-2550 spectrophotometer from Shimadzu, in diffuse reflectance mode, in the range from 190 to 900 nm. The band gap energies were calculated from the absorbance curve according to Wood and Tauc method [35]. Micro-Raman spectra were collected using a Invia micro-Raman from Renishaw operating with an Ar laser (514 nm) and power of 20 mW. Spectra were obtained in the range from 100 to 1000 cm−1, using an objective lens of 50 x. The specific surface area of the catalysts was determined in an ASAP 2420 analyser from Micromeritics, using N2 adsorption/desorption curves at 77 K. Samples were previously treated at 150 °C for 2 h under ultra-dry N2 flow.
2.4 Catalytic Activity
Materials calcined at 800 °C were evaluated in the catalytic reduction of NO by CO. During the catalytic test, 100 mg of catalyst (previously treated at 500 °C for 1 h under He flow) were diluted in 100 mg of amorphous silica and placed in a fixed bed tubular reactor coupled to a vertical furnace with controlled heating under atmospheric pressure. Reaction was performed between 300 and 700 °C, with space speed of 17,000 h−1. The reactor was powered with a continuous gaseous flow (25 mL min−1), regulated with the use of mass flow meters, with a mixture of 0.5% of NO and 0.5% of CO under He balance, both as a pattern mixture of 1% mol/mol (gas/He). Reaction products were identified in a GC-17A gas chromatograph from Shimadzu, equipped with a thermal conductivity detector. NO, N2, CO and CO2 were separated at 50 °C, using two packed columns operating in series (Porapack N and Molecular Sieve 13X).
3 Results and Discussion
Figure 2 displays the TG and DTA curves of the pristine SrSnO3 precursor. Four mass loss steps were observed in the TG curve. In the first step, water and adsorbed gases were eliminated from the material surface, corresponding to endothermic transitions in the DTA curve, around 80 °C. The second step was assigned to the combustion reaction of the organic material with elimination of CO2 and H2O, corresponding to an exothermic peak in the DTA curve, between 300 and 600 °C. Mass loss assigned to endothermic peaks above 710 °C (third step) and above 1000 °C were assigned to the carbonate decomposition.

TG and DTA curves of the SrSnO3 precursor
A careful evaluation of the crystallization process of SrSnO3 was performed after calcination between 400 and 800 °C, and characterization by FTIR and XRD, as displayed in Fig. 3.

Infrared spectra (a) and XRD patterns (b) of SrSnO3 calcined at different temperatures
According to Nakamoto [36], the hydroxyl group is distinguished from the aqueous group, which displays H–O–H bending modes around 1600 cm−1, while the hydroxyl-metal bond, M–OH, displays a vibrational mode between 1000 and 1200 cm−1. These two bands were observed in the spectra of Fig. 3a—one band assigned to adsorbed water was observed at 1630 cm−1, while one band assigned to M–OH group was observed around 1070 cm−1.
Bands assigned to chelated esters were also observed at 1389 and 1547 cm−1 [36] after calcination at 400 and 500 °C and their intensities decrease after calcination at and above 600 °C, confirming that the exothermic peak between 300 and 600 °C is related to the combustion of the ester.
According to Nyquist and Kagel [37], absorption bands assigned to carbonates are observed at 1320–1530 cm−1 (strong), at 1040–1100 cm−1 (weak) and at 800–890 cm−1 (medium). In the present work, these bands are observed at 1450, 1080 e 860 cm−1 due to the presence of unidentate carbonate, according to Hodjati et al. [8]. The intensity of these bands decrease after calcination above 700 °C, but they are not completely eliminated, as the highest calcination temperature was 800 °C and TG/DTA curves indicate the carbonate elimination only above 1200 °C. The high intensity of the band at 1080 cm−1 is due to the superposition of the hydroxyl band.
The orthorhombic SrSnO3 perovskite phase has 57 normal modes, with 25 infrared active modes, Γiv = 9B1u + 7B2u + 9B3u [38]. The perovskite structure has bands assigned to three triply degenerate active modes: the lattice mode ν1 (A-BO3) between 80 and 140 cm−1; the bending mode ν2 (O-B-O) between 170 and 280 cm−1; and the stretching mode ν3 (B-O), between 490 and 680 cm−1; while the torsional mode F2u may become active for perovskites with structure distortion from the ideal cubic symmetry, with the appearance of more bands in the IR spectrum. The ν3 mode is observed around 500 cm−1 for cubic perovskites, and around 660 cm−1 for orthorhombic ones, due to the octahedral distortion [39]. For instance, Pfaff et al. [40] observed the Sn–O band around 550 cm−1 for the cubic BaSnO3 (cúbico), while Last [39] observed this same band around 666 cm−1, for the orthorhombic SrSnO3. These data agree to Moreira et al. [38], who reported that bands assigned to the stretching of the Sn–O bond are observed around 674 and 530 cm−1, while vibrations of the stannate group (SnO32−) are observed around 300–400 and 600–700 cm−1.
In the present work, υ2 and υ3 modes were observed around 400 and 670 cm−1, respectively. The υ3 mode was observed around 540 cm−1, for samples calcined at 400 and 500 °C. As temperature increases, the intensity of this band decreases, while a high intensity band is observed at 670 cm−1. It should be observed that the band previously assigned to the undistorted SnO6 octahedra of BaSnO3, at 540 cm−1, was observed at low temperaturas, while the band assigned to the distorted SnO6 octahedra of SrSnO3, at 670 cm−1, was observed at higher temperatures. This behaviour indicates that the octahedral distortion probably takes place simultaneously to the crystallization process.
This behaviour is in agreement with the XRD patterns of the pure SrSnO3 (Fig. 3b, which displays an increase of the peaks assigned to the perovskite structure at and above 600 °C, simultaneously to the SnO6 band at 670 cm−1, while no peaks assigned to this phase are observed at 400 and 500 °C. All peaks indicated in Fig. 3b were indexed according to the ICDD index card 77-1798 and confirm that an orthorhombic structure was obtained. Peaks assigned to SrCO3 were observed after calcination at 400, 500 and 600 °C, in agreement to FTIR spectra.
Azad et al. [41] synthesized SrSnO3 by solid state reaction (SSR) and self-sustained synthesis (SHS) and observed the formation of the perovskite structure at 1100 °C and 800 °C, respectively. A single-phase material was obtained by the synthesis using the SRS method, while Sr2SnO4 was obtained as a secondary phase when the SHS method was used.
In the present work, the perovskite phase was obtained with good crystallinity at 800 °C. This is an interesting result for catalysis as low calcination temperatures usually lead to materials with greater surface area, which favours the catalytic processes. As all doped SrSnO3 presented a similar behaviour, the calcination temperature of 800 °C was used for all other samples presented hereafter.
3.1 Influence of Doping on the Catalytic Activity of SrSnO3: Me (Me: Fe, Ni, Cu)
3.1.1 Characterization of the Catalysts
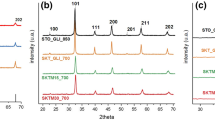
The XRD patterns of doped SrSnO3 are presented in Fig. 4. All samples displayed an orthorhombic perovskite structure, according to the ICDD 01-77-1798 index card. For all of the samples, no peaks assigned to the dopants were observed, which indicate that the dopants were incorporated into the perovskite lattice. Moreover, according to the inset in Fig. 4, doping decreases the crystallinity of the perovskite phase, as indicated by the smaller intensities of the (200) peaks. A displacement of these peaks is also observed, which is another indication that dopant got into the perovskite lattice. Similarly to other works published in literature, strontium carbonate (SrCO3) was observed as a secondary phase for most samples [21].

XRD patterns of the pure and doped SrSnO3 calcined at 800 °C
The lattice parameters were calculated for all samples, as displayed in Table 2. Theoretical data displayed in the ICDD 01-77-1798 index card are displayed for comparison. For SrSnO3, very similar values were obtained compared to the theoretical one. After doping, no meaningful change in the lattice parameters ‘a’ and ‘b’ was observed, while an increase in the parameter ‘c’ was noticed for samples doped with copper and iron, with a small increase of the unit cell volume.
According to literature, the ionic radii of Sn4+, Ni2+, Fe3+ and Cu2+ are 69 pm, 69 pm, 65 pm and 73 pm, respectively [42]. The small difference of lattice parameters observed after Ni doping may be assigned to the similar ionic radii of Sn4+ and Ni2+. Curiously, even when Sn4+ replacement was made with a larger size cation, as Fe3+, an increase of the unit cell volume was observed. This behaviour may be due to the lattice distortion related not only to the size difference of the cations, but also due to the formation of oxygen vacancies as Sn4+ was replaced by cations with smaller oxidation state [43, 44]. This distortion may also be responsible for the smaller crystallite size after doping, as displayed in Table 2, because distortion can make diffusion more difficult.
The infrared spectra of pure and doped SrSnO3 samples, calcined at 800 °C for 4 h are displayed in Fig. 5. For all samples, the Sn–O stretching band was observed around 670 cm−1 besides a weak band around 540 cm−1, which indicated that different octahedral symmetries are observed in the perovskites. After doping with Ni and Fe, an increase of the intensity of the band assigned to undistorted octahedra at 540 cm−1 was observed. An absorption band below 400 cm−1 was assigned to the O–Sn–O bending mode. The presence of carbonate was also observed, as indicated by bands at 860, 1070 and 1470 cm−1, as described before.

Infrared spectra of pure and doped SrSnO3 samples, calcined at 800 °C for 4 h
Raman spectra of the pure and doped SrSnO3 samples, calcined at 800 °C for 4 h are displayed in Fig. 6. Orthorhombic perovskites display 24 Raman active modes, according to the irreducible representation, ΓRaman = 7Ag + 5B1g + 7B2g + 5B3g. Not all modes are always observed, as superposition of some modes may occur, as well as a low polarizability of some chemical bonds. Literature data concerning Raman modes for the orthorhombic SrSnO3 are displayed in Table 3 and the bands observed in the present work are indicated in Fig. 6.

According to [45], bands assigned to strontium carbonate (SrCO3) are observed at 148, 180, 214, 236, 244, 258, 511, 690 and 1064 cm−1. In the present work, these were observed at 149, 179, 700 and 1070 cm−1, while bands at 214, 236, 244 and 258 cm−1 may be superposed to the bands assigned to SrSnO3.
After doping, a symmetry breakage was clearly observed in the Raman spectra. For the sample SrSn0.9Ni0.1O3, no peaks assigned to the perovskite structure were observed, while small intensity bans were observed for SrSn0.9Cu0.1O3. A broad band around 570 cm−1, was observed for this sample, which could be related to the formation of oxygen vacancies.
UV–vis spectra of the pure and doped SrSnO3 samples, calcined at 800 °C for 4 h are displayed in Fig. 7.

UV–vis spectra of the pure and doped SrSnO3 samples, calcined at 800 °C for 4 h
The spectra have similar profiles with a high intensity absorption between 200 and 350 nm, which is assigned to the ligand–metal charge transfer (LMCT), O2− → Sn4+, as observed by Liu et al. [46] for Sn4+ in octahedral environment. No other bands were observed for undoped SrSnO3 above 400 nm, as currently observed for light color materials.
For doped materials, different absorption bands are observed between 300 and 900 nm, depending on the dopant added into the lattice. These bands also depend on the oxidation state and coordination of the transition metal and give an indication of redox reactions, during synthesis. Band positions were determined for all of the doped samples and assignments are displayed in Table 4, considering an octahedral environment. For doping with nickel and iron, only bands assigned to Ni2+ and Fe3+ were observed, but this result does not exclude the possibility of the presence of other species, since UV–vis spectroscopy does not have high sensitivity. For copper doped SrSnO3, Cu+ and Cu2+ were observed in a distorted environment, as reported by Rao et al. [47].
SrSnO3 is a semiconductor with band gap values varying between 3.0 and 4.0 eV [21, 53]. In the present work, a band gap value of 3.7 eV was obtained, similarly to literature data. After doping, a meaningful decrease of this value was observed, as displayed in Table 5, which was assigned to distortions in the perovskite lattice besides the presence of oxygen vacancies and of the dopant itself, which can form intermediate levels inside the band gap.
Considering the results obtained, the Kroger Vink notation was used to evaluate the defects formed in the SrSnO3 lattice due to doping with Ni, Fe or Cu, as displayed in Eqs. (13) to (16). All the charge differences due to doping are probably compensated by the formation of oxygen vacancies, as usually shown in literature.
The specific surface areas of the pure and doped SrSnO3 calcined at 800 °C for 4 h were determined by the BET equation, as displayed in Table 6. The undoped SrSnO3 had a slightly larger surface area than the material synthesized by Hodjati et al. [8] by the sol–gel method (16 m2 g−1). After doping, a small decrease of surface area was observed for SrSn0.9Ni0.1O3, while a growing trend was observed for SrSn0.9Fe0.1O3 and SrSn0.90Cu0.1O3.
3.1.2 Catalytic Tests of NO Reduction by CO
The results of NO conversion into N2 and CO conversion into CO2, using doped and undoped SrSnO3 as catalysts are displayed in Fig. 8. All samples were catalytic active for the NO reduction and for CO oxidation. The smallest conversion was obtained by the undoped SrSnO3, which displayed 34.4% NO conversion into N2 and 38.1% of conversion of CO into CO2, at 600 °C. These values are not much higher than those obtained at 500 °C, with less than 2% of increase. This behavior changed after doping, as a continuous increase of conversion was observed as temperature increased, reaching values above 75% for both reactions at 600 °C, confirming the positive effect of doping on the catalytic activity of SrSnO3. This is a very interesting result as Brazil is an important producer of SnO2 and doped SrSnO3 can be used as a new catalyst for reaction applications in this temperature range.

Conversion of NO to N2 (a) and CO to CO2 (b) of the undoped and doped SrSnO3, calcined at 800 °C for 4 h
Comparing the different dopants, the biggest activity was obtained using SrSn0.9Cu0.1O3 as catalyst. For NO reduction, there is no meaningful difference between this catalyst and SrSn0.9Ni0.1O3, as conversions around 90% were obtained with both of them. But for CO oxidation, 100% of CO oxidation was obtained using SrSn0.9Cu0.1O3 as catalyst, while 90% of conversion was obtained using SrSn0.9Ni0.1O3.
We believe that the greatest catalytic activity of SrSn0.9Cu0.1O3 is related to the redox ability of Cu, making the oxidation and the reduction processes involved in the catalysis easier. Moreover, Cu-coping leads to the greater amount of oxygen vacancies, as indicated by Eqs. (13) to (16). On the other hand, the sample with smaller amount of oxygen vacancies, SrSn0.9Fe0.1O3, had the smallest conversion for all temperatures of the catalytic test. When oxygen vacancies are formed, Sn4+ coordination changes, as fivefold coordinations or even fourfold coordinations may be present in the lattice. Moreover, oxygen vacancies may have three different charges: neutral complexes, \([SnO_{5} .V_{O}^{x} ]_{complex}\); double positive complexes, \([SnO_{5} .V_{O}^{{\ddot{}}} ]_{complex}\), which capture electrons; and monopositive complexes, \([SnO_{5} .V_{O}^{\dot{}} ]_{complex}\), which may capture or donate electrons. The \([SnO_{5} .V_{O}^{{\ddot{}}} ]_{complex}\) or the \([SnO_{5} .V_{O}^{x} ]_{complex}\) may donate an electron to the adsorbed NO to form NO−, which is the active species for NO decomposition [54].
3.2 Influence of the Synthesis Procedure on the Catalytic Results of SrSnO3: Fe
3.2.1 Characterization of the Catalysts
Due to the lowest catalytic activity of SrSn0.9Fe0.1O3, this material was chosen to improve the synthesis method in order to evaluate its influence on the catalytic activity. This sample was synthesized using two steps to obtain the polymeric resin, as previously described; and using only one step to obtain the polymeric resin, as described in this section. The XRD patterns of the samples synthesized by the two different routes are presented in Fig. 9.

XRD patterns of SrSnO3 and SrSn0.9Fe0.1O3-x, synthesized by two different routes and calcined at 800 °C
All samples were identified using the ICDD 01-77-1798 index card, which confirms the formation of the orthorhombic SrSnO3 with Pbnm space group. The samples synthesized using the 1 step route do not have SrCO3 as secondary phase, probably due to a more homogeneous behaviour of the polymeric resin. The lattice parameters were calculated as displayed in Table 7.
According to the lattice parameters displayed in Table 7, Fe3+ addition into the SrSnO3 lattice did not take to meaningful variation of the lattice parameters and very similar unit cell volumes were obtained. Comparing the synthesis route, it was observed that smaller variations were obtained using the 1 step route, probably due to a greater homogeneity of the cations distribution in the polymeric resin, on an atomic scale.
The infrared and Raman spectra of the samples synthesized by the two different routes are displayed in Fig. 10. The infrared spectra were similar to those presented before and showed the Sn–O stretching band around 670 cm−1 besides a weak band around 540 cm−1 and an absorption band below 400 cm−1, assigned to the O–Sn–O bending mode. The presence of carbonate was also observed, as indicated by bands at 860, 1070 and 1470 cm−1, as described before.

Infrared spectra (a) and Raman spectra (b) of SrSnO3 and SrSn0.9Fe0.1O3-x, synthesized by two different routes and calcined at 800 °C
Although similar absorption bands were observed for samples obtained by the two different routes, some meaningful differences may be observed, especially concerning the carbonate bands, which had a higher intensity when the 2 steps route was used during synthesis, in agreement with the XRD patterns. Moreover, the band assigned to undistorted octahedra at 540 cm−1 had also a higher intensity for the 2 steps route, indicating the change of the Sn4+ symmetry and confirming the lower homogeneity of the polymeric resin.
The Raman spectra of the samples obtained by the one step route with and without Fe-doping (Fig. 10b) display bands at 113 cm−1, 146 cm−1 and 170 cm−1 assigned to the Sr-SnO3 mode; an intense band at 220 cm−1 (Ag) assigned to the scissor movement of the Sn-O-Sn group along the c-axis; one shoulder at 256 cm−1 (Ag) assigned to O-Sn-O and Sn-O-Sn groups along ab plane, perpendicular to the c-axis [38]. Bands at 332 cm‑1 (B2g) and 397 cm‑1 (Ag) are assigned to Sn-O3 bonds and to the torsional Sn-O; the symmetric stretching of the Sn–O bond was observed at 565 cm−1. Bands assigned to SrCO3 were observed at 146 cm‑1, 181 cm‑1and 700 cm−1 [55], while one small band assigned to SnO2 was observed at 646 cm−1 [21].
Comparing the undoped and doped samples, a small decrease of the intensity of the bands at 113 cm−1, 146 cm−1 and 170 cm−1 was observed, while the greatest difference was observed in the Sn–O-Sn band at 220 cm−1. Besides the decrease of intensity the broadening of this band was also observed as FWHM increased from 15.8 cm−1 to 23.6 cm−1, determined by deconvolution of the peaks. This behavior indicates that the octahedral site was more affected by doping, since Fe3+ replaces Sn4+ in the lattice, as expected considering the site preference of Fe3+.
The UV–vis spectra of the samples synthesized by the two different routes are displayed in Fig. 11.

UV–vis spectra of SrSnO3 and SrSn0.9Fe0.1O3-x, synthesized by two different routes and calcined at 800 °C
For undoped SrSnO3, the UV–vis spectrum was very similar to the sample obtained by the 2 steps route, as displayed in Fig. 6, with a high energy absorption between 200 and 350 nm due to the LMCT [46] and almost no absorption above this wavelength. After doping with Fe, absorption bands above 350 nm were observed for both samples, which were deconvolved to identify the electronic transitions.
The Fe3+ cation has a d5 electronic configuration in high spin state in octahedral configuration, which means that all electrons are unpaired, as displayed in Fig. 12. Therefore, any electronic transition inside the d energy level will only occur if coupled to a spin inversion [51]. Consequently, only spin-forbidden electronic transitions are expected [50]. According to literature [50, 51], these transitions occur from the fundamental state 6A1g (S) to the excited states 4T1g (G), 4T2g (G), 4A1g (G), 4Eg (G). All assignments are displayed in Table 8 and confirm that spin-forbidden transitions due to the presence of Fe3+ are present in the UV–vis spectra.

Splitting of the octahedral crystal field (high spin) for Fe3+ [56]
The band gap values of undoped and Fe-doped SrSnO3 are displayed in Table 9. The band gap value obtained using the 1-step synthesis route was higher than the value obtained by the 2-steps route (3.7 eV), displayed in Table 5. This is probably due to a higher short-range order in the final material, due to a more homogeneous polymeric resin, as indicated by the XRD patterns and the IR spectra. After doping, a meaningful decrease of the band gap was observed for both samples, due to the formation of structural defects as displayed in Eq. (14), which form electronic levels inside the band gap. Moreover, d-d electronic transitions assigned to Fe3+ may also influence the final band gap.
The specific surface area of the undoped and Fe-doped SrSnO3 are displayed in Table 9. The surface area of the undoped SrSnO3 obtained by the 1-step route was smaller than the material obtained by the 2-steps route (18.1 m2 g−1), displayed in Table 6, while doping leads to a tendency to increase the surface area, for both routes. The modified Pechini method uses a large amount of organic material, which goes under combustion reaction during heat treatment. As this reaction is highly exothermic, an increase of temperature may occur forming aggregated particles, which decrease the surface area. For this reason, a partial elimination of the organic material is done under oxygen atmosphere, at low temperatures (around 300 °C), but this control is not easy and these surface area variations may occur, with no clear relation to variations of the synthesis routes.
3.2.2 Catalytic Tests of NO Reduction by CO
Undoped and Fe-doped SrSnO3 were applied as catalysts for the NO reduction by CO, as displayed in Fig. 13. Quartz (SiO2) was used to dilute the catalyst and its conversion was subtracted from the present results, before plotting.

Conversion of NO to N2 (a) and CO to CO2 (b) using the undoped and Fe-doped SrSnO3, as catalysts, obtained by two synthesis routes
Conversion of NO to N2 and CO to CO2 was favoured by Fe-doping, with a meaningful increase of activity, especially at 600 °C. At this temperature, a 3.2-fold increase was observed for SrSn0.9Fe0.1O3 synthesized by the 1-step route, while a 6.9-fold increase was observed for the 2-steps route, for the NO to N2 conversion. At this same temperature, no meaningful variation was observed comparing SrSnO3, SrSn0.9Fe0.1O3 synthesized by the 1-step route, while a 1.9-fold increase was observed for SrSn0.9Fe0.1O3 synthesized by the 2-step route. This behaviour is due to a higher activity of undoped SrSnO3 towards CO oxidation.
It is well known that oxygen vacancies are very important for NO reduction due to the formation of active sites, in addition to promoting the dissociation of adsorbed NO species. For instance, Zhang et al. [14] evaluated the catalytic activity of LaCoO3 in the NO reduction and observed an increase of 20% in the conversion when Co3+ was partially replaced by Cu2+. This improvement was assigned to the formation of oxygen vacancies in the lattice.
A similar behaviour was observed in the present work, when Sn4+ was replaced by Fe3+ in the perovskite lattice. Curiously, it seems that the higher short-range disorder induced by the 2-steps synthesis route favoured the catalytic activity. This behaviour may be related to an easier electron transfer of the highly distorted octahedrons favouring the redox reactions involved in the current system.
3.3 Influence of the Dopant Amount on the Catalytic Properties of SrSnO3: Cu
3.3.1 Characterization of the Catalysts
Due to the higher catalytic activity among all dopants, different percentages of Cu were added into the SrSnO3 lattice, using the 2-steps route, to evaluate its influence in the structural and catalytic properties. The XRD patterns of the Cu-doped materials are displayed in Fig. 14.

XRD patterns of the samples SrSn1-xCuxO3 (x = 0, 1, 5 and 10 mol %), calcined at 800 °C for 4 h
Peaks assigned to carbonates were observed at 25.3° and 36.3° with very low intensity except for the sample with 5% of Cu. Peaks assigned to SnO2 (cassiterite) are usually observed at 26.9°, 33.6°, 51.8° e 64.2°, as reported by Dazhi et al. [59]. Small peaks assigned to SnO2 were observed for the samples with 5 and 10% of Cu, while peaks assigned to Cu compounds were not identified, which indicated that the dopant was incorporated into the SrSnO3 lattice.
Pure and Cu-doped SrSnO3 samples displayed the orthorhombic perovskite with space group Pbnm as main phase, according to the ICDD 01-77-1798 index card. Based on this system, lattice parameters of the samples SrSn1-xCuxO3 (x = 0, 1, 5 and 10 mol %) were calculated, as displayed in Table 10.
The a and b lattice parameters were very similar to the theoretical data, calculated from the ICDD 01-77-1798 index card, while c parameter was slightly higher and led to a higher unit cell volume. No meaningful change was observed after doping whatever the amount of Cu added into the lattice. Inclination among the octahedra was also calculated and indicated similar absolute values comparing the undoped SrSnO3 with the theoretical value, while random variations were observed after doping. The greatest distortion was observed for SrSn0.95Cu0.05O3.
Infrared spectra of the samples SrSn1-xCuxO3 (x = 0, 1, 5 and 10 mol %), calcined at 800 °C for 4 h are displayed in Fig. 15.

Infrared spectra of the samples SrSn1-xCuxO3 (x = 0, 1, 5 and 10 mol %), calcined at 800 °C for 4 h
Bands assigned to SrCO3 were observed at 1460 cm−1 (strong), 1070 cm−1 (weak) and around 860 cm−1 (medium), confirming the formation of strontium carbonate as secondary phase, in agreement to XRD patterns. According to Mesíková et al. [60], decomposition of SrCO3 starts at 937 °C and its total decomposition occurs at ~1240 °C. In the present case, as calcination was done at 800 °C, this phase could not be eliminated. Furthermore, high temperature thermal treatments decrease the specific surface area, which is harmful to catalytic purposes.
All the infrared spectra displayed in Fig. 15 displayed bands at about 666 and 330 cm−1, assigned to the υ3 stretching mode (Sn–O) and to the υ2 bending mode (O–Sn–O), respectively. The υ3 band is characteristic of orthorhombic structures with distortion of the octahedra. These two bands were also present after Cu-doping, with no displacements. On the other hand, a small intensity band was observed at around 550 cm−1, after doping with 5 and 10 mol% of Cu, which was also assigned to the υ3 stretching mode (Sn–O) but currently observed in cubic perovskites, as discussed before. These two bands were observed for measurements in the FAR and in the MID infrared and indicated that different symmetries of the octahedral sites occur after Cu-doping. These results are in agreement with the XRD tilting angles, which indicated a higher distortion among the octahedra after Cu-doping.
Raman spectra of the samples SrSn1-xCuxO3 (x = 0, 1, 5 and 10 mol %), calcined at 800 °C for 4 h are displayed in Fig. 16.

Raman spectra of the samples SrSn1-xCuxO3 (x = 0, 1, 5 and 10 mol %), calcined at 800 °C for 4 h
Bands assigned to SrCO3 were observed in all of the Raman spectra at 148, 180 and 701 cm−1. The Raman spectrum of the undoped SrSnO3 displayed 6 active modes assigned to the orthorhombic perovskite, at 113, 167, 223, 255, 311 and 401 cm−1 [38]. Bands at 113 and 167 cm−1 are assigned to the B2g a Ag modes, respectively and correspond to the Sr-SnO3 lattice mode; the high intensity bands at 223 and 253 cm−1 are assigned to the Ag mode and correspond to the Sn-O-Sn bond; bands at 311 and 401 cm−1 are assigned to the B1g, Ag and B2g modes, respectively and correspond to the torsional Sn-O3 mode. A second order broad band was observed around 570 cm−1, which may be related to the presence of oxygen vacancies.
After Cu-doping, a meaningful decrease in the intensity of the perovskite bands of the Raman spectra was observed, compared to the undoped SrSnO3. A broadening of these bands was also observed besides a low definition especially for the sample with 10% of Cu. On the other hand, the band around 570 cm−1, assigned to oxygen vacancies is clearly observed for all Cu-doped samples. This behavior is a clear indication of the formation of defects after Cu-doping, which leads to a higher short-range disorder, with symmetry loss of the octahedra.
The UV–vis spectra of the samples SrSn1-xCuxO3 (x = 0, 1, 5 and 10 mol %), calcined at 800 °C for 4 h are displayed in Fig. 17. The electronic transitions were identified considering the deconvolution of the absorption bands.

UV–vis spectra of the samples SrSn1-xCuxO3 (x = 0, 1, 5 and 10 mol %), calcined at 800 °C for 4 h
All of the UV–vis absorption spectra display the same profile, as observed in Fig. 17, with a high intensity absorption band around 200 and 300 nm due to O2− → Sn4+ LMCT transitions [46] and low intensity absorption bands between 300 and 900 nm, assigned to d-d transitions of Cu species. For undoped SrSnO3, absorption is almost zero at this region, as expected.
It is well known that Cu2+, with a d9 electronic configuration, displays a breaking of orbital degeneracy in octahedral crystal-fields, with an eg−t2g splitting. Cu2+ is rarely observed in a regular octahedral symmetry, due to the Jahn Teller effect. When a tetragonal distortion occurs, the eg level splits into 2A1g and 2B1g levels, while the t2g level splits into 2Eg e 2B2g [52, 57]. The 2B1g is the fundamental state and a broad absorption band is observed between 650 and 900 nm, corresponding to the 2B1g → 2B2g transition of Cu2+ ions. This broad band may be assigned to the superposition of the three d-d electronic transitions, corresponding to the 2B1g → 2Eg, 2B1g → 2A1g and 2B1g → 2B2g transitions [52, 57].
When copper is added into the SrSnO3 lattice (Fig. 17), a gradual increase of the absorbance is observed with the formation of absorption bands around 340 and 440 nm due to the 3d10 → 3d9 4s1 electronic transitions of Cu+. As more Cu is added into the lattice, these bands become more intense and one more band is observed around 500 nm, also assigned to a forbidden transition of Cu+. Absorption bands were also observed around 650 and 780 nm, which were assigned to the electronic transitions of Cu2+ in tetragonally distorted octahedral symmetry.
The band gap energies of SrSn1-xCuxO3 (x = 0, 1, 5 and 10 mol %) are displayed in Table 11. The band gap value of undoped SrSnO3 was 3.8 eV, in agreement to literature data [20, 61]. After Cu-doping, smaller band gap values were observed, especially when 10 mol% of Cu was added into the SrSnO3 lattice. This behavior may be related to a greater number of defects with formation of intermediate levels inside the band gap.
The specific surface area of the samples SrSn1-xCuxO3 (x = 0, 1, 5 and 10 mol %) are displayed in Table 11. These data were used to calculate the particle size, while crystallite sizes were calculated from XRD patterns, according to the Scherrer equation. As stated before, the control of the particle size is rather difficult in the modified Pechini method due to the large amount of organic matter, eliminated by an exothermic combustion reaction. This may also influence the crystallite size, which has a random variation.
All of the results displayed in this section indicate that copper gets into the SrSnO3 lattice, with the formation of defects, as oxygen vacancies, which change the symmetry of the octahedra. The formation of the defects is displayed in Eqs. (15) and (16), according to the Kroger Vink notation and considers the presence of reduced Cu+, as indicated by the UV–vis spectra. Moreover, the desired perovskite structure was obtained at a relatively low temperature.
3.3.2 Catalytic Tests of NO Reduction by CO
Undoped and Cu-doped SrSnO3 were applied as catalysts for the NO reduction by CO, as displayed in Fig. 18. All samples were active in the NO reduction and in the CO oxidation. For undoped SrSnO3, the greatest catalytic conversion of NO into N2 was 38% and of CO into CO2 was 34%, both at 600 °C.

Conversion of NO to N2 (a) and CO to CO2 (b) using SrSn1-xCuxO3 (x = 0, 1, 5 and 10 mol %) as catalysts
Temperature increase had a positive effect in the catalytic activity for all samples, while CO oxidation was slightly greater than NO reduction. In relation to the Cu-doping, its addition into the SrSnO3 lattice clearly improved the catalytic activity for both reactions, even for the sample with only 1 mol% of Cu. This behaviour may be assigned to the formation of oxygen vacancies as charge compensation species when Cu replaces Sn4+ in the perovskite lattice. The formation of Cu+ species promotes an even greater amount of oxygen vacancies, as displayed in Eqs. (15) and (16).
The highest conversions were obtained when the sample SrSn0.95Cu0.05O3 was used as a catalyst, between 350 and 550 °C, while similar conversions were obtained for samples SrSn0.95Cu0.05O3 and SrSn0.9Cu0.1O3 at low temperatures (300 and 350 °C) and at 600 °C, which was the highest conversion temperature. At this condition, N2 and CO2 percentages of conversion were 91% and 100%, respectively. This behaviour may be assigned to a very high deformation of the lattice when 10% of Cu is added to SrSnO3, as indicated in the Raman spectra.
According to literature, oxygen vacancies have a very important role in the catalytic activity of perovskites, especially considering the NO reduction. For instance, Zhang et al. [14] also observed an improvement of the catalytic activity after Cu-doping. In their work, the perovskite LaCo1-xCuxO3 was synthesized by solid state reaction and conversion has been improved by 20% after Cu2+ addition. According to the authors, the high catalytic activity was assigned to the high density of oxygen vacancies, as they form more active site for NO and CO adsorption, as well to the presence of Cu+ ions and to the Cu+/Cu2+ redox reaction, which favour the NO reduction by CO.
As previously discussed, when NO adsorbs on the perovskite surface, an electron is transferred from the metal (active site), the nitrosyl species (NO−) is formed and the bond dissociation is favoured. The oxygen from the NO dissociation is trapped by the oxygen vacancy and the material surface is oxidized. Simultaneously, the metal is reduced by CO, by transfer of the oxygen bonded to the metal to the carbon. As a consequence, the behavior of the catalyst is related to the oxidation state of the metal before reaction (preferably reduced), its affinity with NO molecule and its redox property, which determines the tendency of the metal in the activity site to be oxidized by NO and reduced by CO during the reaction [62].
The SrSn0.95Cu0.05O3 sample was characterized by Raman spectroscopy and XRD after the catalytic reaction, as displayed in Fig. 19.

XRD pattern (a) and Raman spectrum (b) of the sample SrSn0.95Cu0.05O3 after catalytic reaction
The perovskite structure was maintained after the catalytic reaction, which confirmed the stability of the material. The formation of SrCO3 was clearly observed in the Raman spectrum and in the XRD pattern and indicated that Sr had an important role in the catalytic mechanism. This element easily forms carbonates, and it may be an important adsorption site for CO. Raman spectrum also suggested an increase of the short-range disorder after the catalytic reaction, as indicated by the smaller intensity and displacement of the Sn-O-Sn peak, from 221 cm−1 to 214 cm−1. The other bands assigned to the perovskite phase were not observed after reaction, and even the high broad band assigned to oxygen vacancies may not be distinguished.
Based on these data, a reaction mechanism was suggested for the NO reduction by CO using Cu-doped SrSnO3 perovskite as catalyst. In the mechanism, oxygen vacancies cannot be thought as isolated species, as they are associated to the cation in the lattice, changing the symmetry, which is especially important during the catalytic process, as oxygen vacancies may also undergo redox reactions, as discussed before, acting as Lewis acids or bases. During reaction NO probably adsorbs on the \([SnO_{5} .V_{O}^{\dot{}} ]_{c}\) cluster, which donate an electron to NO, forming the NO− species. The N–O bond is weakened, broken and N2 desorbs from the perovskite surface, while oxygen diffuses to the cluster, where it is trapped, probably by another oxygen vacancy. The CO species adsorbs on the \([SrO_{11} .V_{O}^{\dot{}} ]_{c}\) cluster, interacts with the oxygen from the lattice and forms CO2 or even \(CO_{3}^{2 - }\), as indicated by the characterization post-catalysis. The symmetry of the octahedra is changed during the reaction, but the overall structure is maintained. Reaction mechanism is displayed in Eqs. (17) to (22).
For Cu-doped samples, a similar mechanism may occur for NO reduction and electron transfers to and from the adsorbed molecules are favoured by the presence of Cu+/Cu2+ in the perovskite lattice, as displayed in Eqs. (24) and (25).
The reaction mechanism is schematically summarized in Fig. 20, which shows the electron transfer during the catalytic reduction of NO by CO. Cu-doping has a very important role in the process, due to the ability of this cation to easily change its oxidation state, favoring the electron transfer between the surface and the adsorbed species and also inside the perovskite lattice. Oxygen vacancies also have an important role in the process, as they favor the NO adsorption on the perovskite surface. Moreover, they also favor electron transfer, as vacancies may be present as neutral, single positive or double positive species.

Scheme of the reaction mechanism of NO reduction by CO in the presence of Cu-doped SrSnO3 as catalyst
4 Conclusions
SrSnO3 was synthesized by the modified-Pechini method, without doping and doped with different transition metals (Fe, Ni and Cu), using relatively low temperatures (800 °C), with high crystallinity. Strontium carbonate was observed for almost all synthesis and change in the octahedral symmetry was observed after doping. Specific surface area did not show a meaningful variation, as well as the lattice parameters. The catalytic tests confirmed the importance of doping in the activity of SrSnO3 towards NO reduction by CO. The highest activity was obtained using 5% of Cu as dopant, which was related to its ability to change its oxidation state, besides the formation of oxygen vacancies due to charge compensation, when it replaces Sn4+ in the perovskite lattice.
References
Song, J., Wang, Z., Cheng, X., Wang, X.: State-of-art review of NO reduction technologies by CO, CH4 and H2. Process 9, 563(1–28) (2021). https://doi.org/10.3390/pr9030563
Han, L., Cai, S., Gao, M., Hasegawa, J., Wang, P., Zhang, J., Shi, L., Zhang, D.: Selective catalytic reduction of NOx with NH3 by using novel catalysts: state of the art and future prospects. Chem. Rev. 119(19), 10916–10976 (2019). https://doi.org/10.1021/acs.chemrev.9b00202
Erickson, L.M., Newwark, G., Higgins, M.J., Wang, Z.: Nitrogen oxides and ozone in urban air: a review of 50 plusyears of progress. Environ. Progr. Sustain. Energy 39(e13484), 1–9 (2020). https://doi.org/10.1002/ep.13484
Lin, Y., Cao, Y., Yao, Q., Chai, O.J.H., Xie, J.: Engineering noble metal nanomaterials for pollutant decomposition. Ind. Eng. Chem. Res. 59, 20561–20581 (2020). https://doi.org/10.1021/acs.iecr.0c04258
Liu, C., Kubota, H., Amanda, T., Toyao, T., Maeno, Z., Ogura, M., Nakazawa, N., Inagaki, S., Kubota, Y., Shimizu, K.: Selective catalytic reduction of NO over Cu-AFX zeolites: mechanistic insights from in situ/operando spectroscopic and DFT studies. Catal. Sci. Technol. 11, 4459–4470 (2021). https://doi.org/10.1039/D1CY00282A
Shen, Q., Dong, S., Li, S., Yang, G., Pan, X.: A review on the catalytic decomposition of NO by perovskite-type oxides. Catalyts 11, 622(1–12) (2021). https://doi.org/10.3390/catal11050622
Zhang, R., Villanueva, A., Alamdari, H., Kaliaguine, S.: Cu and Pd-substituted nanoscale Fe-based perovskites for selective catalytic reduction of NO by propene. J. Catal. 237(2), 368–380 (2006). https://doi.org/10.1016/j.jcat.2005.11.019
Hodjati, S., Vaezzadeh, K., Petit, C., Pitchon, V., Kiennemann, A.: Absorption/desorption of NOx process on perovskites: performances to remove NOx from a lean exhaust gas. Appl. Catal. B 26(1), 5–16 (2000). https://doi.org/10.1016/S0926-3373(99)00143-5
Smith, J.M., Ness, H.C.V., Abbott, M.M.: Introdução à Termodinâmica da Engenharia Química. ed. 7, Rio de Janeiro: LTC (2007)
Deng, C., Huang, Q., Zhu, X., Hu, Q., Su, W., Qian, J., Dong, L., Li, B., Fan, M., Liang, C.: The influence of Mn-doped CeO2 on the activity of CuO/CeO2 in CO oxidation and NO + CO model reaction. Appl. Surf. Sci. 389, 1033–1049 (2016). https://doi.org/10.1016/j.apsusc.2016.08.035
Tanabe, E.Y., Assaf, E.E.: Óxidos do tipo perovskita para reação de NO com CO. Quim. Nova 32(5), 1129–1133 (2009). https://doi.org/10.1590/S0100-40422009000500009
Castillo, S., Pineda, M.M., Gómez, R.: Reduction of NO by CO under oxidizing conditions over Pt and Rh supported on Al2O3-ZrO2 binary oxides. Catal. Commun. 2(10), 295–300 (2001). https://doi.org/10.1016/s1566-7367(01)00049-8
Leontiou, A.A., Ladavos, A.K., Pomonis, P.J.: Catalytic NO reduction with CO on La1−xSrx(Fe3+/Fe4+)O3± δ perovskite-type mixed oxides (x = 0.00, 0.15, 0.30, 0.40, 0.60, 0.70, 0.80, and 0.90). Appl. Catal. A 241(1–2), 133–141 (2003). https://doi.org/10.1016/S0926-860X(02)00457-X
Zhang, R., Villanueva, A., Alamdari, H., Kaliaguine, S.: Reduction of NO by CO over nanoscale LaCo1−xCuxO3 and LaMn1−xCuxO3 perovskites. J. Mol. Catal. A: Chem. 258(1–2), 22–34 (2006). https://doi.org/10.1016/j.molcata.2006.05.008
Teraoka, Y., Nii, H., Kagawa, S., Jansson, K., Nygren, M.: Influence of the simultaneous substitution of Cu and Ru in the perovskite-type (La, Sr)MO3 (M=Al, Mn, Fe, Co) on the catalytic activity for CO oxidation and CO–NO reactions. Appl. Catal. A 194–195, 35–41 (2000). https://doi.org/10.1016/S0926-860X(99)00351-8
Hadjarab, B., Bouguelia, A., Trati, M.: Synthesis, physical and photo electrochemical characterization of La-doped SrSnO3. J. Phys. Chem. Solids 68(8), 1491–1499 (2007). https://doi.org/10.1016/j.jpcs.2007.03.013
Azad, A.M., Hon, N.C.: Characterization of BaSnO3-based ceramics: Part 1. Synthesis, processing and microstructural development. J. Alloy. Compd. 270(1–2), 95–106 (1998). https://doi.org/10.1016/S0925-8388(98)00370-3
Honório, L.M.C., Santos, M.V.B., Filho, E.C.S., Osajima, J.A., Maia, A.S., Santos, I.M.G.: Alkaline earth stannates applied in photocatalysis: prospection and review of literature. Cerâmica 64, 559–569 (2018). https://doi.org/10.1590/0366-69132018643722480
Zidi, N., Omeiri, S., Hadjarab, B., Bouguelia, A., Akroun, A., Trari, M.: Transport properties and photoelectrochemical characterization of oxygen-deficient ASnO3 (A= Ca, Sr and Ba). Physica B 405(16), 3355–3359 (2010). https://doi.org/10.1016/j.physb.2010.05.004
Mizoguchi, H., Eng, H.W., Woodward, P.M.: Probing the electronic structures of ternary perovskite and pyrochlore oxides containing Sn4++ or Sb5+. Inorg. Chem. 43(5), 1667–1680 (2004). https://doi.org/10.1021/ic034551c
Alves, M.C.F., Souza, S.C., Silva, M.R.S., Paris, E.C., Lima, S.J.G., Gomes, R.M., Longo, E., Souza, A.G., Santos, I.M.G.: Thermal analysis applied in the crystallization study of SrSnO3. J. Therm. Anal. Calorim. 97, 179–183 (2009). https://doi.org/10.1007/s10973-009-0242-x
Bohnemann, J., Libanori, R., Moreira, M.L., Longo, E.: High-efficient microwave synthesis and characterisation of SrSnO3. Chem. Eng. J. 155(3), 905–909 (2009). https://doi.org/10.1016/j.cej.2009.09.004
Peña, M.A., Fierro, J.L.: Chemical structures and performance of perovskite oxides. Chem. Rev. 101(7), 1981–2017 (2001). https://doi.org/10.1021/cr980129f
Song, S., Xu, L., He, Z., Ying, H., Chen, J., Xiao, X., Yan, B.: Photocatalytic degradation of C.I. Direct Red 23 in aqueous solutions under UV irradiation using SrTiO3/CeO2 composite as the catalyst. J. Hazard. Mater. 152(3), 1301−1308 (2008). https://doi.org/10.1016/j.jhazmat.2007.08.004
Ferri, D., Forni, L.: Methane combustion on some perovskite-like mixed oxides. Appl. Catal. B 16(2), 119–126 (1998). https://doi.org/10.1016/S0926-3373(97)00065-9
Zhu, J., Thomas, A.: Perovskite-type mixed oxides as catalytic material for NO removal. Appl. Catal. B 92(3–4), 225–233 (2009). https://doi.org/10.1016/j.apcatb.2009.08.008
Chen, L., Si, Z., Wu, X., Weng, D.: DRIFT study of CuO–CeO–TiO2 mixed oxides for NOx reduction with NH3 at low temperatures. ACS Appl. Mater. Interf. 6(6), 8134–8145 (2014). https://doi.org/10.1021/am5004969
Tien-Thao, N., Alamdari, H., Kaliaguine, S.: Characterization and reactivity of nanoscale La(Co, Cu)O3 perovskite catalyst precursors for CO hydrogenation. J. Solid State Chem. 181(8), 2006–2019 (2008). https://doi.org/10.1016/j.jssc.2007.11.016
Zhu, J., Zhao, Z., Xiao, D., Li, J., Yang, X., Wu, Y.: Study of La2−xSrxCuO4 (x = 0.0, 0.5, 1.0) catalysts for NO+CO reaction from the measurements of O2-TPD, H2–TPR and cyclic voltammetry. J. Mol. Catal. A: Chem. 238(1–2), 35–40 (2005). https://doi.org/10.1016/j.molcata.2005.03.036
Giannakas, A.E., Leontiou, A.A., Ladavos, A.K., Pomonis, P.J.: Characterization and catalytic investigation of NO + CO reaction on perovskites of the general formula LaxM1-xFeO3 (M = Sr and/or Ce) prepared via a reverse micelles microemulsion route. Appl. Catal. A 309(2), 254–262 (2006). https://doi.org/10.1016/j.apcata.2006.05.016
Zhang, R., Alamdari, H., Kaliaguine, S.: Fe-based perovskites substituted by copper and palladium for NO+ CO reaction. J. Catal. 242(2), 241–253 (2006). https://doi.org/10.1016/j.jcat.2006.05.033
He, H., Liu, M., Dai, H., Qiu, W., Zi, X.: An investigation of NO/CO reaction over perovskite-type oxide La0.8Ce0.2B0.4Mn0.6O3 (B = Cu or Ag) catalysts synthesized by reverse microemulsion. Catal. Today 126(3–4), 290–295 (2007). https://doi.org/10.1016/j.cattod.2007.06.004
Dai, H., He, H., Li, P., Gao, L., Au, C.H.: The relationship of structural defect–redox property–catalytic performance of perovskites and their related compounds for CO and NOx removal. Catal. Today 90(3–4), 231–244 (2004). https://doi.org/10.1016/j.cattod.2004.04.031
Wu, Y., Li, L., Chu, B., Yi, Y., Qin, Z., Fan, M., Qin, Q., He, H., Zhang, L., Dong, L., Li, B., Dong, L., Li, B., Dong, L.: Catalytic reduction of NO by CO over B-site partially substituted LaM0.25Co0.75O3 (M = Cu, Mn, Fe) perovskite oxide catalysts: the correlation between physicochemical properties and catalytic performance. Appl. Catal. A 568, 43–53 (2018). https://doi.org/10.1016/j.apcata.2018.09.022
Wood, D.L., Tauc, J.: Weak absorption tails in amorphous semiconductors. Phys. Rev. B 5(8), 3144–3151 (1972). https://doi.org/10.1103/PhysRevB
Nakamoto, K.: Infrared and Raman Spectra of Inorganic and Coordination Compounds. John Wiley & Sons, New York, Inc (1986)
Nyquist, R.A., Kagel, R.O.: Infrared Spectra Inorganic Compounds. Academic Press, New York, Inc (1971)
Moreira, E., Henriques, J.M., Azevedo, D.L., Caetano, E.W.S., Freire, V.N., Albuquerque, E.L.: Structural, optoelectronic, infrared and Raman spectra of orthorhombic SrSnO3 from DFTcalculations. J. Solid State Chem. 184(4), 921–928 (2011). https://doi.org/10.1016/j.jssc.2011.02.009
Last, J.T.: Infrared-absorption studies on barium titanate and related materials. Phys. Rev. 105(6), 1740–1750 (1956). https://doi.org/10.1103/PhysRev.105.1740
Pfaff, G., Hildenbrand, V.D., Fuess, H.: Spectroscopic study of amorphous precursors for alkaline-earth titanates and stannates. J. Mater. Sci. Lett. 17(23), 1983–1985 (1998). https://doi.org/10.1023/A:1006652405086
Azad, A.-M., Shyan, L.L.W., Yen, P.T.: Synthesis, processing and microstructural characterization of CaSnO3 and SrSnO3 ceramics. J. Alloy. Compd. 282(1–2), 109–124 (1999). https://doi.org/10.1016/S0925-8388(98)00808-1
Chiang, Y.M., Bimie III, D.P., Kingery, W.D.: Physical Ceramics. John Wiley & Sons, New York (1997)
Tarrida, M., Larguem, H., Madon, M.: Structural investigations of (Ca, Sr)ZrO3 and Ca(Sn, Zr)O3 perovskite compounds. Phys. Chem. Miner. 36, 403–413 (2009). https://doi.org/10.1007/s00269-008-0286-7
Doroftei, C., Popa, P.D., Iacomi, F.: Study of the influence of nickel ions substitutes in barium stannates used as humidity resistive sensors. Sens. Actuators, A 173(1), 24–29 (2012). https://doi.org/10.1016/j.sna.2011.10.007
Kaabar, W., Botta, S., Devonshire, R.: Raman spectroscopic study of mixed carbonate materials. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 78(1), 136–141 (2011). https://doi.org/10.1016/j.saa.2010.09.011
Liu, Z.C., Chen, H.R., Huang, W.M., Gu, J.L., Bu, W.B., Hua, Z.L., Shi, J.L.: Synthesis of a new SnO2/mesoporous silica composite with room-temperature photoluminescence. Microporous Mesoporous Mater. 89, 270–275 (2006). https://doi.org/10.1016/j.micromeso.2005.10.037
Rao, L.S., Reddy, M.S., Rao, D.K., Veeraiah, N.: Influence of redox behavior of copper ions on dielectric and spectroscopic properties of Li2O–MoO3–B2O3:CuO glass system. Solid State Sci. 11, 578–587 (2009). https://doi.org/10.1016/j.solidstatesciences.2008.06.022
Sorlí, S.M.A., Tena, J.A., Badenes, J.C., Calbo, M., Llusar, G.M., Monrós, G.: Structure and color of NixA1–3xB2xO2 (A=Ti, Sn; B=Sb, Nb) solid solutions. J. Eur. Ceram. Soc. 24(8), 2425–2432 (2004). https://doi.org/10.1016/j.jeurceramsoc.2003.07.012
Dondi, M., Cruciani, G., Guarini, G., Matteucci, F., Raimondo, M.: The role of counterions (Mo, Nb, Sb, W) in Cr-, Mn-, Ni- and V-doped rutile ceramic pigments: Part 2. Colour and technological properties. Ceram. Int. 32(4), 393–405 (2006). https://doi.org/10.1016/j.ceramint.2005.03.015
Reddy, K.N., Reddy, G.S., Reddy, S.L., Rao, P.S.: Optical absorption and EPR spectral studies of vauquelinite. Cryst. Res. Technol. 41(8), 818–821 (2006). https://doi.org/10.1080/10420150008211834
Lee, J.D.: Concise Inorganic Chemistry, 5th edn. Chapman & Hall, London, Inc (1996)
Rajyasree, Ch., Rao, D.K.: Spectroscopic investigations on alkali earth bismuth borate glasses doped with CuO. J. Non-Cryst. Solids 357(3), 836–841 (2011). https://doi.org/10.1016/j.jnoncrysol.2010.11.008
Liu, Q., Dai, J., Zhang, X., Zhu, G., Liu, Z., Ding, G.: Perovskite-type transparent and conductive oxide films: Sb- and Nd-doped SrSnO3. Thin Solid Films 519(18), 6059–6063 (2011). https://doi.org/10.1016/j.tsf.2011.03.038
Fritz, A., Pitchon, V.: The current state of research on automotive lean NOx catalysis. Appl. Catal. B 113(1), 1–5 (1997). https://doi.org/10.1016/S0926-3373(96)00102-6
Lin, C.-H., Liu, L.-G.: Post-aragonite phase transitions in strontianite and cerussite—A high-pressure raman spectroscopy study. J. Phys. Chem. Solids 58(6), 977–987 (1997). https://doi.org/10.1016/S0022-3697(96)00201-6
House, J.E.: Inorganic Chemistry. Academic Press, Canada (2008)
Rao, J.L., Murali, A., Rao, E.D.: Electron paramagnetic resonance and optical absorption spectra of Fe(III) ions in alkali zinc boro sulphate glasses. J. Non-Cryst. Solids 202(3), 215–221 (1996). https://doi.org/10.1016/0022-3093(96)00199-8
Taran, M.N., Langer, K.: Electronic absorption spectra of Fe3+ in andradite and epidote at different temperatures and pressures. Eur. J. Miner. 12(1), 7–15 (2000). https://doi.org/10.1127/0935-1221/2000/0012-0007
Dazhi, W., Shulin, W., Jun, C., Suyuan, Z., Fangqing, L.: Microstructure of SnO2. Phys. Rev. B 49(20), 14282–14285 (1994). https://doi.org/10.1103/PhysRevB.49.14282
Mesíková, Z., Sulcová, P., Trojan, M.: Synthesis and description of SrSn0.6In0.4O3 perovskite pigments. J. Therm. Anal. Calorim. 91(1), 163–166 (2008). https://doi.org/10.1007/s10973-007-8312-4
Zidi, N., Omeiri, S., Hadjarab, B., Bouguelia, A., Akroun, A., Trari, M.: Transport properties and photoelectrochemical characterization of oxygen-deficient ASnO3-δ (A= Ca, Sr and Ba). Physica B 405(16), 3355–3359 (2010). https://doi.org/10.1016/j.physb.2010.05.004
Lima, R.K.C., Batista, M.S., Wallau, M., Sanches, E.A., Mascarenhas, Y.P., Urquieta-González, E.A.: High specific surface area LaFeCo perovskites—Synthesis by nanocasting and catalytic behavior in the reduction of NO with CO. Appl. Catal. B 90(3–4), 441–450 (2009). https://doi.org/10.1016/j.apcatb.2009.04.004
Acknowledgements
The authors acknowledge Petrobras (Process Number 2014/00327-2), for the financing of this project and CNPq Proc. 151055/2012-2.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Lucena, G.L. et al. (2022). SrSnO3 Applied in the Reduction of NO by CO: Influence of Transition Metal Doping on the Catalytic Activity. In: Taft, C.A., de Lazaro, S.R. (eds) Research Topics in Bioactivity, Environment and Energy. Engineering Materials. Springer, Cham. https://doi.org/10.1007/978-3-031-07622-0_4
Download citation
DOI: https://doi.org/10.1007/978-3-031-07622-0_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-07621-3
Online ISBN: 978-3-031-07622-0
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)