
Abstract
Bispecific antibodies are composed of two monoclonal antibodies that engage T cells with tumor cell antigens and lead to tumor cell lysis. The most common types fall into the category of bispecific T cell engagers, or BiTEs, that have the canonical CD3-CD19 bispecific construct. Blinatumomab is the first bispecific antibody that received FDA approval for relapsed refractory B cell precursor acute lymphoblastic leukemia. Blinatumomab has been shown to have robust clinical outcomes and is associated with adverse events such as cytokine release syndrome and neurotoxicity. Other bispecific antibodies are under clinical investigation for multiple myeloma and acute myeloid leukemia. Along with immune checkpoint inhibitors and chimeric antigen T cell receptor therapies, bispecific antibodies are considered a mainstay as a therapeutic option for cancer immunotherapies for Hematologic malignancies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Bispecific antibodies
- Bispecific T cell engagers
- Acute lymphoblastic leukemia
- Blinatumomab
- Tumor-associated antigens
11.1 Introduction
Immune checkpoint inhibitors and chimeric antigen receptor T cells have been discussed in great depth in this volume. As shown, they form a class of T cell-based cancer immunotherapies that focus on immunosuppressive factors and immunostimulatory pathways, respectively [1]. ICIs have demonstrated clinical efficacy against solid tumors such as melanoma and NSCLC by releasing the blockade of T cells from PD-1/PD-L1 and CTLA-4 immunosuppressive molecules in “hot” tumors. CAR-T cells therapies, also discussed in this volume, are genetically engineered T cells generated ex-vivo from patients for re-infusion to direct T cell activity for tumor cell destruction.
This chapter focuses on a third class of T cell-based cancer immunotherapy known as bispecific antibodies, which are composed of two monoclonal antibodies that link cell surface molecules on T cells to tumor-associated antigens to lead to cancer cell lysis. This class of therapies has shown clinical efficacy in Hematologic malignancies mainly and is discussed in this chapter.
11.2 The Construction of Bispecific Antibodies and Their Cellular Properties
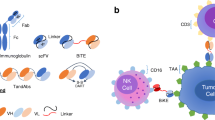
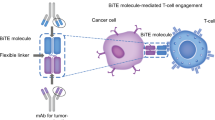
Bispecific antibodies comprise a therapeutic class of agents that are designed to target tumor cells by directing T cells to the antigens on these tumor cells. They recognize and bind to two distinct antigens. The majority of bispecific antibodies fall into the category of bispecific T cell engagers or BiTEs. One of the first FDA-approved agents is blinatumomab, a bispecific T cell engager with CD19 and CD3 epitopes. CD3 is invariably utilized as a surface epitope on the T cell receptor as a target, and BsAbs are developed with CD3 targets, which are in turn categorized as BsAbs with or without Fc domains. Bispecific antibodies are constructed to bring two different antigens on different cells together and bring cytotoxic T cells in contact with tumor cells, thus destroying them. More than 100 different BsAb formats have been invented, making them much more complex than monoclonal antibodies as a result of innovative advances in protein and gene engineering [2]. Based on the structure of the five classes of antibodies, IgG, IgM, IgA, IgD, and IgE, these antibodies are composed of the antigen-binding fragments (Fab) and the fragment crystallizable region (Fc). The Fc domain confers stability, high half-life, and a relatively uncomplicated purification process, but can lead to non-specific immune response from interaction of the Fc domain receptors with other cytotoxic immune cells such as natural killer cells, monocytes, and macrophages [1]. According to Huang, the various BsAb formats can be distinguished into two categories depending on the presence of an Fc domain. They can also be divided into the Fc architecture and the Fc less architecture. The latter include BiTE, DART, and TandAbm, which hold benefits of high yield and are more able to penetrate tissues. However, they have short in vivo half-lives and decreased stability.
Tumor-associated antigens are presented by MHC molecules expressed on tumor cells, allowing for T cells to be activated and destroy these cells. ICIs restriction is due to MHC restriction impairment, inhibiting T cell presentation to tumor cells. BiTE, on the other hand, can lead to interaction between cytotoxic T cells and tumor cells independently of MHC restriction, which leads to immunological synapse and the secretion of perforins and granzymes. BiTE can overcome the limitations of CAR T-cells, being produced in a simple and fast process. The coupling of tumor-associated antigen with the CD3 complex of T cells leads to T cell engagement with malignant cells and a T cell response. Tumor lysis results since the BiTE design bypasses the MHC barrier, thus bypassing this “common evasion mechanism” of tumor cells [3].
11.3 Clinical Outcomes of BiTE in Hematologic Malignancies
Hematologic malignancies benefited from the development of BsAbs since many of these cancers are amenable to treatment of BsAbs. In 2005, a clinical trial established blinatumomab as an effective therapeutic agent for non-Hodgkin’s lymphoma [4]. BiTEs therapy depends on the identification of an antigen that is tumor cell-specific; CD19 has abnormal expression on malignant cells of B cell lineage and is selected as a target, being a glycoprotein that has stable expression on B cell precursor cells, particularly malignant cells of B cell origin [3].
As of 2020, 123 BsAbs are being clinically evaluated, of which bispecific T cell engagers or BiTEs, remain the largest category, which targets the two different surfaces of the immune cell and tumor cell and thereby engaging them toward tumor cell destruction. Among B cell malignancies, acute lymphoblastic leukemia, multiple myeloma, chronic lymphocytic leukemia, non-Hodgkin lymphoma, BiTEs are being evaluated for their treatment strategies for these malignancies. Standard of care approaches includes the anti-CD20 monoclonal Ab rituximab and the Bruton tyrosine kinase ibrutinib, and autologous stem-cell transplantation, with considerations for minimal residual disease for determining efficacy [4] (Fig. 11.1).

The schematic representation of structure and mechanism of action of canonical bispecific T-cell engager (BiTE). mAb: Monoclonal antibody; CH: heavy chain variable region; VL: light chain variable region; TAA: tumor-associated antigen. Adapted from Zhou (2021)
“The agent blinatumomab is considered the “CD19-CD3” canonical BiTE construct” with a clinical efficacy for ALL [1].
11.4 Development of Blinatumomab
Blinatumomab was first evaluated in R/R NHL and CLL patients and intravenously administered in phase I studies. Dosage was 0.5–90.0181 ug/m2/day to accommodate its short half-life (2 h). CRS and neurological sequelae were observed. BCP-ALL patients were also among the first to be evaluated as well and achieved complete remission with minimal residual disease. Orphan drug designation in 2008 and Breakthrough Therapy (2014) and Priority Review (2014) designations soon followed in the US. Marketing authorization was initially given for Ph-R/R BCP-ALL as a result of two phase II studies that were open-label, single-arm, and multicenter. In turn, the drug received accelerated approval pathway (2014) followed by regular FDA approval granted for patients as a result of the clinical benefit demonstrated as a result of the TOWER study, as discussed below. The ALCANTRA trial, also discussed below, provided the clinical data for full FDA approval for both Ph- and Ph+ R/R BCP-ALL. According to Stein et al., as of 2017, the agent was approved in 53 countries in R/R patients, and as of 2018, BCP-ALL pediatric patients were included for blinatumomab treatment in the phase II BLAST study who are in remission but have MRD [5, 6].
11.5 Acute Lymphoblastic Leukemia
11.5.1 Administration and Dosing Schedules
Blinatumomab dosing is dependent on the type of malignancy and evidence of tumor burden. Short-term infusion scheduled 2–4 h for 1–3 times/week demonstrates no clinical response as measured by B cell depletion as observed in ALL. Recommendations include two induction cycles followed by maintenance and consolidation cycles with hospitalization for 9 days in the first cycle and first 2 days of the second cycle for R/R ALL patients. An induction cycle of 28 days is followed by a 14-day treatment-free interval and accompanied by hematologic CR and MRD. To minimize CRS, patients are premedicated with dexamethasone or a similar corticosteroid, especially in R/R cases with a dosing regimen consisting of step-up blinatumomab in R/R disease >25% blasts in the bone marrow especially for NHL. A phase 2 study showed that a stepwise dosing schedule was tolerated to also avoid adverse neurological side effects. After a period of four treatment-free weeks, patients experiencing CR, PR, or stable disease were administered further consolidation cycle; efficacy was monitored during these weeks as progression occurs rapidly [7,8,9]. Some disadvantages of IV perfusion are lack of convenience for patients and costs. BsAbs such as blinatumomab are known for their short half-lives that lead to the need for more frequent infusion. The BsAbs with longer half-lives could have the potential for greater toxicity, which is being corroborated by ongoing clinical trials [10,11,12].
11.5.1.1 Clinical Efficacy of Blinatumomab in B-ALL
Blinatumomab, the anti-CD19-CD3 bispecific T cell engager, was approved by the FDA in 2014 and the EMA in December 2015 for the treatment of relapsed refractory B cell precursor ALL [13,14,15]. Katarajan et al. showed in a multi-institutional phase 3 trial that blinatumomab had better outcomes when compared with chemotherapy. Out of 405 evaluable patients, 271 received blinatumomab while 134 received chemotherapy. OS was the primary endpoint and blinatumomab OS was 7.7 months, compared to 4.0 months for chemotherapy (HR: 0.71). Grade 3 or higher adverse events were 87% versus 92% for blinatumomab and chemotherapy, respectively [16]. In a phase 2 single-group trial, Foa et al. evaluated dasatinib, a tyrosine kinase inhibitor, with glucocorticoids followed by two cycles of blinatumomab in 63 patients with (Ph)-positive ALL with no upper age limit. Sustained molecular response was the primary endpoint. At day 85, 29% had a molecular response and this percentage increased to 60% after two cycles of blinatumomab at median follow-up of 18 months; OS was 95% and disease-free survival was 88% (Table 11.1) [17].
11.5.2 BCP-ALL in CR with MRD and R/R BCP-ALL [3]
A series of trials established the clinical utility of blinatumomab for BCP-ALL. The phase 2 BLAST study evaluated adults with BCP-ALL in hematologic CR with MRD. Median OS was 38.9 months versus 12.5 months (P = 0.002) in patients who did or did not have complete MRD response within one cycle of treatment, respectively [18]. However, cure was eventually achieved by these patients after five years, as median OS was not reached for patients who experienced MRD in cycle 1 [19]. Since BLAST was a single-arm study, control data was provided through a historical comparator that led to an analysis of Hematologic relapse-free survival [20] and eventual approval in this patient population.
The phase III TOWER study examined a randomized Ph- R/R BCP-ALL patient population that compared blinatumomab (n = 271) with standard treatment chemotherapy (n = 134) [21], and was considered requisite since the drug received accelerated approval by the FDA. These patients were heavily pretreated with intensive combination therapy for initial or subsequent salvage treatment [22]. Deep, durable outcomes were achieved by the blinatumomab cohort compared to chemotherapy to the point that the trial was halted due to robust OS benefits [21]. OS served as the primary endpoint with CR with complete Hematologic recovery as secondary endpoints. The blinatumomab cohort was found to have superior OS relative to SOC, with median OS at 8 and 4 months, respectively. [HR 0.71 (95% CI 0.55–0.93); p = 0.01]. According to Stein et al., “[r]emission within 12 weeks following initiation of treatment was also significantly higher in the blinatumomab group versus the SOC group: CR with full hematologic recovery (34% vs 16%, p < 0.001) and CR with full, partial, or incomplete hematologic recovery (44% vs. 25%; p < 0.001)” [5]. In terms of salvage therapy, blinatumomab was effective, especially in first salvage, and led to doubling of the median survival compared to standard of care chemotherapy. Salvage status was the driving factor for survival in these responders independent of subsequent allogeneic stem-cell transplantation. Additionally, blinatumomab also led to significant quality of life that was health-related [23,24,25]. Adverse events included pyrexia, CRS, and infusion site reactions at a greater than 5% incidence, and serious neurotoxicity was associated in the blinatumomab arm. After adjusting treatment exposure, however, the overall incidence of these events in the blinatumomab was significantly lower (349 vs. 642 events per 100 patient-years of exposure) [22]. After adjusting for time on treatment, Grade ≥ exposure adjusted event rates were less for blinatumomab compared with the SOC arm (11 vs. 45 events per patient-year; p < 0.001). “For specific Grade ≥ 3 events of clinical interest, the exposure adjusted event rates for blinatumomab versus SOC were lower for infections (2 vs. 6 events per patient-year; p < 0.001), cytopenias (4 vs. 20 events per patient-year; p < 0.001), and neurologic events (0.4 vs. 1 event per patient-year; p = 0.008), and higher for CRS (0.2 vs. 0 events per patient-year; p = 0.038)” [5].
Ph+ positive with ALL is associated with poor prognosis. TKIs in combination with chemotherapy is considered the standard frontline treatment for Ph+ BCP-ALL in adults. The phase 2 ALCANTRA study was a single-arm trial that evaluated blinatumomab therapy in Ph+ BCP-ALL patients that were unresponsive to second-generation TKIs or imatinib [26, 27]. The results were that CR or CR with partial hematologic recovery was achieved in 16/45 of 36% of patients. Additionally, blinatumomab treatment was seen to be highly effective in leading to the elimination of detectable MRD in 12/14 or 86% of responders that had complete MRD response [27].
11.5.3 Predictive Indicators for Blinatumomab Treatment in R/R B-ALL
Blinatumomab has been the most intensively studied example of BiTE with substantial clinical outcomes demonstrated, especially for high tumor burden disease as represented by ≥50% bone marrow blasts R/R B-ALL. As of 2020, it is the only FDA and EMA-approved BiTE therapy [28,29,30,31]. Tumor burden or percentage of bone marrow blasts is predictive of CD19 BiTE therapy. In Ph-BCP-ALL patients from previous trials underwent subgroup analysis and those populations with <50% bone marrow blasts were observed to have the greatest OS and remission rates when treated with blinatumomab [21].
According to one phase 3 study that showed statistical significance, CR for these patients in terms of exhibiting full or partial hematologic recovery with the percentage of bone marrow blasts served as a predictive indicator: 65.5% for less than 50% and 34.4% for ≥50%. (P = 0.039) [28, 31]. As mentioned earlier, dexamethasone has a cytoreductive effect for blinatumomab therapy [32]. Extramedullary disease or EMD can serve as a “surrogate for disease burden”, being indicative of progressive disease. A retrospective historical study evaluated baseline and treatment measures of EMD and demonstrated lower CR rates associated with EMD (P = 0.005 and P = 0.05, respectively). MRD has a similar role and one study showed at the day 15, bone marrow MRD in children receiving blinatumomab could predict “complete MRD response” with significant accuracy (up to 95% for the first two treatment cycles) [33]. At day 15, 59 patients were evaluated for complete MRD response: “among 46 MRD positive patients, 44/46 patients had no complete MRD response with an accuracy of 96%, meanwhile, 12/13 patients achieved complete MRD response with an accuracy of 92% for 13 MRD negative patients.” [33].
Wei et al. examined prognostic and predictive biomarkers associated with blinatumomab or chemotherapy in adults with Ph-negative R/R ALL. Patients were randomized 2:1 and administered blinatumomab or chemotherapy. After evaluating baseline blood samples, platelets, tumor burden, and T cell percentage were found to be prognostic markers: platelets were associated with improved 6-month survival, decreased tumor burden was prognostic for remission, and CD3+ T cell percentage was prognostic for minimal residual disease. CD45+, CD3+, and CD8+ T cells were found to be associated with Hematologic remission after receiving blinatumomab [34].
11.5.4 Adverse Events Associated with Blinatumomab in ALL
Blinatumomab treatment is associated with AEs, and the most concerning are CRS and neurotoxicity, which have been observed as Grade ≥3, which range from 0 to 6% for B cell malignancies, can occur within the first several days and are dose-limiting. Higher tumor burden and disease are associated with higher incidence of CRS [28, 29, 35,36,37,38,39]. CRS in particular can range from mild symptoms resembling the flu to fatal multi-organ failure. The mechanism of CRS is not completely understood and is mainly thought of as a product of distinct immune signatures such as T lymphocytes, monocyte and macrophage activation, leading to the massive release of inflammatory cytokines such as IL-6 and Interferon-gamma that is initiated by T cell activation [39]. The systemic production of these toxic cytokines is massive that is facilitated by monocyte and macrophage activation. T cell interferon-gamma, IL-6, IL-10, and tumor necrosis factor-alpha facilitate this cytokine production [40]. Symptomology presents as fever, fatigue, chills, headache, and more serious events such as hypotension, tachycardia, and other cardiovascular events such as vascular leaks and circulatory collapse and during and post-administration of the medication that appears mainly in the first cycle whose severity does not impact response. CRS is generally managed by steroids and IL-6 blockade, and more complicated management strategy involves disassociating tumor cell lysis from cytokine release based on the “two distinct thresholds for T cell activation based on the number of TCR peptide MHC complexes formed:” [4, 41, 42]
An alternative way to avoid CRS-related problems is to dissociate tumor cell destruction and cytokine release. There are two distinct thresholds for T cell activation based on the number of TCR- peptide-MHC (pMHC) complexes formed. The formation of two TCR-pMHC complexes is sufficient between a T cell and an Ag-presenting cell, to trigger T cell-mediated cell lysis. On the other hand, 10 TCR-pMHC complexes are required for the formation of a complete immune synapse and cytokine secretion. Thus, adjusting the binding characteristics for the CD3-binding arm, a BsAb could more closely mimic the natural TCR-pMHC induced T cell activation. Consequently, new CD3-binding Abs have been generated that bind to multiple epitopes on CD3 with a wide range of affinities and agonist activities. Functional studies were realized with BsAbs that integrated the different CD3-binding domains. A BsAb with a new T cell-engaging domain could be created that elicited strong in vivo tumor cell killing and low levels of cytokine release [4].
The second most common event associated with BsAbs is neurotoxicity, for which symptomology ranges from personality changes, tremors, confusion, and focal neurological episodes. More serious episodes such as ataxia, encephalopathy, convulsions, and delirium may also result. As in CRS, these neurotoxicity episodes may be precipitated by inflammatory cytokines. 10–20% of patients treated with blinatumomab experience Grade 3 or higher adverse events, which are considered reversible after stopping the perfusion and corticosteroid initiation. Additionally, these events may be avoided by implementing a progressive dosing regimen and prophylactic administration of dexamethasone, but this constitutes a double-edged sword as the application of steroids could potentially lead to mitigated immune response. However, no inhibition of the cytotoxic capabilities of T cells was observed when reduced levels of inflammatory cytokines were produced as a result of dexamethasone-treated T cells, indicating that dexamethasone does not interfere with therapeutic efficacy of BsAbs [43].
Neurotoxicity can also lead to death. Both are usually managed with corticosteroids and supportive therapy and in severe cases of CRS, the interleukin (IL)-6 receptor inhibitor tocilizumab. More milder cases are treated with dexamethasone as prophylaxis “combined with stepwise administration of blinatumomab is useful to decrease the risk of severe CRS” [1]. Other adverse events are neutropenia, elevated liver enzymes, and infection [28, 35, 44,45,46].
Immune-effector cell-associated neurotoxicity syndrome, or ICANS, is also associated with T cell engaging therapies. Grade ≥3 events range from 5.5 to 24% for blinatumomab [28, 29, 35, 36, 38, 47,48,49,50]. Neurotoxicity, in general, occurs in treatment cycle 1 and its risk is increased when higher dosage of blinatumomab administration. Most common symptoms manifest as dizziness, tremor, confusion, and encephalopathy [45]. Administration of blinatumomab has also led to other adverse effects, such as tumor lysis syndrome, cytopenias, pyrexia, and anemia [51]. CTCAE or Common Terminology Criteria for Adverse Events apply to blinatumomab as well and approximately 5% of R/R BCP-ALL or MRD-positive BCP-ALL experienced a serious CRS event with (CTCAE Grade ≥3) [52, 53]. During phase 2 studies, BCP-ALL patients receiving blinatumomab, close to 53% experienced neurological events of any Grade, and up to 13% had Grade ≥3/4, with no associated deaths [18, 54]. However, these events are manageable as a result of blinatumomab’s pharmacokinetics (high clearance rate) and interruption of treatment is sufficient [55].
The pathogenic mechanisms behind neurotoxicity remain unclear and are characterized as “complex and incompletely understood” [55]. An analysis of five clinical trials showed that selected patients exhibited adhesion of T cells to endothelial cells, leading to neurotoxicity, which was supported by in vitro experiments and preclinical evidence [56]. According to Zhou et al., blinatumomab led to peripheral T cell recruitment to the brain through this process: T cells attached to the cerebral microvascular endothelium, endothelial cells were activated leading to an increased level of Ang-2 (a marker of endothelial cell activation), T cells transmigrated across the blood-brain barrier into the brain that in turn led to the release of cytokines and severe immunological response and neurotoxicity as a result of these T cells destroying resident B cells [56]. Perhaps then agents that inhibit this adhesion between T cells and blood vessel endothelium could potentially be developed to mitigate neurotoxicity.
Other avenues are being pursued in clinical trials to reduce risk of the systemic toxicity of CRS such as developing novel routes of administration for B-ALL, such as subcutaneous administration which could improve convenience and compliance and reduce overall costs versus intravenous infusion [57, 58]. Management strategies include pretreatment with steroids and dose adjustments [53, 58].
Other adverse events, such as medication errors, elevated liver enzymes, and infections have been reported, especially in clinical trial settings. Medication errors usually result from incorrect setting of the infusion rate and malfunction of the flow rate in the pump leading to accidental increase of dose, and usually occurred at Grade 1 or 2 in severity. Additionally in this immunocompromised patient population, treatment-related infections occurred, such as sepsis and pneumonia and opportunistic infections. As a result of B cell depletion and associated decrease in serum immunoglobulins, risk for infection is higher. In the MT103-211 phase 2 study 32% or 60/189 had serious infections including sepsis, pneumonia, and catheter site infections with 9% or 17/189 leading to death [58]. In the TOWER study, transient elevation of liver enzymes during cycle 1 was observed in both cohorts, with 22% in the blinatumomab, arm and 25% in the chemotherapy arm; Grade ≥3 TEAEs were reported for 13% and 15%, respectively. Three serious elevated liver enzyme events and one treatment discontinuation were reported in the blinatumomab arm. No fatal events due to elevation of liver enzymes were reported during this trial [5].
11.5.5 Resistance to Blinatumomab and BITEs
Non-responders form a significant portion of patients receiving BiTEs that implicate loss of CD19 antigen and immunosuppressive factors. The PD-1/PD-L1 axis plays a role in the suppression of anti-tumor activity, as their blockade through antibodies led to significant clinical outcomes. One case study demonstrated positivity to this action in a patient receiving blinatumomab, leading to less tumor cell destruction accompanied by lower levels of interferon-gamma [59]. As a result, ICI administration has been proposed as a way of overcoming this resistance [60]. The immune environment with Tregs also contributes to non-response, since increased levels of Tregs have been observed in R/R ALL [61, 62]. As CR was observed in blinatumomab this also accompanied by relapse, approximately 8–50% experience CD19-negative relapse as a result of antigen loss, which can be “interpreted as the loss of antigen expression and the loss of antigen-binding to targeted antibodies or cells, the presence of either situation or both can lead to the CD19-negative relapse. A study analyzed data from four B-ALL patients who had been treated with blinatumomab and experienced CD19-negative relapse and found that CD19 trafficking from the intracellular space to the membrane of B cells was prevented with the lack of CD81 that provided docking sites for CD19 signal transduction, resulting in absent CD19 expression” [31, 63, 64]. This is as a result of CD19 mutations such as in-frame deletions, SNVs, and nonsense mutation, which were observed in R/R ALL patients with CD19-negative relapse. Other mechanisms are responsive for loss of CD19 expressions such as CD19 mutant allele-specific expression and low CD19 expression of mRNA [32]. Alternative splicing led to CD19 release “which caused antigen escape by changing CD19 epitopes and ultimately disrupting the binding of blinatumomab to CD19 molecule rather than reducing CD19 expression” [1].
These mutations and alternative splicing were observed to occur in parallel leading to antigen loss. Lineage transformation has also been observed where B lymphocytes turn into cells of myeloid lineage, as myeloid marker levels “upregulate”, including CD33 [65, 66]. This lineage switch was thought to be associated with the existence of subclones that had significant selective advantage that did not express CD19 and were shown to have KMT2A/AFF1 and ZNF384 gene rearrangements, generating the need for multitargeted therapies to overcome antigen loss such as one drug that can concomitantly target multiple tumor antigens or in combination with other immunotherapies [67, 68].
11.6 Resistance Mechanisms
11.6.1 T Cell Exhaustion/Dysfunction
Other causes of resistance may occur such as T cell exhaustion or dysfunction as a result of persistent antigen exposure. Their proliferation and cytotoxicity are impacted, and inhibitor receptors such as PD-1, CTLA-4, and LAG-3 (discussed in this volume) become overexpressed in tumor cells, the most central on being the PD-1/PD-L1 axis. This inhibitory pathway is targeted for blocking immunosuppressive signals and leads to more enduring T cell activation. T cells do not become completely inactive, but are not as effective in promoting cell lysis [69,70,71].
The mechanism of T cell activation and proliferation requires antigen recognition by the T cell receptor, co-stimulation, and consequent release of cytokines by the T cells, and then followed by T cell expansion. BsAb meets the first requirement, but the development of BiTEs may be enhanced to trigger “effective immunological synapse” obviating the need for co-stimulation. [72]. Additionally, CD28 or 4-1BB could lead to co-activation and further affect T cell activation as a result of the bispecific T cell engager [73, 74]. Other BsAbs have been constructed to include the IL-15 cytokine [75]. Further, blocking the PD-1/PD-L1 axis can reactivate T cells, but sustainable responses have not been observed in patients for this therapy since other inhibitory pathways are present. A balance must be achieved between enabling sufficient BsAb targeting activities and mitigating lethal autoimmune adverse events, as resistance and evasion mechanisms that sustain dysfunction among T cells are a major concern, but must still be considered in the development and clinical utility of BiTEs [76]. Since by design BsAbs lead to T cell activation, ancillary T cells may also be activated, such as regulatory T lymphocytes or Tregs, which is predictive of treatment resistance and preventing of tumor cell lysis. T cell depletion pre-therapy is suggested [77].
11.6.1.1 CD20 BsAbs
A number of CD20-based BsAbs are in development including REGN1979, Mosunetuzumab, and RG6026. REGN1979 is a fully-humanized IgG4 Ab that since it is has a similarity with natural human Abs is conferred with stability and stable pharmacokinetics and low immunogenicity. In a phase I trial on R/R NHL patients, a 100% overall response was observed in follicular lymphoma and demonstrated CR in CAR-T nonresponders [78]. Mosunetuzumab also has similarities with native B structure being a full-length humanized IgG molecule. In aggressive NHL, an ORR of 37.1% with a CR rate of 19.4% was observed, which was even higher in indolent NHL, with an ORR of 62.7% and CR of 43.3% [79]. RG6026 is unique in that it was constructed in a 2:1 format, providing better TAA binding affinity. A short flexible linker ties the CD3 binding arm with the CD20 binding arm. An extended half-life is conferred through its modified heterodimeric Fc region that prevents binding to FcgRs, which leads to an extended circulatory half-life. Substantive clinical activity was shown in in vitro and in vivo models, even on cells that have low CD20 expression. It has the further advantage of bypassing rituximab resistance since it remains active in the presence of competing for anti-CD20 monoclonal antibodies. Its safety profile is also significant with low cytotoxicity activity. Each of these compounds is undergoing initial clinical investigation to evaluate efficacy [80, 81].
11.7 Acute Myeloid Leukemia
The development of bispecific antibodies for the treatment of AML has been limited due to the lack of tumor-associated antigens on leukemic cells for targeting, (that would be selectively expressed on leukemic cells but spare healthy hematopoietic cells, similar to blinatumomab for B cell destruction) [82]. CD33 and CD123 have been implicated in acute myeloid leukemia, being a mediator of myeloid cell proliferation and differentiation. CD33 is also known as sialic acid-binding Ig-like lectin 3 and is a 7-kDa transmembrane cell surface glycoprotein with expression on leukemic cells [83, 84]. In the initial stages of development of BsAbs for AML, four agents were under investigation one being AMG330 human BiTE tandem single-chain antibody with the N-terminal specific for human CD33 and C-terminal directed toward CD3 [85]. This agent showed anti-leukemic activity in in vitro and in clinical models. Once daily IV infusion was conducted in a phase I study for R/R AML. GEM333 is a CD3 × CD33 BsAb in a phase I study for R/R AML. GEM333 is a humanized antibody with a single-chain bispecific antibody with variable light and heavy chains targeting both CD3 and CD33 that is linked in unique fashion through a tandem format arrangement [86]. In preclinical models, the GEM333 construct efficiently redirected cytotoxic T cells toward CD33+ AML blasts and led to the destruction of AML cell lines and AML blasts in patients. Of interest is that this agent spared normal human CD34+ hematopoietic and progenitor stem cells in vitro [87]. A CD33 × CD3 bispecific antibody was constructed and evaluated in AML patients. 55 patients were dosed at 0.5–720 μg/d continuously through infusion; among 42 evaluable patients, 3 CR and 4 CRis (incomplete Hematologic recovery) were observed the dose of ≥120 μg/d [88, 89]. Grade 3 or higher CRS at a rate of 13% was also observed [88].
CD123 BsAb targets have also been developed for AML. CD123, also known as IL-3 receptor alpha chain, is considered the low-affinity binding subunit of the IL3 receptor. Its mechanism of action is that it “triggers CD123 heterodimerization with the granulocyte-macrophage stimulating factor and IL5 receptor complex” leading to PI3 kinase activity and anti-apoptotic protein upregulation [90, 91]. CD123 expression on AML blasts is associated with lower CR rates and poor prognosis concurrent with higher blast counts. JNJ-63709178 is a CD3 × CD123 construct that contains a bispecific IgG1 antibody created through Genmab DuoBody technology which employs a process termed Fab-arm exchange [82]. Since they retain the Fc region, their effector functions and in vivo stability are enhanced. In murine models, the compound exerted anti-tumor effects and led to tumor regression in a human peripheral blood T cell environment [92]. This compound is undergoing phase I trials for relapsed and refractory patients. XmAb14045 is another CD123 BsAb that also possesses a unique Fc region and undergoes spontaneous formation of stable heterodimers facilitating its manufacturing. In a preclinical monkey model, this agent strongly activated T cells to stimulate CD123+ cell destruction [93]. Other BsAbs for AML are undergoing evaluation. MCLA-117 is a human full-length IgG1 BsAb that targets CLEC12A, a myeloid antigen expressed on AML cells [82]. CLEC12A has selective expression, being expressed on leukemic stem cells but sparing normal hematopoietic cells [56, 57]. In an HL-60 cell line, MCLA-117 led to efficient CLEC12A antigen-dependent T cell activity and targeted tumor cell lysis. The agent also induced “T cell-mediated lysis of AML blasts in an ex vivo culture system and is currently being investigated in a phase I clinical study” to assess safety, tolerability, and efficacy in AML adult patients [82]. These compounds have associated adverse events as a result of their T cell redirecting therapy, which is similar to blinatumomab, including CRS, with high levels of inflammatory cytokines such as IL-6 and IL-2, and associated with flu-like symptoms and quite possibly elevated fever, end-organ dysfunction, or even more threatening complications such as renal failure, hepatic failure and cardiac dysfunction [94]. Neurotoxicity may also result including mild confusion, headaches, to even severe encephalopathy, aphasia, seizures, and delirium [95,96,97].
11.8 Multiple Myeloma
B cell maturation antigen or BCMA is likewise expressed on multiple myeloma tumor cells, with very little expression on normal cells, leading to development of anti-BCMA bispecific antibodies [1]. BCMA is a membrane antigen that has selective expression on malignant cells but it is not expressed in naïve B cells nor other normal tissue cells. It is considered a crucial target of study for the development of a BsAb for multiple myeloma. It is prognostic of poor clinical outcomes and is highly expressed on multiple myeloma cells. According to Lejeune et al., “a rapid re-emergence of B cell immunity after the end of the anti-BCMA treatment would be possible since this [antigen] is not expressed early in B cell development” and “the lack of BCMA expression in other bone marrow populations prevents off-tumor toxicities.” [4].
One such example, AMG420 showed in a clinical study favorable efficacy and safety profiles in R/R MM patients. In this trial, 42 R/R MM patients with more than two lines of prior therapies “were enrolled and received 6-week cycles of AMG 420 at the dose of 0.2–800 μg/d.” [1]. Objective response rate was robust, being 70% with 5 CR (MRD-negative), 1 very good PR, and 1 PR. Infections constituted the most common adverse events, with a rate of 33%. CNS toxicity was not observed; Grade ≥3 CRS was 2%, leading to FDA consideration for approval [98]. AMG420 and 701 are BCMA-CD3 BiTEs that have short-life and IV infusion administration like blinatumomab, being administered for 4 weeks followed by 2 weeks treatment-free. AMG420 targets BCMA-positive MM selectively while avoiding BCMA-negative MM cells in both in vitro and in vivo models. In clinical trials evaluating 42 refractory MM patients, a 70% response rate was observed with 70% MRD-negativity; adverse events included infections and neuropathy [99]. AMG701 has single-chain variable fragments of AMG420 with a half-life extension and is undergoing evaluation for toxicity and response through once-weekly dosing.
11.8.1 Clinical Development for MM (CD38-CD3)
A number of tumor-associated antigen BsAbs have been studied for MM. BsAbs that target CD38, such as humanized anti-CD38/CD3 XmAbs with differing affinities for CD38 and CD3. AGM424 has been studied in in vitro and in vivo models and led to significant tumor cell destruction in the presence of soluble CD38. It has lower affinity for CD3 and is associated with uncontrolled CRS. It is currently in phase I studies to evaluate safety and tolerability, and PK, PD and efficacy in R/R MM [100]. GBR 1342 is another anti-CD38/CD3 BsAb that has a complete Fc domain and was shown in preclinical models to have superiority to the anti-CD38 monoclonal antibody daratumumab. T cells and CD38+ T cell depletion were induced in both blood and bone marrow. A phase 1 study began to evaluate its tolerability.
IgG2a-based BCMA-CD3 (PF-06863135) is also a humanized BsAb that has an IgG2a backbone with mutations in the Fc region that lead to heavy chain heterodimer formation which reduces FcG receptor binding [48]. This compound is undergoing a phase I study for safety but has shown anti-myeloma activity in in vivo models [101].
11.8.2 Clinical Development for MM (FcRL5-CD3 and GPRC5D-CD3)
These anti-myeloma compounds comprise two new targets developed as part of MM-related target: Fc Receptor-Like 5 (FcRL5) and G-protein coupled receptor family C group 5 member D (GPRC5D). FcRH5 contains an exclusive surface marker from B cell lineage but is detected starting from early pre-B cell stage development [102]. It remains unique among other B cell-specific surface proteins in that FcRL5 is preserved in both normal and tumor B cells, which enables further activity in other B cell tumors, such as CLL, DLBCL, and follicular lymphoma [102, 103].
GPRC5D on the other hand is “expressed on the surface of malignant cells involved in multiple myeloma without being expressed at appreciable levels by normal hematopoietic cells, such as T cells, NK cells, monocytes, granulocytes and bone marrow progenitors, including hematopoietic stem cells” [104]. Additionally, mRNA expression of the marker was only expressed in MM patients with low expression in normal tissues, which was associated with poor outcomes [105]. This profile lends itself as a suitable target for MM patients. Two BsAbs are in development against these targets and are currently in phase I clinical studies: RG6160 which targets FcRL5 and the DuoBody JNJ-64407564. Both target GPRC5D, and led to encouraging results from in vitro and in vivo models that demonstrated B cell depletion and tumor growth suppression in myeloma models [104, 106].
Teclistamab, a B cell maturation antigen × CD3 bispecific antibody showed clinical efficacy in R/R MM patients in an open-label phase 1 multicenter trial. Teclistamab was initially considered an investigational bispecific T cell engager that has structural differences from AMG 420 with “promising efficacy” (Table 11.2).
11.9 Non-Hodgkin Lymphoma
Blinatumomab has demonstrated clinical efficacy for R/R DLBCL, as shown in Table 11.3. Other bispecific antibodies are being studied for determining safety and tolerability in DLBCL and NHL.
Pharmacokinetic and pharmacodynamic analysis revealed B cell depletion rates for NHL patients receiving blinatumomab. B cell depletion took place within 48 h after continuous IVD infusion doses of greater than 5 ug/m2/day that took place in first-order kinetics. A 50% reduction in tumor size was a result of dosage of 47 ug/m2/day for 28 days. The authors concluded that B-lymphocyte depletion was dependent on exposure while adverse cytokine elevation was transient but also increased with dose. Overall a PK/PD relationship was established in this medication dose selection [108].
11.10 Dual Affinity BsAbs and Tandem Diabodies
Dual affinity bispecific antibodies and tandem diabodies, other types of BsAb constructs have been discussed earlier, but are worth specifying further here. MGD011 (duvortuxizumab) is a CD19 × CD3 DART with a silenced, human IgG1 Fc domain that confers it with a relatively long circulating half-life (approximately 14.3–20.6 days), which is similar to conventional mAbs that allow for a every 2-weeks administration. The benefit of this DART is that its humanized Ab arms have a much greater affinity for CD19 than for CD3 which lends itself to preferential binding to targets. However, due to high neurotoxicity in phase I studies for B cell malignancies such as NHL and CLL, its clinical development was stopped [109].
“AFM11 is a tetravalent bispecific TandAb with two binding sites for CD3 and two for CD19”. With its increased binding affinity, its high potential for treatment efficacy was anticipated but phase I studies for ALL and R/R NHL was suspended as it was further revealed that AFM11 potency was not correlated with the CD19 density on target cell surfaces and additionally severe neurotoxicity with one fatality occurred [110].
AMV564 is a tetravalent anti-CD33-anti-CD3 tandem diabody construct that forms a homodimer from two VH and VL chains that are composed of antigen-binding single-chain variable fragments (scFvs). Two binding sites for each epitope are created, thus increasing the avidity of the antibody to its targets. It also has a longer half-life compared to BiTEs owing to its molecular weight of 106 kDa [111]. This tandem diabody is being studied in a first-in-human phase I trial in R/R AML patients at a 14-day continuous infusion every 28 days and may hold potential over the two constructs discussed above since preliminary evidence has shown evidence of T cell activation by increased cytokine levels. 13–38% reduction in bone marrow blasts was evidence of biological activity in 10/16 patients. Safety profiles were also favorable with no Grade 3–4 toxicities present. A Grade 2 CRS at the 50 ug/day dose in a single patient was observed [112].
Table 11.4 is a summary of bispecific antibodies for Hematologic malignancies.
11.11 Combination and Sequential Therapies [3]
PD-1 expression has been associated with resistance to blinatumomab [113] since T cell exhaustion is observed when PD-1 is overexpressed. This association led to studies combining blinatumomab with immune checkpoint inhibitors, case in point being a 12-year patient with refractory ALL achieved remission was administered pembrolizumab and blinatumomab since pembrolizumab enhanced T cell function [113]. Table 11.5 from Lejeuene et al. shows the clinical studies as of 2019 combining BsAbs with immune checkpoint inhibitors. These studies have been extrapolated to design BsAbs that concomitantly target two immune checkpoints such as the dual blockade of PD-1 and LAG-3 (also discussed in this volume). MGD013 is an anti-PD-1/anti-LAG-3 DART that binds specifically to both PD-1 and LAG-3 (142) and enhances T cell pathways. However, these positive outcomes have been deterred to some extent by increases in adverse events due to over-activation of the immune system, which investigators are studying to overcome. A new approach was developed that “consists in the deletion of the PD-1 pathway via high-affinity PD-1 binding while inhibiting CTLA-4 with a low-affinity binding arm. This construct inhibits CTLA-4 in double-positive T cells while reducing the binding to peripheral T lymphocytes expressing CTLA-4, resulting in better tolerability” [114].
In R/R BCP-ALL patients, blinatumomab is combined with PD-1 and CTLA-4 inhibitors to determine efficacy and tolerability. Preliminary findings from a study evaluating nivolumab and blinatumomab have demonstrated feasibility with acceptable toxicity. Heavily pretreated patients showed a 80% MRD complete response rate [115]. Deep and durable remissions are also anticipated from a study combining pembrolizumab with blinatumomab in a phase 1/2 study in R/R BCP-ALL patients with a high percentage of bone marrow blasts as a result of resistance to blinatumomab [116]. Limited data are also arriving from studies combining TKIs such as ponatinib, dasatinib, and bosatinib, achieving 50% hematologic response rates [117].
Bispecific antibodies have made enormous progress in the treatment of Hematologic malignancies, the most prominent being the bispecific T cell engager blinatumomab for acute lymphoblastic leukemia. Acute myeloid leukemia and multiple myeloma are also benefiting from BsAb treatment. They have proved to be a formidable alternative to immune checkpoint inhibitors and adoptive cellular therapies, and are vying with each for clinical and commercial viability. They are most often compared with chimeric antigen receptor therapies in terms of clinical efficacy and response, toxicity and manufacture, and several studies are underway for combining both of them for more deep and durable responses. Other types of bispecific antibodies other than the canonical CD3 T cell construct are in development, such as those for directing natural killer cells to leukemic and myeloid targets through their cell surface antigens and mitigating tumor escape as tumor cells lose their antigens. Others are being developed with longer half-lives so that the intravenous administration of these compounds is more facile and there is less possibility of medication errors happening. Also, one of the major advances of bispecific antibody-drug development is that their clinical effectivity runs the gamut for all patient populations: pediatric, adult, and elderly. As regulatory bodies are continuously receiving data for the hundreds of bispecific antibodies, particularly bispecific T cell engagers, under study and ongoing development, the landscape for the treatment of hematologic malignancies by these agents will expand most certainly.
Abbreviations
- Ab:
-
Antibody
- AE:
-
Adverse event
- ALL:
-
Acute lymphocytic leukemia
- AML:
-
Acute myeloid leukemia
- BCMA:
-
B-cell maturation antigen
- BCP-ALL:
-
B-cell precursor acute lymphocytic leukemia
- Ph-R/R:
-
Philadelphia relapsed/recurrence
- BiTEs:
-
Bispecific T cell engagers
- BsAbs:
-
Bispecific antibodies
- CAR-T cells:
-
Chimeric antigen receptor-T cells
- CD:
-
Cluster of differentiation
- CLL:
-
Chronic lymphocytic leukemia
- CR:
-
Complete response
- CRS:
-
Cytokine release syndrome
- CTCAE:
-
Common terminology criteria for adverse events
- CTLA-4:
-
Cytotoxic T-lymphocyte associated antigen-4
- DART:
-
Dual-affinity re-targeting antibody
- DLBCL:
-
Diffuse large B-cell lymphoma
- EMA:
-
European Medicines Agency
- Fc:
-
Fragment crystallizable
- FDA:
-
Food and Drug Administration
- HR:
-
Hazard ratio
- ICANs:
-
Immune effector cell-associated neurotoxicity syndrome
- ICIs:
-
Immune checkpoint inhibitors
- Ig:
-
Immunoglobulin
- IL:
-
Interleukin
- IV:
-
Intravenous
- kDa:
-
Kilodalton
- LAG-3:
-
Lymphocyte-activation gene 3
- MHC:
-
Major histocompatibility complex
- MM:
-
Multiple myeloma
- MRD:
-
Minimal residual disease
- NK:
-
Natural killer
- NSCLC:
-
Non-small cell lung cancer
- ORR:
-
Objective response rate
- OS:
-
Overall survival
- PD:
-
Pharmacodynamic
- PD-1/PD-L1:
-
Programmed death-1/programmed death ligand-1
- Ph+ R/R BCP-ALL:
-
Philadelphia chromosome positive relapsed/recurrence B-cell precursor acute lymphocytic leukemia
- PI3:
-
Phosphoinositol-3
- PK:
-
Pharmacokinetic
- R/R MM:
-
Relapsed/recurrent multiple myeloma
- R/R NHL:
-
Relapsed/recurrence non-Hodgkin lymphoma
- scRvs:
-
Single-chain variable fragments
- SNVs:
-
Single nucleotide variations
- SOC:
-
Standard of care
- TandAb:
-
Tandem diabody
- TCR:
-
T-cell receptor
- TEAEs:
-
Treatment emergent adverse events
- TKIs:
-
Tyrosine kinase inhibitors
- VH:
-
Heavy chain
- VL:
-
Light chain
References
Zhou S, Liu M, Ren F, Meng X, Yu J (2021) The landscape of bispecific T cell engager in cancer treatment. Biomarker Res 9:38
Huang S, van Duijnhoven SMJ, Sijtx AJA, van Elsas A (2020) Bispecific antibodies targeting dual tumor-associated antigens in cancer therapy. J Cancer Res Clin Oncol 146:3111–3122
Viardot A, Locatelli F, Stieglmaier J, Zaman F, Jabbour E (2020) Concepts in immune-oncology: tackling B cell malignancies with CD19-directed bispecific T cell engager therapies. Ann Hematol 999:2215–2229
Lejeune M, Cem Kose M, Duray E, Einsele H, Beguin Y, Caers J (2020) Bispecific, T-cell recruiting antibodies in B-cell malignancies. Front Immunol 11(762)
Stein A, Franklin JL, Chia VM, Arrindell D, Kormany W et al (2019) Benefit-risk assessment of blinatumomab in the treatment of relapsed/refractory B-cell precursor acute lymphoblastic leukemia. Drug Saf 42:587–601
Poussin M, Sereno A, Wu X, Huang F, Manro J et al (2021) Dichotomous impact of affinity on the function of T cell engaging bispecific antibodies. J Immunother Cancer 9:e002444
Nagorsen D, Kufer P, Baeuerle PA, Bargou R (2012) Blinatumomab: a historical perspective. Pharmacol Ther 136(3):334–342
BLINCYTO® (blinatumomab) [prescribing information] (2019) Amgen, Thousand Oaks, CA
Viardot A, Goebeler ME, Hess G, Neumann S, Pfreundschuh M, Adrian N, Zettl F, Libicher M, Sayehli C, Stieglmaier J, Zhang A, Nagorsen D, Bargou RC (2016) Phase 2 study of bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B cell lymphoma. Blood 127(11):1410–1416
Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS et al (2012) Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood 119:6226–6233
De Gast GC, Van Houten AA, Haagen IA, Klein S, De Weger RA, Van Dijk A et al (1995) Clinical experience with CD13 x CD19 bispecific antibodies in patients with B cell malignancies. J Hematother 4:433–437
Kontermann RE, Brinkmann U (2015) Bispecific antibodies. Drug Discov Today 7
Topp MS, Gokbuget N, Stein AS, Zugmaier G, O’Brien S, Bargou RC et al (2015) Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol 16:57–66
Topp MS, Gokbuget N, Zugmaier G, Klappers P, Stelljes M, Neumann S et al (2014) Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol 32:4134–4140
Topp MS, Kufer P, Gokbuget N, Goebeler M, Klinger M, Neumann S et al (2011) Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol 29:2493–2498
Kantarjian H, Stein A, Gokbuget N, Fielding AK, Schuh AC et al (2017) Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med 376(9):836–847
Foa R, Bassan R, Vitale A, Elia L, Piciocchi A (2020) Dasatinib-blinatumomab for Ph-positive acute lymphoblastic leukemia in adults. N Engl J Med 383(17):1613–1623
Gökbuget N, Dombret H, Bonifacio M, Reichle A, Graux C, Faul C, Diedrich H, Topp MS, Brüggemann M, Horst HA, Havelange V, Stieglmaier J, Wessels H, Haddad V, Benjamin JE, Zugmaier G, Nagorsen D, Bargou RC (2018) Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 131(14):1522–1531
Gökbuget N, KelshM, Chia V, Advani A, Bassan R, Dombret H, Doubek M, Fielding AK, Giebel S, Haddad V, Hoelzer D, Holland C, Ifrah N, Katz A, Maniar T, Martinelli G, Morgades, M, O'Brien S, Ribera JM, Rowe JM, Stein A, Topp M, Wadleigh, M, Kantarjian H (2016) Blinatumomab vs historical standard therapy of adult relapsed/refractory acute lymphoblastic leukemia. Blood Cancer J 6(9):e473
Jen EY, Xu Q, Schetter A, Przepiorka D, Shen YL, Roscoe D, Sridhara R, Deisseroth A, Philip R, Farrell AT, Pazdur R (2019) FDA approval: blinatumomab for patients with B-cell precursor acute lymphoblastic leukemia in morphologic remission with minimal residual disease. Clin Cancer Res 25(2):473–477
Dombret H, Topp MS, Schuh AC, Wei AH, Durrant S, Bacon CL, Tran Q, Zimmerman Z, Kantarjian H (2019) Blinatumomab versus chemotherapy in first salvage or in later salvage for B-cell precursor acute lymphoblastic leukemia. Leuk Lymphoma 60(9):2214–2222
Kantarjian HM, Thomas D, Ravandi F, Faderl S, Jabbour E, Garcia-Manero G et al (2010) Defining the course and prognosis of adults with acute lymphocytic leukemia in first salvage after induction failure or short first remission duration. Cancer 116(24):5568–5574
Minson KA, Prasad P, Vear S, Borinstein S, Ho R, Domm J, Frangoul H (2013) t(17;19) in children with acute lymphocytic leukemia: a report of 3 cases and a review of the literature. Case Rep Hematol 563291:1–4
Jabbour EJ, Gokbuget N, Kantarjian HM, Thomas X, Larson RA, Yoon SS, Ghobadi A, Topp MS, Tran Q, Franklin JL, Forman SJ, Stein AS (2019) Transplantation in adults with relapsed/refractory acute lymphoblastic leukemia who are treated with blinatumomab from a phase 3 study. Cancer 125(23):4181–4192
Topp MS, Zimmerman Z, Cannell P, Dombret H, Maertens J, Stein A, Franklin J, Tran Q, Cong Z, Schuh AC (2018) Health related quality of life in adults with relapsed/refractory acute lymphoblastic leukemia treated with blinatumomab. Blood 131(26):2906–2914
Pulte ED, Vallejo J, Przepiorka D, Nie L, Farrell AT, Goldberg KB, McKee AE, Pazdur R (2018) FDA supplemental approval: blinatumomab for treatment of relapsed and refractory precursor B-cell acute lymphoblastic leukemia. Oncologist 23(11):1366–1371
Martinelli G, Boissel N, Chevallier P, Ottmann O, Gökbuget N, ToppMS, FieldingAK,Rambaldi A, Ritchie EK, Papayannidis C, Sterling LR, Benjamin J, Stein A (2017) Complete hematologic and molecular response in adult patients with relapsed/refractory Philadelphia chromosome–positive B-precursor acute lymphoblastic leukemia following treatment with blinatumomab: results from a phase II, single-arm, multicenter study. J Clin Oncol 35(16):1795–1802
Martinelli G, Boissel N, Chevallier P, Ottmann O, Gökbuget N, Topp MS et al (2017) Complete hematologic and molecular response in adult patients with relapsed/refractory Philadelphia chromosome–positive B-precursor acute lymphoblastic leukemia following treatment with blinatumomab: results from a phase II, single-arm, multicenter study. J Clin Oncol 35:1795–1802
Gökbuget N, Zugmaier G, Dombret H, Stein A, Bonifacio M, Graux C et al (2020) Curative outcomes following blinatumomab in adults with minimal residual disease B cell precursor acute lymphoblastic leukemia. Leuk Lymphoma 61:2665–2673
von Stackelberg A, Locatelli F, Zugmaier G, Handgretinger R, Trippett TM, Rizzari C et al (2016) Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol 34:4381–4389
Aldoss I, Song J, Stiller T, Nguyen T, Palmer J, O’Donnell M et al (2017) Correlates of resistance and relapse during blinatumomab therapy for relapsed/refractory acute lymphoblastic leukemia. Am J Hematol 92:858–865
Zhao Y, Aldoss I, Qu C, Crawford JC, Gu Z, Allen EK et al (2021) Tumor-intrinsic and -extrinsic determinants of response to blinatumomab in adults with BALL. Blood 137:471–484
King AC, Bolanos R, Velasco K, Tu H, Zaman F, Geyer MB et al (2019) Real world chart review of blinatumomab to treat patients with high disease burden of relapsed or refractory B cell precursor acute lymphoblastic leukemia. Blood 134:5079
Brown P, Zugmaier G, Gore L, Tuglus CA, Stackelberg A (2019) Day 15 bone marrow minimal residual disease predicts response to blinatumomab in relapsed/refractory pediatric B-ALL. Brit J Haematol. 188:e36–e39
Gökbuget N, Zugmaier G, Klinger M, Kufer P, Stelljes M, Viardot A et al (2017) Long-term relapse-free survival in a phase 2 study of blinatumomab for the treatment of patients with minimal residual disease in B-lineage acute lymphoblastic leukemia. Haematologica 102:e132–e135
Topp MS, Gökbuget N, Zugmaier G, Klappers P, Stelljes M, Neumann S et al (2014) Phase II trial of the anti-CD19 bispecific T cell–engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol 32:4134–4140
Kujawski M, Li L, Bhattacharya S, Wong P, Lee W-H et al (2019) Generation of dual specific bivalent BiTEs (dbBIspecific T cell engaging antibodies) for cellular immunotherapy. BMC Cancer 19:882
Coyle L, Morley NJ, Rambaldi A, Mason KD, Verhoef G, Furness CL et al (2020) Open-label, phase 2 study of blinatumomab as second salvage therapy in adults with relapsed/refractory aggressive B cell non-Hodgkin lymphoma. Leuk Lymphoma 61:2103–2112
Viardot A, Goebeler M, Hess G, Neumann S, Pfreundschuh M, Adrian N et al (2016) Phase 2 study of the bispecific T cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B cell lymphoma. Blood 127:1410–1416
Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M et al (2014) Current concepts in the diagnosis and management of cytokine release syndrome. Blood 124:188–195. https://doi.org/10.1182/blood-2014-05-552729
Li J, Piskol R, Ybarra R, Chen Y-JJ, Li J, Slaga D et al (2019) CD3 bispecific antibody induced cytokine release is dispensable for cytotoxic T cell activity. Sci Transl Med 11:eaax8861
Barrett DM, Teachey DT, Grupp SA (2014) Toxicity management for patients receiving novel T cell engaging therapies. Curr Opin Pediatr 26:43–49
Shimabukuro-Vornhagen A, Godel P, Subklewe M, Stemmler HJ, Schlosser HA, Schlaak M et al (2018) Cytokine release syndrome. J Immunother Cancer 6:56
Brandl C, Haas C, d’Argouges S, Fisch T, Kufer P, Brischwein K et al (2007) The effect of dexamethasone on polyclonal T cell activation and redirected target cell lysis as induced by a CD19/CD3-bispecific single-chain antibody construct. Cancer Immunol Immunother 56:1551–1563
Seckinger A, Delgado JA, Moser S, Moreno L, Neuber B, Grab A et al (2017) Target expression, generation, preclinical activity, and pharmacokinetics of the BCMA-T cell bispecific antibody EM801 for multiple myeloma treatment. Cancer Cell 31:396–410
Costa LJ, Wong SW, Bermúdez A, de la Rubia J, Mateos M-V, Ocio EM et al (2019) First clinical study of the B cell maturation antigen (BCMA) 2+1 T cell engager (TCE) CC-93269 in patients (Pts) with relapsed/refractory multiple myeloma (RRMM): interim results of a phase 1 multicenter trial. Blood 134(Suppl. 1):143
Wei AH, Ribera J-M, Larson RA, Ritchie D, Ghobadi A (2021) Biomarkers associated with blinatumomab outcomes in acute lymphoblastic leukemia. Leukemia 35:2220–2231
Panowski SH, Kuo TC, Zhang Y, Chen A, Geng T, Aschenbrenner L et al (2019) Preclinical efficacy and safety comparison of CD3 bispecific and ADC modalities targeting BCMA for the treatment of multiple myeloma. Mol Cancer Ther 18:2008–2020
Liu D, Zhao J, Song Y, Luo X, Yang T (2019) Clinical trial update on bispecific antibodies, antibody-drug conjugates, and antibody-containing regimens for acute lymphoblastic leukemia. J Hematol Oncol 12:15
Locatelli F, Zugmaier G, Mergen N, Bader P, Jeha S, Schlegel P et al (2020) Blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia: results of the RIALTO trial, an expanded access study. Blood Cancer J 10:1–5
Goebeler M, Knop S, Viardot A, Kufer P, Topp MS, Einsele H et al (2016) Bispecific T cell engager (BiTE) antibody construct blinatumomab for the treatment of patients with relapsed/refractory non-Hodgkin lymphoma: final results from a phase I study. J Clin Oncol 34:1104–1111
Stein AS, Larson RA, Schuh AC, StevensonW L-M, Tran Q, Zimmerman Z, KormanyW TMS (2018) Exposure adjusted adverse events comparing blinatumomab with chemotherapy in advanced acute lymphoblastic leukemia. Blood Adv 2(13):1522–1531
Frey N, Porter D (2016) Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. Hematol Am Soc Hematol Educ Program 2016(1):567–572
von Stackelberg A, Locatelli F, Zugmaier G, Handgretinger R, Trippett TM, Rizzari C, Bader P, O’Brien MM, Brethon B, Bhojwani D, Schlegel PG, Borkhardt A, Rheingold SR, Cooper TM, Zwaan CM, Barnette P, Messina C, Michel G, DuBois SG, Hu K, ZhuM WJA, Gore L (2016) Phase I/Phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol 34(36):4381–4389
Jain T, Litzow MR (2018) No free rides: management of toxicities of novel immunotherapies in ALL, including financial. Blood Adv 2(22):3393–3403
Klinger M, Zugmaier G, Nägele V, Goebeler M, Brandl C, Stelljes M et al (2020) Adhesion of T cells to endothelial cells facilitates blinatumomab-associated neurologic adverse events. Cancer Res 80:91–101
Matasar MJ, Cheah CY, Yoon DH, Assouline SE, Bartlett NL, Ku M et al (2020) Subcutaneous mosunetuzumab in relapsed or refractory B cell lymphoma: promising safety and encouraging efficacy in dose escalation cohorts. Blood 136:45–46
Lesokhin AM, Levy MY, Dalovisio AP, Bahlis NJ, Solh M, Sebag M et al (2020) Preliminary safety, efficacy, pharmacokinetics, and pharmacodynamics of subcutaneously (SC) administered PF-06863135, a B cell maturation antigen (BCMA)-CD3 bispecific antibody, in patients with relapsed/refractory multiple myeloma (RRMM). Blood 136:8–9
Ribas A, Wolchok JD (2018) Cancer immunotherapy using checkpoint blockade. Science 359:1350–1355
Feucht J, Kayser S, Gorodezki D, Hamieh M, Döring M, Blaeschke F et al (2016) Tcell responses against CD19+ pediatric acute lymphoblastic leukemia mediated by bispecific T cell engager (BiTE) are regulated contrarily by PDL1 and CD80/CD86 on leukemic blasts. Oncotarget 7:76902–76919
Duell J, Dittrich M, Bedke T, Mueller T, Eisele F, Rosenwald A et al (2017) Frequency of regulatory T cells determines the outcome of the T-cell engaging antibody blinatumomab in patients with B-precursor ALL. Leukemia 31:2181–2190
Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C et al (2004) CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol 34:336–344
Jabbour E, Düll J, Yilmaz M, Khoury JD, Ravandi F, Jain N et al (2018) Outcome of patients with relapsed/refractory acute lymphoblastic leukemia after blinatumomab failure: no change in the level of CD19 expression. Am J Hematol 93:371–374
Braig F, Brandt A, Goebeler M, Tony H, Kurze A, Nollau P et al (2017) Resistance to anti-CD19/CD3 BiTE in acute lymphoblastic leukemia may be mediated by disrupted CD19 membrane trafficking. Blood 129:100–104
Gardner R, Wu D, Cherian S, Fang M, Hanafi L, Finney O et al (2016) Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 127:2406–2410
Rayes A, McMasters RL, O’Brien MM (2016) Lineage switch in MLL-rearranged infant leukemia following CD19-directed therapy. Pediatr Blood Cancer 63:1113–1115
Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J et al (2016) Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest 126:3814–3826
Dai H, Wu Z, Jia H, Tong C, Guo Y, Ti D et al (2020) Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J Hematol Oncol 13:30
Woo S-R, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ et al (2012) Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T cell function to promote tumoral immune escape. Cancer Res 72:917–927
Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y et al (2014) The immunoreceptor TIGIT regulates anti-tumor and antiviral CD8(+) T cell effector function. Cancer Cell 26:923–937
Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S et al (2006) PD-1 expression on HIV-specific T cells is associated with T cell exhaustion and disease progression. Nature 443:350–354
Ruella M, Maus MV (2016) Catch me if you can: leukemia escape after CD19-directed T cell immunotherapies. Comput Struct Biotechnol J 14:357–362
Arndt C, Feldmann A, von Bonin M, Cartellieri M, Ewen E-M, Koristka S et al (2014) Costimulation improves the killing capability of T cells redirected to tumor cells expressing low levels of CD33: description of a novel modular targeting system. Leukemia 28:59–69
Liu R, Jiang W, Yang M, Guo H, Zhang Y, Wang J et al (2010) Efficient inhibition of human B cell lymphoma in SCID mice by synergistic anti-tumor effect of human 4–1BB ligand/anti-CD20 fusion proteins and anti-CD3/anti-CD20 diabodies. J Immunother 33:500–509
Schmohl JU, Felices M, Oh F, Lenvik AJ, Lebeau AM, Panyam J et al (2017) Engineering of Anti-CD133 trispecific molecule capable of inducing NK expansion and driving antibody-dependent cell-mediated cytotoxicity. Cancer Res Treat 49:1140–1152
Khan S, Gerber DE (2019) Autoimmunity, checkpoint inhibitor therapy and immune-related adverse events: a review. Semin Cancer Biol S1044-579X(19)30019-7
Duell J, Dittrich M, Bedke T, Mueller T, Eisele F, Rosenwald A et al (2017) Frequency of regulatory T cells determines the outcome of the T cell engaging antibody blinatumomab in patients with B-precursor ALL. Leukemia 31:2181–2190
Bannerji R, Brown JR, Advani RH, Arnason J, O’Brien SM, Allan JN et al (2016) Phase 1 study of REGN1979, an anti-CD20 x anti-CD3 bispecific monoclonal antibody, in patients with CD20+ B cell malignancies previously treated with CD20-directed antibody therapy. Blood 128:621
Schuster SJ, Bartlett NL, Assouline S, Yoon S-S, Bosch F, Sehn LH et al (2019) Mosunetuzumab induces complete remissions in poor prognosis Non-Hodgkin lymphoma patients, including those who are resistant to or relapsing after chimeric antigen receptor T cell (CAR-T) therapies, and is active in treatment through multiple lines. Blood 134(Suppl. 1):6
Reusch U, Burkhardt C, Fucek I, Le Gall F, Le Gall M, Hoffmann K et al (2014) A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. MAbs 6:728–739
Bacac M, Colombetti S, Herter S, Sam J, Perro M, Chen S et al (2018) CD20-TCB with obinutuzumab pretreatment as next-generation treatment of hematologic malignancies. Clin Cancer Res 24:4785–4797
Guy DG, Uy GL (2018) Bispecific antibodies for the treatment of acute myeloid leukemia. Curr Hematol Malig Rep 13:417–425
Dinndorf PA, Andrews RG, Benjamin D, Ridgway D, Wolff L, Bernstein ID (1986) Expression of normal myeloid-associated antigens by acute leukemia cells. Blood 67(4):1048–1053
Hauswirth AW, Florian S, Printz D, Sotlar K, KrauthMT, Fritsch G et al (2007) Expression of the target receptor CD33 in CD34+/CD38-/CD123+ AML stem cells. Eur J Clin Inv 37(1):73–82
Krupka C, Kufer P, Kischel R, Zugmaier G, Bogeholz J, Kohnke T et al (2014) CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T cell-engaging antibody AMG 330. Blood 123(3):356–365
Stamova S, Cartellieri M, Feldmann A, Arndt C, Koristka S, Bartsch H et al (2011) Unexpected recombinations in single-chain bispecific anti-CD3-anti-CD33 antibodies can be avoided by a novel linker module. Mol Immunol 49(3):474–482
Arndt C, von Bonin M, Cartellieri M, Feldmann A, Koristka S, Michalk I et al (2013) Redirection of T cells with a first fully humanized bispecific CD33-CD3 antibody efficiently eliminates AML blasts without harming hematopoietic stem cells. Leukemia 27(4):964–967
Crocker PR, Paulson JC, Varki A (2007) Siglecs and their roles in the immune system. Nat Rev Immunol 7:255–266
Ravandi F, Walter RB, Subklewe M, Buecklein V, Jongen-Lavrencic M, Paschka P et al (2020) Updated results from phase I dose-escalation study of AMG 330, a bispecific T cell engager molecule, in patients with relapsed/refractory acute myeloid leukemia (R/R AML). J Clin Oncol 38:7508
Reddy EP, Korapati A, Chaturvedi P, Rane S (2000) IL-3 signaling and the role of Src kinases, JAKs and STATs: a covert liaison unveiled. Oncogene 19(21):2532–2547
BlalockWL,Weinstein-Oppenheimer C, Chang F, Hoyle PE, Wang XY, Algate PA et al (1999) Signal transduction, cell cycle regulatory, and anti-apoptotic pathways regulated by IL-3 in hematopoietic cells: possible sites for intervention with anti-neoplastic drugs. Leukemia 13(8):1109–1166
Gaudet FNJ et al (2016) Development of a CD123xCD3 bispecific antibody (JNJ-63709178) for the treatment of acute myeloid leukemia (AML). Blood 128(22):2824
Chu SY, Pong E, Chen H, Phung S, Chan EW, Endo NA et al (2014) Immunotherapy with long-lived anti-CD123 × anti-CD3 bispecific antibodies stimulates potent Tcell-mediated killing of human AML cell lines and of CD123+ cells in monkeys: a potential therapy for acute myelogenous leukemia. Blood 124(21):2316
Billiau AD, Roskams T, Van Damme-Lombaerts R, Matthys P, Wouters C (2005) Macrophage activation syndrome: characteristic findings on liver biopsy illustrating the key role of activated, IFN-g producing lymphocytes and IL-6- and TNF-alpha-producing macrophages. Blood 105(4):1648–1651
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K et al (2014) Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 6(224):224ra25
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA et al (2015) T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukemia in children and young adults: a phase 1 dose-escalation trial. Lancet (London, England). 385(9967):517–528
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ et al (2014) Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371(16):1507–1517
Topp MS, Duell J, Zugmaier G, Attal M, Moreau P, Langer C et al (2020) Anti-B cell maturation antigen BiTE molecule AMG 420 induces responses in multiple myeloma. J Clin Oncol 38:775–783
Hipp S, Tai Y-T, Blanset D, Deegen P, Wahl J, Thomas O et al (2017) A novel BCMA/CD3 bispecific T cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia 31:1743–1751
Zuch de Zafra CL, Fajardo F, Zhong W, Bernett MJ, Muchhal US, Moore GL et al (2019) Targeting multiple myeloma with AMG 424, a novel Anti-CD38/CD3 bispecific t cell-recruiting antibody optimized for cytotoxicity and cytokine release. Clin Cancer Res 25:3921–3933
Lesokhin AM, Raje N, Gasparetto CJ, Walker J, Krupka HI, Joh T et al (2018) A phase I, open-label study to evaluate the safety, pharmacokinetic, pharmacodynamic, and clinical activity of PF-06863135, a B cell maturation antigen/CD3 bispecific antibody, in patients with relapsed/refractory advanced multiple myeloma. Blood 132(Suppl. 1):3229
Polson AG, Zheng B, Elkins K, Chang W, Du C, Dowd P et al (2006) Expression pattern of the human FcRH/IRTA receptors in normal tissue and in B-chronic lymphocytic leukemia. Int Immunol 18:1363–1373
Ise T, Nagata S, Kreitman RJ, Wilson WH, Wayne AS, Stetler-Stevenson M et al (2007) Elevation of soluble CD307 (IRTA2/FcRH5) protein in the blood and expression on malignant cells of patients with multiple myeloma, chronic lymphocytic leukemia, and mantle cell lymphoma. Leukemia 21:169–174
Kodama T, Kochi Y, Nakai W, Mizuno H, Baba T, Habu K et al (2019) Anti-GPRC5D/CD3 bispecific T cell redirecting antibody for the treatment of multiple myeloma. Mol Cancer Ther 18:1555–1564
Atamaniuk J, Gleiss A, Porpaczy E, Kainz B, Grunt TW, Raderer M et al (2012) Overexpression of G protein-coupled receptor 5D in the bone marrow is associated with poor prognosis in patients with multiple myeloma. Eur J Clin Invest 42:953–960
Li J, Stagg NJ, Johnston J, Harris MJ, Menzies SA, DiCara D et al (2017) Membrane proximal epitope facilitates efficient T cell synapse formation by anti-FcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell 31:383–395
Viardot A, Goebeler ME, Hess G, Neumann S, Pfreundschuh M, Adrian N et al (2016) Phase 2 study of the bispecific Tcell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuselarge B-cell lymphoma. Blood 127:1410–6
Hijazi Y, Klinger M, Kratzer A, Wu B, Baeuerle PA et al (2018) Pharmacokinetic and pharmacodynamic relationship of blinatumomab in patients with non-hodgkin lymphoma. Curr Clin Pharm 13:55–64
Liu L, Lam C-YK, Long V, Widjaja L, Yang Y, Li H et al (2017) MGD011, A CD19 x CD3 dual-affinity retargeting bi-specific molecule incorporating extended circulating half-life for the treatment of B cell malignancies. Clin Cancer Res 23:1506–1518
Reusch U, Duell J, Ellwanger K, Herbrecht C, Knackmuss SH, Fucek I et al (2015) A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19(+) tumor cells. MAbs 7:584–604
Reusch U, Harrington KH, Gudgeon CJ, Fucek I, Ellwanger K, Weichel M et al (2016) Characterization of CD33/CD3 tetravalent bispecific tandem diabodies (TandAbs) for the treatment of acute myeloid leukemia. Clin Cancer Res 22(23):5829–5838
Westervelt P, Roboz, GJ et al (2018) Phase 1 first-in-human trial of AMV564, a bivalent bispecific (2x2) CD33/CD3 T cell engager, in patients with relapsed/refractory acute myeloid leukemia (AML). Presented at the 23rd Congress of the European Hematology Association (EHA), 14–17 June 2018, Stockholm, Sweden
Feucht J, Kayser S, Gorodezki D, Hamieh M, Doring M, Blaeschke F et al (2016) T cell responses against CD19+ pediatric acute lymphoblastic leukemia mediated by bispecific T cell engager (BiTE) are regulated contrarily by PDL1 and CD80/CD86 on leukemic blasts. Oncotarget 7:76902–76919
Dovedi SJ, Mazor Y, Elder M, Hasani S, Wang B, Mosely S et al (2018) Abstract 2776: MEDI5752: a novel bispecific antibody that preferentially targets CTLA-4 on PD-1 expressing T cells. Cancer Res 78(13 Suppl.):2776
Webster J, Luskin MR, Prince GT, DeZern AE, DeAngelo DJ, Levis MJ, Blackford A, Sharon E, Streicher H, Luznik L, Gojo I (2018) Blinatumomab in combination with immune checkpoint inhibitors of PD-1 and CTLA-4 in adult patients with relapsed/refractory (R/R) CD19 positive B cell acute lymphoblastic leukemia (ALL): preliminary results of a phase I study. Blood 132(Suppl. 1):557
Schwartz MS, Jeyakumar D, Damon LE, Schiller GJ, Wieduwilt MJ (2019) A phase I/II study of blinatumomab in combination with pembrolizumab for adults with relapsed refractory B-lineage acute lymphoblastic leukemia: University of California Hematologic Malignancies Consortium Study 1504. J Clin Oncol 37(15_suppl):TPS7064
Assi R, Kantarjian H, Short NJ, Daver N, Takahashi K, Garcia-Manero G, DiNardo C, Burger J, Cortes J, Jain N, Wierda W, Chamoun S, Konopleva M, Jabbour E (2017) Safety and efficacy of blinatumomab in combination with a tyrosine kinase inhibitor for the treatment of relapsed Philadelphia chromosome-positive leukemia. Clin Lymphoma Myeloma Leuk 17(12):897–901
Usmani SZ et al (2021) Teclistamab, a B cell maturation antigen x CD3 bispecific antibody, in patients with relapsed or refractory multiple myeloma (MajesTEC-1): a multicenter, open label, single-arm, phase 1 study. Lancet
Tian Z, Liu M, Zhang Y, Wan X (2021) Bispecific T cell engagers: an emerging therapy for management of hematologic malignancies. J Hematol Oncol 14:75
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Hays, P. (2022). Clinical Development and Therapeutic Applications of Bispecific Antibodies for Hematologic Malignancies. In: Hays, P. (eds) Cancer Immunotherapies. Cancer Treatment and Research, vol 183. Springer, Cham. https://doi.org/10.1007/978-3-030-96376-7_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-96376-7_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-96375-0
Online ISBN: 978-3-030-96376-7
eBook Packages: MedicineMedicine (R0)