
Abstract
A wide range of advanced experimental methods has been used in catalysis science to understand chemical transformations at molecular level. Among these, neutron scattering not only gives catalytic information that is highly complementary to other microscopic scattering techniques such as electrons (microscopy and diffraction) and photons from visible light to synchrotron X-rays but also often provides unique insights into catalysis. This chapter aims to provide a general overview of neutron scattering and its applications for heterogeneous catalysis, emphasizing chemistry at the gas/solid interface. It starts with an introduction to the theory of neutron scattering, with pros and cons for catalyst research, followed by a description of three main neutron scattering techniques: neutron diffraction (ND), inelastic neutron spectroscopy (INS), and quasi-elastic neutron scattering (QENS). It includes recent applications in each technique and then a short introduction of other neutron techniques that are less frequently used in the catalysis field. It ends with a summary and a future outlook.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Neutron scattering
- Catalysis
- Instrumentation
- Modeling
- In situ
- Inelastic neutron spectroscopy
- Neutron diffraction
- Quasi-elastic neutron scattering
- Neutron imaging
- Small-angle neutron scattering
1 Introduction
Neutron scattering has been applied in heterogeneous catalysis and has a widely recognized role in structural studies of catalysts, especially of light elements such as hydrogen, oxygen, and nitrogen [1,2,3,4,5,6,7,8,9,10,11,12]. Neutrons are electrically neutral and only interact with the nucleus; thus, they can penetrate deeply into a target material. Although neutrons are weakly scattered by matter, neutron scattering’s unique features allow us to obtain various types of information such as magnetic [13] and vibrational excitations [1, 14,15,16,17] and molecular dynamics (MD) of catalysts [10, 12, 18,19,20,21]. Additionally, each isotope has a unique feature known as neutron scattering cross section, and this isotopic sensitivity allows contrast variation, which could discriminate elements with similar atomic numbers [22,23,24]. The application of neutron scattering to heterogeneous catalysis can be dated back to the 1970s when Stockmeyer et al. reported H2 chemisorption on Raney nickel using inelastic neutron scattering (INS) [25].
Several types of neutron scattering techniques have been applied to the research of catalytic systems. Neutron methods are frequently split into elastic (diffraction and imaging) and inelastic (spectroscopic) techniques and used to investigate where atoms are or how atoms move, respectively. Neutron diffraction (ND) has long made a significant contribution to catalysis combined with X-ray diffraction, especially determining the location of light elements in the structure of the catalyst [24, 26,27,28]. INS gives access to vibrational modes, providing information that is difficult or even impossible to obtain from conventional spectroscopy techniques. Quasi-elastic neutron scattering (QENS) studies allow us to understand molecular motions (diffusion and rotational) on a range of timescales, mostly of a stochastic nature. The application of neutron scattering to heterogeneous catalysis has been discussed in excellent reviews [3, 16, 29,30,31,32,33,34] and books [9, 15, 35,36,37,38], and we refer readers to these references for more detailed information of different types of neutron scattering techniques. This chapter intends to give a general overview of neutron scattering including background, instrumentation, and the applications of neutron scattering in the field of heterogeneous catalysis.
2 Theory of Neutron Scattering
2.1 Properties of Neutrons and Neutron Sources
Bombarding a material with particles to probe its internal structure and dynamics is a standard scientific research approach. For X-ray diffraction and Raman and infrared spectroscopy, such probing particles are photons (X-ray or laser); for transmission electron microscopy and electron energy loss spectroscopy, such probing particles are electrons. To many, a less familiar probing particle is the neutron. Compared to photons and electrons, neutrons have unique properties and can perform certain measurements with information that is difficult or impossible with other techniques (see details in Sect. 24.3). A list of fundamental properties of neutrons, in comparison with photons and electrons, is given in Table 24.1.
A neutron beam is relatively difficult (and expensive) to produce. There are two ways to make neutrons: one is reactor-based fission, and the other is accelerator-based spallation. Reactor-based neutron sources generate a continuous neutron beam with high average power, whereas accelerator-based neutron sources generate a pulsed neutron beam with high peak brightness. The generated neutrons need to be slowed down by neutron moderators to be used in materials research. Depending on the neutrons’ energy range, the source can be classified into three categories, as listed in Table 24.2.
An example of a standard neutron moderator is the “water moderator.” As its name implies, it uses light water at room temperature (293 K). Therefore, neutrons coming out of the moderator follow a Maxwellian distribution with its peak corresponding to 293 K. A list of properties of “room temperature” neutrons, which are representative of the neutrons most useful for the present research, is given in Table 24.3.
The combination of these properties of thermal neutrons in Tables 24.1 and 24.2 renders them unique advantages in materials characterization. A major bottleneck, however, is the flux and brightness of the neutron beam, which is more than ten orders of magnitude lower than synchrotron X-ray sources. The maximum neutron flux for a spallation neutron source (SNS) is around 109 neutrons ∙ s−1 ∙ cm−2 [40], and the brilliance of a synchrotron can be as high as 1018photons ∙ s−1 ∙ mm−2 ∙ mrad−2 (0.1 % bandwidth) [41].
This limits the ability of neutrons to study very small quantity of samples or collect data quickly. Presently, the highest flux neutron sources include the accelerator-based SNS operated by Oak Ridge National Laboratory, US, ISIS neutron source, England, and J-PARC, Japan, and the reactor-based high flux isotope reactor (HFIR), operated by Oak Ridge National Laboratory, US. The next-generation neutron sources such as European Spallation Source (ESS) and the Second Target Station (STS) at SNS are being built or planned. A timeline summarizing the evolution of neutron sources can be found in Fig. 24.1.

Evolution of neutron sources. (Adapted from Ref. [42], Copyright 2006, with permission from AIP publishing). Note that this is the total flux of neutrons produced by the source, not at sample position
2.2 How Neutron Scattering Works
Neutrons interact with nuclei through the strong nuclear forces. The capability of a nucleus to scatter neutrons can be characterized by its neutron scattering length, or cross section. Scattering from different nuclei can be coherent or incoherent, depending on whether the relative phase of the neutron wave function is preserved or not. The neutron scattering properties of some common elements in chemical systems are given in Table 24.4, and more can be found at the reference [43] or NIST website [44].
Despite the fundamental differences between neutrons and X-rays in their ways of interacting with matter, the measurement mechanism is similar. Specifically, for diffraction measurement of the atomic structure, they both follow Bragg’s law. Coherent elastic scattering is what we are interested in solving the positions of atoms. The study of dynamical processes is usually called spectroscopy. Neutrons can also exchange energy with the target material, causing quasi-elastic and inelastic scattering. These neutron spectroscopies measure the diffusive and vibrational dynamics of atoms, as illustrated in Fig. 24.2. For chemical systems, the coherent effects are often negligible, and the scattering can be evaluated following the incoherent approximation with the total scattering cross section.

Neutron spectroscopy methods and the energy/time/length scales they probe. (Adapted from Ref. [30], Copyright 2016, with permission from IOP Publishing Ltd)
2.3 Instrumentation
Most catalysis experiments using neutrons, especially in recent years, were performed at accelerator-based neutron sources, mainly for their high peak brightness, the availability of epithermal neutrons, and time-of-flight (ToF) techniques that are more amenable to pulsed sources. With pulsed neutron beam (e.g., 60 Hz at SNS), each pulse contains many neutrons with different energies compressed in the very short time window (~1 μs). As these neutrons travel toward the samples, their different speeds spread them out, and after interacting with the samples, they eventually arrive at the detector at different times. A ToF neutron instrument measures the arrival time and location of the received neutrons, then calculates what happened to the neutrons, and puts them in the correct momentum (Q) and energy transfer. Under this overall mechanism, the design of the instrument can be optimized for different purposes.
Since the vast majority of neutrons will be scattered elastically under most circumstances, a neutron diffractometer intended to measure elastic scattering does not require energy resolution and thus does not need a Fermi chopper or an analyzer. The wavelength of the neutron scales with its speed and therefore the total ToF. From the wavelength and scattering angle, one can calculate the momentum transfer. The Q range and resolution of a diffractometer are thus determined by flight path, detector coverage, and the accuracy of the ToF.
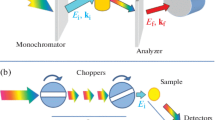
To achieve energy resolution, it is necessary to know the incident and final energy of the neutron. Neutron spectrometers can be designed in two different ways. One is to fix the incident neutron energy (using a Fermi chopper) so that one can derive the final energy from the total ToF. This design is called direct geometry. In contrast, in an indirect geometry spectrometer, the final energy is fixed (by an analyzer) so that from the total ToF, one can calculate the initial energy. A schematic illustrating how a ToF instrument works is shown in Fig. 24.3, with more detailed discussion afterward. For a direct geometry instrument, the energy resolution is often expressed as a fraction of the incident energy (e.g., 2% of Ei), whereas for indirect geometry instrument, the energy resolution is usually a fraction of the energy transfer (e.g., 1.5% of ΔE). The consequences of this difference are best shown in Fig. 24.4.

A schematic illustration of how a neutron ToF instrument works. (a) Direct geometry instrument has a fixed incident energy, and (b) indirect geometry instrument has a fixed final energy. L1 is the length of the primary flight path from the moderator to the sample, and L2 is the length of the secondary flight path from the sample to the detector

INS spectra of toluene, at 20 K using MARI (direct geometry) with (a) Ei = 4000 cm−1, (b) Ei = 2000 cm−1, (c) Ei = 800 cm−1, and (d) TOSCA (indirect geometry). (Adapted from Ref. [15], Copyright 2005, with permission from World Scientific Publishing Co Pte Ltd)
For an inelastic process, the arrival time of the neutron at the detector and either the source-to-sample flight time or the sample-to-detector flight time is required. This amounts to choosing either the incident neutron energy through a direct geometry or the final neutron energy through indirect geometry. In other words, the direct geometry is the instrument that fixes the incident energy, whether the indirect geometry is an instrument that fixes the final energy.
The direct geometry instruments at the spallation source use the chopper to fix the incident energy. The monochromation process causes a low incident flux, and this requires a large area of the detector. Due to the size of the detector, the resolution of the instrument can be improved by increasing the distance between the sample and detector and requiring a larger vacuum tank. This geometry usually includes several banks with different ranges of scattering angles, and thus it can measure Q and ω, respectively.
For indirect geometry spectrometer, the sample is exposed to a white beam of neutrons. After the neutrons scattering off the samples, the neutrons travel through a secondary spectrometer located at angles of 45° and 135°. The secondary spectrometer comprises highly orientated pyrolytic graphite. This selects the final energy of the neutrons as ~30 cm−1. The resolution of the indirect geometry spectrometer is determined by the characteristics of the secondary spectrometer and the distance between the sample and the moderator. As the initial flight path increases, the resolution increases [45].
In addition to the conventional diffractometers measuring the atomic structure, there are other diffractometers optimized for specific applications. These are small-angle neutron scattering (SANS) for nanometer to submicron structure and neutron reflectometers for surface structure [46, 47].
With catalysis experiments, it is often necessary to load gases and to control the reaction temperature and pressure. The development of sample environment is therefore essential. One thing we can take advantage of is the high penetration of neutrons, which means we can choose from a lot of materials, including the very hard ones such as steel or copper-beryllium to make the reactors. Another major advantage is that there is no need to have optically transparent materials such as glass and quartz for the reactors so that high-pressure experiments can be easily achieved. However, in the case of indirect geometry spectrometers, one often needs to make neutron measurements at very low temperatures (<50 K). Otherwise, the Debye-Waller factor could significantly suppress the signal. It is challenging for some in situ reactions (which may require >500 °C) without changing the sample environment equipment. In these cases, an alternate option is to pull the sample stick out of the instrument to perform ex situ reactions and then place it back for neutron scattering measurement. This limitation is less of a problem for diffractometers and direct geometry spectrometers.
2.4 Modeling
Neutrons interact directly with nuclei, and the scattering can be expressed in a rigorous analytical formula. This means from an atomistic model one can accurately simulate the neutron scattering spectra. For example, at low temperature under incoherent approximation, the inelastic neutron spectroscopy (INS) intensity from a vibrating atom can be calculated with a very simple equation [15]:
where \( {\boldsymbol{U}}_{\mathrm{s}}=\sqrt{\frac{\hslash }{2M{\omega}_{\mathrm{s}}}}{\boldsymbol{e}}_{\mathrm{s}} \) is essentially the zero-point displacement of the atom due to mode s, ωs is the frequency (of mode s), M is the atomic mass, es is the polarization vector, n in the excitation order, and Utotal is the total displacement of the atom with all modes. This simple equation governs the spectra measured at TOSCA/VISION on hydrogen-containing materials at base temperature.
The atomistic model needed for neutron scattering simulation can be either classical or quantum. For catalysis experiments, the density functional theory (DFT) [48, 49] and reactive force field method [50] are often used for the modeling. To compare with neutron scattering data, various tools can be used. For example, for pair distribution functions (PDF) measured in a diffraction experiment, reverse Monte Carlo method [51] can help to reconstruct the real-space atomic level structure and compare with theoretical predictions from DFT models. For QENS, MD can be used to simulate the diffusive dynamics and extract the corresponding parameters as obtained from the experimental data. To interpret INS data, an atomistic model is often needed to calculate the vibrational (phonon) behavior. The OCLIMAX software [52] developed at ORNL can be used to convert the vibrational information from various atomistic simulation tools to simulated INS spectra for various neutron spectrometers (Fig. 24.5). There are other software packages like aClimax [53] and ABIns [54] that mostly do the incoherent approximation for indirect geometry spectrometers.

The integrated approach at the VISION beamline for catalysis research. Background shows the crystal analyzers of the instrument. (Credit: Jill Hemman@ORNL). The OCLIMAX software serves as the link between the atomistic models and the INS experiments
3 Pros and Cons of Neutron Scattering for Catalysis Research
Many different problems in catalysis research can be tackled using neutron scattering. There are advantages and disadvantages associated with the technique and purpose of the experimental measurements. In this section, the pros and cons of neutron scattering will be discussed in general terms, and the details regarding each technique will be discussed in each of the following section.
For catalysis research, the major advantages of neutron scattering are as follows:
-
High penetration of neutrons: Neutrons do not have an electrical charge, and thus it does not ionize atoms in the same way that charged particles such as protons and electrons do. Consequently, neutrons have high penetration with low adsorption for most of the elements and allow probing the bulk structure of the samples. Thus, many commonly used catalysis reactor materials, such as stainless steel, aluminum, quartz, and sapphire, can be used in sample environments with a small loss of neutron signals.
-
Different neutron scattering lengths and cross sections with atomic number: Neutron scattering lengths are independent from atomic number, and the scattering lengths of different isotopes of a given element can be different. Particularly, there is a large difference between hydrogen and deuterium. Hydrogen has Hbcoh = −3.7406 fm, Hbinc = 25.274 fm, resulting in a scattering cross sections of Hσcoh = 1.7583 barn, Hσinc = 80.27 barn, while deuterium has Dbcoh = 6.671 fm, Dbinc = 4.04 fm, resulting in cross sections of Dσcoh = 5.592 barn, Hσinc = 2.05 barn. In other words, because of different signs of neutron scattering, by manipulating the H/D ratio in a molecule, one can vary the contrast within the sample or reactants and differentiate the source of hydrogen. In the case of ND, D is the choice, while for spectroscopy, H is usually better.
-
Right momentum and energy transfer for analysis: Neutron has the right momentum transfer and energy transfer to investigate both structure and dynamics in condensed matter such as catalysts.
-
A wide range of wavelengths: A wide range of wavelengths is available with various neutron sources and moderators. Probed length scale covers from the near angstrom sizes to the near micron sizes, thus covering structural information of catalysts from atomic to mesoscale and above.
The major limitations of neutron scattering are as follows:
-
Limited neutron sources: Neutron sources are very expensive to build and maintain. As a result, neutron scattering experiments can only be performed at limited reactor sources or spallation sources through user proposals. The experiment is also limited by the approved beamtime.
-
Long acquisition time: Most of neutron scattering techniques require longer acquisition time (up to hours) to get better statistics due to its low flux compared to X-ray techniques. Thus, is it difficult to carry out time-resolved experiments to follow the dynamic evolution of catalysts and surface reactions.
-
Limited application of operando/in situ experiments: The Debye-Waller factor is used to describe the attenuation of coherent neutron scattering caused by thermal motion. This factor can be suppressed when the lower temperature (<30 K) were applied. Most of the catalytic reactions require higher temperature, and thus the measurement especially for INS experiment should be first conducted at the reaction condition and then quenched to measurement temperature to avoid the effect by the Debye-Waller factor.
-
Large sample amounts: For the same reason (low flux), a neutron scattering experiment, especially neutron spectroscopy, typically requires relatively large amount of sample (~ grams). This is not an issue for studying industry catalysts but can be challenging for studying new catalytic materials that are difficult to synthesize at large quantity.
4 Inelastic Neutron Spectroscopy (INS)
4.1 Basic Principles of INS
Vibrational spectroscopy is a commonly used analysis tool in the field of catalyst for direct measurement of specific chemical bonds of adsorbed molecules and atoms on the surface of the catalyst. INS is a form of vibrational spectroscopy that is a complementary tool to conventional methods such as Raman and infrared spectroscopies.
The main difference between INS and conventional methods can be considered through the intensity of an INS mode as follows:
where \( {U}_d^i \) is the amplitude of the vibration of the dth atom in the ith mode and σd and md are neutron scattering cross section and mass, respectively. The total intensity is the summation of all individual atomic contributions.
The scattering intensity, which scales with the dynamic structure factor S(Q, ω), depends on both the transition energy, ω, and the momentum transfer, Q. The exponential term is the Debye-Waller factor, and UTot2 is the mean square displacement of the atom. The magnitude of the displacement is the part determined by the thermal motion of the molecule. Generally, the Debye-Waller factor can be suppressed by applying low temperature, which is why the INS spectra at indirect geometry spectrometers are typically measured at below 30 K. This however leads to limitations to in situ/operando studies for the catalysis researches. Thus, historically, most of the catalytic reactions for INS measurements were first conducted at the reaction condition and then quenched to measurement temperature to avoid the effect by the Debye-Waller factor [36]. The equation shows the dependence of momentum transfer and the amplitude of vibration of the atoms.
Additionally, the intensity is weighted by the total cross section of the element under incoherent approximation. Normally, the total scattering cross section of hydrogen (1H) is 82 barns, whereas that of deuterium (2H) or most other elements is less than 5 barns (Table 24.4). The combination of a large cross section and the low mass of hydrogen allows strong signals in INS spectra. Thus, the scattering from all other elements other than hydrogen can sometimes be ignored/minimized in hydrogenous materials and obtain useful information about the hydrogen species in the structure.
Since neutron is a particle with zero charge with high permeability of the sample, INS spectrum is the sum of the signals from neutrons scattered in the bulk of a substance and partially from the surface [15]. The surface scattering only can be obtained when a catalyst possesses a high surface-to-bulk signal ratio, for example, the surface possesses enough amounts of hydrogen compared with the bulk structure. For catalysts, the total active surface to obtain a reasonable INS signal is required to be about 100 m2. As a result, high loading of catalyst is commonly adapted, and generally up to 10 g of catalyst is used. The amount of the catalyst also depends on the instrumentation background level, and normally the amount of hydrogen required to obtain over the background level is of the order of 1 mmol of hydrogen atoms in the beam [38]. Because of the required large quantity of catalyst, it is a challenge for new materials but not a problem for industrial catalysts.
Catalytic reaction schemes often go through the first step of adsorption of at least one of the reactants. To understand the reaction through the INS experiment, the background spectrum has to be measured. The background here is the spectrum of the fresh sample that is not exposed to any reactants but after the needed treatment. If it is possible, the extra strong scattering source such as water should be removed before the measurement. This is generally conducted in the INS reactor cell with inert gas flow and evacuating while monitoring the desorbed products with a mass spectrometer. Depending on the sample properties, this can be a time-consuming step.
Several features of an INS spectrum have to be considered while analyzing the data: scattering intensity, energy range, transitions, peak positions, peak intensity, and peak shape. The scattering intensity is proportional to the number of scattered neutrons, against the energy that is transferred from the neutrons to the scatterer. The spectrum is a neutron energy loss spectrum; the neutrons deposit energy on to the sample. IR and Raman spectroscopy have limitations in the energy range due to the instrumental considerations (cutoff is 200–400 cm−1). The INS experiment can obtain the information in the low-energy region (less than 300 cm−1). However, it becomes weak and poorly resolved beyond 1600 cm−1 due to the Debye-Waller factor, particularly for indirect geometry instrumentation; this can be sometimes circumvented by using a direct geometry spectrometer with suitable incident energies, as shown in Fig. 24.4. In some cases, IR and Raman could be measured before the experiment in order to obtain complementary data prior to the INS experiment. The peak positions of INS spectra are signatures of a molecule’s structure and intramolecular forces. The intramolecular forces and the structure of the molecule determine atomic displacements, and thus different transitions show different intensities. The corresponding modes are observed in the INS, IR, and Raman at the same energies, and thus one could get complementary information through different types of vibrational spectroscopy. The peak intensity shown as the integrated area under the peak is proportional to the amplitude of motion during the vibration. INS is not limited by selection rules, and consequently the shape of the INS peak shows additional structural and dynamic information and has quantitative nature measured by the integrated spectral intensity [38].
In order to analyze the obtained spectra, theoretical modeling of the INS spectrum is often required. Neutron cannot see a local site at atomic level, but it shows the cumulative effect on the energies and numbers of the scattered neutrons of all collisions with the catalyst and adsorbed species. The challenge here is to separate the spectrum into the different scattering species using modeling. Using computer simulations, the information of phonons can be obtained from either quantum or classical calculations such as DFT and MD. For calculation, experimental perspectives such as the instrument geometry, resolution, and nature of the samples are considered. The model not only predicts the correct active phase of the structure but also gives the right vibrational dynamics. OCLIMAX [52] program allows users to obtain the full calculation of INS spectra including coherent effects and temperature effect for various INS instruments and arbitrary trajectories in the momentum and energy transfer (Q-ω) space.
4.2 Application of INS to Heterogeneous Catalysis
To date, INS is the mostly utilized neutron technique for catalysis research and has been mainly used on systems that are difficult or impossible to study by other spectroscopic techniques. When the catalyst is completely black or opaque, IR is incapable in studying either the surface or the bulk (without dilution) structures, while Raman spectroscopy only allows limited bulk structural information. Furthermore, the sample can decompose or exhibit fluorescence under a laser beam irradiation in Raman measurements. Additionally, there are very limited commercial techniques that can measure hydrogenous species. Unlike IR or Raman, most of the catalysts are transparent to neutrons when the sample includes a small quantity of hydrogen. INS has been applied in a wide range of catalyst studies including both ex situ and in situ conditions such as hydrogen adsorption [17, 55,56,57,58,59,60,61], hydrogen storage [24, 32, 62,63,64], hydrogenation reactions [13, 65, 66], CO2 methanation [14], hydrocarbons [67,68,69], and water adsorption [70,71,72] on different types of catalysts.
Hydride species are common intermediates in a variety of homo- and heterogeneous catalytic cycles and are involved in many important processes, from H2 storage to catalytic processes such as hydrogenation, dehydrogenation, hydrolysis, or reforming [73, 74]. Raman, IR, and 1H NMR have been adapted to characterize the structure of hydride in a variety of catalysts, but it is not well understood regarding their structure and catalytic functions due to the limitation in detecting the hydride in the lattice structure through conventional techniques. Neutron scattering, however, is a unique technique to detect hydride due to the large incoherent neutron cross section of hydrogen. It has been comprehensively used to study the bulk metallic materials such as palladium black [75,76,77,78,79], platinum black [80,81,82], Raney nickel [25, 83], Raney palladium [84], and bulk hydride such as CaH2, MgH2, BaH2, NiH2, and complex hydride [1, 4, 6, 85]. For example, Parker et al. studied Pd black through INS and computational approaches to elucidate the effects of particle size, support, and morphology on the formation of palladium hydride [78]. They reported that the particle size and state of aggregation influenced hydride formation. Additionally, the support affects the hydrogen storage capacity of the catalyst and the rate of hydrogen release. Chinta et al. employed INS to identify surface species formed during the H2-O2 reaction on Au/TiO2 catalysts [86]. They reported the formation of hydrogen peroxide and hydroperoxyl radical during the reaction of H2 and O2 over supported Au nanocluster, which is the first direct experimental evidence that shows hydrogen and oxygen react to form one or more hydrogen peroxide/surface hydroperoxy species, which then further react with propylene to form propylene oxide.
However, many catalysts of interest consist of supported active metal phase, which can achieve high surface area by exposing the large amounts of active sites. Specifically, for supported metal catalysts, hydrogen spillover occurs from a metal catalyst particle onto the catalyst support. The phenomenon has been widely studied , but questions remain about whether hydrogen spillover occurs on the non-reducible support. Mitchell et al. [17] investigated hydrogen spillover on carbon-supported precious metal with INS spectroscopy. The INS spectra showed characteristic riding modes of H on carbon and Pt or Ru. Riding modes are frequently observed when surface vibrational modes of the substrate are amplified by neutron scattering from surface-bound hydrogen atoms [17]. Thus, this surface mode can be distinguished from bulk modes of the substrate.
Metal oxides have often been used either as supports for metals or as catalysts themselves for hydrogenation reactions and can dissociate hydrogen either homo- or heterolytically. For some oxides where IR/Raman cannot detect hydride species, question still remains on whether the dominant mechanism is a homolytic path forming two new OH groups or a heterolytic path forming one new OH group and one metal hydride species. This was the case for ceria before INS study was attempted. Wu et al. [11] reported the direct observation of both surface and bulk cerium hydrides in addition to a new hydroxyl group through the INS experiment of H2 adsorption and dissociation over ceria. Coupling with other in situ spectroscopy including Raman, IR, and AP-XPS, it is revealed that H2 dissociates via homolytic pathway to form OH products on the O-vacancy-free ceria surface, while the dissociation is caused by a heterolytic pathway resulting in Ce-H and Ce-OH when O-vacancies are generated in ceria. The capability of INS in detecting and differentiating surface and bulk hydrides in ceria provides a unique opportunity to differentiate the active hydrogen species in similar phenomena over other oxide catalysts and in hydrogenation reactions. The follow-up study by Moon et al. [65] showed the reactivity of both surface atomic hydrogen and Ce-H in semi-hydrogenation of acetylene over reduced ceria using in situ INS study (Fig. 24.6). Interestingly, in this study, the surface atomic hydrogen with an effective mass of 3 amu has been confirmed at 32 cm−1 and was suggested as a precursor to OH groups from H2 dissociation. After this species is exposed to C2D2 for acetylene hydrogenation reaction, it is consumed during the reaction (Fig. 24.6a). Likewise, the Ce-H is also consumed after being exposed to C2D2 (Fig. 24.6b). The species left on the surface were confirmed as adsorbed ethylene species from the comparison with the computed spectra from ethane and ethylene (Fig. 24.6c), suggesting the selective hydrogenation capability of the two H species. In situ DRIFT result showed that bridging OH also participates in the selective hydrogenation of acetylene but is less active than the surface hydride species. Capitalizing on the complementary nature of INS (for hydrides) and DRIFTS (for OHs), the reaction mechanism of acetylene hydrogenation has been revealed in details. In particular, it underscores the critical role of hydride species involved in the hydrogenation reactions over ceria-based catalysts.

INS spectra of (a) CeO2-533K-H and (b) CeO2-623K-H collected at 5 K before and after reaction with C2D2. The DFT-calculated INS spectra of ethylene and ethane are shown in (c). The sample was activated at 673 K under an oxygen flow for 2 h, denoted as CeO2-673K-O. The sample was further exposed to 1 bar of H2 at various temperatures (533, 623, and 673 K in sequence), resulting in different degrees of reduction and oxygen vacancy formations, and such treated samples are denoted as CeO2-523K-H, CeO2-623K-H, and CeO2-673K-H. (Adapted from Ref. [65], Copyright 2020, with permission from American Chemical Society)
Zeolites are important materials used in industrial applications as adsorbents, ion exchangers, and catalysts. Specifically, INS has been widely applied to understand the location and energetics for H2 adsorbed in zeolites [55, 87,88,89,90] and other types of porous materials including metal-organic framework (MOFs) [64, 91, 92], fullerenes [93,94,95,96,97], and clathrate hydrates [98, 99]. For example, Ramirez-Cuesta et al. studied hydrogen adsorption in cation-substituted zeolite X [90] and copper-doped ZSM-5 [55] through INS . They reported the rotational-vibrational spectrum of adsorbed hydrogen in the low energy less than 200 cm−1 and showed the polarization of the H2 molecules dominates the interaction of H2 with the binding site for the first time. Additionally, they demonstrated the interaction of H2 molecule bounded in a particular microporous material, Cu/ZSM-5, and revealed H2 molecule is bound as a single species lying parallel with the surface. Lateral interactions between the adsorbed H2 molecules become apparent as H2 dosing increases. INS studies also have been applied study reactions over zeolites. For example, Hawkins et al. [69] studied the oligomerization of propene in ZSM-5 at 293 K by INS. The oligomerization of olefin over zeolite generally occurred via protonation of the olefin to a carbenium ion, which reacts with other olefin molecules to give alkyl chains. The selectivity and exact composition have been determined by shape-selective effects from the zeolite pore. It has confirmed that the oligomerization is particularly important at low temperature and showed the small-pore zeolite ZSM-5 can convert propene at room temperature. Figure 24.7 shows the INS spectra of propene adsorbed in HZSM-5 with dehydrated zeolite and pure propene spectra. The propene spectrum shows multiple clear peaks from following modes: methyl torsion (220 cm−1), C=C-C scissors (429 cm−1), =CH2 twist (585 cm−1), and C-H rocking (915 cm−1) and C-H stretch mode (broad peak centered at 3000 cm−1). For the spectrum of the catalyst after reacting with propene, the C=C modes are completely suppressed and are replaced by a broad band of overlapping methylene modes in the 700–1100 cm−1 range. The three peaks at 1312, 1376, and 1464 cm−1 are assigned to a new methylene scissors peak and the symmetric and asymmetric deformation of the terminal methyl groups, respectively. Additionally, the shift in location and narrowing of the C-H stretch peak to a new center at 2945 cm−1 represents the removal of essentially all sp2 carbon center by the chain formation reaction. No sign of O-H stretching also indicates the consumption of a majority of the acid sites to initiate the oligomerization reaction.

INS spectra of propene adsorbed in HZSM-5 at room temperature (upper blue) compared with pure propene (middle red) and the spectrum of the dehydrated zeolite (lower black). Spectra were measured at 20 K on the MAPS spectrometer. Arrow marked at the 736 cm−1 band is due to an in-phase rocking mode of CH2 groups in a linear oligomer. (Adapted from Ref. [69], Copyright 2019, with permission from The Royal Society of Chemistry)
5 Quasi-Elastic Neutron Scattering (QENS)
5.1 Basic Principles of QENS
QENS is one of neutron scattering techniques for catalysis. It is a very-low-energy inelastic process, which usually exhibits as a broadening of the elastic line at ω = 0 and probes whole-body motions such as the diffusional (translational or rotational) motion of atoms. There are excellent reviews in the literature about QENS technique [100]. It can be seen as similar to a Doppler broadening of the elastic scattered signal from a sample. Examples of new generation QENS instruments can be found in references [101,102,103,104].
QENS is sensitive to the diffusion and reorientation of atoms and molecules in the structure on a pico- and nanosecond timescale (10−13 to 10−7 second) with energies of 104 to 10−2 micro-electronvolt (μeV). The length scale of the QENS experiment can cover the inter- and intramolecular distance so that the diffusion of molecules in the lattice could be elucidated. The faster the diffusional motion, the larger the quasi-elastic linewidth. The classical instruments to measure QENS are direct geometry of ToF, indirect geometry so-called backscattering (BS) spectrometers, non-ToF indirect geometry, and neutron spin echo neutronics.
A typical QENS spectrum, at a given momentum transfer, Q, consists of a single peak centered at ω = 0. A resolution function of the sample is measured, R(Q, ω), at certain Q values; usually, this resolution function is measured at cryogenic temperatures. A series of QENS spectra as function of temperature, for the same sample, are then measured. The measured signal, SMeasured(Q, ω), is the convolution of the resolution function of the instrument with the quasi-elastic response, S(Q, ω), plus a linear background:
The extraction of the quasi-elastic response requires a deconvolution of the measured spectra; the symbol ⊗ represents convolution in ω. The deconvolution process requires the assumption of a functional form of the QENS response. The response can have more than one component, depending on the nature of the motion. A delta function δ(ω − ω0) indicates an elastic component, which is the fraction of the population that is not changing. The terms in the summation are usually assumed to be Lorentzian functions, with a full width half maximum (FWHM), Γi. The IE factor is the weight of the elastic component, while the \( {I}_{{\mathrm{QE}}_i} \) factors are the relative weights of each quasi-elastic component:
There is software designed to perform this analysis [105, 106]. By studying the dependence of Γ vs Q and using some simple models, it is possible to characterize parameters of the motion, for example, diffusion coefficients, residence times, characteristic lengths, etc. It is however challenging to analyze a system with more than two inelastic components unambiguously.
From the functional form of Γi(Q), it is possible to determine the nature of the process and characteristic parameters such as diffusion coefficients, residence times, characteristic lengths, etc.
By analyzing the dependence of the parameters on temperature, it is possible to determine quantities such as activation energies of the associated motions.
There is another important derived quantity, the elastic incoherent structure factor (EISF), which is a function defined as the intensity of the elastic response divided the total intensity:
In general, the functional representation EISF(Q) as function of Q is essentially the probability that a particle can be found in the same volume of space at some subsequent time. This is used to determine overall fraction of mobile atoms and the geometry of the molecular reorientations [100].
5.2 Application of QENS to Heterogeneous Catalysis
Hydrogen has the largest incoherent cross section, and thus the motion of hydrogen species can be well investigated through QENS analysis. The early QENS methods have been applied to measure the transport process of hydrogen over zeolite in the mid-1980s [19]. Afterward, hydrocarbon dynamics in zeolite has been heavily studied due to its importance in the petrochemical industry. Combined experimental QENS studies and theoretical studies have gained important insights into linear and branched hydrocarbon dynamics in different types of zeolites or porous materials [10, 20, 107,108,109,110,111,112,113,114,115].
The methanol-to-gasoline (MTG) or methanol-to-light olefin (MTO) reaction is one example of producing hydrocarbons from natural gas or coal, and the zeolite catalyst ZSM-5 (MFI) was first used in the Mobil MTG process commercialized in 1985. The chemistry beyond the carbon chain growth in the MTG process is a debated topic, but there is widespread agreement on so-called hydrocarbon pool created in the zeolite pores. To understand the reaction mechanism, the diffusion of the reactant into pores of the zeolite and the extent which can be influenced by the presence of a hydrocarbon and coke deposits is an important issue that needs to be explained. Matam et al. [116] investigated the dynamics of methanol in ZSM-5 catalysts both with and without the hydrocarbon pool resulting from MTG reaction at 623 and 673 K. Three zeolite catalysts were studied: ZSM-5-F (clean unreacted), ZSM-5-623 (subjected to MTG at 623 K for 3 days), and ZSM-5-673 K (MTG at 673 K for 3 days). QENS spectra for each sample were obtained as a function of Q at 325 K. This study showed a notable decrease in the elastic intensity as increase Q, and the decay of relative elastic intensity does not become significant until Q is higher than 1 Å−1. However, the peak broadens at higher Q value, as shown in Fig. 24.8, when comparing ZSM-5-F with ZSM-5-623. The lack of any broadening of the elastic peak is attributed to the presence of strongly hydrogen-bonded methanol, or the absence of mobility of the methanol is due to dissociation to form methoxy group. ZSM-5-623 is indistinguishable from the fresh ZSM-5-F as far as the QENS of adsorbed methanol is concerned, other than a reduced intensity due to the lower methanol uptake/loading. The obtained number from EISF fittings allows one to characterize the localized motion of methanol, related to the geometrical motions of the proton in the molecule. The analysis, combined with EISF, isotropic rotation, and translational motion of methanol, suggests that methanol is immobile on the QENS instrumental timescale in the fresh catalyst and in the sample that was exposed to MTG condition for 3 days at 623 K. However, for the MTG reaction at 673 K for 3 days, isotropic methanol rotation is visible.

QENS spectra at Q = 1.7 Å−1 for methanol at 325 K in ZSM-5-F, ZSM-5-623, and ZSM-5-673. Dashed blue line is the total fit to the data points; red dashed line is the resolution function representing the elastic component. (Adapted from Ref. [116], Copyright 2018, with permission from The Royal Society of Chemistry)
QENS analysis is also adapted to investigate hydrogen diffusion over an iron-based Fischer-Tropsch synthesis catalyst. Previous studies using XRD, Raman, temperature-programmed oxidation and TEM, and INS showed the evolutionary profile of the active phase of the catalyst during FT in addition to the development of a hydrocarbon overlayer. Although the active phase for the FTS has been elucidated, the role of hydrocarbonaceous overlayer on the iron-based FTS catalysts during CO hydrogenation is still under debate. Earlier study from Warringham et al. showed that a hydrocarbonaceous overlayer was formed during the catalyst conditioning period [68]. The evolving nature of the catalyst modifies the proportion of phase presented within the catalyst which can influence the transport of hydrogen within the reaction system. Davidson et al. [12] examined the hydrogen diffusion using a QENS experiment to understand the role of the formed overlayer on the FTS catalyst. An unpromoted iron FTS catalyst was exposed to various levels of time-on-stream (0, 12, and 24 h) of CO hydrogenation at ambient pressure. The intensity of an elastic peak changed as a function of reaction temperature range from 20 to 100 K and showed the hydrogen incorporation into the catalyst surface structure. The data at 50 and 70 K showed the diffusion within condensed hydrogen, but the diffusion rate varied with time-on-stream, which is consistent with the hydrogen associating with the evolving catalyst matrix. From the obtained diffusion constant for Fickian diffusion, one can suggest that hydrogen diffusion within the overlayer is physically feasible, and the longer the reaction period, the greater the diffusion rate.
Recently, QENS was also applied to understand the properties of a new material called oxyhydride, particularly the one that possesses the structure of perovskite oxide. An oxyhydride is a mixed anion compound containing both oxide anion, O2−, and hydride ions, H−. Generally, these two ions do not coexist, but it is stable when the anions are electropositive, and the conditions are sufficiently reducing. Oxyhydride in the perovskite oxide structure includes a sizable amount of hydride in the lattice and has shown the potential for various applications such as ammonia synthesis [117] and CO2 hydrogenation [5]. Although it showed good reactivity for different reactions, the dynamics beyond the diffusion of hydride in the structure are still being investigated. Bridges et al. [2] conducted a QENS experiment on LaSrCoO3H0.7 and observed hydride-ion mobility through hopping along the direction of vacancies in the hydride ion sub-lattice. The reported jumping distance is close to the distance between adjacent hydride-ion sites. Additionally, it has been elucidated that temperature is an important factor to initiate the hopping in the lattice structure and it causes the loss of hydrogen from the sample. Similarly, Carin et al. [21] studied the dynamics of hydride ions in a perovskite oxyhydride material that possesses a BaTiO3 perovskite structure. QENS analysis measured in a lower temperature region (<250 K) showed that hydride ion jumps between the nearest-neighbor oxygen vacancies with a mean residence time of the order of 0.1 ns. For a higher temperature region (>400 K), hydride ion jumps between the next nearest-neighbor oxygen vacancies with the order of picoseconds, but there is no strong dependence on the concentration of hydride ions and oxygen vacancies. The diffusion coefficient was obtained through two jump-diffusion models: the Chudley-Elliott model and the Gaussian model (Fig. 24.9). The diffusion coefficient follows the Arrhenius law, and, thus, one could obtain activation energy. The derived activation energy Ea shows a slight increase with increasing oxygen vacancy concentration. This is useful new information for the comparison with future experimental and computation results for different hydrogenation/dehydrogenation reactions to determine the defect nature and transport properties of the materials.

Arrhenius plot of the diffusion coefficient D extracted from the Chudley-Elliott and Gaussian models of the CA1 and CA2. CA1 represents BaTiO2.82H0.1 which include more hydride than oxygen vacancies. CA2 possesses more oxygen vacancies than hydride with a formula of BaTiO2.65H0.087. NAB includes a large amount of oxygen vacancies compared with hydride (BaTiO2.3H0.04). The fits correspond to activation energies of 85(2) and 94(2) meV for the Chudley-Elliott model and 105(18) and 118(42) meV for the Gaussian model for CA2 and NAB, respectively. (Adapted from Ref. [21], Copyright 2018, with permission from American Chemical Society)
6 Neutron Diffraction (ND)
6.1 Basic Principles of ND
In a diffraction experiment , as the incident neutron wave reaches an atom, the atomic site becomes the center of a scattered spherical wave that has a definite phase. As the waves expand from a regular array of sites in a crystal, the individual interference will amplify each other only in particular directions. The direction is closely related to the spacing of the scattering sites and the symmetry of the crystal. As a result, the direction in which interference occurs can be used to infer the lattice parameter and symmetry of the crystal.
ND peaks appear when the neutron wavelength λ and the angle θ satisfied as Bragg’s law, nλ = 2d sinθ. To obtain diffraction from a set of planes, the crystal must be rotated to the correct orientation, and thus the scattering vector is perpendicular to the scattering planes. The observed signal during this crystal rotation is called a Bragg peak, and this signal only occurs when the crystal is in the correct orientation. The intensity of the scattered neutrons is proportional to the square of the density of atoms in the atomic planes responsible for the scattering. Thus, the observation and analysis of those Bragg peaks allow one to understand the spacing and the density of atoms in the planes.
For powder diffractometer at a reactor neutron source, a beam of the neutron with a single wavelength can be selected by a device called a monochromator. The monochromator is an assembly of single crystals that are correctly oriented to reflect a mono-energetic beam of neutrons toward the scattering samples. It is usually made with either pyrolytic graphite, silicon, or copper. The scattered neutron from the sample is counted by suitable neutron detectors and recorded as a function of an angle. Bragg peaks from atomic planes in a typical ND pattern correlate with different interplanar spacing, d. However, for diffraction pattern from a pulsed neutron source, the sample is irradiated by pulsed beams of neutrons with a wide spectrum of energies. The scattered neutrons from the samples are recorded in banks of detectors located at different scattering angles. Additionally, the time that scattered neutrons arrive at the detector is recorded. Diffraction patterns from reactor source and pulsed neutron source are very similar at a certain scattering angle, but the independent variable is a time of flight from the neutron, not the scattering angle for pulsed neutron sources. Normally, several banks of position-sensitive detectors are located in the beamline, and this can cover the wide range of the d-spacing allowing accurate determination of scale factors such as phase fraction through refinement.
ND is a complementary technique to X-ray diffraction. XRD is a well-established analysis tool that gives information on the crystalline phase (fingerprint), crystal structure, and quantitative phase analysis through the Rietveld refinement and size/strain effects from line profile analysis. However, the crystalline structure of light elements cannot be elucidated with XRD only; instead, ND is powerful to enlighten the structure of light elements in the lattice structure. Additionally, scattering amplitude of neutron shows significant variations in the same range of scattering vector, although scattering amplitude decays greatly with the scattering vector for X-ray [118]. In other words, intensities of Bragg peaks at higher Bragg angles from XRD decay quickly, and it is hard to access information located at higher angle, but for ND, it can resolve fine structure and textural details of nanocatalysts, allowing access to a large number of reflections to very short d-spacing (high Bragg angles).
Traditional diffraction shows the information on an average scale, and the diffraction is not able to recognize nonequivalent sites and slight distortions from local interactions and connectivity. The local atomic structure of materials can be elucidated through PDF analysis without invoking crystalline symmetry. Although diffraction patterns of nanoscale, amorphous, and disordered materials do not have well-defined Bragg peaks, PDF analysis combined computational modeling and simulation allow us to observe detailed descriptions and their application in structural changes in the field of catalysis [119,120,121,122]. The PDF, or G(r), is obtained from the structure factor, S(Q), via a Fourier transformation:
The obtained analysis from this quantity gives information of the local structure of the materials. The Fourier transform requires data over a large Q range to avoid truncation effects, so the technique typically uses rather short-wavelength neutrons at spallation source or the hot end of reactor source.
6.2 Application of ND to Heterogeneous Catalysis
Generally, structural investigations on crystalline catalysts are often performed by X-ray diffraction. In fact, it is required to obtain X-ray diffraction pattern to confirm the phase of synthesized catalysts. However, as we mentioned above, it is hard to determine the structure that includes atoms with similar atomic number or light elements with X-ray. ND fills the gap and provides complementary structural information. Mawdsley et al. [123] studied nickel-containing perovskite oxide catalysts used to catalyze the autothermal reforming (ATR) of isooctane (C8H18) into a hydrogen-rich gas. ND offers the advantage of being particularly sensitive to nickel, because nickel has one of the largest neutron scattering lengths of all of the elements, so that even small amounts can be easily detected [124]. The catalyst was analyzed through XRD and ND together to determine the phase stability of catalyst in the reducing environment. Perovskite oxide has the general formula of ABO3, where A is an alkaline-earth metal and lanthanide and B is a transition metal. Both A and B can be substituted with different cations to introduce structural and electronic defects which may increase catalytic performance. In this study, the catalytic performance of perovskite oxide with general formula La1-xSrxM0.9Ni0.1O3±δ (M=Cr, Mn, or Fe) has been tested. For perovskite oxide that includes nickel, a hysteresis has been observed in the hydrogen and CO yields, with the decreasing-temperature yields being higher than the increasing-temperature yield. This is believed to be due to the formation of metallic nickel excluded from the perovskite structure after the high-temperature reaction, which promotes steam reforming. XRD has been used to analyze the phase of the catalyst; however, the sensitivity is not enough to confirm the presence of metal phase of nickel in this study. The high sensitivity of ND allows detecting the presence of the nickel metal phase. The segregation of the nickel metal phase has been confirmed by ND.
ND also can be used as a complementary tool to other in situ techniques used in catalysis research. In contrast to XRD, the scattering power is independent of the diffraction angle providing diffraction peaks at higher angles with higher intensity. This in particular useful for the investigation of nanomaterials with highly symmetric crystal structures that often suffer from few and weak XRD peaks available for structural analysis. Kandemir et al. utilized ND as a method for in situ analysis of heterogeneous catalysts, especially over methanol synthesis catalyst, Cu/ZnO/Al2O3 [125]. Figure 24.10 shows the high-resolution ND patterns of the reduced catalyst in 0.1 MPa Ar at 523 K right before the methanol synthesis and during the synthesis under 6 MPa of syngas after 3 and 34 h time-on-stream. The Rietveld fit of the fresh catalyst (Fig. 24.10, top) reveals the presence of fcc-Cu as the major phase at 523 K. No major changes of the Cu phase are obvious from a first comparison of the three ND patterns recorded at different conditions. This result showed that bulk inclusion of C or H species from the gas phase in the Cu lattice of working catalysts can be readily excluded. Additionally, the Rietveld fitting showed the crystalline domain size increment due to sintering of the Cu particles which proves as a relevant deactivation mechanism of methanol synthesis catalysts.

The Rietveld fits of the catalyst before (0.1 MPa Ar, top panel), at the beginning (center), and after 24 h of methanol synthesis (bottom) at 523 K and 6 MPa. The data with gray is experimental data and with black line is the calculated pattern. The thin gray line is the difference between experimental and calculated pattern. The contribution of the Cu phase and ZnO is marked as red and green lines with tick marks at the positions of Bragg reflections. Additional strong peaks from the Al reactor wall were treated as peak phase during the Rietveld analysis and are excluded from the overall calculated profile shown here. (Adapted from Ref. [125], Copyright 2013, with permission from WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim)
Additionally, ND has been used to elucidate the location of hydrogen species in the structure of catalysts. ND is sensitive to deuterium, an isotope of hydrogen, which has a large coherent scattering length. Oftentimes, ND has been used to understand the position of hydrogen in the structure of hydrogen storage materials [126] including metal hydride or complex hydride [6, 7, 127,128,129,130,131], carbon-based materials [132, 133], MOF [134,135,136], ammonia borane [137], etc. In the field of catalysis, knowledge of the exact active sites during the reaction and how they work is primitive. The development of high flux neutron instruments at both reactor and spallation sources allows studying dynamic process using in situ or operando experiments. Kammert et al. studied electride material using in situ ND and INS for ammonia synthesis [24]. Ru-loaded C12A7:e- (12CaO·7Al2O3-type electride with an anti-zeolite nano-porous structure) was used to synthesize ammonia at the atmospheric condition at 400 °C. Although this catalyst shows better reactivity, the catalytic mechanism has not been determined due to the lack of information on the formation and reactivity of hydride species during ammonia synthesis. In this study, in situ diffraction incorporated with difference Fourier map analysis confirms the formation of hydride in the cage of the electride and stability of this hydride species during different gas environments (D2, N2, and the mixture of D2 and N2 with a ratio of 3:1) (Fig. 24.11). An isolated atomic species in the center of the cage was observed in the cage lattice during the D2/N2 reaction. The measured neutron scattering from this Fourier map analysis was 65 pm which is close to the scattering length density of D (66.7 pm). The chemical nature of the newly formed species was confirmed by INS to be hydride (deuteride) which was found both thermally and chemically stable under ammonia synthesis condition. The ND and INS work suggest that it is the surface hydrogen, not the bulk hydrides, that plays the major role in ammonia synthesis over the electride-based catalyst. The findings from this work give insights for understanding catalysis by electride-based materials for ammonia synthesis and hydrogenation reactions in general. Additionally, Kandemir et al. studied the real structure and stability of ammonia iron under Haber-Bosch process using in situ ND [138]. The iron catalyst was exposed close to the conditions of the Haber-Bosch process (75 bar and 425 °C) during in situ ND measurement, and no major dynamic phase change due to the nitridation step was observed on an industrial ammonia synthesis catalyst. The stability of α-Fe phase in the catalyst could not be expected based on the thermodynamics of pure iron at such ammonia pressure. Compared to reference Fe, the industrial catalysts showed a complex and defective microstructure that arises in the preparation and activation steps. This unique feature was stable, and hardly any structural healing was observed after 88 h of ammonia synthesis operations. The complex real structure of the catalyst after activation and its stability suggests a prerequisite for high performance and should be target of any alternative synthesis approaches for ammonia synthesis catalysts for a simple preparation.

Difference Fourier maps created from results obtained using from ND. All maps are representations of the (001) planes in C12A7:e-. Diffraction patterns were obtained at 673 K after continuous reaction under conditions in the order (a) to (f) in 0.1 MPa gas pressure. (a) He treatment for 30 min, (b) D2:N2 = 3:1 for 3 h, (c) D2 for 2 h, (d) N2 for 2 h, (e) D2:N2 = 3:1 for 2 h, and (f) D2:15N2 = 3:1 for 2 h. Note that the scale for neutron scattering length density is the same in all maps. (Adapted from Ref. [24], Copyright 2020, with permission from American Chemical Society)
ND total scattering methods spanning a wide Q range allows to measure time-resolved catalytic chemistry to probe the reaction within the pore of the catalyst. This method gives access to the reaction rates on both nanometric and atomic length scales, providing an atomistic structural viewpoint on the reaction mechanism. Youngs et al. utilized this method to study kinetics of benzene hydrogenation over Pt/SiO2 [139]. The evolution of F(Q), formed from a weighted sum of the partial structure factor, is recorded as a function of time (Fig. 24.12) during benzene hydrogenation (reaction scheme in Fig. 24.12). It illustrates the changes within structural features across atomic, molecular, and mesoscopic length scales simultaneously. At specific Q values with respect to time, the kinetics corresponding to different length scales in the system can be analyzed further (Fig. 24.12b). The first diffraction peak at Q = 0.19 Å−1 showed both a fast and a slow component with a rate constant of 2.146 h−1 and 0.138 h−1, respectively, while at higher Q > 1.2 Å−1, it revealed an intermediate rate (k2,3,4 ≈ 0.35 h−1). The first diffraction peak is related to the change in contrast between the mesoporous substrate and absorbed material, but the second process also contributes to this change on a different timescale. Therefore, the first process is attributed to the dissociative adsorption of D2, and this is consistent with the rapid and relatively large increase in temperature over the first 30 minutes following introduction of the D2. In Fig. 24.12c, the slow component k1 does not reflect a chemical change and is likely to be related to the mass transport of the products within the pore. The change at Q = 1.22 Å−1 reflects nearest-neighbor molecular interactions, while those at 3.1 and 4.3 Å−1 are associated with atomistic chemical changes within the system. Additionally, these features evolve with similar time constants and thus are correlated with the reduction of benzene. This information gives the kinetic information of the hydrogenation of benzene reaction and elucidates the process scheme for the reaction at conditions of room temperature with 250 mBar of D2 (Fig. 24.12e). This study provides a method of examining structure-reactivity correlations for these complex systems in detail, thus allowing the effects of mass transport within the catalyst and surface reaction to be decoupled. Additionally, the technique takes a step to allow both structure and spatial data to be tied effectively with the kinetics of the underlying processes.

(a) F(Q)/time domain data over the course of the reaction with the benzene hydrogenation reaction scheme on the left panel. (b) Slices taken through (a) at specific Q values and corresponding exponential fits to the data. (c) Sample temperature as measured at the top and bottom extremes of the TiZr cell (d) pressure of D2 gas present in 4 L supply reservoir. (e) Reaction scheme elucidated from the experiment. (Adapted from Ref. [139], Copyright 2013, with permission from The Royal Society of Chemistry)
7 Other Neutron Scattering Techniques for Heterogeneous Catalysis
In addition to INS, QENS, and ND, SANS and neutron imaging have also been used in catalysis.
SANS is an elastic neutron scattering at a small angle to investigate the structure covering mainly the nanometer (10−9 m) to micrometer scale (10−6 m) [46]. Scattering measurements are performed in the Fourier space (also called reciprocal space) but not in real space like microscopy. Obtained scattering data needs to be converted to real space or fitted to various models to describe structures in reciprocal space. Normally, SANS is complementary to small-angle X-ray scattering (SAXS) which uses X-ray instead of the neutron. Due to the high penetration of thermal neutrons, the bulk structure of the catalyst may be studied, and sample environments are easily varied over a wide range of pressure and temperature. Additional advantages of SANS over SAXS include minimum beam damage and contrast variation achieved through isotopic labeling. The isotope labeling is particularly useful for the study of hydrogen-rich organic and biological materials. However, the low flux of neutron sources compared to the X-ray source means it requires longer data acquisition time.
One application of SANS in catalysis is to study fuel cell electrode, focused on electrode layers of high-temperature PEM fuel cells. Holderer et al. utilized neutron scattering experiment where simultaneously data in the small-angle and diffraction regime are taken on an electrode layer with different Pt loading with and without phosphoric acid (Fig. 24.13) [140]. It probes the ensemble average of the sample and is in the sense complementary to the local real space information obtained with microscopy techniques. Both SANS and wide-angle neutron scattering (WANS) have been applied in this study, and thus it can cover the Q range from 1 to 1000 Å−1 including diffraction part of the scattering curve with q > 1 Å−1. The characteristic length scale with the two instances of the Beaucage model with Rg,0 of about 10 Å from Pt particles and a large length scale Rg,1 ≈ 500 Å of the supporting structure indicating Porod scattering of a flat interface such as no surface fractal structure. This work showed that scattering experiments over a large range of length scales provide an insight into the individual components such as catalyst particles and carbon structure and investigate at the same time on larger scale to understand the fractal structure of the electrode material and its evolution upon filling with electrolyte. SANS analysis covering a large range in reciprocal space provides the potential to reveal structural properties on length scale from sub-nm to micrometers with unique contrast variation properties useful for samples with light elements and heterogeneous multicomponent environment.

(a) SANS-WANS diffraction data for the empty 20% platinum coating electrode layer, (b) only the high-q region on a linear scale, (c) the same sample filled with phosphoric acid, (d) high-q region of the phosphoric acid-doped electrode. The hydrogen content is responsible for the higher incoherent background. (Adapted from Ref. [140], Copyright 2020, with permission from MDPI, Basel, Switzerland)
Neutron image is an imaging process based on the neutron attenuation properties of the objects. It is a complementary tool to neutron scattering; it can directly probe the samples in real space on macroscopic length scales within the range from micrometers to decimeters. It is however rarely used to characterize catalysts due to the length scale it probes. Presently, neutron imaging in catalysis is mainly used to understand the flow pattern in the reactor or porous media. For example, Borgschulte et al. [141] studied the CO2 methanation process in a sorption fixed bed reactor using time-resolved neutron imaging. Thanks to the high neutron attenuation coefficient of hydrogen, the adsorbed water in the sorption catalyst, such as LTA zeolite, gives a high contrast allowing researchers to follow the formation and distribution map. LTA zeolite is known to absorb large quantities of water. The removal of water from the reaction centers is the key to enhance the conversion yield while suppressing side products such as CO. Normally, sorption reactor includes significantly large amount of catalyst to improve the sorption to make water remains in the bed. Figure 24.14 shows a typical neutron imaging result of the relative neutron absorbance of the reactor during the reaction and regeneration. Dark regions normally occurred when the neutron absorbance increased, and thus it represents the higher water contents in the sorption reactor. The water forms from the Sabatier reaction (the hydrogenation of CO2), which is adsorbed by the zeolite while the methane leaves the reactor. The neutron images show the formation of a reaction front running through the reactor. The gas product from the outline was also continuously monitored, and the water contents of the sorbent and catalytic activity can be correlated. There is no CO2 leaving from the reactor as long as the total water contents increase linearly. The neutron imaging may be applied to a similar process with sorption-enhanced reactions such as reversed water-gas shift reaction and purification of gases by adsorption under static and dynamic conditions.

(a) Top: simplified experimental setup used to measure the water content in sorption reactors at the NEUTRA beamline at SINQ, PSI Switzerland. (a) Bottom left: drawing of the reactor with dimension and gas fluxes and gas analysis. (b) Hydrogen flux is kept constant, and during the reaction and regeneration phase, the CO2 flux is switched on and off, respectively. (Adapted from Ref. [141], Copyright 2016, with permission from the Owner Societies)
8 Summary
The applications of neutron scattering to catalysis have been mainly focused on the systems which are difficult to study by other available techniques. It has a major recognized role in structural studies including light atoms and made a significant contribution to understanding the role of hydrogen or hydrogen-containing molecules over the catalysts. Throughout this chapter, theories and the applications for catalyst researches of main neutron scattering techniques, INS, QENS, and ND, along with other neutron techniques have been reviewed. INS has provided a different perspective of adsorbates on catalysts and sometimes complementary information on surface reactions over catalysts to other techniques such as IR and Raman. QENS has been used to understand transport motions of hydrogen-containing species on catalyst surfaces and in catalyst pores. This technique deserves more attention as it reveals the significance of transport processes in controlling catalytic environments. ND allows one to locate the light element in the lattice structure of the catalyst and gives better insights at a higher diffraction angle with stronger intensity. Overall, the unique power of neutron scattering in probing the vibration modes of adsorbed atoms and dynamics of molecular processes in catalytic systems has been demonstrated from the case studies with these neutron techniques.
Neutron scattering provides observable characteristic results that can be straightforward calculated, and thus neutron scattering combined with modeling allows one to obtain better understating of the observed neutron scattering results. Due to the recent development of computer science and quantum chemistry, we believe that the coupling of modeling and data science with neutron scattering experiments could revolutionize the field of the catalyst research.
With further developments in neutron sources, instrumentation, detectors, and more techniques and methodologies, neutron scattering is becoming available for the advanced characterization of catalysis materials.
Although catalysis research is a well-established area, the fundamental properties and the nature of the catalytically active site of heterogeneous catalysts under realistic reaction conditions have remained largely unknown. Having access to such information is of utmost importance to understand the rate-determining processes and steps of many heterogeneous reactions and identify important structure-activity/selectivity relationships. In situ and operando methods have become available to identify the structural and morphological properties of the catalyst under reaction conditions, but only limited techniques of neutron scattering such as ND can be combined with in situ/operando methods. It is challenging to improve spatial and time resolution of neutron scattering; however, with further development of instrumentation and detection methods under working condition, we believe neutron scattering could revolutionize the field of in situ/operando characterization of catalyst and thus plays an important role in understanding the catalysis in the future.
References
Schimmel, H.G., Johnson, M.R., Kearley, G.J., Ramirez-Cuesta, A.J., Huot, J., Mulder, F.M.: The vibrational spectrum of magnesium hydride from inelastic neutron scattering and density functional theory. Mater. Sci. Eng. B. 108, 38–41 (2004)
Bridges, C.A., Fernandez-Alonso, F., Goff, J.P., Rosseinsky, M.J.: Observation of hydride mobility in the transition-metal oxide hydride Lasrcoo3h0.7. Adv. Mater. 18, 3304–3308 (2006)
Polo-Garzon, F., Luo, S., Cheng, Y., Page, K.L., Ramirez-Cuesta, A.J., Britt, P.F., Wu, Z.: Neutron scattering investigations of hydride species in heterogeneous catalysis. ChemSusChem. 12, 93–103 (2019)
Fu, Q.J., Ramirez-Cuesta, A.J., Tsang, S.C.: Molecular aluminum hydrides identified by inelastic neutron scattering during H2 regeneration of catalyst-doped Naalh4. J. Phys. Chem. B. 110, 711–715 (2006)
Tang, Y., Kobayashi, Y., Tassel, C., Yamamoto, T., Kageyama, H.: Hydride-enhanced Co2 methanation: water-stable Batio2.4h0.6 as a new support. Adv. Energy Mater. 8, 1800800 (2018)
Sato, T., Ramirez-Cuesta, A.J., Daemen, L.L., Cheng, Y., Orimo, S.-i.: Evidence of intermediate hydrogen states in the formation of a complex hydride. Inorg. Chem. 57, 867–872 (2018)
Noritake, T., Aoki, M., Towata, S., Ninomiya, A., Nakamori, Y., Orimo, S.: Crystal structure analysis of novel complex hydrides formed by the combination of Libh4 and Linh2. Appl. Phys. A. 83, 277–279 (2006)
Masakuni, O., Chun-Keung, L.: Neutron studies of nanostructured Cuo□Al2o3 Nox removal catalysts. Phys. B Condens. Matter. 241-243, 269–275 (1997)
Huq, A., Chen, W.-R.: Neutron scattering for in-situ characterization of heterogeneous catalysis. In: In-Situ Characterization of Heterogeneous Catalysts, pp. 169–189 (2013). https://doi.org/10.1002/9781118355923.ch6
Omojola, T., Silverwood, I.P., O’Malley, A.J.: Molecular behaviour of methanol and dimethyl ether in H-Zsm-5 catalysts as a function of Si/Al ratio: a Quasielastic neutron scattering study. Catal. Sci. Technol. 10, 4305 (2020). https://doi.org/10.1039/D0CY00670J
Wu, Z., Cheng, Y., Tao, F., Daemen, L., Foo, G.S., Nguyen, L., Zhang, X., Beste, A., Ramirez-Cuesta, A.J.: Direct neutron spectroscopy observation of cerium hydride species on a cerium oxide catalyst. J. Am. Chem. Soc. 139, 9721–9727 (2017)
Davidson, A.L., Webb, P.B., Silverwood, I.P., Lennon, D.: The application of quasi-elastic neutron scattering to investigate hydrogen diffusion in an iron-based Fischer–Tropsch synthesis catalyst. Top. Catal. 63, 378 (2020). https://doi.org/10.1007/s11244-020-01259-2
Warringham, R., McFarlane, A.R., MacLaren, D.A., Webb, P.B., Tooze, R.P., Taylor, J., Ewings, R.A., Parker, S.F., Lennon, D.: The application of inelastic neutron scattering to explore the significance of a magnetic transition in an iron based Fischer-Tropsch catalyst that is active for the hydrogenation of Co. J. Chem. Phys. 143, 174703 (2015)
Terreni, J., Sambalova, O., Borgschulte, A., Rudić, S., Parker, S.F., Ramirez-Cuesta, A.J.: Volatile hydrogen intermediates of Co2 methanation by inelastic neutron scattering. Catalysts. 10 (2020)
Mitchell, P.C.H.: Tomkinson, J., Parker, S.F., Ramirez-Cuesta, A.J.: Vibrational Spectroscopy with Neutrons: With Applications in Chemistry, Biology, Materials Science and Catalysis, vol. 3. World Scientific, Singapore (2005)
Parker, S.F., Lennon, D., Albers, P.W.: Vibrational spectroscopy with neutrons: a review of new directions. Appl. Spectrosc. 65, 1325–1341 (2011)
Mitchell, P.C.H., Ramirez-Cuesta, A.J., Parker, S.F., Tomkinson, J., Thompsett, D.: Hydrogen spillover on carbon-supported metal catalysts studied by inelastic neutron scattering. Surface vibrational states and hydrogen riding modes. J. Phys. Chem. B. 107, 6838–6845 (2003)
Jobic, H., Renouprez, A., Bee, M., Poinsignon, C.: Quasi-elastic neutron scattering study of the molecular motions of methanol adsorbed on H-Zsm-5. J. Phys. Chem. 90, 1059–1065 (1986)
Jobic, H., Bee, M., Renouprez, A.: Quasi-elastic neutron scattering of benzene in Na-Mordenite. Surf. Sci. 140, 307–320 (1984)
Silverwood, I.P., Sakai, V.G.: Propane diffusion in Zsm-5 pores measured by quasielastic neutron scattering under macroscopic flow. Chem. Eng. Sci. 186, 116–121 (2018)
Eklöf-Österberg, C., Nedumkandathil, R., Häussermann, U., Jaworski, A., Pell, A.J., Tyagi, M., Jalarvo, N.H., Frick, B., Faraone, A., Karlsson, M.: Dynamics of hydride ions in metal hydride-reduced Batio3 samples investigated with quasielastic neutron scattering. J. Phys. Chem. C. 123, 2019–2030 (2019)
Turner, J.F.C., Benmore, C.J., Barker, C.M., Kaltsoyannis, N., Thomas, J.M., David, W.I.F., Catlow, C.R.A.: Probing the nature of acetylene bound to the active site of a Nina−zeolite Y catalyst by in situ neutron scattering. J. Phys. Chem. B. 104, 7570–7573 (2000)
Thomas, J.M.: High-resolution electron microscopy, neutron diffraction with isotopic substitution and X-ray absorption fine structure for the characterisation of active sites in oxide catalysts. In: Gamba, A., Colella, C., Coluccia, S. (eds.) Studies in Surface Science and Catalysis, 140, pp. 1–12. Elsevier, Amsterdam (2001)
Kammert, J., Moon, J., Cheng, Y., Daemen, L., Irle, S., Fung, V., Liu, J., Page, K., Ma, X., Phaneuf, V., Tong, J., Ramirez-Cuesta, A.J., Wu, Z.: Nature of reactive hydrogen for ammonia synthesis over a Ru/C12a7 electride catalyst. J. Am. Chem. Soc. 142, 7655–7667 (2020)
Stockmeyer, R., Conrad, H.M., Renouprez, A., Fouilloux, P.: Study of hydrogen chemisorbed on Raney-nickel by neutron inelastic spectroscopy. Surf. Sci. 49, 549–566 (1975)
Wright, P.A., Thomas, J.M., Cheetham, A.K., Nowak, A.K.: Localizing active sites in zeolitic catalysts: neutron powder profile analysis and computer simulation of deuteropyridine bound to gallozeolite-L. Nature. 318, 611–614 (1985)
Goyal, R., Fitch, A.N., Jobic, H.: Powder neutron and X-ray diffraction studies of benzene adsorbed in zeolite Zsm-5. J. Phys. Chem. B. 104, 2878–2884 (2000)
Falkowska, M., Chansai, S., Manyar, H.G., Gladden, L.F., Bowron, D.T., Youngs, T.G.A., Hardacre, C.: Determination of toluene hydrogenation kinetics with neutron diffraction. Phys. Chem. Chem. Phys. 18, 17237–17243 (2016)
Neumann, D.A.: Neutron scattering and hydrogenous materials. Mater. Today. 9, 34–41 (2006)
Parker, S.F., Lennon, D.: Applications of neutron scattering to heterogeneous catalysis. J. Phys. Conf. Ser. 746, 012066 (2016)
Lennon, D., Parker, S.F.: Inelastic neutron scattering studies of methyl chloride synthesis over alumina. Acc. Chem. Res. 47, 1220–1227 (2014)
Ramirez-Cuesta, A.J., Jones, M.O., David, W.I.F.: Neutron scattering and hydrogen storage. Mater. Today. 12, 54–61 (2009)
O’Malley, A.J., Parker, S.F., Catlow, C.R.A.: Neutron spectroscopy as a tool in catalytic science. Chem. Commun. 53, 12164–12176 (2017)
Xue, Z.-L., Ramirez-Cuesta, A.J., Brown, C.M., Calder, S., Cao, H., Chakoumakos, B.C., Daemen, L.L., Huq, A., Kolesnikov, A.I., Mamontov, E., Podlesnyak, A.A., Wang, X.: Neutron instruments for research in coordination chemistry. Eur. J. Inorg. Chem. 2019, 1065–1089 (2019)
Albers, P.W., Parker, S.F.: Inelastic incoherent neutron scattering in catalysis research. In: Gates, B.C., Knözinger, H. (eds.) Advances in Catalysis, vol. 51, pp. 99–132. Academic Press, Amsterdam (2007)
Albers, P.W., Lennon, D., Parker, S.F.: Chapter 5: catalysis. In: Fernandez-Alonso, F., Price, D.L. (eds.) Experimental Methods in the Physical Sciences, vol. 49, pp. 279–348. Academic Press, Cambridge (2017)
Mittal, R., Chaplot, S.L., Choudhury, N.: Inelastic neutron scattering, lattice dynamics, computer simulation and thermodynamic properties. In: Thermodynamic Properties of Solids, pp. 75–121 (2010). https://doi.org/10.1002/9783527630417.ch3
Ramirez-Cuesta, A.J., Mitchell, P.C.H.: Neutrons and neutron spectroscopy. In: Local Structural Characterisation, pp. 173–224. Wiley, Chichester (2013). https://doi.org/10.1002/9781118681909.ch3
Squires, G.L.: Introduction to the Theory of Thermal Neutron Scattering. Courier Corporation (1996)
Andersen, K.H., Argyriou, D.N., Jackson, A.J., Houston, J., Henry, P.F., Deen, P.P., Toft-Petersen, R., Beran, P., Strobl, M., Arnold, T., Wacklin-Knecht, H., Tsapatsaris, N., Oksanen, E., Woracek, R., Schweika, W., Mannix, D., Hiess, A., Kennedy, S., Kirstein, O., Petersson Årsköld, S., Taylor, J., Hagen, M.E., Laszlo, G., Kanaki, K., Piscitelli, F., Khaplanov, A., Stefanescu, I., Kittelmann, T., Pfeiffer, D., Hall-Wilton, R., Lopez, C.I., Aprigliano, G., Whitelegg, L., Moreira, F.Y., Olsson, M., Bordallo, H.N., Martín-Rodríguez, D., Schneider, H., Sharp, M., Hartl, M., Nagy, G., Ansell, S., Pullen, S., Vickery, A., Fedrigo, A., Mezei, F., Arai, M., Heenan, R.K., Halcrow, W., Turner, D., Raspino, D., Orszulik, A., Cooper, J., Webb, N., Galsworthy, P., Nightingale, J., Langridge, S., Elmer, J., Frielinghaus, H., Hanslik, R., Gussen, A., Jaksch, S., Engels, R., Kozielewski, T., Butterweck, S., Feygenson, M., Harbott, P., Poqué, A., Schwaab, A., Lieutenant, K., Violini, N., Voigt, J., Brückel, T., Koenen, M., Kämmerling, H., Babcock, E., Salhi, Z., Wischnewski, A., Heynen, A., Désert, S., Jestin, J., Porcher, F., Fabrèges, X., Fabrèges, G., Annighöfer, B., Klimko, S., Dupont, T., Robillard, T., Goukassov, A., Longeville, S., Alba-Simionesco, C., Bourges, P., Le Bouffy, J.G., Lavie, P., Rodrigues, S., Calzada, E., Lerche, M., Schillinger, B., Schmakat, P., Schulz, M., Seifert, M., Lohstroh, W., Petry, W., Neuhaus, J., Loaiza, L., Tartaglione, A., Glavic, A., Schütz, S., Stahn, J., Lehmann, E., Morgano, M., Schefer, J., Filges, U., Klauser, C., Niedermayer, C., Fenske, J., Nowak, G., Rouijaa, M., Siemers, D.J., Kiehn, R., Müller, M., Carlsen, H., Udby, L., Lefmann, K., Birk, J.O., Holm-Dahlin, S., Bertelsen, M., Hansen, U.B., Olsen, M.A., Christensen, M., Iversen, K., Christensen, N.B., Rønnow, H.M., Freeman, P.G., Hauback, B.C., Kolevatov, R., Llamas-Jansa, I., Orecchini, A., Sacchetti, F., Petrillo, C., Paciaroni, A., Tozzi, P., Zanatta, M., Luna, P., Herranz, I., del Moral, O.G., Huerta, M., Magán, M., Mosconi, M., Abad, E., Aguilar, J., Stepanyan, S., Bakedano, G., Vivanco, R., Bustinduy, I., Sordo, F., Martínez, J.L., Lechner, R.E., Villacorta, F.J., Šaroun, J., Lukáš, P., Markó, M., Zanetti, M., Bellissima, S., del Rosso, L., Masi, F., Bovo, C., Chowdhury, M., De Bonis, A., Di Fresco, L., Scatigno, C., Parker, S.F., Fernandez-Alonso, F., Colognesi, D., Senesi, R., Andreani, C., Gorini, G., Scionti, G., Schreyer, A.: The instrument suite of the European spallation source. Nucl. Instrum. Methods Phys. Res., Sect. A. 957, 163402 (2020)
The Advanced Photon Source - a National Synchrotron Radiation Research Facility at Argonne National Laboratory, Technical Bulletins Series Tb-25. (1997)
Mason, T.E.: Pulsed neutron scattering for the 21st century. Phys. Today. 59, 44–49 (2006)
Sears, V.F.: Neutron scattering lengths and cross sections. Neutron News. 3, 26–37 (1992)
Neutron Scattering Lengths and Cross Sections. https://www.ncnr.nist.gov/resources/n-lengths/
Colognesi, D., Celli, M., Cilloco, F., Newport, R.J., Parker, S.F., Rossi-Albertini, V., Sacchetti, F., Tomkinson, J., Zoppi, M.: Tosca neutron spectrometer: the final configuration. Appl. Phys. A. 74, s64–s66 (2002)
Feigin, L., Svergun, D.I.: Structure Analysis by Small-Angle X-Ray and Neutron Scattering, vol. 1. Plenum Press, New York (1987)
Zhou, X.-L., Chen, S.-H.: Theoretical foundation of X-ray and neutron reflectometry. Phys. Rep. 257, 223–348 (1995)
Kohn, W., Sham, L.J.: Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965)
Hedin, L., Lundqvist, B.I.: Explicit local exchange-correlation potentials. J. Phys. C Solid State Phys. 4, 2064 (1971)
Senftle, T.P., Hong, S., Islam, M.M., Kylasa, S.B., Zheng, Y., Shin, Y.K., Junkermeier, C., Engel-Herbert, R., Janik, M.J., Aktulga, H.M.: The Reaxff reactive force-field: development, applications and future directions. npj Comput. Mater. 2, 1–14 (2016)
McGreevy, R.L., Pusztai, L.: Reverse Monte Carlo simulation: a new technique for the determination of disordered structures. Mol. Simul. 1, 359–367 (1988)
Cheng, Y.Q., Daemen, L.L., Kolesnikov, A.I., Ramirez-Cuesta, A.J.: Simulation of inelastic neutron scattering spectra using Oclimax. J. Chem. Theory Comput. 15, 1974–1982 (2019)
Ramirez-Cuesta, A.J.: aCLIMAX 4.0.1, The new version of the software for analyzing and interpreting INS spectra. Comput. Phys. Commun. 157(3), 226–238 (2004)
Dymkowski, K., et al.: AbINS: The modern software for INS interpretation. Phys. B Condens. Matter. 551, 443–448 (2018)
Ramirez-Cuesta, A.J., Mitchell, P.C.H.: Hydrogen adsorption in a copper Zsm5 zeolite: an inelastic neutron scattering study. Catal. Today. 120, 368–373 (2007)
Mojet, B.L., Eckert, J., van Santen, R.A., Albinati, A., Lechner, R.E.: Evidence for chemisorbed molecular hydrogen in Fe-Zsm5 from inelastic neutron scattering. J. Am. Chem. Soc. 123, 8147–8148 (2001)
Juárez, R., Parker, S.F., Concepción, P., Corma, A., García, H.: Heterolytic and heterotopic dissociation of hydrogen on ceria-supported gold nanoparticles. Combined inelastic neutron scattering and Ft-Ir spectroscopic study on the nature and reactivity of surface hydrogen species. Chem. Sci. 1, 731–738 (2010)
Jobic, H., Clugnet, G., Lacroix, M., Yuan, S., Mirodatos, C., Breysse, M.: Identification of new hydrogen species adsorbed on ruthenium sulfide by neutron spectroscopy. J. Am. Chem. Soc. 115, 3654–3657 (1993)
Lacroix, M., Jobic, H., Dumonteil, C., Afanasiev, P., Breysse, M., Kasztelan, S.: Role of adsorbed hydrogen species on ruthenium and molybdenum sulfides. Characterization by inelastic neutron scattering, thermoanalysis methods and model reactions. In: Hightower, J.W., Nicholas Delgass, W., Iglesia, E., Bell, A.T. (eds.) Studies in Surface Science and Catalysis, vol. 101, pp. 117–126. Elsevier, Amsterdam (1996)
Cavallari, C., Pontiroli, D., Jiménez-Ruiz, M., Ivanov, A., Mazzani, M., Gaboardi, M., Aramini, M., Brunelli, M., Riccò, M., Rols, S.: Hydrogen on graphene investigated by inelastic neutron scattering. J. Phys. Conf. Ser. 554, 012009 (2014)
Tsao, C.-S., Liu, Y., Chuang, H.-Y., Tseng, H.-H., Chen, T.-Y., Chen, C.-H., Yu, M.-S., Li, Q., Lueking, A., Chen, S.-H.: Hydrogen spillover effect of Pt-doped activated carbon studied by inelastic neutron scattering. J Phys. Chem. Lett. 2, 2322–2325 (2011)
Itoh, K., Miyahara, Y., Orimo, S.-i., Fujii, H., Kamiyama, T., Fukunaga, T.: The local structure of hydrogen storage nanocrystalline graphite by neutron scattering. J. Alloys Compd. 356–357, 608–611 (2003)
Colognesi, D., Ulivi, L., Zoppi, M., Ramirez-Cuesta, A.J., Orecchini, A., Karkamkar, A.J., Fichtner, M., Gil Bardají, E., Zhao-Karger, Z.: Hydrogen-storage materials dispersed into nanoporous substrates studied through incoherent inelastic neutron scattering. J. Alloys Compd. 538, 91–99 (2012)
Rowsell, J.L.C., Eckert, J., Yaghi, O.M.: Characterization of H2 binding sites in prototypical metal−organic frameworks by inelastic neutron scattering. J. Am. Chem. Soc. 127, 14904–14910 (2005)
Moon, J., Cheng, Y., Daemen, L.L., Li, M., Polo-Garzon, F., Ramirez-Cuesta, A.J., Wu, Z.: Discriminating the role of surface hydride and hydroxyl for acetylene semihydrogenation over ceria through in situ neutron and infrared spectroscopy. ACS Catal. 10, 5278–5287 (2020)
Hochard, F., Jobic, H., Massardier, J., Renouprez, A.J.: Gas phase hydrogenation of acetonitrile on Raney nickel catalysts: reactive hydrogen. J. Mol. Catal. A Chem. 95, 165–172 (1995)
Hawkins, A.P., Zachariou, A., Parker, S.F., Collier, P., Barrow, N., Silverwood, I.P., Howe, R.F., Lennon, D.: Effect of steam de-alumination on the interactions of propene with H-Zsm-5 zeolites. RSC Adv. 10, 23136–23147 (2020)
Warringham, R., Davidson, A.L., Webb, P.B., Tooze, R.P., Ewings, R.A., Parker, S.F., Lennon, D.: Examining the temporal behavior of the hydrocarbonaceous overlayer on an iron based Fischer–Tropsch catalyst. RSC Adv. 9, 2608–2617 (2019)
Hawkins, A.P., Zachariou, A., Collier, P., Ewings, R.A., Howe, R.F., Parker, S.F., Lennon, D.: Low-temperature studies of propene oligomerization in Zsm-5 by inelastic neutron scattering spectroscopy. RSC Adv. 9, 18785–18790 (2019)
Hall, P.G., Pidduck, A., Wright, C.J.: Inelastic neutron scattering by water adsorbed on silica. J. Colloid Interface Sci. 79, 339–349 (1981)
Beta, I.A., Böhlig, H., Hunger, B.: Structure of adsorption complexes of water in zeolites of different types studied by infrared spectroscopy and inelastic neutron scattering. Phys. Chem. Chem. Phys. 6, 1975–1981 (2004)
Jobic, H., Tuel, A., Krossner, M., Sauer, J.: Water in interaction with acid sites in H-Zsm-5 zeolite does not form hydroxonium ions. A comparison between neutron scattering results and ab Initio calculations. J. Phys. Chem. 100, 19545–19550 (1996)
Copéret, C., Estes, D.P., Larmier, K., Searles, K.: Isolated surface hydrides: formation, structure, and reactivity. Chem. Rev. 116, 8463–8505 (2016)
Norton, J.R., Sowa, J.R.: Introduction: metal hydrides. Chem. Rev. 116, 8315–8317 (2016)
Nicol, J.M., Rush, J.J., Kelley, R.D.: Inelastic neutron scattering studies of the interaction of hydrogen with palladium black. Surf. Sci. 197, 67–80 (1988)
Albers, P.W., Krauter, J.G.E., Ross, D.K., Heidenreich, R.G., Köhler, K., Parker, S.F.: Identification of surface states on finely divided supported palladium catalysts by means of inelastic incoherent neutron scattering. Langmuir. 20, 8254–8260 (2004)
Albers, P., Burmeister, R., Seibold, K., Prescher, G., Parker, S.F., Ross, D.K.: Investigations of palladium catalysts on different carbon supports. J. Catal. 181, 145–154 (1999)
Parker, S.F., Walker, H.C., Callear, S.K., Grünewald, E., Petzold, T., Wolf, D., Möbus, K., Adam, J., Wieland, S.D., Jiménez-Ruiz, M., Albers, P.W.: The effect of particle size, morphology and support on the formation of palladium hydride in commercial catalysts. Chem. Sci. 10, 480–489 (2019)
Parker, S.F., Adroja, D., Jiménez-Ruiz, M., Tischer, M., Möbus, K., Wieland, S.D., Albers, P.: Characterisation of the surface of freshly prepared precious metal catalysts. Phys. Chem. Chem. Phys. 18, 17196–17201 (2016)
Albers, P., Auer, E., Ruth, K., Parker, S.F.: Inelastic neutron scattering investigation of the nature of surface sites occupied by hydrogen on highly dispersed platinum on commercial carbon black supports. J. Catal. 196, 174–179 (2000)
Albers, P.W., Lopez, M., Sextl, G., Jeske, G., Parker, S.F.: Inelastic neutron scattering investigation on the site occupation of atomic hydrogen on platinum particles of different size. J. Catal. 223, 44–53 (2004)
Rush, J.J., Cavanagh, R.R., Kelley, R.D.: Summary abstract: neutron scattering from adsorbates on platinum black. J. Vac. Sci. Technol. A. 1, 1245–1246 (1983)
Wright, C.J.: Alternative explanation of the inelastic neutron scattering from hydrogen adsorbed by Raney nickel. J. Chem. Soc., Faraday Trans. 2. 73, 1497–1500 (1977)
Jobic, H., Candy, J.-P., Perrichon, V., Renouprez, A.: Neutron-scattering and volumetric study of hydrogen adsorbed and absorbed on Raney palladium. J. Chem. Soc., Faraday Trans. 1. 81, 1955–1961 (1985)
Barrera, G.D., Colognesi, D., Mitchell, P.C.H., Ramirez-Cuesta, A.J.: Lda or Gga? A combined experimental inelastic neutron scattering and ab initio lattice dynamics study of alkali metal hydrides. Chem. Phys. 317, 119–129 (2005)
Sivadinarayana, C., Choudhary, T.V., Daemen, L.L., Eckert, J., Goodman, D.W.: The nature of the surface species formed on Au/Tio2 during the reaction of H2 and O2: an inelastic neutron scattering study. J. Am. Chem. Soc. 126, 38–39 (2004)
Breck, D.W.: Zeolite molecular sieves: structure, chemistry, and use D. W. Breck (Union Carbide Corporation, Tarrytown, New York) John Wiley and Sons, New York, London, Sydney, and Toronto. 1974. 771. J. Chromatogr. Sci. 13, 18A (1975)
Weitkamp, J., Fritz, M., Ernst, S.: Zeolites as media for hydrogen storage. Int. J. Hydrog. Energy. 20, 967–970 (1995)
Yang, R.T.: Adsorbents: Fundamentals and Applications. Wiley, New Jersey (2003)
Ramirez-Cuesta, A.J., Mitchell, P.C.H., Ross, D.K., Georgiev, P.A., Anderson, P.A., Langmi, H.W., Book, D.: Dihydrogen in cation-substituted zeolites X—an inelastic neutron scattering study. J. Mater. Chem. 17, 2533–2539 (2007)
Rosi, N.L., Eckert, J., Eddaoudi, M., Vodak, D.T., Kim, J., O’Keeffe, M., Yaghi, O.M.J.S.: Hydrogen storage in microporous metal-organic frameworks. Science. 300, 1127–1129 (2003)
Rowsell, J.L.C., Millward, A.R., Park, K.S., Yaghi, O.M.: Hydrogen sorption in functionalized metal−organic frameworks. J. Am. Chem. Soc. 126, 5666–5667 (2004)
Kroto, H.W., Fischer, J.E., Cox, D.: The Fullerenes. Newnes, Oxford (2012)
Schimmel, H.G., Kearley, G.J., Nijkamp, M.G., Visser, C.T., de Jong, K.P., Mulder, F.M.: Hydrogen adsorption in carbon nanostructures: comparison of nanotubes, fibers, and coals. Chem. Eur. J. 9, 4764–4770 (2003)
Georgiev, P.A., Ross, D.K., Monte, A.D., Montaretto-Marullo, U., Edwards, R.A.H., Ramirez-Cuesta, A.J., Colognesi, D.: Hydrogen site occupancies in single-walled carbon nanotubes studied by inelastic neutron scattering. J. Phys. Condens. Matter. 16, L73–L78 (2004)
Horsewill, A.J., Panesar, K.S., Rols, S., Johnson, M.R., Murata, Y., Komatsu, K., Mamone, S., Danquigny, A., Cuda, F., Maltsev, S., Grossel, M.C., Carravetta, M., Levitt, M.H.: Quantum translator-rotator: inelastic neutron scattering of dihydrogen molecules trapped inside anisotropic fullerene cages. Phys. Rev. Lett. 102, 013001 (2009)
Horsewill, A.J., Panesar, K.S., Rols, S., Ollivier, J., Johnson, M.R., Carravetta, M., Mamone, S., Levitt, M.H., Murata, Y., Komatsu, K., Chen, J.Y.C., Johnson, J.A., Lei, X., Turro, N.J.: Inelastic neutron scattering investigations of the quantum molecular dynamics of a H${}_{2}$ molecule entrapped inside a fullerene cage. Phys. Rev. B. 85, 205440 (2012)
Mao, W.L., Mao, H.-k., Goncharov, A.F., Struzhkin, V.V., Guo, Q., Hu, J., Shu, J., Hemley, R.J., Somayazulu, M., Zhao, Y.: Hydrogen clusters in clathrate hydrate. Science. 297, 2247 (2002)
Lee, H., Lee, J.-w., Kim, D.Y., Park, J., Seo, Y.-T., Zeng, H., Moudrakovski, I.L., Ratcliffe, C.I., Ripmeester, J.A.: Tuning clathrate hydrates for hydrogen storage. Nature. 434, 743–746 (2005)
Bée, M.: Localized and Long-range diffusion in condensed matter: state of the art of Qens studies and future prospects. Chem. Phys. 292, 121–141 (2003)
Mamontov, E., Herwig, K.W.: A time-of-flight backscattering spectrometer at the spallation neutron source, BASIS. Rev. Sci. Instrum. 82, 085109 (2011)
Shibata, K., Takahashi, N., Kawakita, Y., Matsuura, M., Yamada, T., Tominaga, T., Kambara, W., Kobayashi, M., Inamura, Y., Nakatani, T., Nakajima, K., Arai, M.: The performance of Tof near backscattering spectrometer DNA in Mlf, J-Parc. In: Proceedings of the 2nd International Symposium on Science at J-Parc? Unlocking the Mysteries of Life, Matter and the Universe ? Jps Conference Proceedings vol 8, vol. 8. Journal of the Physical Society of Japan (2015)
Bernhard, F., Eugene, M., van Lambert, E., Tilo, S.: Recent backscattering instrument developments at the Ill and Sns. Z. Phys. Chem. 224, 33–60 (2010)
Tsapatsaris, N., Lechner, R.E., Markó, M., Bordallo, H.N.: Conceptual design of the time-of-flight backscattering spectrometer, miracles, at the European spallation source. Rev. Sci. Instrum. 87, 085118 (2016)
Azuah, R.T., Kneller, L.R., Qiu, Y., Tregenna-Piggott, P.L., Brown, C.M., Copley, J.R., Dimeo, R.M.: Dave: a comprehensive software suite for the reduction, visualization, and analysis of low energy neutron spectroscopic data. J. Res. Natl. Inst. Stand. Technol. 114, 341–358 (2009)
Mamontov, E., Smith, R.W., Billings, J.J., Ramirez-Cuesta, A.J.: Simple analytical model for fitting Qens data from liquids. Phys. B Condens. Matter. 566, 50–54 (2019)
Gautam, S., Le, T.T.B., Rother, G., Jalarvo, N., Liu, T., Mamontov, E., Dai, S., Qiao, Z.-A., Striolo, A., Cole, D.: Effects of water on the stochastic motions of propane confined in Mcm-41-S pores. Phys. Chem. Chem. Phys. 21, 25035–25046 (2019)
Ruthven, D.M., Eic, M., Richard, E.: Diffusion of C8 aromatic hydrocarbons in silicalite. Zeolites. 11, 647–653 (1991)
Teixeira, A.R., Chang, C.-C., Coogan, T., Kendall, R., Fan, W., Dauenhauer, P.J.: Dominance of surface barriers in molecular transport through silicalite-1. J. Phys. Chem. C. 117, 25545–25555 (2013)
Song, L., Sun, Z.-L., Rees, L.V.C.: Experimental and molecular simulation studies of adsorption and diffusion of cyclic hydrocarbons in silicalite-1. Microporous Mesoporous Mater. 55, 31–49 (2002)
Fried, J.R., Weaver, S.: Atomistic simulation of hydrocarbon diffusion in silicalite. Comput. Mater. Sci. 11, 277–293 (1998)
Koriabkina, A.O., de Jong, A.M., Schuring, D., van Grondelle, J., van Santen, R.A.: Influence of the acid sites on the intracrystalline diffusion of hexanes and their mixtures within Mfi-zeolites. J. Phys. Chem. B. 106, 9559–9566 (2002)
Krishna, R., van Baten, J.M.: Diffusion of hydrocarbon mixtures in Mfi zeolite: influence of intersection blocking. Chem. Eng. J. 140, 614–620 (2008)
Leroy, F., Rousseau, B., Fuchs, A.H.: Self-diffusion of N-alkanes in silicalite using molecular dynamics simulation: a comparison between rigid and flexible frameworks. Phys. Chem. Chem. Phys. 6, 775–783 (2004)
Jiang, M., Eic, M., Miachon, S., Dalmon, J.-A., Kocirik, M.: Diffusion of N-butane, isobutane and ethane in a Mfi-zeolite membrane investigated by gas permeation and Zlc measurements. Sep. Purif. Technol. 25, 287–295 (2001)
Matam, S.K., O’Malley, A.J., Catlow, C.R.A., Suwardiyanto, Collier, P., Hawkins, A.P., Zachariou, A., Lennon, D., Silverwood, I., Parker, S.F., Howe, R.F.: The effects of Mtg catalysis on methanol mobility in Zsm-5. Cat. Sci. Technol. 8, 3304–3312 (2018)
Tang, Y., Kobayashi, Y., Masuda, N., Uchida, Y., Okamoto, H., Kageyama, T., Hosokawa, S., Loyer, F., Mitsuhara, K., Yamanaka, K., Tamenori, Y., Tassel, C., Yamamoto, T., Tanaka, T., Kageyama, H.: Metal-dependent support effects of oxyhydride-supported Ru, Fe, Co catalysts for ammonia synthesis. Adv. Energy Mater. 8, 1801772 (2018)
Shull, C.G., Wollan, E.O.: X-ray, electron, and neutron diffraction. Science. 108, 69 (1948)
Shoemaker, D.P., Li, J., Seshadri, R.: Unraveling atomic positions in an oxide spinel with two Jahn−Teller ions: local structure investigation of Cumn2o4. J. Am. Chem. Soc. 131, 11450–11457 (2009)
Page, K., Stoltzfus, M.W., Kim, Y.-I., Proffen, T., Woodward, P.M., Cheetham, A.K., Seshadri, R.: Local atomic ordering in Batao2n studied by neutron pair distribution function analysis and density functional theory. Chem. Mater. 19, 4037–4042 (2007)
Page, K., Hood, T.C., Proffen, T., Neder, R.B.: Building and refining complete nanoparticle structures with total scattering data. J. Appl. Crystallogr. 44, 327–336 (2011)
Liu, J., Olds, D., Peng, R., Yu, L., Foo, G.S., Qian, S., Keum, J., Guiton, B.S., Wu, Z., Page, K.: Quantitative analysis of the morphology of {101} and {001} faceted anatase Tio2 nanocrystals and its implication on photocatalytic activity. Chem. Mater. 29, 5591–5604 (2017)
Mawdsley, J.R., Vaughey, J.T., Krause, T.R.: Neutron diffraction studies of nickel-containing perovskite oxide catalysts exposed to autothermal reforming environments. Chem. Mater. 21, 4830–4838 (2009)
Rauch, H., Waschkowski, W., Neutron scattering lengths. In: Dianoux, A.-J., Lander, G. (eds.) Neutron Data Booklet, 2nd ed, pp. 11–15. Institut Laue-Langevin, Grenoble (2003)
Kandemir, T., Girgsdies, F., Hansen, T.C., Liss, K.-D., Kasatkin, I., Kunkes, E.L., Wowsnick, G., Jacobsen, N., Schlögl, R., Behrens, M.: In situ study of catalytic processes: neutron diffraction of a methanol synthesis catalyst at industrially relevant pressure. Angew. Chem. Int. Ed. 52, 5166–5170 (2013)
Otomo, T., Ikeda, K., Honda, T.: Structural studies of hydrogen storage materials with neutron diffraction: a review. J. Phys. Soc. Jpn. 89, 051001 (2020)
Orimo, S.-i., Nakamori, Y., Eliseo, J.R., Züttel, A., Jensen, C.M.: Complex hydrides for hydrogen storage. Chem. Rev. 107, 4111–4132 (2007)
Chater, P.A., David, W.I., Johnson, S.R., Edwards, P.P., Anderson, P.A.: Synthesis and crystal structure of Li 4 Bh 4 (Nh 2). Chem. Commun. 3, 2439–2441 (2006)
Weidenthaler, C.: Crystal structure evolution of complex metal aluminum hydrides upon hydrogen release. J. Energy Chem. 42, 133–143 (2020)
Brinks, H., Langley, W., Jensen, C., Graetz, J., Reilly, J., Hauback, B.: Synthesis and crystal structure of Β-Ald3. J. Alloys Compd. 433(433), 180–183 (2007)
Brinks, H.W., Istad-Lem, A., Hauback, B.C.: Mechanochemical synthesis and crystal structure of Α ‘-Ald3 and Α-Ald3. J. Phys. Chem. B. 110(110), 25833–25837 (2006)
Orimo, S., Majer, G., Fukunaga, T., Züttel, A., Schlapbach, L., Fujii, H.: Hydrogen in the mechanically prepared nanostructured graphite. Appl. Phys. Lett. 75, 3093–3095 (1999)
Yildirim, T., Hartman, M.R.: Direct observation of hydrogen adsorption sites and nanocage formation in metal-organic frameworks. Phys. Rev. Lett. 95, 215504 (2005)
Lee, H., Choi, Y.N., Choi, S.B., Kim, J., Kim, D., Jung, D.H., Park, Y.S., Yoon, K.B.: Liquid-like hydrogen stored in nanoporous materials at 50 K observed by in situ neutron diffraction experiments. J. Phys. Chem. C. 117, 3177–3184 (2013)
Zhao, Y., Xu, H., Daemen, L.L., Lokshin, K., Tait, K.T., Mao, W.L., Luo, J., Currier, R.P., Hickmott, D.D.: High-pressure/low-temperature neutron scattering of gas inclusion compounds: Progress and prospects. Proc. Natl. Acad. Sci. 104, 5727 (2007)
Dincǎ, M., Dailly, A., Liu, Y., Brown, C.M., Neumann, D.A., Long, J.R.: Hydrogen storage in a microporous metal−organic framework with exposed Mn2+ coordination sites. J. Am. Chem. Soc. 128, 16876–16883 (2006)
Parvanov, V.M., Schenter, G.K., Hess, N.J., Daemen, L.L., Hartl, M., Stowe, A.C., Camaioni, D.M., Autrey, T.: Materials for hydrogen storage: structure and dynamics of borane ammonia complex. Dalton Trans., 4514 (2008). https://doi.org/10.1039/B718138H4514-4522
Kandemir, T., Schuster, M.E., Senyshyn, A., Behrens, M., Schlögl, R.: The Haber–Bosch process revisited: on the real structure and stability of “ammonia iron” under working conditions. Angew. Chem. Int. Ed. 52, 12723–12726 (2013)
Youngs, T.G.A., Manyar, H., Bowron, D.T., Gladden, L.F., Hardacre, C.: Probing chemistry and kinetics of reactions in heterogeneous catalysts. Chem. Sci. 4, 3484–3489 (2013)
Holderer, O., Carmo, M., Shviro, M., Lehnert, W., Noda, Y., Koizumi, S., Appavou, M.-S., Appel, M., Frielinghaus, H.: Fuel cell electrode characterization using neutron scattering. Materials (Basel). 13, 1474 (2020)
Borgschulte, A., Delmelle, R., Duarte, R.B., Heel, A., Boillat, P., Lehmann, E.: Water distribution in a sorption enhanced methanation reactor by time resolved neutron imaging. Phys. Chem. Chem. Phys. 18, 17217–17223 (2016)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Moon, J., Cheng, Y., Wu, Z., Ramirez-Cuesta, A.J. (2023). Neutron Scattering (NS) Spectroscopy. In: Wachs, I.E., Bañares, M.A. (eds) Springer Handbook of Advanced Catalyst Characterization. Springer Handbooks. Springer, Cham. https://doi.org/10.1007/978-3-031-07125-6_24
Download citation
DOI: https://doi.org/10.1007/978-3-031-07125-6_24
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-07124-9
Online ISBN: 978-3-031-07125-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)