
Abstract
Computation and simulation is a powerful approach to aid in characterizing, understanding, and ultimately making predictions about materials, processes, and systems for efficient and cost-effective photo-electro-chemical (PEC) conversions of solar energy to fuels, one of the essential elements of broad strategies toward renewable energy. Solar energy-driven water splitting using semi-conductor-based photo-catalysts is perceived as the most desirable opportunity. For robust PEC technologies, viable materials and systems must exhibit good visible light absorption and carrier generation, good carrier transport, and good carrier redox reactivity. Overall conversion efficiencies of systems have improved in recent years. Challenges remain to achieve the needed performance characteristics, including several fundamental science issues toward the discovery and development of semiconductor photo-electrode materials that permit high overall efficiency in devices. Modern first principles-based multi-scale approaches are proving extremely valuable in providing fundamental understanding of many experimental observations in PEC catalysis. Here we highlight illustrative examples of the application of theory and computation to study carrier transport and carrier utilization in semiconducting electrodes. The examples address strategies to enhance charge carrier separation and strategies to mitigate stability and high over-potentials in redox reactivity of carriers. The growing body of computational studies in these areas suggests a bright and impactful future of theory and computation in the field of renewable and sustainable energies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


1 Introduction
Computation and modeling is now playing a major role in essentially all areas of chemistry and materials sciences. Efficient and powerful theories have been developed and methods implemented in the past couple of decades that deal with the atomic, mesoscale, and macroscale levels of characterization and understanding of processes and systems. Beyond characterization and understanding, the ultimate objective of computation and modeling is to predict new molecules, new materials, new processes, and new chemistries that go beyond the current state of synthesis, efficiencies, and capabilities. Photo-electro-chemical conversions of solar energy to fuels comprise one of the essential elements of broad strategies toward renewable energy. In fact, solar energy-driven water splitting using semi-conductor-based photo-catalysts is perceived as the most attractive, challenging, and yet highest pay-off opportunity. In this chapter we highlight from a computation and modeling-driven perspective where computation has become a powerful contributor and can emerge as a driver of progress in solar energy conversion technologies. In Sect. 1 of this chapter, we discuss the fundamentals of PEC and give an overview of where computation has a role. In Sect. 2, we highlight our research involvement in this field in collaborations with others. We end the chapter with an outlook, in particular mentioning areas of computational and theoretical limitations and challenges.
2 Photo-Electro-Chemical Catalysis and the Role of Computation
2.1 Fundamental Science Concepts for Photo-Electro-Chemical Conversions
Efficient and cost-effective conversion of solar energy into electrical and chemical energy is widely accepted as an essential element of a broad strategy toward renewable energy [1,2,3,4,5,6,7]. In fact, solar energy-driven water splitting using semi-conductor-based photo-catalysts is perceived as the most desirable opportunity [8,9,10]. An illustration of the science of photo-electro-chemical conversion, also referred often as photocatalysis, is seen in the schematics of PEC devices in Fig. 1 adapted from Sivula and van de Krol [11], and of the chemical processes taking place in such devices yielding water splitting in Fig. 2, reproduced from Yang et al.. [12] Sunlight absorbed by an anode semiconductor material excite the electrons in the material and create electron e− and hole h+ carriers that migrate to the electrode surface to split water through water oxidation at an anode and proton reduction at a cathode. The overall process converts essentially solar energy into chemical fuels.

Schematic of a PEC device for water splitting. Under the right circumstances, absorption of sunlight in a semiconductor anode electrode gives rise to catalytically active electrons e− and holes h+. Holes go on to oxidize water while electrons go on to the cathode where they reduce protons to evolve H2. The sunlight is the source of the energy required to split water. Illustration adapted from Sivula and van de Krol [11], with permission

Light absorption and charge carrier generation, carrier transport, and carrier redox reactivity are the three critical characteristics for viable photo-catalytic systems. The present proposal addresses issues on charge carrier structure, transport, and reactivity. Illustration from Yang et al. [12], with permission
For robust photo-electro-chemical conversion technologies, viable materials and systems must exhibit good visible light absorption and carrier generation, good carrier transport, and good carrier redox reactivity [9, 11], as depicted in Fig. 2. Overall conversion efficiencies of PEC systems have improved in recent years [13,14,15,16,17]. Challenges remain to achieve the needed performance characteristics [11, 16,17,18,19,20,21] including several fundamental science issues. Among those are (a) understanding and predicting material structure and properties, and their relationships to light absorption, carrier generation, transport, and reactivity efficiencies in single, multi-phase, and/or multi-materials systems; (b) tailoring and optimizing the composition, phase, microstructure, and integration of materials to achieve superior light harvesting, charge carrier transport, and overall energy conversion performance. The biggest challenge to enabling practical PEC science and technology is the discovery and development of semiconductor photo-electrode materials that permit high overall efficiency in devices.
The physical chemistry fundamentals and challenges that come into play in PEC’s are illustrated in Figs. 3 and 4[22]. Absorption of sunlight by the semiconductor electrode is the first step in the creation of charge carriers that ultimately get involved in the redox chemistries of water splitting at the electrode/electrolyte interface. For water oxidation at an anode electrode, the h+ charge carrier states at or near the maximum of the valence band need be below the O2/H2O potential, while, at a cathode, the e− carrier states at or near the conduction band minimum need be higher than the H2/H2O potential. These points make the thermodynamics requirements clearly.

Band structure and potentials of selected semiconductors positioned relative to the Normal Hydrogen Electrode (NHE) at pH = 7, with standard potentials for selected conversion chemistries also indicated (illustration from Zhang et al. [22], with permission

Solar irradiance (Figure reproduced from https://www.fondriest.com). Visible light radiation makes up ~ 42% of light that reaches the earth, indicating that the best PEC’s will absorb light in the visible
To create charge carriers, sunlight photons need have an energy greater than the band gap of the material. The sun radiation spectrum displayed in Fig. 4 shows that ~42% of the light that reaches the earth is visible. Thus, to make good use of solar light on earth, the challenge is to have materials that absorb strongly in the visible region of light AND that have bands edges (valence and conduction bands) well positioned vs the standard redox potentials of the relevant chemistries.
2.2 Fundamental Challenges in Photo-Electro-Chemical Conversions
Based on the fundamentals enunciated above, it is possible to formulate broad strategies to improve PEC efficiency. Those include: a. band structure and band gap engineering of existing materials [15] for improved visible light absorption; b. surface engineering [16] and multi-phase or multi-material junction engineering [16] for improved charge separation; and c. hetero- and homo-junction engineering for improved light absorption, charge separation, and redox band alignment. Beyond engineering existing electrode materials for improved properties, much research is also devoted to the discovery of semiconductors materials with inherently better properties [11, 15] than current materials (TiO2, WO3, Fe2O3, CuWO4, and BiVO4—the highest performing oxide to date).
Light absorption: Band structure and band gap engineering are widely pursued to extend material absorption into the visible region and enhance carrier generation [23,24,25]. Band structure engineering includes cation and anion doping and substitution to reduce the band gap, usually through up-shifting of the valence band edge and down-shifting of the conduction band edge. We note that band structure engineering is very conducive to data science-based discovery, and recent such computational investigations have been reported [23,24,25,26,27]. It must be realized that engineering the band gap shifts also the band edges, thus affecting the redox surface chemistry at the solid/electrolyte interfaces [11, 15].
Charge carrier structure: The generation and dynamics of charge carriers in early times following light absorption has been studied extensively in recent years because of the availability of time-resolved techniques. The impetus for this research arose from the thought that detailed knowledge of charge carrier characteristics (exciton generation and lifetime) would lead to the identification of design principles, and to the design and discovery of intrinsically better photocatalytic materials. The experimental techniques to generate this information are many. They include a variety of time-resolved techniques such as transient UV-vis spectroscopies to detect the changes in UV-vis spectra due carriers’ thermodynamic and structural features [28,29,30] and transient microwave conductivity (TRMC) measurements [31, 32]. Other approaches include time-resolved photo-charge (TRPC) measurements for lifetime of charge carriers [33, 34] and for electron-hole separation distance [35]; and time-resolved terahertz THz spectroscopy (TRTS) for ultrafast carrier dynamics and conductivity mechanisms and measurements [36, 37]. Electron paramagnetic resonance (EPR) is also useful to get information about trapping of photogenerated electron [38]. Transient extreme—ultraviolet (XUV) spectroscopy is an emerging powerful capability. A notable point is that it yields descriptions of exciton and polaron structures and dynamics in terms of ‘localized’ atomistic and chemical concepts as the absorption is interpreted in terms of oxidation states of atoms [39,40,41,42,43]. Theoretical methods have emerged also recently that afford the characterization of exciton structures and lifetimes, based on non-adiabatic molecular dynamics combined with density functional theory and other wavefunction types [44,45,46,47], with recent applications in photovoltaics [48,49,50].
Carrier transport: The overarching issue about ‘carrier transport’ is charge separation. Promoting charge separation means reducing charge recombination and making use of as many e− and h+ carriers as possible that were created by light absorption. This number is one of the quantifiers of conversion efficiency. Physical situations that promote charge separation include single phase facet selective materials [51, 52], multi-phase and/or multi-material junctions [53, 54], single-phase homo-junctions [55,56,57], and the use of cocatalysts [12, 58, 59]. Surface engineering [16] refers to utilization and manipulation of all these situations. Crystal facet engineering and interface engineering are emerging as promising approaches to enhance carrier separation and overall water splitting efficiency, both for hydrogen evolution and oxygen evolution [51, 54, 60, 61]. Facet engineering deals with the growth of crystal facets that exhibit selectively either reduction or oxidation chemistry. Facet selectivity has been reported for a number of semiconductor materials, including bismuth vanadate BiVO4 (BVO) [51], strontium titanate SrTiO3, [62] copper oxide Cu2O [52, 63, 64], and titania TiO2, [61, 65, 66] for example. Other examples include tungsten oxide WO3, gallium oxide Ga2O3, tantalum nitrite Ta3N5, and several tantalates. Interface engineering is the design of mixed-phase interfaces such as in TiO2, [67,68,69] and gallium oxide Ga2O3, [53] or multi-materials interfaces such as BiVO4/ Cu2O [63], WO3/TiO2, [70] WO3/BiVO4, [71] and many other combinations, all interfaces that enhance charge separation.
Redox chemistry at electrode/electrolyte interface: There are numerous studies about solar fuels [9, 17] (water splitting, CO2 and CO reduction and conversion, along with N2 reduction to NH3) but a relatively small number have focused on understanding the details of the chemical reactions at the surfaces in contrast to the more global and effective conversion efficiencies. From the perspective of overall solar conversion efficiency, detailed knowledge of surface reaction mechanisms and kinetics is needed, as importantly as details about light absorption, carrier generation, transport and separation [15]. For surface reaction kinetics, transient IR spectroscopy (TRIR) is playing an increasingly important role when attempting to identify and characterize the reactions intermediates [72,73,74]. It has been used for a number of materials, including BiVO4, TiO2, SrTiO3, and others. For water splitting, the emphasis has been on water oxidation as it is the more challenging process compared to proton reduction. [75].
Water oxidation with O2 evolution (OER) is most often discussed in terms of a four proton-coupled-electron transfer steps with *OH, *O, and *OOH intermediate species [76]. The design of better catalysts relies on Sabatier’s principle of bonding strength [77]. Still, not all cases fall into this framework. Nucleophilic attack of hole-carrying surface lattice oxygen sites have been proposed as mechanism [73]. Other proposals include O-O coupling in adsorbates (adsorbate evolution mechanism AEM) or direct O-O coupling to a lattice oxygen (lattice oxygen oxidation mechanism LOM) [78]. To date, to the best our understanding, it remains challenging to predict which mechanism applies to which class of materials. Even if volcano plots can be derived from universal scaling of binding energies of intermediates *OH and *O, there are large differences in binding strengths within classes of materials (oxide perovskites are an example) [79]. An increased understanding of the structure-redox activity of materials and of the chemical factors that govern the reaction mechanisms is highly desirable.
2.3 Computation and Simulation for Photo-Electro-Chemical Catalysis
The field of photo-electro-chemical catalysis is ripe for the application of existing computation and simulation methods, and the development of new methods for the characterization of the fundamental science in the three phases of PEC operations outlined earlier. The scientific challenges that can be addressed by computational means are summarized in Table 1.
2.3.1 Computational Materials Screening and Discovery
Characterization of processes in the phase of light absorption and carrier generation fall under the umbrella of ‘computational materials screening and discovery’, some aspects of it were mentioned earlier. The objective is to discover improved or new materials that absorb strongly in and near the visible range of light and generate e− and h+ charge carriers with a high degree of efficiency while exhibiting low recombination rates. Systematic calculations of material structure and stability, of their phonons, of their band gaps and band structures, of the effects of doping and defects (such as vacancies), and of their work functions, are widely pursued and found in the literature [23,24,25]. They are aided by modern approaches of machine learning (ML) and artificial intelligence (AI) as extensive databases have become well established and widely used, such as from The Materials Project and from the AFLOW consortium [24,25,26, 80,81,82,83].
A challenge affecting selected transition metals semiconductors deals with d-d transitions. Such transitions occur in semiconductors with partially occupied d bands, such as hematite Fe2O3. Hematite absorbs light in the visible range, around ~2 eV, a range that is highly desirable in the quest for materials that are chemically photo-active while absorbing a large fraction of solar radiation. However, excitations of d electrons within the d band occur in the same range of excitation energies, they are intra-band excitations and do not lead to e−/h+ separation because of the electronic structure of the resulting excited state, in contrast to valence-to-conduction band excitations that lead to carriers that are mobile within the separated bands. In these situations of d-d transitions, the materials are plagued with absorption that is not efficient for photocatalytic e−/h+ activity. The characterization, understanding, and control of d-d transition remains a challenging problem of computational modeling and design of efficient PEC devices.
2.3.2 Computational Modeling of e−/h+ Carrier Dynamics in Crystalline Single-Phase, Multi-Phase, and Multi-Materials Systems
Upon creation of charge carriers, the dynamics of carriers are the essential elements of the phenomena involved in carrier transport and separation. Their computational modeling falls under the umbrella of ‘computation modeling of e−/h+ carrier dynamics in crystalline single-phase, multi-phase, and multi-materials systems.’ Issues are the carrier transport proper, be it by band transport [84] or by polaron hopping with activation barriers [85,86,87,88,89,90]. Transport in crystalline semiconductors is affected by dopants and defects that may lead to trapping (thus reducing transport efficiency) and defect-mediated recombination (thus eliminating e− and h+ and affecting overall efficiency). Trapping energies can be obtained from first-principles calculations. To the best of our knowledge, computational investigations of recombination are not widely carried out. Both trapping (relatively ‘standard’ calculations) and recombination (challenging theoretical models) can be addressed with solid state quantum chemical calculations.
The overarching objective here toward tailoring efficient PEC materials is to enhance charge separation to increase PEC overall efficiency (number of molecules of H2 and O2 that evolve per absorbed photon. Several strategies are being pursued, that fall in three main categories: 1. synthesis of electrodes with designed and tailored structures and that exhibit facet selectivity, whereby oxidation and reduction occur at specific facets; 2. phase separation in junctions of materials that exhibit more than one stable phases. Titania and gallium oxide are two examples mentioned above with experimental and computational evidence that e− carriers are more thermodynamically stable in one phase and the h+ carriers in the other phase. Multi-material junctions and co-catalysts are other design features that are particularly conductive to enhanced charge separation, again owing to the different thermodynamic stability of e− and h+ among materials. Obvious examples are those of e−/h+ transport layers, with junctions between materials enducing charge separation due to differential thermodynamic stability between electron and hole carriers.
2.3.3 Computational Modeling of e−/h− Catalytic Utilization
This domain of modeling encompasses all the aspects of chemical reactivity by the charge carriers e− and/or h+ at the semiconductor/electrolyte interfaces. In the context of water splitting, the hydrogen evolution reaction (HER) occurs at a cathode while the water oxidation reaction (oxygen evolution reaction OER) occurs at an anode. (In the chemical equations below, * represent an empty site for adsorption and H*, or OH*, or X* in general, denotes an H atom adsorbed at that site, or an OH or X group adsorbed at that site).
The multi-step mechanism of HER has been the subject of much research, an informative review of which is given by Lasia [91]. The mechanism is a combination of the Volmer reaction and the Heyrovsky electrochemical reaction or the chemical hydrogen desorption reaction.
In acidic conditions, the steps have the form:
for an overall reaction:
In alkaline conditions, the equivalent steps have the form:
for an overall reaction:
Similarly, the OER mechanism is written as a sequence of four coupled (proton + electron) steps, popularized by the group of Norskov: [76]
for an overall reaction:
The computational modeling of the carrier utilization involves the determination of the reaction energy profiles associated with the formation of the intermediates as listed above. Knowing these profiles gives an indication of the ‘thermodynamic over-potential’, the energy beyond the minimum energy required to dissociate H2O into O2 plus four (proton + electron) pairs (4.92 eV) in water oxidation. Note that, most often computational studies do not focus on determining transition states and activation barriers for the elementary steps, but simply on the thermodynamics stability of the intermediates, keeping in mind that activation energies and thermodynamics stability are connected by the Bell-Evans-Polanyi principle [92, 93].
3 Computation and Modeling of Carrier Structure, Dynamics, and Utilization
In recent years our group has pursued a research program focused on the science of PEC from a computational focus strongly integrated with experimental investigations by collaborators. Our emphasis has been on theory and simulation of ‘carrier transport’ in PEC systems. We highlight below some of this work and the key findings. We also point to new subjects of investigation as we view them timely and of fundamental importance considering current happenings in this field of research. However, ahead of this, we think it is important to make two significant observations that bring a stronger light of impact and timeliness to the research ideas. One is about ‘facet selectivity’ and its potentially far-reaching impact beyond water oxidation and PEC’s. The second is about exciton structure and dynamics, and the far-reaching cross-validation between theory and experiment in this field.
The first remark is about intrinsic facet selectivity. Many materials (as discussed) exhibit the property whereby, within a single crystal, some crystal facets are active in oxidation, and other facets are active in reduction. The property was discovered about water oxidation on BVO as reported in Nature Communications [51]. That report inspired our research. In a very recent paper entitled “Facet-dependent active sites of a single Cu2O particle photocatalyst for CO2 reduction to methanol” in Nature Energy [94], Wu et al. showed that facet (110) of a single Cu2O crystal is active for CO2 conversion to methanol as a photocatalyst, while facet (110) is inert. In addition, Cu2O oxidizes water as it reduces CO2. In other words, Cu2O performs the two functions, with water oxidation as the source of protons for the reduction of CO2. CO2 conversion is arguably the world’s greatest challenge in energy and environment. This report renders the research on facet selectivity in photocatalytic particles even more relevant and potentially impactful.
The second remark is about the ‘chemical’ characterization of exciton structures from XUV spectroscopy [39,40,41]. In brief, the XUV technique permits the identification of an element’s oxidation state in ultra-fast time-resolution. For hematite Fe2O3, [40] a ‘benchmark’ photo-electrode materials that has the merit of very favorable light absorption characteristics and natural abundance, the Baker group describes how charge localization and small polaron formation occur within ~660 fs, and that the electron-hole e−/h+ exciton, created within 100 fs, has a radius of a single Fe-O bond length. For CuFeO2, an earth-abundant metal oxide that is attracting much interest as a photocathode [15, 41, 95] for proton reduction by photo-generated electrons, the Baker group reported that O 2p holes thermalize within ~500 fs into Cu 3d hybridized 3d holes. Beyond the novel insights that come from the ability to elucidate site-specific charge carrier dynamics in real time, the ‘localized chemical’ picture of excitons is particularly noteworthy and exciting for electronic structure theorists. Traditionally, electronic structure in the solid state is discussed in terms of band theory (valence band, conduction band, band gap, gap states, exciton, band bending) most often determined from density functional theory (DFT). The results are not easily translated into ‘localized’ concepts of chemistry. We view the Baker results as a unique opportunity to validate experiment by theory using advanced quantum chemical theories, and, conversely, validate the quantum chemical theories by experiment. We will come back to this point later in Sect. 3 of this chapter on ‘Challenges and outlook’.
3.1 Overview of Our Multiscale Modeling Framework
As indicated above, the overall research ideas and directions about PEC’s deal with the timely societal challenges of renewable energy, specifically the efficient and cost-effective conversion of solar energy to electrical and chemical energies. The scope of this use-inspired fundamental research program centers around fundamental characterization of solar energy-to-fuels conversion systems, with theoretical studies in the three stages of photo-electro-chemical PEC conversion, mainly ‘light absorption’, ‘carrier transport’, and ‘carrier reactivity’ as illustrated in Fig. 5. The research aims to advance fundamental knowledge and understanding in PEC to lead ultimately to the design of systems with enhanced overall conversion efficiencies.

Combining first-principles computation and mesoscale kinetics modeling is critical for qualitative and quantitative understanding of the science and performance of photo-electro-chemical devices accounting for light absorption and carrier generation, carrier transport, and carrier redox reactivity. Initially the modeling will provide the foundation for crystal facet engineering and interface engineering as they pertain to the rational design of efficient solar energy conversion materials
Among the three research thrusts elaborated earlier, one focuses on ‘carrier transport’, with the aim to understand, characterize, and generate design principles to control the factors that lead to enhanced separation of photo-generated electron and holes toward higher solar energy-to-fuel conversion efficiency. To this end we view it as a powerful approach to model the space-charge distribution dynamics of charge carriers by combining first-principles atomistic computation and mesoscale kinetics simulation. The research relies on existing software tools DFT-based quantum mechanical (QM) modules as well as our group’s own developed QM tools and a powerful and versatile lattice based kinetic Monte-Carlo (KMC) capability. The early applications of these tools are starting to provide the theoretical foundations for strategies to enhance carrier transport and conversion efficiency. At the most fundamental level, the research addresses how the flow of charge carriers in complex crystalline environments of single phase, multi-phase, and multi-materials semiconductor systems can be tailored to enhance redox reactivity in photo-electro-chemical conversion. This research aligns well with broad themes addressing ‘charge transport and reactivity’ and ‘chemistry at complex interfaces.’
Our general modeling framework is depicted in Fig. 6. It starts with solid state DFT calculations of single polarons in a supercell, with their localized character and the localized lattice distortion associated with polarons. Then, site-to-site hops get identified and the energy profiles associated with the hops are determined via DFT calculations along a pathway whereby the polaron structure at the initial site is morphed into the polaron structure at the final site. The calculated profile approximates closely the profile displayed in Fig. 6. The hopping rate is determined from Marcus / Holstein theory, based on the rate expression for diabatic hopping or for adiabatic hopping (thermal hopping assisted by phonons). The calculated rates serve as input to the lattice based larger scale model of a solid or particle using the kinetic Monte Carlo methodology to describe the spatial and temporal dynamic distribution of a set of polarons, the number of which can be chosen consistent with experimental concentration of charge carriers. This multiscale methodology has been used and described in many publications by our group and other groups as well [86, 87, 90, 96,97,98,99,100,101,102,103,104].

General framework of multiscale modeling of carrier transport in semiconductor electrodes of PEC systems: from left to right, DFT calculations of polaron structure and hopping via the Marcus/Holstein theory, to KMC modeling of the collective dynamics of multiple polarons at experimentally relevant concentrations of charge carriers. The diagram of the potential energy profile is reproduced from Rettie et al. [85] with permission
About DFT for polarons: From a theoretical standpoint, the ability to obtain qualitatively correct descriptions of localized polarons hinges on overcoming the self-interaction error that plagues current functionals of the density that are the foundation of DFT. Many current polaron calculations by us [87, 97, 105, 106] and others [107,108,109,110,111,112] use the DFT+U methodology to overcome the self-interaction error of DFT. Admittedly, the approach has an ad hoc aspect in the choice of the on-site Hubbard+U parameter, and some prefer the use of hybrid functionals [113,114,115]. Localization of the unpaired electron or hole is the essence of the Marcus/Holstein small polaron model. Practical experience shows that the activation barrier is not overly sensitive to the choice of +U value. Nonetheless, care must be applied. In the same vein, the amount of exact exchange in hybrid functionals varies from one functional to the other. Care must be applied here too. In any case, we note a broad similarity in results between DFT+U and the popular PBE0 and HSE06 hybrid functionals for a number of oxide perovskites (including strontium, barium, and lead titanates, STO, BTO, and PBO) [112]. Our use of a +U parameter on oxygen to ‘create’ localized holes on O atoms is less pervasive in the literature than +U on metal ions, but it is found [74, 101, 107]. While there are examples in the literature that +U on oxygen does not introduce significant artifacts in the DFT wavefunction [101], even with +U(oxygen) ~8 eV, a value used by us [101] and others [74], we recently demonstrated that, in the case of BVO, the +U vs hybrid is not a resolved issue, and that varying the fraction of exact exchange in hybrid functional leads to substantially different descriptions of h+ polarons in BVO [102]. This issue is elaborated upon further down in Sect. 3 of this chapter on ‘Challenges’. ‘Tricks of the trade’ to generate polaronic wavefunctions are described in a recent review [86].
About Lattice based KMC for charge carrier dynamics and transport: We use the kinetic Monte Carlo KMC methodology embodied in a new Python code to characterize the temporal and spatial carrier dynamics in semiconductor electrodes. A flowchart for the KMC code is shown in Fig. 7, as an object-oriented framework written in Python language that is hosted as a repository on GitHub. The code consists of three essential components, a ‘Model Engine’, a ‘KMC Engine’, and a ‘Analysis Engine’. The ‘Model Engine’ includes two object definitions, one called ‘Material’ with all physical parameters related to the material of interest, system size, atomic coordinates etc.; the other object is ‘Neighbors’ with the lists of hopping neighbors. The second component of the code, the ‘KMC Engine’, includes a collection of ‘objects’ driving the execution of the stochastic ‘moves’ corresponding to processes for which the rate constants are provided, and therefore treated in the model. The “Analysis Engine’ includes a collection of objects to analyze the KMC trajectories of the charge carrier distribution.

Flow chart of PyCT, a Python-based charge transport, cross-platform, object-oriented, lattice based KMC code for carrier transport [89]
A simulation starts with a random distribution of excess charges (−1 for electrons, +1 for holes) on lattice sites that are assigned the full oxidation state charges of the constituent ions of the material. The number of excess charges should be consistent with the charge carrier concentration resulting from the absorption of light. The ‘Model Engine’ object includes the species locations and their associated hopping neighbors and generates a list of candidate hops and occupation states. The ‘KMC engine’ uses the variable step size method (VSSM) implementation [116] of KMC with the elementary rates coming from first-principle calculations (preferably) or from experiments.
The interactions governing the system are electrostatic (long range) in nature and individual rates are affected at any given time ‘t’ by the distribution of charges at that time, requiring the evaluation of the change in energy upon a ‘move’. An ‘object’ calculates the change in energy and the concurrent change in the activation barrier and associated process rate (using the Bell-Evans-Polanyi principle [92, 93]), making use of the Ewald summation approach. For example, an electron involved in a set of possible hops (one of which gets randomly selected) will experience an increased hopping barrier (slower rate) when the hop brings the electron closer to other electrons (increased electrostatic repulsion) or a decreased hopping barrier (faster rate) when the hop takes the electron farther away from other electrons (decreased electrostatic repulsion i.e. electrostatic stabilization). The treatment of long-range electrostatics in these charge models for polaron transport required the use of the Ewald summation [100].
Lattice based KMC simulation is particularly well suited [116,117,118] to describe the dynamics of space-charge distribution in complex systems such as those considered here. KMC is a stochastic approach to solving coupled kinetics equations for a set of processes with varied rates (see Fig. 5). Processes of interest here include all processes dealing of the fate of charge carriers: photon absorption, exciton creation, dynamics, and recombination, carrier separation, diffusion, and recombination, and carrier reactivity. They span many orders of magnitude in time scales: a. bulk exciton dynamics, charge carrier formation, carrier separation, carrier recombination, carrier trapping, with time scale from femtoseconds up to milliseconds; b. interfacial charge transfer with time scales from femtoseconds to nanoseconds; and c. surface reaction processes in the case of photocatalytic systems, with time scales from microseconds to seconds. A lattice based KMC model allows also to account for local effects (near surface vs. bulk trapping, vacancy and doping defects at relevant concentrations, phase boundaries, interfaces, and homo-junctions) as it provides a spatial and temporal resolution of space-charge distribution. Our recent KMC studies of BVO and W/Mo-doped BVO highlighted below illustrate the unique insights afforded by our (QM+KMC) model. It is worth noting that hopping barriers for the systems of interest here (BVO, STO, W-doped BVO, TiO2, and Ga2O3) are in the range of ~0.1 eV up to ~0.5 eV, while thermodynamic junction potentials in mixed-phase systems due to band alignment (rutile-anatase mixed-phase TiO2, mixed-phase α-β Ga2O3) are in the range of ~0.2 to 0.4 eV [53, 119,120,121,122]. Accordingly, KMC modeling of mixed-phase interfaces makes sense and ought to provide a good description of space-charge separation dynamics induced by interfaces.
The rate constants used in the KMC model may come from experiment or from theory. A complete characterization of transport and PEC conversions efficiency requires that KMC include all relevant processes, each described at an appropriate level of accuracy. Thus, comparison with experimental data is important. Through a sensitivity analysis, KMC can also reveal which processes affect the overall efficiency most. With lattice based KMC, we can introduce inhomogeneity in the model, due for example to defects such as doping and vacancies. Defects can be readily introduced at concentrations relevant to experiment, including with a concentration gradient (homo-junction [57]). In these cases, it is necessary to calculate the rates for processes that occur in the vicinity of defects and to include them in the list of possible KMC ‘events’. If needed, defect migration could also be included among the KMC processes. Lattice based KMC model is also well suited to capture the spatial inhomogeneity in light absorption, whereby absorption is depth-dependent and strongest near the crystalline surface.
We note that KMC simulations have been used to investigate thermal catalytic processes on surfaces [116] and also carrier transport in organic photo-voltaic systems [123, 124], both of which are distinctly different from transport in crystalline systems due to the nature of the crystalline network and the underlying interactions. To the best of our knowledge, very few studies have used lattice based KMC to study charge and ion transport in crystalline systems [118, 125]. Our objective has been to go beyond that work and build a set of tools that will allow us to increase understanding of photo-electro-chemical conversion systems in all phases of absorption, transport, and redox reactivity. Lastly, we aspire to use ab initio data in preference over empirical data as much as possible to, ultimately, have predictive ability and gain new insights that are hard to extract from experiment.
At present, we continue to address facet selectivity in BVO. We are at a stage where we need to include ‘surface reactions’ among the KMC events and e−/h+ separation in homo-junctions (W-doped BVO), a task that requires a 2D periodic KMC implementation to model gradient doping. Other systems to investigate could include facet-selective STO, and charge-separating mixed-phase interfaces (TiO2 and Ga2O3). In all cases, we will determine the space-charge distribution dynamics of charge carriers in realistic computational models with shapes and sizes relevant to experiment (see Figs. 8 and 9).


Considering the significance of the discovery of facet selectivity in Cu2O for photocatalytic water oxidation concurrently with CO2 reduction, we will want to investigate carrier transport in Cu2O. A number of DFT studies of Cu2O have appeared that discuss the nature and origin of carriers, but none have addressed transport [64, 126,127,128,129]. Conduction in Cu2O is known to be polaronic [126]. Clearly, characterizing computationally h+ polarons in Cu2O is challenging in itself due to the electron-rich 3d states of Cu contributing strongly to the top of the valence band. Electron correlated electronic structure calculations beyond DFT are likely to be required as a prerequisite to periodic DFT calculations.
Nanoparticle sizes in experiments are upward of ~500 nm in size. Our initial simulations dealt with cell sizes of ~50 Ǻ and up to 50 e− or h+ carriers. Parallel implementation and execution of the code should allow simulations up to ~100 to 200 nm in size with ~100 electrons. Such sizes and carrier densities are in regimes relevant to experimental situations. Note that h+ mobility simulations are significantly more demanding than e− mobility simulations owing to the larger number of anions (oxygens) compared to cations (metals). Two capabilities that ought to be implemented to model finite size particles and facet selectivity include the treatment of free-space boundaries (in KMC configurational energy calculation by Ewald summation) and the inclusion of surface reaction events among the KMC events that carriers can undergo.
Regarding surface reaction events, in our lattice based KMC model, carriers occupy lattice sites at any given time. Their next moves are stochastically determined among the possible events for these sites. To include surface reactions in the model, we need to append a new event, a ‘OER redox reaction event’, to the list of events available to carriers at surface exposed sites. We will need reaction rates for these ‘surface reaction events’, quantities that we can determine from the Marcus-Gerischer model for electron transfer at electrodes [130,131,132,133] following the protocol by the Bieberle-Hutter group for water oxidation on hematite Fe2O3, based on the four intermediates of the traditional PCET mechanism [134, 135]. Each step has a Gerisher rate determined from the DFT step free energy and the valence band maximum level. The needed data are available for the (010) and (011) facets of BVO [136], and can be easily determined for STO and other semi-conductors. To insure consistency of data from one surface to another, we can use the facet work function or the bismuth Bi or Vanadium V core levels to align the VB and CB energy levels across facets [101]. We should be able to extract a single effective OER rate from the four elementary rates to be used in the KMC model.
An important question to address is accuracy in KMC modeling. By in large, mobility data extracted from KMC simulations are most sensitive to the activation barriers of the processes, owing to the exponential dependence of hopping rates, both in the non-adiabatic and in the adiabatic regimes. It is worth noting that kBT at 300 °K is ~0.026 eV. Accordingly, a change in ΔG* by ~0.060 eV brings a factor of ~10 change in mobility. Reaching this level of accuracy (nearly chemical accuracy in chemistry) is a tall order but it is not out of reach. DFT calculations mentioned earlier for BVO yielded activation barriers of ~0.37 eV for electron polarons (without the electronic coupling adjustment), compared to ~0.30 eV experimentally [100]. Previous calculations on Fe2O3 and TiO2 yielded similar or closer agreement [87, 97, 105]. Nonetheless validation of this type is constantly useful and required.
In a brief conclusion of where we are to date, we note that we have are able to model separately e− and h+ polarons and their dynamics in semiconductors like bismuth vanadate BiVO4. [89, 90] The methodology is however general and can be extended to more complex modeling that include other elementary processes, such as polaron generation, polaron recombination, and polaron surface redox reactivity, as long as we have rates. Extending the mesoscale modeling of the PEC systems, through combined QM and lattice based KMC studies has the potential to lead us to solid theoretical foundations of facet selectivity in nanoparticles. This effort will take us closer to a more global simulation model of PEC’s from the level of atomic-scale description, all the way to microscale description, for example in nanoparticles of BiVO4 and SrTiO3 for which experimental data is available. We should remark that there are a great many number of materials that are being evaluated, experimentally mostly, for use as anode and cathode materials [15]. There might be merit in investigating their transport properties systematically by computation and simulation. Nonetheless, resolving our understanding of the phenomenon of facet selectivity is likely to be uniquely impactful with great prospects in technology.
3.2 Mesoscale Modeling in Photo-Electro-Catalysis
As indicated earlier, our vision, depicted in Fig. 5 above, is to have the ability to simulate the overall efficiency of solar-to-fuel conversion in photo-electro-chemical devices [137]. The modeling starts from first-principle characterization of all essential elementary processes in light absorption, carrier transport, and redox reactivity (across several orders of magnitude in time scales). The modeling continues with the determination of the complex and collective kinetics of carrier transport and activity in mesoscale models [125, 138]. The ultimate outcome is the conversion efficiency, e.g. the number of product molecules per absorbed photons, a number which depends on the three phases of PEC conversion (absorption, transport, and redox reactivity). For a given system, one or more of these phases could be the limiting factor.
Our initial efforts have been on ‘carrier transport’ broadly speaking, including the development of necessary tools to model ‘transport’ qualitatively and quantitatively. We stipulated that both quantum chemical tools and mesoscale kinetic tools would be required and that the ability to combine first-principles characterization with mesoscale kinetics modeling would prove to be critically important for both qualitative and quantitative understanding. Our BiVO4 studies to date bear this point [90, 100]. The necessity to combine scales is likely to be pervasive due to the complexity of materials and device architectures for PEC conversion.
With regard to ‘carrier transport’, physical situations that promote charge separation and, as a consequence, conversion efficiency include multi-phase and/or multi-material junctions [53, 54], single-phase homo-junctions [55,56,57], and the use of cocatalysts [12, 58, 59]. Crystal facet engineering and interface engineering are emerging as highly promising approaches to enhance carrier separation and overall water splitting efficiency, both for hydrogen evolution and oxygen evolution [51, 54, 60, 61]. At the present, synthesis efforts in this field appear Edisonian. We envision that our efforts with multi-scale computation and simulation will be an important contribution toward reaching rational design.
Facet engineering (Fig. 8) involves the synthesis of crystals with exquisite control of facet growth, and a concomitant separation of photo-excited electrons and holes [51, 60]. For example, in bismuth vanadate BiVO4 (BVO) photo-generated electrons accumulate mainly on (010) facets and holes on (110) facets as inferred from noble metals (Au, Ag, and Pt) depositing selectively on exposed (010) facets (reduction) and metal oxide (PbO2 and MnOx) particles on (110) facets (oxidation) [51]. Similar observations exist for several other semiconductors [61, 139]. Perhaps a most dramatic example is found in the observation of facet selectivity for 18-facet cubic strontium titanate SrTiO3 (STO) and the absence of facet selectivity for 6-facet cubic STO [60, 140] as illustrated in Fig. 9.
Several explanations have been put forward to rationalize the observed selective photo-deposition: a. preferred migration of holes and electrons to specific crystal facets under photo-illumination [12]. b. preferred selective sorption of metal and oxides on surface sites [13]. c. different conduction band and valence band levels for different facets. d. different internal electric fields along particular directions [11]. In catalysis, corners, edges, kinks are often thought of as sites with higher activity because of the stronger electrostatic potentials and fields at these sites. While such an argument may apply here as well, it does not explain why deposition/precipitation from oxidation or reduction occurs all over the flat surfaces, including away from corners.
Below, we will highlight progress we have made in addressing facet selectivity in BVO. To date we showed that there is no directional anisotropy in e− or h+ transport (no kinetic factor of facet selectivity). We established that h+ transport is bimodal, with a rattling mode that is not transport-efficient and a slower transport-efficient mode [100]. We showed also that the near-surface thermodynamic stability of e− or h+ is qualitatively similar for different facets (no thermodynamic factors of facet selectivity) [101]. These findings point to the need to account for the kinetics of the interfacial redox reactions to ‘see’ facet selectivity in simulation. In addition, we considered sulfur-doped BVO and established that sulfur-to-sulfur hopping transport of h+ replaces the ineffective bimodal oxygen-to-oxygen transport. These observations are very significant with regards to modeling PEC systems. Indeed, they are a clear demonstration that mesoscale modeling is essential to characterize and understand the attributes of PEC systems. Mesoscale modeling captures the critical connection between elementary hopping rates and crystal structure and topology with regards to transport. Interpretations based on elementary hop characterization, even if at the quantum level of theory, have the potential to be misleading.
Multi-phase junctions and homo-junctions: multi-phase junctions have long been thought as promoting redox activity. Indeed mixed-phase titania TiO2 is known to be more active than single phase titania [141,142,143]. It is also well established that the degree of interconnectivity between crystallites and the structure of grain boundaries affects transport of carriers in materials [144,145,146,147,148,149,150]. Characterizing, quantifying, and understanding the fundamental parameters that control the diffusive e−/h+ transport across structurally complex interfaces is essential for an overall assessment of transport. Homo-junctions are ‘interfaces’ within a material that occur at the boundaries of layers with different levels of doping (more about homo-junctions below). While doping is generally used to increase the concentration of charge carriers and tune band edges, doping with a gradient concentration has been shown to enhance carrier separation by creating homo-junctions, and leading to increased overall catalytic efficiency [55,56,57]. It is noteworthy that doping levels in homo-junctions are low and yet, they affect carrier transport, separation, and conversion efficiency at the larger scale. The characterization of these effects requires mesoscale modeling. Their physics are traditionally described in terms of band bending and carrier depletion regions. In our work, we like to give a chemistry-based, atomistic-derived description and characterization at the mesoscale of carrier transport across multi-phase junctions and homo-junctions with doping levels relevant to experiment.
In summary, both facet engineering and junction engineering share the common trait regarding carrier transport simulation: they require combined atomistic and mesoscale kinetic modeling to provide a fundamental understanding of the factors that create and affect space-charge distributions and their temporal evolutions. The combined modeling can give important insights on what the bottlenecks for ‘good’ transport are. The ability to characterize and predict these effects will ultimately enable the control of carrier transport, separation, and redox reactivity.
3.3 Highlights
The overarching theme of our PEC research is to characterize and model the transport of photo-generated electrons and holes in complex crystalline materials to establish the theoretical foundation for facet and interface engineering, two strategies used to tailor charge separation and to promote redox activity. The enabling elements of these strategies originate in the dynamics of charge carriers at the mesoscale. The leading factors that control charge separation can be several, thermodynamics, dynamics, or surface reactivity. Manipulating carrier separation in facet-selective, mixed-phase, and doped systems offers opportunities to control and enhance solar energy conversion efficiency.
Describing the mesoscale behavior of carriers is a challenge that requires modeling beyond traditional ways of single species (e− or h+ polaron) and individual processes (polaron stability, trapping, and hopping), even if done at the quantum chemical (QM) level of theory. Our approach to investigating facet and phase selectivity has been to create lattice based kinetic Monte Carlo (KMC) models of space-charge distribution dynamics and reactivity based on individual processes characterized at first principles levels of theory (Fig. 6). Note that it is critically important to validate the models using relevant, available, computed and/or experimental data for well-characterized materials. To accomplish these tasks, we developed, as needed, selected original QM and KMC tools and methodologies.
To date, we have made major progress toward this goal, to the point that we are getting closer to being able to use the model to formulate and predict design points for absorption, transport, and reactivity for enhanced overall photocatalytic performance. We are at a point where we need to include surface reaction processes in our mesoscale models. This is a major objective at present. In addition, we have identified selected other important fundamental contributions that we can make and that will advance the state-of-the-art in studies of condensed phase and interfacial molecular science in photo-electro-chemical conversions.
3.3.1 Carrier Transport: The Case of Bismuth Vanadate BVO
Our goal was to model e−/h+ transport in doped and undoped materials such as bismuth vanadate BiVO4 (BVO), tantalum nitrite Ta3N5, and other materials (like strontium titanate SrTiO3 (STO)) that exhibit the intriguing phenomenon of facet selectivity in oxidation and reduction chemistry (Fig. 8 above). Oxidation occurs on some facets, reduction on other facets. We aimed to establish the theoretical foundation for facet selectivity and other strategies to enhance carrier separation and improve solar energy conversion performance. The combined QM+KMC mesoscale approach that we used has proven essential to understand the nature of intrinsic carrier transport and the role of cation and anion doping in affecting transport. The insights from mesoscale modeling are a step toward screening of photo-active materials with superior photocatalytic performance. Structural and chemical descriptors of ‘good’ transport and carrier separation ability are starting to emerge.
-
Mesoscale transport dynamics in BiVO4: We recently completed a study of carrier transport in BVO. DFT calculations indicated that thermodynamic stability is not a factor in facet selectivity. A thorough characterization of e− and h+ hopping pathways in BVO yielded activation energies ~0.36 eV for electrons, and as low as ~0.17 eV for holes. Mesoscale KMC modeling revealed that hole transport is not nearly as efficient as the low barrier would suggest. Hole transport is bi-modal, with very fast but not transport-efficient hops (‘rattling’ motion) and slower but transport-efficient hops [100], as depicted in Fig. 10. It emerged from this work that strategies to eliminate hole ‘rattling’ will improve hole transport efficiency and water oxidation performance.
Fig. 10 
Bimodal h+ transport in BVO. e− transport gates BVO conductivity [89]

This work illustrates how mesoscale modeling is critically important to reveal fundamental characteristics of carrier transport. KMC captures the connection between elementary hopping rates and material structure and topology. Interpretations based on elementary hop characterization alone, even if at the quantum level of theory, have the potential to be misleading.
Toward modeling facet selectivity in BVO, we have investigated the thermodynamic stability of e−/h+ on ‘bulk’ sites compared to ‘near-surface’ or ‘surface’ sites [101]. A cursory look at the data plotted in Fig. 11 does not appear to reveal striking preferences in stability from one facet over the others. e− polarons prefer sub-surface sites over exposed surface sites and bulk sites. The stronger stability for the (011) surface may affect the space-charge distribution dynamics and, perhaps, the facet selectivity of e− polarons. New KMC simulations are in progress and are expected to reveal such effects. h+ polarons appear to have a more homogeneous behavior across facets.

Bulk versus surface stability of e−/h+ polaron in BVO [101]
Cation and anion doping in BiVO4: cation doping of BVO with W/Mo has been shown to enhance conversion efficiency in BVO-based devices. We studied how W/Mo doping of BVO affects carrier mobility and electric conductivity [90]. W/Mo doping results in a small decrease in electron mobility. However, the increase in carrier density upon doping overshadows the marginal decrease in electron mobility, resulting into an increased electronic conductivity. Work in progress deals with modeling homo-junctions, layered systems with a gradient of W-doping concentration and anion-doped BVO with ~25% level of sulfur doping, that have been shown to enhance charge separation. Simulations show that hole ‘rattling’ is eliminated by S-doping, as hole transport is reduced to S-to-S hoping.
3.3.2 Carrier Utilization: Solar-To-Fuels Surface Reactions
We pointed out in Sect. 1 that improved detailed knowledge and understanding of the interface reactions is needed to design better performing solar fuels catalysts [9, 15, 17]. Time-resolved IR spectroscopy is useful to characterize the reactions intermediates [72,73,74] and it has been used for a number of materials, including BiVO4, TiO2, SrTiO3, and others. For water splitting, the emphasis has been more on water oxidation than on proton reduction as it is the bottleneck process in overall water splitting [75, 151,152,153,154].
The mechanism prevalently considered for water oxidation is the four proton-coupled-electron transfer (PCET) mechanism with *OH, *O, and *OOH intermediate species [76]. Underlying the design of improved catalysts in general and in particular those based on the thermodynamics of the 4-step PCET model (also called ‘adsorbate evolution mechanism’ with cationic active sites), is the reliance on Sabatier’s principle of bonding strength [77] in conjunction with the Bell-Evans-Polanyi principle that ties kinetic barriers and step energies [92, 93]. Not all cases of water oxidation catalysts fall into the four-step PCET framework and other mechanisms have been proposed [73, 78]. One such mechanism is the ‘lattice oxygen mechanism’ (LOM) whereby lattice oxygens appear to be active specie [155,156,157]. To the best our knowledge, it remains challenging to predict which mechanism applies to which class of materials. An increased understanding of the structure-redox activity of materials and of the chemical factors that govern the reaction mechanisms is highly desirable. Improved understanding can come from computation of reaction energy profiles and of properties of the intermediates (IR frequencies, charge and spin populations, and other quantities) as described below.
OER takes place on semiconductors that are not defect-free in general, and oxygen vacancies (Ovac’s) are the most common defects [158,159,160,161,162]. The overall effect of Ovac’s is broadly understood as improving carrier concentration and conductivity in the bulk, but it is not fully clear why and how they might improve surface reactivity and PEC conversion efficiency, for a number of materials including BVO [163,164,165], WO3, and hematite Fe2O3 [166] and other materials. This is an important question as experimentalists are making progress in synthesizing materials with varying levels of vacancy concentrations [167]. From an experimental point of view, the features associated with excess electron arising from Ovac’s are often described in the solid-state terminology of gap states, deep trap states, and overlap of the gap state densities with the electronic states of the reactive water molecule. There is also some discussion of the degree of ionicity on the metal-oxygen bonds of the semiconductor when attempting to explain the trapping and reacting potentials of the defects [156, 163, 166].
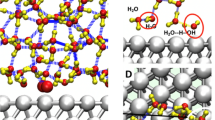
We have been interested, for some time, in the role of excess electrons on the surface chemistry of oxides. As we did for charge carriers, our approach to the question of the effect of Ovac’s on reactivity is to provide a chemistry-oriented understanding. It started with a ‘universal’ role of excess electrons on the surface chemistry of TiO2 that we verbalized [168]. In this work we noted that excess electrons arising from Ovac’s or interstitial Ti atoms can participate in charge transfer from the solid to an adsorbate, depicted in Fig. 12. The charge transfer makes the metal-adsorbate bond more ionic, and therefore stronger. The amount of stabilization depends on the electronegativity of the solid surface and of the adsorbate. Accordingly, the stabilization energy is different for *OH, *O, and *OOH along the OER reaction coordinate. We confirmed this hypothesis in several of our studies: a. OER on oxygen-deficient BaTiO3; [169] b. OER on Ga2O3; [170] c. comparison of 1e-, 2e-, and 4e- processes on oxygen-deficient rutile, anatase, and brookite [171].

Illustration of charge transfer of excess electrons from Ovac from the surface to the adsorbate, depending on the relative electronegativity χ of the surface and the adsorbate [167]
For these systems, we investigated the stability of Ovac’s on the surface versus sub-surface, the work function of the oxygen-deficient surfaces, the relative position of the gap states. We analyzed the structural data (bond lengths), the electron atomic populations on the cationic sites and adsorbates, and the spin atomic populations using Bader analysis. These data led us to characterize the amount of charge transfer from the solid to the adsorbate on the pristine surfaces compared to the reduced surfaces. Strong charge transfer correlate with strong free energy stabilization. The OER energy diagrams exhibit variations in step free energies consistent with the amount of charge transfer and charge transfer, as seen in Fig. 16.
The presence of Ovac’s may or may not lower the overpotential, depending on how much stabilization is gained in one intermediate over the other ones. Too strong a stabilization in one intermediate is not desirable as it makes a step free energy too large and increases the overpotential (Sabatier principle). Among the intermediates *OH has a strong electronegativity. So has *O in its dangling structure. In contrast, *O as a peroxo species (O atoms inserted in a metal-O-metal motif to give a metal-O-O-metal motif) does not have a strong electronegativity since all of its valencies are full. Lastly, *OOH has a weaker electronegativity. These differences lead to strongly changing reaction energy profiles and overpotentials, as depicted in Fig. 13 for rutile. Thus, knowing and predicting the charge transfer ability of the material is key to manipulating the overpotential.

OER free energy reaction profile and associated overpotential for pristine and oxygen-deficient rutile. O* is a peroxo species for the clean surface and a dangling O* species for the oxygen-deficient surface [171]
We uncovered two types of charge transfer: a. in some cases the pristine surface transfers electron density to the adsorbate, and the excess electrons from Ovac stabilize the electron-deficient solid. b. in other cases the pristine surface transfers no or little charge density to the adsorbate, and the excess Ovac electrons are directly involved in the charge transfer [171]. These studies were carried with the VASP code [172, 173] with charge analyses performed with the Bader theory of atoms in molecules [174].
We plan to carry out similar studies for BVO and Fe2O3 in the presence of oxygen vacancies. With the recent availability of detailed experimental measurements about the gap states [163], we will be in a position to compare work functions and gap state levels, to correlate these quantities with charge transfer, and to assess theory vs. experiment. The charge transfer analysis will be the connection between data on surfaces and the reaction thermodynamics. Of special interest will be comparisons between the traditional 4 PCET-step mechanism and the lattice oxygen mechanism that has been suggested for Fe2O3. We will make use of the extensive study by Hegner et al. of the structure of Ovac’s in BVO [165]. These authors have identified two distinctive structural motifs of Ovac’s in BVO with comparable formation energies. Our work will characterize these structures from the point of view of their charge transfer ability to the OER intermediates. For the key intermediates on pristine surfaces and on oxygen-deficient surfaces, we will go on to determine their spectroscopic IR signature.
4 Challenges in Computation and Modeling in Photo-Electro-Catalysis
While computation and modelling in photo-electro-catalysis has been broadly successful in providing fundamental characterization and understanding of the physics and chemistry of PEC’s, there remain some important challenges about the consistent reliability of predictions. It is well understood and accepted for example that functionals of the density that account for the ‘local’ density only (LDA), or those of the ‘generalized gradient approximation’ type (GGA) under-estimate consistently band gaps. A correction to the inherent ‘self-interaction’ of electrons in DFT is required, and this is commonly achieved by the use of the DFT+U formalism or the use of hybrid functionals. For the description of excitons and polarons the challenge remains. The two approaches yield qualitatively different descriptions of polarons as illustrated below. This observation underscores the need for theorists to develop functionals of density for DFT and/or other advanced theories, perhaps based on wavefunction theories, that provide computational characterizations that are more consistently reliable. Careful cross-validation of theory and experiment is critically important. In what follows we highlight work in our group that points to this type of challenges in functionals of the density, and other that is underway about validation.
4.1 Challenge to DFT: Accurate and Reliable Functionals for Polarons and Excitons
As mentioned, when modelling the polaron in materials, corrections to electron self-interaction need be included in the DFT formalism, in the form of DFT+U [175] or hybrid functionals [176] like PBE0 [177] and HSE06 [178]. These approaches have ad hoc parameters that researchers adjust often, mainly by choosing the+U values or the fraction of the exact exchange mixed into the functional.
About polarons in BiVO4 (BVO), that e− and h+ polarons exist is confirmed by experiment [179,180,181]. For e− polarons, there is a general concurrence that they are localized on the V sites in BVO, and that they form small polarons. The DFT+U approach [89, 182,183,184] and the hybrid functional approach [115, 185, 186] yield the same picture of e− polarons. For the h+ polarons in BVO, there are very notable differences between these theories. Using HSE06 functional (with 25% fraction of the HF exchange), Kweon and Hwang [187, 188] reported that the charge of the excess hole spreads over one BiO8 dodecahedron or across many Bi and O atoms, depending on the BVO phase. Using PBE0 functional (with 22% fraction of HF exchange) and molecular dynamics (MD) Wiktor [185] et al. found that the hole charge is distributed between one bismuth and eight oxygen atoms, and in about 20% of the MD configurations the hole appears localized on a single oxygen atom. In contrast, using a DFT + U level of theory with Ueff = 9 eV, Pasumarthi [184] et al. and Liu [183] et al. found that an excess hole localizes strongly on a single oxygen atom. The optimized structure exhibits a Bi-O bond lengthening in the range of ∼0.12 to ∼0.18 Å. Recently, Liu [189] et al. re-investigated the structure of hole polarons in BVO using hybrid DFT with varying fractions (α) of the exact exchange interaction that enters hybrid functionals. For values of α, from α = 0.25 to α = 0.45, both the h+(BiO8) hole structure and the h+(O) hole structure were obtained. For the smaller values of α, the h+(BiO8) structure was found to be lower in energy, while for the larger values of α, the h+(O) structure was found to be more stable as shown in Fig. 14. For α = 0.25, the diffusivity of h+(BiO8) holes determined from the Marcus/Holstein two-state model was determined to be in close agreement with published THz experimental data.

Relative energies of the two holes polaron structures h+(BiO8) and h+(O) obtained with HSE(α) for each value of α when α varies from 0.25 to 0.40: square symbols are for the hole distributed over one Bi atom and the neighboring oxygen atoms, h+(BiO8); circle symbols are for the hole localized on two or one oxygen atom, h+(O) [102]
Another example of contrasting findings is about e− and h+ polarons in oxynitrides and nitrides. The charge transport mechanism is these materials proved to be more complex than in metal oxides, as the N 2p state is less strongly localized than the O 2p orbital. Using time resolved microwave conductivity (TRMC) measurements, Respinis [190] et al. observed the carrier mobility increased with the nitrogen content from 1 × 10−5 cm−2 V−1 s−1 in Ta2O5, to 1 × 10−2 cm−2 V−1 s−1 in β-TaON, up to 1 × 10−1 cm−2 V−1 s−1 in Ta3N5. Lee [191] et al. predicted the formation of spin-polarized polarons in Ta2O5 with oxygen vacancies using hybrid functionals. Morbec and Galli [192] reported charge transport properties of the Ta3N5 from first principles calculations. They found that small e− polarons may occur but h+ polarons are not energetically stable according to the DFT+U approach. The estimated polaronic mobility for electron calculated by DFT+U approach is at least three orders of magnitude smaller than that of the measured value. Accordingly, the authors suggested that the main transport mechanism for both e− and h+ is band-like. Dey [193] et al. studied small polaron formation in β-TaON using DFT + U approach. They found that an excess electron tends to form a localized small polaron on a Ta site. The calculated diffusion barrier was ~0.3 eV, and the calculated mobility ~9.41 × 10−5 cm2V−1 s−1 in pristine TaON, calculated values that are at least two orders of magnitude smaller than the measured values. Liu [194] et al. investigated the charge transport properties in Ta2O5, TaON, and Ta3N5 by polaron hopping and band-like models. From the polaron binding energies and hybrid functional calculations, they found the charge transport mechanism to be small polaron hopping in Ta2O5, whereas in TaON and Ta3N5 hopping may not occur. Furthermore, the calculated mobility from the band-like model was not found consistent with experimental results neither.
These examples underscore the challenges with DFT approaches when investigating polarons in semiconductor. Which method to use (DFT+U or hybrid functional), and what values of +U values or of the fraction of HF exchange, are unsettled questions. There is a clear need for more accurate theoretical treatments, possibly some derived from molecular wavefunction formalisms and extended to the solid state.
4.2 Exciton Structure and Dynamics: Cross-Validation of Theory and Experiment.
In the same vein of validation of theory and experiment, recent experimental findings and chemical descriptions of excitons and polarons in selected oxides can be viewed as challenges to theory! The theoretical characterization of these ‘species’ with the most advanced tools of electronic structure of molecules and solids should prove a fertile ground for cross-validation of theory and experiment.
Exciton in Fe2O3: Emerging XUV experimental techniques are providing remarkable descriptions of the electronic structure of excited states (excitons) in short times following the event of light absorption [40]. By following the oxidation states of the elements through XUZ spectroscopy, the Baker group assigned a generation time of less than 100 fs to e−/h+ excitons in Fe2O3, Co3O4, and NiO, and a small polaron relaxation time of ~660 fs. Perhaps most striking is the assignment of the exciton radius of a single metal-oxygen bond length, as schematically displayed in Fig. 15. To the best of our knowledge, this is the first atomic scale ‘localized’ description of an exciton (excited state) in a ‘strongly correlated’ extended system. In fact, discussions of excitons are most often in terms of band theory, rarely in terms of ‘localized’ concepts as in the present cases.

Illustation of exciton in Fe2O, based on XUV characterization of Biswas et al. [39]. The exciton radius is found to be of a asingle metal oxygen bond length. An electron is excited from an O 2p lone pair state into the 3d state of an Fe atom directly linked to the O atom. High-level excited state calculations will be carries out to validate the experiment-derived description, while the experimental data will help to validate the theories

h+ polaron structure in CuFeO2 adapted from Ref. [41]. The hole thermalizes into a Cu 3d state, with potential mobility anisotropy in the Cu layer versus across the FeO2 layer
Highly accurate excited-state methods have been developed in recent years in chemistry, but, typically, they are not benchmarked against metal oxide data. Time-dependent DFT (TDDFT) is also extensively used in chemistry as it permits to investigate larger systems at reduced computational costs. The availability of the ‘localized’ atomistic pictures for the materials mentioned above offers the opportunity to validate experiment against ‘molecular-based theories of excited states while validating excited states theories applied to strongly correlated systems against experiment. Beyond Fe2O3, we could consider similar investigations of Co3O4, and NiO for which XUV characterization is available. Perhaps most intriguing would be similar exciton studies of BVO, a material in which the configuration of the metal cation is formally V 3d0 and whether the exciton radius might be or not a single metal-oxygen bond length.
The significance of such studies cannot be over-emphasized. They are far-reaching in that address a fundamental issue heretofore not enunciated, mainly the description as ‘localized’ of excitons in oxides such as hematite Fe2O3. At the same time, such studies would serve as stringent validation, as they’d provide a critical validation of excited state theories against experiment for strongly correlated systems like metal oxides.
h+ polaron in CuFeO2: delafossite CuFeO2 is another material of interest. It is an earth-abundant metal oxide with good stability in aqueous environments and favorable light absorption properties. It is attracting very strong attention as a photocathode [15, 41] for proton reduction. Electron-hole separation efficiency (excition lifetime), carrier mobility, and existence of surface states promoting carrier recombination, are issues that can benefit from theoretical characterization [15, 95]. The XUV spectroscopy-based description by Baker et al. of the ‘localized’, site-specific, structure and dynamics of holes that evolve from O 2p holes into hybridized Cu 3d holes within ~500 fs is interesting and challenging [41]. The structure is depicted in Fig. 16. It shows a hole localized in a Cu 3d state rather than an O 2p state. From a fundamental understanding, it is important to know why holes localize on Cu. Few DFT studies of CuFeO2 have been reported. The density of states suggests that O 2p states and Cu 3d states contribute to the top of the valence band, indicative of a strong mixing of the atomic states [195]. We note the symmetric environment around Cu sites (Fig. 16), so that the super-exchange interaction of O 2p holes may turn a double-well oxygen hole into a single-well Cu hole [196]. We are in a unique position to be able to validate the XUV picture, as well as to establish the characteristics of e− and h+ transport in CuFeO2. Beyond the electronic structure issue, there is the potential for a strong anisotropy in mobility in the Cu layer vs. across the FeO6 layers, clearly seen in Fig. 16.
5 Conclusion and Outlook
In this chapter we presented a broad overview of photo-electro-catalysis, a high risk, high-payoff domain of renewable energy. Efficient and cost-effective inter-conversion of electrical and chemical energy is widely accepted as an essential element of a broad strategy toward renewable energy. Current system conversion efficiencies, including for solar water splitting, are however far from the level needed for practical applications. From the standpoint of computation and modeling, there is already a wide body of research that has been instrumental in leading to new understanding and predictions of improved electrode materials for PECs with enhanced conversion efficiencies. The research encompasses computation and modeling for material structure, carrier transport, and redox reactivity. Modern DFT capabilities and multiscale models have already led to the characterization of new materials, of small polaron structures and dynamics [97, 105, 168], and new redox-active semiconductors, all contributions that are impactful in the field [107, 109]. Continuing developments and application, supported by cross-validation of experiment and theory, are expected to significantly advance the field further in years to come.
References
Nocera, D.G.: The artificial leaf. Acc. Chem. Res. 45, 767–776 (2012)
USDOE/BES. Basic Research Needs for Catalysis Science (2017). https://www.science.energy.gov/bes/community-resources/reports/
NSF. Catalytic Chemistry Workshop on Defining Critical Directions for the Future (2011). http://www.faculty.chemistry.harvard.edu/files/friend-lab/files/nsf_catreport_final_august_2011_mar2013_1.pdf
USDOE/BES. Computational Materials Science and Chemistry: Accelerating Discovery and Innovation through Simulation-based Engineering and Science (2010). https://www.science.energy.gov/bes/community-resources/reports/
USDOE/BES. Science for Energy Technology: Strengthening the Link Between Basic Research and Industry (2010). https://www.science.energy.gov/bes/community-resources/reports/
USDOE/BES. Basic Research Needs for Solar Energy Utilization (2005). https://www.science.energy.gov/bes/community-resources/reports/
Cook, T.R., Dogutan, D.K., Reece, S.Y., Surendranath, Y., Teets, T.S., Nocera, D.G.: Solar energy supply and storage for the legacy and non legacy worlds. Chem. Rev. 110, 6474–6502 (2010)
Maeda, K., Teramura, K., Lu, D.L., Takata, T., Saito, N., Inoue, Y., Domen, K.: Photocatalyst releasing hydrogen from water—enhancing catalytic performance holds promise for hydrogen production by water splitting in sunlight. Nature 440, 295 (2006)
Han, H., Li, C.: Photocatalysis in solar fuel production. Natl. Sci. Rev. 2, 145–147 (2015)
Shi, J., Li, C.: Photoelectrochemical approach using photocatalysts. In: Sugiyama, M., Fujii, K., Nakamura, S. (Eds.) Solar to Chemical Energy Conversion: Theory and Application. Lecture Notes in Energy, Vol. 32, pp. 319–344 (2016)
Sivula, K., van de Krol, R.: Semiconducting materials for photoelectrochemical energy conversion. Nat. Rev. Mater. 1 (2016)
Yang, J.H., Wang, D.G., Han, H.X., Li, C.: Roles of cocatalysts in photocatalysis and photoelectrocatalysis. Acc. Chem. Res. 46, 1900–1909 (2013)
Kudo, A., Miseki, Y.: Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 38, 253–278 (2009)
Osterloh, F.E.: Inorganic materials as catalysts for photochemical splitting of water. Chem. Mater. 20, 35–54 (2008)
Abdi, F.F., Berglund, S.P.: Recent developments in complex metal oxide photoelectrodes. J. Phys. D-Appl. Phys. 50, 193002–193024 (2017)
Wang, Z., Li, C., Domen, K.: Recent developments in heterogeneous photocatalysts for solar-driven overall water splitting. Chem. Soc. Rev. 48, 2109–2125 (2019)
Montoya, J.H., Seitz, L.C., Chakthranont, P., Vojvodic, A., Jaramillo, T.F., Norskov, J.K.: Materials for solar fuels and chemicals. Nat. Mater. 16, 70–81 (2017)
Yao, T.T., An, X.R., Han, H.X., Chen, J.Q., Li, C.: Photoelectrocatalytic materials for solar water splitting. Adv. Energy Mater. 8 (2018)
Chen, S.S., Qi, Y., Li, C., Domen, K., Zhang, F.X.: Surface strategies for particulate photocatalysts toward artificial photosynthesis. Joule 2, 2260–2288 (2018)
Sivula, K.: A step toward economically viable solar fuel production. Chem 4, 2490–2492 (2018)
Chu, S., Li, W., Yan, Y., Hamaan, T., Shih, I., Wang, D., Mi, Z.: Roadmap on solar water splitting: current status and future prospects. Nano Fut. 1, 2399 (2017)
Zhang, N., Long, R., Gao, C., Xiong, Y.J.: Recent progress on advanced design for photoelectrochemical reduction of CO2 to fuels. Sci. China Mater. 61, 771–805 (2018)
Yin, W.J., Wei, S.H., Al-Jassim, M.M., Yan, Y.: Double-hole-mediated coupling of dopants and its impact on band gap engineering in TiO2. Phys. Rev. Lett. 106, 066801–066804 (2011)
Dey, P., Bible, J., Datta, S., Broderick, S., Jasinski, J., Sunkara, M., Menon, M., Rajan, K.: Informatics-aided bandgap engineering for solar materials. Comput. Mater. Sci. 83, 185–195 (2014)
Castelli, I.E., Huser, F., Pandey, M., Li, H., Thygesen, K.S., Seger, B., Jain, A., Persson, K.A., Ceder, G., Jacobsen, K.W.: New light-harvesting materials using accurate and efficient bandgap calculations. Adv. Energy Mater. 5, 1400915–1400921 (2015)
Jain, A., Hautier, G., Moore, C.J., Ong, S.P., Fischer, C.C., Mueller, T., Persson, K.A., Ceder, G.: A high-throughput infrastructure for density functional theory calculations. Comput. Mater. Sci. 50, 2295–2310 (2011)
Wu, Y., Lazic, P., Hautier, G., Persson, K., Ceder, G.: First principles high throughput screening of oxynitrides for water-splitting photocatalysts. Energy Environ. Sci. 6, 157–168 (2013)
Godin, R., Wang, Y., Zwijnenburg, M.A., Tang, J.W., Durrant, J.R.: Time-resolved spectroscopic investigation of charge trapping in carbon nitrides photocatalysts for hydrogen generation. J. Am. Chem. Soc. 139, 5216–5224 (2017)
Godin, R., Kafizas, A., Durrant, J.R.: Electron transfer dynamics in fuel producing photosystems. Curr. Opin. Electrochem. 2, 136–143 (2017)
Kafizas, A., Godin, R., Durrant, J.R.: Charge carrier dynamics in metal oxide photoelectrodes for water oxidation. In: Mi, Z., Wang, L., Jagadish, C. (Eds.) Semiconductors for Photocatalysis . Semiconductors and Semimetals, Vol. 97, pp. 3–46 (2017)
Colbeau-Justin, C., Valenzuela, M.A.: Time-resolved microwave conductivity (TRMC) a useful characterization tool for charge carrier transfer in photocatalysis: a short review. Rev. Mex. Fis. 59, 191–200 (2013)
Zarifi, M.H., Mohammadpour, A., Farsinezhad, S., Wiltshire, B.D., Nosrati, M., Askar, A.M., Daneshmand, M., Shankar, K.: Time-resolved microwave photoconductivity (TRMC) using planar microwave resonators: application to the study of long-lived charge pairs in photoexcited titania nanotube arrays. J. Phys. Chem. C 119, 14358–14365 (2015)
Paulus, C., Wilke, K., Breuer, H.D.: Time resolved photocharge (TRPC) measurements on titanium dioxide: charge carrier dynamics and photocatalytic activity. J. Inf. Record. 24, 299–306 (1998)
Wilke, K., Breuer, H.D.: The influence of transition metal doping on the physical and photocatalytic properties of titania. J. Photochem. Photobiol. Chem. 121, 49–53 (1999)
Sadeghi, M., Liu, W., Zhang, T.G., Stavropoulos, P., Levy, B.: Role of photoinduced charge carrier separation distance in heterogeneous photocatalysis: oxidative degradation of CH3OH vapor in contact with Pt/TiO2 and cofumed TiO2-Fe2O3. J. Phys. Chem. 100, 19466–19474 (1996)
Butler, K.T., Dringoli, B.J., Zhou, L., Rao, P.M., Walsh, A., Titova, L.V.: Ultrafast carrier dynamics in BiVO4 thin film photoanode material: interplay between free carriers, trapped carriers and low-frequency lattice vibrations. J. Mater. Chem. A 4, 18516–18523 (2016)
Dringoli, B.J., Butler, K.T., Zhou, L., Giri, B., Rao, P.M., Walsh, A., Titova, L.V.: Ultrafast carrier dynamics in BiVO4: interplay between free carriers, trapped carriers and low-frequency lattice vibrations. In: 2016 41st International Conference on Infrared, Millimeter, and Terahertz Waves; International Conference on Infrared Millimeter and Terahertz Waves (2016)
Berger, T., Sterrer, M., Diwald, O., Knozinger, E., Panayotov, D., Thompson, T.L., Yates, J.T.: Light-induced charge separation in anatase TiO2 particles. J. Phys. Chem. B 109, 6061–6068 (2005)
Husek, J., Cirri, A., Biswas, S., Baker, L.R.: Surface electron dynamics in hematite (alpha-Fe2O3): correlation between ultrafast surface electron trapping and small polaron formation. Chem. Sci. 8, 8170–8178 (2017)
Biswas, S., Husek, J., Londo, S., Baker, L.R.: Highly localized charge transfer excitons in metal oxide semiconductors. Nano Lett. 18, 1228–1233 (2018)
Husek, J., Cirri, A., Biswas, S., Asthagiri, A., Baker, L.R.: Hole thermalization dynamics facilitate ultrafast spatial charge separation in CuFeO2 solar photocathodes. J. Phys. Chem. C 122, 11300–11304 (2018)
Carneiro, L., Cushing, S., Liu, C., Leone, S.: Evidence for small polaron formation leading to intrinsic photoexcited charge trapping in alpha-Fe2O3. Abstr. Papers Am. Chem. Soc. 253 (2017)
Carneiro, L.M., Cushing, S.K., Liu, C., Su, Y.D., Yang, P.D., Alivisatos, A.P., Leone, S.R.: Excitation-wavelength-dependent small polaron trapping of photoexcited carriers in alpha-Fe2O3. Nat. Mater. 16, 819-+ (2017)
Akimov, A.V., Prezhdo, O.V.: The PYXAID program for non-adiabatic molecular dynamics in condensed matter systems. J. Chem. Theory Comput. 9, 4959–4972 (2013)
Akimov, A.V., Prezhdo, O.V.: Advanced capabilities of the PYXAID program: integration schemes, decoherenc: e effects, multiexcitonic states, and field-matter interaction. J. Chem. Theory Comput. 10, 789–804 (2014)
Akimov, A.V.: Nonadiabatic molecular dynamics with tight-binding fragment molecular orbitals. J. Chem. Theory Comput. 12, 5719–5736 (2016)
Akimov, A.V.: Libra: an open-source “methodology discovery” library for quantum and classical dynamics simulations. J. Comput. Chem. 37, 1626–1649 (2016)
Long, R., Fang, W., Akimov, A.V.: Nonradiative electron-hole recombination rate is greatly reduced by defects in monolayer black phosphorus: ab initio time domain study. J. Phys. Chem. Lett. 7, 653–659 (2016)
Madjet, M.E.-A., Akimov, A.V., El-Mellouhi, F., Berdiyorov, G.R., Ashhab, S., Tabet, N., Kais, S.: Enhancing the carrier thermalization time in organometallic perovskites by halide mixing. Phys. Chem. Chem. Phys. 18, 5219–5231 (2016)
Nijamudheen, A., Akimov, A.V.: Criticality of symmetry in rational design of chalcogenide perovskites. J. Phys. Chem. Lett. 9, 248–257 (2018)
Li, R., Zhang, F., Wang, D., Yang, J., Li, M., Zhu, J., Zhou, X., Han, H., Li, C.: Spatial separation of photogenerated electrons and holes among 010 and 110 crystal facets of BiVO4. Nat Commun 4, 1432–1438 (2013)
Chen, R.T., Zhu, J., An, H.Y., Fan, F.T., Li, C.: Unravelling charge separation via surface built-in electric fields within single particulate photocatalysts. Faraday Discuss. 198, 473–479 (2017)
Wang, X., Xu, Q., Li, M., Shen, S., Wang, X., Wang, Y., Feng, Z., Shi, J., Han, H., Li, C.: Photocatalytic overall water splitting promoted by an alpha-beta phase junction on Ga2O3. Angew. Chem. Int. Ed. 51, 13089–13092 (2012)
Marschall, R.: Semiconductor composites: strategies for enhancing charge carrier separation to improve photocatalytic activity. Adv. Func. Mater. 24, 2421–2440 (2014)
Liu, C., Li, X.B., Su, J.Z., Guo, L.J.: Enhanced charge separation in copper incorporated BiVO4 with gradient doping concentration profile for photoelectrochemical water splitting. Int. J. Hydrogen Energy 41, 12842–12851 (2016)
Han, L.H., Abdi, F.F., van de Krol, R., Liu, R., Huang, Z.Q., Lewerenz, H.J., Dam, B., Zeman, M., Smets, A.H.M.: Efficient water-splitting device based on a bismuth vanadate photoanode and thin-film silicon solar cells. Chemsuschem 7, 2832–2838 (2014)
Abdi, F.F., Han, L.H., Smets, A.H.M., Zeman, M., Dam, B., van de Krol, R.: Efficient solar water splitting by enhanced charge separation in a bismuth vanadate-silicon tandem photoelectrode. Nat. Commun. 4, 2195–2201 (2013)
Maeda, K., Xiong, A., Yoshinaga, T., Ikeda, T., Sakamoto, N., Hisatomi, T., Takashima, M., Lu, D., Kanehara, M., Setoyama, T., Teranishi, T., Domen, K.: Photocatalytic overall water splitting promoted by two different cocatalysts for hydrogen and oxygen evolution under visible light. Angewandte Chemie-International Edition 49, 4096–4099 (2010)
Zong, X., Yan, H., Wu, G., Ma, G., Wen, F., Wang, L., Li, C.: Enhancement of photocatalytic H-2 evolution on CdS by loading MOS2 as cocatalyst under visible light irradiation. J. Am. Chem. Soc. 130, 7176–7177 (2008)
Mu, L., Yue, Z., Li, A., Wang, S., Wang, Z., Yang, J., Wang, Y., Liu, T., Chen, R., Zhu, J., Fan, F., Li, R., Li, C.: Enhancing charge separation on high symmetry SrTiO3 exposed with anisotropic facets for photocatalytic water splitting. Energy Environ. Sci. 9, 2463–2469 (2016)
Selcuk, S., Selloni, A.: Facet-dependent trapping and dynamics of excess electrons at anatase TiO2 surfaces and aqueous interfaces. Nat. Mater. 15, 1107–1113 (2016)
Mu, L.C., Zhao, Y., Li, A.L., Wang, S.Y., Wang, Z.L., Yang, J.X., Wang, Y., Liu, T.F., Chen, R.T., Zhu, J., Fan, F.T., Li, R.G., Li, C.: Enhancing charge separation on high symmetry SrTiO3 exposed with anisotropic facets for photocatalytic water splitting. Energy Environ. Sci. 9, 2463–2469 (2016)
Li, R., Tao, X., Chen, R., Fan, F., Li, C.: Synergetic effect of dual co-catalysts on the activity of p-type Cu2O crystals with anisotropic facets. Chem. Eur. J. 21, 14337–14341 (2015)
Wang, L., Ge, J., Wang, A., Deng, M., Wang, X., Bai, S., Li, R., Jiang, J., Zhang, Q., Luo, Y., Xiong, Y.: Designing p-type semiconductor-metal hybrid structures for improved photocatalysis. Angew. Chem. Int. Ed. Engl. 53, 5107–5111 (2014)
Liu, C., Han, X.G., Xie, S.F., Kuang, Q., Wang, X., Jin, M.S., Xie, Z.X., Zheng, L.S.: Enhancing the photocatalytic activity of anatase TiO2 by improving the specific facet-induced spontaneous separation of photogenerated electrons and holes. Chem. Asian J. 8, 282–289 (2013)
Roy, N., Sohn, Y., Pradhan, D.: Synergy of low-energy 101 and high-energy 001 TiO2 crystal facets for enhanced photocatalysis. ACS Nano 7, 2532–2540 (2013)
Gao, Y., Zhu, D., An, H., Yan, P., Huang, B., Chen, R., Fan, F., Li, C.: Directly probing charge separation at interface of TiO2 phase junction. J. Phys. Chem. Lett. 8, 1419–1423 (2017)
Zhang, X.R., Lin, Y.H., He, D.Q., Zhang, J.F., Fan, Z.Y., Xie, T.F.: Interface junction at anatase/rutile in mixed-phase TiO2:formation and photo-generated charge carriers properties. Chem. Phys. Lett. 504, 71–75 (2011)
Garcia, J.C., Nolan, M., Deskins, N.A.: The nature of interfaces and charge trapping sites in photocatalytic mixed-phase TiO2 from first principles modeling. J. Chem. Phys. 142, 024708–024717 (2015)
Sotelo-Vazquez, C., Quesada-Cabrera, R., Ling, M., Scanlon, D.O., Kafizas, A., Thakur, P.K., Lee, T.L., Taylor, A., Watson, G.W., Palgrave, R.G., Durrant, J.R., Blackman, C.S., Parkin, I.P.: Evidence and effect of photogenerated charge transfer for enhanced photocatalysis in WO3/TiO2 heterojunction films: a computational and experimental study. Adv. Funct. Mater. 27 (2017)
Selim, S., Francas, L., Garcia-Tecedor, M., Corby, S., Blackman, C., Gimenez, S., Durrant, J.R., Kafizas, A.: WO3/BiVO4: impact of charge separation at the timescale of water oxidation. Chem. Sci. 10, 2643–2652 (2019)
Paz, Y.: Transient IR scpectroscopy as a tool for studying photocatalytic materials. J. Phys. Condens. Matter 31, 503004–503025 (2019)
Nakamura, R., Nakato, Y.: Primary intermediates of oxygen photoevolution reaction on TiO2 (rutile) particles, revealed by in situ FTIR absorption and photoluminescence measurements. J. Am. Chem. Soc. 126, 1290–1298 (2004)
Herlihy, D.M., Waegele, M.M., Chen, X.H., Pemmaraju, C.D., Prendergast, D., Cuk, T.: Detecting the oxyl radical of photocatalytic water oxidation at an n-SrTiO3/aqueous interface through its subsurface vibration. Nat. Chem. 8, 549–555 (2016)
Ye, S., Ding, C.M., Liu, M.Y., Wang, A.Q., Huang, Q.G., Li, C.: Water oxidation catalysts for artificial photosynthesis. Adv. Mater.
Valdes, A., Qu, Z.W., Kroes, G.J., Rossmeisl, J., Norskov, J.K.: Oxidation and photo-oxidation of water on TiO2 surface. J. Phys. Chem. C 112, 9872–9879 (2008)
Medford, A.J., Vojvodic, A., Hummelshoj, J.S., Voss, J., Abild-Pedersen, F., Studt, F., Bligaard, T., Nilsson, A., Norskov, J.K.: From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis. J. Catal. 328, 36–42 (2015)
Huang, Z.F., Song, J.J., Du, Y.H., Xi, S.B., Dou, S., Nsanzimana, J.M.V., Wang, C., Xu, Z.C.J., Wang, X.: Chemical and structural origin of lattice oxygen oxidation in Co-Zn oxyhydroxide oxygen evolution electrocatalysts. Nat. Energy 4, 329–338 (2019)
Man, I.C., Su, H.Y., Calle-Vallejo, F., Hansen, H.A., Martinez, J.I., Inoglu, N.G., Kitchin, J., Jaramillo, T.F., Norskov, J.K., Rossmeisl, J.: Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem 3, 1159–1165 (2011)
Jain, A., Shyue Ping, O., Hautier, G., Chen, W., Richards, W.D., Dacek, S., Cholia, S., Gunter, D., Skinner, D., Ceder, G., Persson, K.A.: Commentary: the materials project: a materials genome approach to accelerating materials innovation. Appl. Mater. 1 (2013)
Ong, S.P., Cholia, S., Jain, A., Brafman, M., Gunter, D., Ceder, G., Persson, K.A.: The materials application programming interface (API): a simple, flexible and efficient API for materials data based on representational state transfer (REST) principles. Comput. Mater. Sci. 97, 209–215 (2015)
Curtarolo, S., Setyawan, W., Hart, G.L.W., Jahnatek, M., Chepulskii, R.V., Taylor, R.H., Wanga, S., Xue, J., Yang, K., Levy, O., Mehl, M.J., Stokes, H.T., Demchenko, D.O., Morgan, D.: AFLOW: an automatic framework for high-throughput materials discovery. Comput. Mater. Sci. 58, 218–226 (2012)
Curtarolo, S., Setyawan, W., Wang, S., Xue, J., Yang, K., Taylor, R.H., Nelson, L.J., Hart, G.L.W., Sanvito, S., Buongiorno-Nardelli, M., Mingo, N., Levy, O.: AFLOWLIB.ORG: A distributed materials properties repository from high-throughput ab initio calculations. Comput. Mater. Sci. 58, 227–235 (2012)
Hautier, G., Miglio, A., Ceder, G., Rignanese, G.M., Gonze, X.: Identification and design principles of low hole effective mass p-type transparent conducting oxides. Nat. Commun. 4, 2292–2298 (2013)
Rettie, A.J.E., Chemelewski, W.D., Emin, D., Mullins, C.B.: Unravelling small-polaron transport in metal oxide photoelectrodes. J. Phys. Chem. Lett. 7, 471–479 (2016)
Deskins, N.A., Rao, P.M., Dupuis, M.: Charge carrier management in semiconductors: modeling charge transport and recombination. Springer Handbook of Inorganic Photochemistry (2022)
Rosso, K.M., Smith, D.M.A., Dupuis, M.: An ab initio model of electron transport in hematite (α-Fe2O3) basal planes. J. Chem. Phys. 118, 6455–6466 (2003)
Rosso, K.M., Dupuis, M.: Reorganization energy associated with small polaron mobility in iron oxide. J. Chem. Phys. 120, 7050–7054 (2004)
Liu, T.F., Pasumarthi, V., LaPorte, C., Feng, Z.C., Li, Q.Y., Yang, J.J., Li, C., Dupuis, M.: Bimodal hole transport in bulk BiVO4 from computation. J. Mater. Chem. A 6, 3714–3723 (2018)
Pasumarthi, V., Liu, T., Dupuis, M., Li, C.: Charge carrier transport dynamics in W/Mo-doped BiVO4: first principles-based mesoscale characterization. J. Mater. Chem. A. 7, 3054–3065 (2019)
Lasia, A.: Mechanism and kinetics of the hydrogen evolution reaction. Int. J. Hydrogen Energy 44, 19484–19518 (2019)
Evans, M.G., Polanyi, M.: J. Chem. Soc. Faraday Trans. 32, 1340 (1936)
Bell, R.P.: The theory of reactions involving proton transfers. Proc. R. Soc. Lond. Ser. A 154, 414 (1936)
Wu, Y.A., McNulty, I., Liu, C., Lau, K.C., Liu, Q., Paulikas, A.P., Sun, C.-J., Cai, Z., Guest, J.R., Ren, Y., Stamenkovic, V., Curtiss, L.A., Liu, Y.: Facet-dependent active sites of a single Cu2O particle photocatalyst for CO2 reduction to methanol. Nat. Energy 4, 957–968 (2019)
Prevot, M.S., Jeanbourquin, X.A., Bouree, W.S., Abdi, F., Friedrich, D., van de Krol, R., Guijarro, N., Le Formal, F., Sivula, K.: Evaluating charge carrier transport and surface states in CuFeO2 photocathodes. Chem. Mater. 29, 4952–4962 (2017)
Rosso, K.M., Dupuis, M.: Electron transfer in environmental systems: a frontier for theoretical chemistry. Theoret. Chem. Acc. 116, 124–136 (2006)
Deskins, N.A., Dupuis, M.: Electron transport via polaron hopping in bulk TiO2: a density functional theory characterization. Phys. Rev. B 75, 195212–195221 (2007)
Deskins, N.A., Dupuis, M.: Intrinsic hole migration rates in TiO2 from density functional theory. J. Phys. Chem. C 113, 346–358 (2009)
Liu, T., Dupuis, M., Li, C.: Band structure engineering: insights from defects, band gap, and electron mobility, from study of magnesium tantalate. J. Phys. Chem. C 120, 6930–6937 (2016)
Liu, T., Pasumarthi, V., LaPorte, C., Feng, Z., Li, Q., Yang, J., Li, C., Dupuis, M.: Bimodal hole transport in bulk BiVO4 from computation. J. Mater. Chem. 6, 3714–3723 (2018)
Liu, T.F., Zhao, Q.Y., Li, C., Lyu, Y., Dupuis, M.: Photocatalytic facet selectivity in BiVO4 nanoparticles: polaron electronic structure and thermodynamic stability considerations for photocatalysis. J. Phys. Chem. C 123, 20142–20151 (2019)
Liu, T.F., Cui, M.S., Dupuis, M.: Hole polaron transport in bismuth vanadate BiVO4 from hybrid density functional theory. J. Phys. Chem. C 124, 23038–23044 (2020)
Morbec, J.M., Narkeviciute, I., Jaramillo, T.F., Galli, G.: Optoelectronic properties of Ta3N5: a joint theoretical and experimental study. Phys. Rev. B 90 (2014)
Plata, J.J., Marquez, A.M., Sanz, J.F.: Transport properties in the CeO2-x(111) surface: from charge distribution to ion-electron collaborative migration. J. Phys. Chem. C 117, 25497–25503 (2013)
Iordanova, N., Dupuis, M., Rosso, K.M.: Charge transport in metal oxides: a theoretical study of hematite alpha-Fe2O3. J. Chem. Phys. 122, 144305 (2005)
Morbec, J.M., Galli, G.: Charge transport properties of bulk Ta3N5 from first principles. Phys. Rev. B 93, 035201–035206 (2016)
Plata, J.J., Marquez, A.M., Fdez Sanz, J.: Electron mobility via polaron hopping in bulk ceria: a first-principles study. J. Phys. Chem. C 117, 14502–14509 (2013)
Shepidchenko, A., Sanyal, B., Klintenberg, M., Mirbt, S.: Small hole polaron in CdTe: Cd-vacancy revisited. Sci. Rep. 5, 14509–14514 (2015)
Adelstein, N., Neaton, J.B., Asta, M., De Jonghe, L.C.: Density functional theory based calculation of small-polaron mobility in hematite. Phys. Rev. B 89, 245115–245123 (2014)
Kanan, D.K., Carter, E.A.: Ab initio study of electron and hole transport in pure and doped MnO and MnO:ZnO alloy. J. Mater. Chem. A 1, 9246–9256 (2013)
Erhart, P., Klein, A., Åberg, D., Sadigh, B.: Efficacy of the DFT plus U formalism for modeling hole polarons in perovskite oxides. Phys. Rev. B 90, 035204 (2014)
Kweon, K.E., Hwang, G.S.: Surface structure and hole localization in bismuth vanadate: a first principles study. Appl. Phys. Lett. 103, 131603–131607 (2013)
Kweon, K.E., Hwang, G.S.: Structural phase-dependent hole localization and transport in bismuth vanadate. Phys. Rev. B 87, 205202–205207 (2013)
Kweon, K.E., Hwang, G.S., Kim, J., Kim, S., Kim, S.: Electron small polarons and their transport in bismuth vanadate: a first principles study. Phys. Chem. Chem. Phys. 17, 256–260 (2015)
Hoffmann, M.J., Matera, S., Reuter, K.: kmos: a lattice kinetic Monte Carlo framework. Comput. Phys. Commun. 185, 2138–2150 (2014)
Leetmaa, M., Skorodumova, N.V.: KMCLib: a general framework for lattice kinetic Monte Carlo (KMC) simulations. Comput. Phys. Commun. 185, 2340–2349 (2014)
Yu, J., Sushko, M.L., Kerisit, S., Rosso, K.M., Liu, J.: Kinetic Monte Carlo study of ambipolar lithium ion and electron-polaron diffusion into nanostructured TiO2. J. Phys. Chem. Lett. 3, 2076–2081 (2012)
Mi, Y., Weng, Y.X.: Band alignment and controllable electron migration between rutile and anatase TiO2. Sci. Rep. 5, 11482–11491 (2015)
Wang, X., Kafizas, A., Li, X., Moniz, S.J.A., Reardon, P.J.T., Tang, J., Parkin, I.P., Durrant, J.R.: Transient absorption spectroscopy of anatase and rutile: the impact of morphology and phase on photocatalytic activity. J. Phys. Chem. C 119, 10439–10447 (2015)
Li, A., Wang, Z., Yin, H., Wang, S., Yan, P., Huang, B., Wang, X., Li, R., Zong, X., Han, H., Li, C.: Understanding the anatase–rutile phase junction in charge separation and transfer in a TiO2 electrode for photoelectrochemical water splitting. Chem. Sci. 7, 6076–6082 (2016)
Jin, S.Q., Wang, X., Wang, X.L., Ju, M.G., Shen, S., Liang, W.Z., Zhao, Y., Feng, Z.C., Playford, H.Y., Walton, R.I., Li, C.: Effect of phase junction structure on the photocatalytic performance in overall water splitting: Ga2O3 photocatalyst as an example. J. Phys. Chem. C 119, 18221–18228 (2015)
Oberhofer, H., Reuter, K., Blumberger, J.: Charge transport in molecular materials: an assessment of computational methods. Chem. Rev. 117, 10319–10357 (2017)
van der Kaap, N.J., Koster, L.J.A.: Massively parallel kinetic Monte Carlo simulations of charge carrier transport in organic semiconductors. J. Comput. Phys. 307, 321–332 (2016)
Kerisit, S., Rosso, K.M.: Kinetic Monte Carlo model of charge transport in hematite (alpha-Fe2O3). J. Chem. Phys. 127, 124706–124715 (2007)
Scanlon, D.O., Morgan, B.J., Watson, G.W.: Modeling the polaronic nature of p-type defects in Cu2O: The failure of GGA and GGA plus U. J. Chem. Phys. 131 (2009)
Scanlon, D.O., Morgan, B.J., Watson, G.W., Walsh, A.: Acceptor levels in p-type Cu2O: rationalizing theory and experiment. Phys. Rev. Lett. 103 (2009)
Scanlon, D.O., Watson, G.W.: Undoped n-type Cu2O: fact or fiction? J. Phys. Chem. Lett. 1, 2582–2585 (2010)
Yu, X., Zhang, X., Wang, S., Feng, G.: A first principle study on the magnetic properties of Cu2O surfaces. Curr. Appl. Phys. 15, 1303–1311 (2015)
Marcus, R.A.: Electron transfer at electrodes and in solution - comparison of theory and experiment. Electrochim. Acta 13, 995–1000 (1968)
Marcus, R.A.: Electron-transfer reactions in chemistry—theory and experiment. Rev. Mod. Phys. 65, 599–610 (1993)
Gerischer, H.: Charge transfer processes at semiconductor-electrolyte interfaces in connection with problems of catalysis. Surf. Sci. 18, 97 (1969)
Gerischer, H.: On role of electrons and holes in surface reactions on semiconductors. Surf. Sci. 13, 265 (1969)
Zhang, X.Q., Bieberle-Hutter, A.: Modeling and simulations in photoelectrochemical water oxidation: from single level to multiscale modeling. Chemsuschem 9, 1223–1242 (2016)
George, K., van Berkel, M., Zhang, X.Q., Sinha, R., Bieberle-Huller, A.: Impedance spectra and surface coverages simulated directly from the electrochemical reaction mechanism: a nonlinear state-space approach. J. Phys. Chem. C 123, 9981–9992 (2019)
Yang, J., Wang, D., Zhou, X., Li, C.: A theoretical study on the mechanism of photocatalytic oxygen evolution on BiVO4 in aqueous solution. Chemistry 19, 1320–1326 (2013)
Coridan, R.H., Nielander, A.C., Francis, S.A., McDowell, M.T., Dix, V., Chatman, S.M., Lewis, N.S.: Methods for comparing the performance of energy-conversion systems for use in solar fuels and solar electricity generation. Energy Environ. Sci. 8, 2886–2901 (2015)
Liu, D.J., Garcia, A., Wang, J., Ackerman, D.M., Wang, C.J., Evans, J.W.: Kinetic Monte Carlo simulation of statistical mechanical models and coarse-grained mesoscale descriptions of catalytic reaction-diffusion processes: 1D nanoporous and 2D surface systems. Chem. Rev. 115, 5979–6050 (2015)
Chen, X., Liu, L., Yu, P.Y., Mao, S.S.: Increasing solar absorption for photocatalysis with black hydrogenated titanium dioxide nanocrystals. Science 331, 746–750 (2011)
Li, D., Liu, Y., Shi, W.W., Shao, C.Y., Wang, S.Y., Ding, C.M., Liu, T.F., Fan, F.T., Shi, J.Y., Li, C.: Crystallographic-orientation-dependent charge separation of BiVO4 for solar water oxidation. ACS Energy Lett. 4, 825–831 (2019)
Ohno, T., Sarukawa, K., Tokieda, K., Matsumura, M.: Morphology of a TiO2 photocatalyst (Degussa, P-25) consisting of anatase and rutile crystalline phases. J. Catal. 203, 82–86 (2001)
Ohno, T., Tokieda, K., Higashida, S., Matsumura, M.: Synergism between rutile and anatase TiO2 particles in photocatalytic oxidation of naphthalene. Appl. Catal. General 244, 383–391 (2003)
Wu, C.Y., Yue, Y.H., Deng, X.Y., Hua, W.M., Gao, Z.: Investigation on the synergetic effect between anatase and rutile nanoparticles in gas-phase photocatalytic oxidations. Catal. Today 93–5, 863–869 (2004)
Frank, A.J., Kopidakis, N., van de Lagemaat, J.: Electrons in nanostructured TiO2 solar cells: transport, recombination and photovoltaic properties. Coord. Chem. Rev. 248, 1165–1179 (2004)
Kopidakis, N., Benkstein, K.D., van de Lagemaat, J., Frank, A.J., Yuan, Q., Schiff, E.A.: Temperature dependence of the electron diffusion coefficient in electrolyte-filled TiO2 nanoparticle films: evidence against multiple trapping in exponential conduction-band tails. Phys. Rev. B 73, 045326–045332 (2006)
Green, A.N.M., Chandler, R.E., Haque, S.A., Nelson, J., Durrant, J.R.: Transient absorption studies and numerical modeling of iodine photoreduction by nanocrystalline TiO2 films. J. Phys. Chem. B 109, 142–150 (2005)
Hendry, E., Wang, F., Shan, J., Heinz, T.F., Bonn, M.: Electron transport in TiO2 probed by THz time-domain spectroscopy. Phys. Rev. B 69, 081101–081104 (2004)
Ulbricht, R., Hendry, E., Shan, J., Heinz, T.F., Bonn, M.: Carrier dynamics in semiconductors studied with time-resolved terahertz spectroscopy. Rev. Mod. Phys. 83, 543–586 (2011)
Hurum, D.C., Agrios, A.G., Gray, K.A., Rajh, T., Thurnauer, M.C.: Explaining the enhanced photocatalytic activity of Degussa P25 mixed-phase TiO2 using EPR. J. Phys. Chem. B 107, 4545–4549 (2003)
Kawahara, T., Konishi, Y., Tada, H., Tohge, N., Nishii, J., Ito, S.: A patterned TiO2(anatase)/TiO2(rutile) bilayer-type photocatalyst: effect of the anatase/rutile junction on the photocatalytic activity. Angew. Chem. Int. Ed. 41, 2811–2813 (2002)
Zou, Z.G., Ye, J.H., Sayama, K., Arakawa, H.: Direct splitting of water under visible light irradiation with an oxide semiconductor photocatalyst. Nature 414, 625–627 (2001)
Young, K.J., Martini, L.A., Milot, R.L., Snoeberger, R.C., Batista, V.S., Schmuttenmaer, C.A., Crabtree, R.H., Brudvig, G.W.: Light-driven water oxidation for solar fuels. Coord. Chem. Rev. 256, 2503–2520 (2012)
Kanan, M.W., Nocera, D.G.: In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+. Science 321, 1072–1075 (2008)
Karkas, M.D., Verho, O., Johnston, E.V., Akermark, B.: Artificial photosynthesis: molecular systems for catalytic water oxidation. Chem. Rev. 114, 11863–12001 (2014)
Mueller, D.N., Machala, M.L., Bluhm, H., Chueh, W.C.: Redox activity of surface oxygen anions in oxygen-deficient perovskite oxides during electrochemical reactions. Nat. Commun. 6 (2015)
Grimaud, A., Hong, W.T., Shao-Horn, Y., Tarascon, J.M.: Anionic redox processes for electrochemical devices. Nat. Mater. 15, 121–126 (2016)
Mefford, J.T., Rong, X., Abakumov, A.M., Hardin, W.G., Dai, S., Kolpak, A.M., Johnston, K.P., Stevenson, K.J.: Water electrolysis on La1-xSrxCoO3-delta perovskite electrocatalysts. Nat. Commun. 7 (2016)
Di Valentin, C., Pacchioni, G., Selloni, A.: Electronic structure of defect states in hydroxylated and reduced rutile TiO2(110) surfaces. Phys. Rev. Lett. 97 (2006)
Scheiber, P., Fidler, M., Dulub, O., Schmid, M., Diebold, U., Hou, W.Y., Aschauer, U., Selloni, A.: (Sub)Surface mobility of oxygen vacancies at the TiO2 anatase (101) surface. Phys. Rev. Lett. 109 (2012)
Brawand, N.P., Goldey, M.B., Voros, M., Galli, G.: Defect states and charge transport in quantum dot solids. Chem. Mater. 29, 1255–1262 (2017)
Fu, Q., Wu, T., Fu, G., Gao, T.L., Han, J.C., Yao, T., Zhang, Y.M., Zhong, W.W., Wang, X.J., Song, B.: Skutterudite-type ternary Co1-xNixP3 nanoneedle array electrocatalysts for enhanced hydrogen and oxygen evolution. ACS Energy Lett. 3, 1744–1752 (2018)
Huang, J.Z., Han, J.C., Wang, R., Zhang, Y.Y., Wang, X.J., Zhang, X.H., Zhang, Z.H., Zhang, Y.M., Song, B., Jin, S.: Improving electrocatalysts for oxygen evolution using NixFe3-xO(4)/Ni hybrid nanostructures formed by solvothermal synthesis. ACS Energy Lett. 3, 1698–1707 (2018)
Selim, S., Pastor, E., Garcia-Tecedor, M., Morris, M.R., Francas, L., Sachs, M., Moss, B., Corby, S., Mesa, C.A., Gimenez, S., Kafizas, A., Bakulin, A.A., Durrant, J.R.: Impact of oxygen vacancy occupancy on charge carrier dynamics in BiVO4 photoanodes. J. Am. Chem. Soc. 141, 18791–18798 (2019)
Wu, J.M., Chen, Y., Pan, L., Wang, P.H., Cui, Y., Kong, D.C., Wang, L., Zhang, X.W., Zou, J.J.: Multi-layer monoclinic BiVO4 with oxygen vacancies and V4+ species for highly efficient visible-light photoelectrochemical applications. Appl. Catal. B Environ. 221, 187–195 (2018)
Hegner, F.S., Forrer, D., Galan-Mascaros, J.R., Lopez, N., Selloni, A.: Versatile nature of oxygen vacancies in bismuth vanadate bulk and (001) surface. J. Phys. Chem. Lett. 10, 6672–6678 (2019)
Wang, Z.L., Mao, X., Chen, P., Xiao, M., Monny, S.A., Wang, S.C., Konarova, M., Du, A.J., Wang, L.Z.: Understanding the roles of oxygen vacancies in hematite-based photoelectrochemical processes. Angew. Chem. Int. Ed. 58, 1030–1034 (2019)
Chen, C.-F., King, G., Dickerson, R.M., Papin, P.A., Gupta, S., Kellogg, W.R., Wu, G.: Oxygen-deficient BaTiO3−x perovskite as an efficient bifunctional oxygen electrocatalyst. Nano Energy 13, 423–432 (2015)
Deskins, N.A., Rousseau, R., Dupuis, M.: Defining the role of excess electrons in the surface chemistry of TiO2. J. Phys. Chem. C 114, 5891–5897 (2010)
Tyminska, N., Wu, G., Dupuis, M.: Water oxidation on oxygen-deficient barium titanate: a first-principles study. J. Phys. Chem. C 121, 8378–8389 (2017)
Liu, T.F., Feng, Z.C., Li, Q.Y., Yang, J.J., Li, C., Dupuis, M.: Role of oxygen vacancies on oxygen evolution reaction activity: beta-Ga2O3 as a case study. Chem. Mater. 30, 7714–7726 (2018)
Malik, A.S., Liu, T., Dupuis, M., Li, R., Li, C.: Water oxidation on TiO2: a comparative study of 1e- , 2e-, and 4e- processes on rutile, anatase, and brookite. II the role of oxygen vacancies Acs Catal. (2020)
Kresse, G., Furthmuller, J.: Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996)
Kresse, G., Furthmuller, J.: Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996)
Bader, R.F.W.: Atoms in Molecules—A Quantum Theory. Oxford Universty Press, New York (1990)
Dudarev, S.L., Botton, G.A., Savrasov, S.Y., Humphreys, C.J., Sutton, A.P.: Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998)
Becke, A.D.: A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 98, 1372–1377 (1993)
Adamo, C., Barone, V.: Toward reliable density functional methods without adjustable parameters: the PBE0 model. J. Chem. Phys. 110, 6158–6170 (1999)
Heyd, J., Scuseria, G.E., Ernzerhof, M.: Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 118, 8207–8215 (2003)
Rettie, A.J., Chemelewski, W.D., Lindemuth, J., McCloy, J.S., Marshall, L.G., Zhou, J., Emin, D., Mullins, C.B.: Anisotropic small-polaron hopping in W: BiVO4 single crystals. Appl. Phys. Lett. 106, 022106 (2015)
Rettie, A.J., Lee, H.C., Marshall, L.G., Lin, J.-F., Capan, C., Lindemuth, J., McCloy, J.S., Zhou, J., Bard, A.J., Mullins, C.B.: Combined charge carrier transport and photoelectrochemical characterization of BiVO4 single crystals: intrinsic behavior of a complex metal oxide. J. Am. Chem. Soc. 135, 11389–11396 (2013)
Ziwritsch, M., Müller, S.n., Hempel, H., Unold, T., Abdi, F.F., van de Krol, R., Friedrich, D., Eichberger, R.: Direct time-resolved observation of carrier trapping and polaron conductivity in BiVO4. ACS Energy Lett. 1, 888–894 (2016)
Liu, T., Zhou, X., Dupuis, M., Li, C.: The nature of photogenerated charge separation among different crystal facets of BiVO4 studied by density functional theory. Phys. Chem. Chem. Phys. 17, 23503–23510 (2015)
Pasumarthi, V., Liu, T., Dupuis, M., Li, C.: Charge carrier transport dynamics in W/Mo-doped BiVO4: first principles-based mesoscale characterization. J. Mater. Chem. A 7, 3054–3065 (2019)
Wiktor, J., Ambrosio, F., Pasquarello, A.: Role of polarons in water splitting: the case of BiVO4. ACS Energy Lett. 3, 1693–1697 (2018)
Wu, F., Ping, Y.: Combining Landau-Zener theory and kinetic Monte Carlo sampling for small polaron mobility of doped BiVO4 from first-principles. J. Mater. Chem. A 6, 20025–20036 (2018)
Kweon, K.E., Hwang, G.S.: Surface structure and hole localization in bismuth vanadate: a first principles study. Appl. Phys. Lett. 103, 131603 (2013)
Kweon, K.E., Hwang, G.S.: Structural phase-dependent hole localization and transport in bismuth vanadate. Phys. Rev. B 87, 205202 (2013)
de Respinis, M., Fravventura, M., Abdi, F.F., Schreuders, H., Savenije, T.J., Smith, W.A., Dam, B., van de Krol, R.: Oxynitrogenography: controlled synthesis of single-phase tantalum oxynitride photoabsorbers. Chem. Mater. 27, 7091–7099 (2015)
Lee, J., Lu, W.D., Kioupakis, E.: Electronic and optical properties of oxygen vacancies in amorphous Ta2O5 from first principles. Nanoscale 9, 1120–1127 (2017)
Morbec, J.M., Galli, G.: Charge transport properties of bulk Ta3N5 from first principles. Phys. Rev. B 93, 035201 (2016)
Dey, M., Singh, A., Singh, A.K.: Formation of a small electron polaron in tantalum oxynitride: origin of low mobility. J. Phys. Chem. C 125, 11548–11554 (2021)
Zhao, Q., Cui, M., Liu, T.: Charge carrier transport mechanism in Ta2O5, TaON, and Ta3N5 studied by applying polaron hopping and bandlike models. ChemPhysChem e202100859
Shi, L.R., Yan, X.W., Ju, L., Tian, J.L., Xia, Z.C.: The effect of oxygen vacancy on magnetism of geometrically frustrated triangular lattice CuFeO2: Ab initio study. J. Magnet. Magnet. Mater. 486 (2019)
Gajdos, F., Oberhofer, H., Dupuis, M., Blumberger, J.: On the inapplicability of electron-hopping models for the organic semiconductor phenyl-C61-butyric acid methyl ester (PCBM). J. Phys. Chem. Lett. 4, 1012–1017 (2013)
Behara, P.K., Dupuis, M.: Electron transfer in extended systems by density functional theory: calculation of the electronic coupling. Phys. Chem. Chem. Phys. 22, 10609–10623 (2019)
Green, M.A., Ho-Baillie, A., Snaith, H.J.: The emergence of perovskite solar cells. Nat. Photonics 8, 506–514 (2014)
Park, N.G.: Perovskite solar cells: an emerging photovoltaic technology. Mater. Today 18, 65–72 (2015)
Acknowledgements
MD gratefully acknowledges support from the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award Number DE-SC0019086. TL acknowledges support from the National Natural Science Foundation of China (grants # 21703054 and #22173026). The authors gratefully acknowledge many stimulating and guiding discussions with Professor Can Li, Director, and many staff members in the Solar Energy Division of the Dalian Laboratory for Clean Energy at the Dalian Institute for Chemical Physics in Dalian, China, where much of the work described in this chapter was initiated.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Liu, T., Dupuis, M. (2022). Theory and Computation in Photo-Electro-Chemical Catalysis: Highlights, Challenges, and Prospects. In: Taft, C.A., de Lazaro, S.R. (eds) Research Topics in Bioactivity, Environment and Energy. Engineering Materials. Springer, Cham. https://doi.org/10.1007/978-3-031-07622-0_1
Download citation
DOI: https://doi.org/10.1007/978-3-031-07622-0_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-07621-3
Online ISBN: 978-3-031-07622-0
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)