
Abstract
Purpose of review
Bispecific antibodies combine antigen recognition sites from two or more antibodies into a single construct allowing simultaneous binding to multiple targets. Bispecific antibodies exist which can redirect immune effector cells against acute myeloid leukemia (AML) targets. This review will highlight the progress to date and the challenges in developing bispecific antibodies for the treatment of AML.
Recent findings
Currently, a number of bispecific antibody formats including bispecific T cell engagers, dual affinity retargeting proteins, and tandem diabodies are in clinical development for AML. These antibodies target antigens present on AML blasts, including CD33, and the low affinity IL3 receptor, CD123. T cell redirecting bispecific antibodies in early phase clinical trials for AML include AG330, flotetuzumab, JNJ-63709178, and AMV564.
Summary
Bispecific antibodies represent a promising immunotherapeutic approach for the treatment of cancer. The results of ongoing studies in AML will elucidate the potential for these agents in AML.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bispecific antibodies combine antigen recognition sites from two or more antibodies into a single construct allowing simultaneous binding to multiple targets. Bispecific antibodies to redirect the immune system against tumor cells have been theorized since the 1980s [1, 2]. In its most commonly used configuration, one segment has affinity for a tumor cell antigen, while the other binds an effector cell, most commonly T cells via the epsilon subunit of the T cell receptor (CD3ε). When used in this way, bispecific antibody redirects effector immune cells against tumor targets in a major histocompatibility complex-independent manner, bypassing immune escape mechanisms of MHC downregulation by tumor cells. In the case of T cell redirecting bispecific antibodies, these constructs facilitate the formation of immunologic synapses between cytotoxic T cells and tumor cells leading to granzyme B/perforin-mediated target cell death (Fig. 1b).

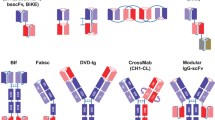
a Bispecific antibody constructs. Single-chain variable fragments (scFv) are derived from the Fab fragment of the IgG immunoglobulin and are composed of the VH and VL domains attached with a linker. Bispecific T cell engagers (BiTEs) consist of two scFVs, one directed against T cell antigen and one against a chosen tumor antigen, connected via a single-chain linker. Tandem diabodies (TandAbs) combine two scFVs for each target connected with a single polypeptide target. This allows them to maintain the avidity of a bivalent antibody as well as have a molecular weight that exceeds the renal clearance threshold. Dual affinity retargeting antibodies (DARTs) consist of variable domains of two antigen-binding specificities linked to two independent polypeptide chains. Each variable domain is formed by associating one VL segment on one chain with a VH segment on a second chain. The chains are covalently linked via disulfide bridge. b Mechanisms of action. Bispecific antibodies have variable fragments with affinities for both a selected tumor-associated antigen (TAA) and a selected target on an effector immune cell. Bispecific T cell engagers (BiTEs) bind to both TAA and the T cell receptor CD3. Bispecific killer cell engagers (BiKEs) bind a TAA with an NK cell antigen, usually CD16. Binding between effector cells and tumor cells facilitates the formation of cytolytic synapses leading to tumor cell destruction
Compared to monoclonal antibodies and antibody-drug conjugates, bispecific antibodies possess several unique advantages. First, these constructs can be engineered to recruit not only T cells but potentially other components of the immune system such as NK cells or immunomodulatory proteins such as PD-1 or CD47. Also, in contrast to drug-antibody conjugates, bispecific antibodies do not require receptor internalization for their therapeutic effect potentially allowing targeting of a more diverse population of tumor antigens. Blinatumomab, a bispecific antibody against CD3 and CD19, was developed and received accelerated approved from the FDA for the treatment of relapsed or refractory B cell acute lymphoblastic leukemia [3, 4]. Given the success of blinatumomab, there has been a high level of interest in developing bispecific antibodies for the treatment of acute myeloid leukemia (AML). This review will focus on those efforts and the challenges of developing bispecific antibodies for the treatment of AML.
Engineering of Bispecific Ab
The initial bispecific antibodies were made by fusing hybridoma cells or chemical cross-linking of antibodies [5, 6]. These first attempts suffered from low yields and immunogenicity with the development of neutralizing human anti-mouse antibodies. Advances in antibody engineering have resulted in dozens of different bispecific antibody formats each with unique characteristics affecting the ability to recruit different components of the immune system as well as other factors including tissue penetrance, circulating half-life, and ease of manufacturing (Fig. 1a) [7•].
Bispecific T cell engagers (BiTEs) are recombinant fusion antibody constructs created by using two single-chain variable fragments (scFvs), which are tandemly arranged on a single polypeptide linker [4]. The classical BiTE construct lacks the Fc domain of the antibody, which decreases its half-life by the lack of the neonatal Fc receptor recycling mechanism that prevents lysosomal degradation of antibodies [8]. Furthermore, BiTEs lack the potential for Fc-mediated effector functions such as antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC) [9, 10]. The low molecular weight of approximately 55 kD for a BiTE means that these constructs are renally excreted and possess a short half-life of 1–4 h which can necessitate treatment by a continuous intravenous infusion. Addition of an Fc domain to a BiTE construct lead to an increase in the molecular weight and an extended half-life of ~ 7 days [11].
A competing format to the tandem scFv format of the BiTE is a diabody. A diabody consists of a dimer of two antibody fragments which are covalently linked to each other. Dual affinity retargeting (DART) antibodies are a type of diabody which consists of heavy-and light-chain variable domains of two antigen-binding specificities linked to two independent polypeptide chains [12]. Similar to a BiTE, DARTs lack an Fc domain. Each Fv is formed by associating one VL segment on one chain with a VH segment on a second chain. The chains are covalently linked via disulfide bridges to limit the freedom of component Fv domains resulting in a high degree of stability [13]. Other diabody formats include tandem diabodies, providing two binding sites for each antigen to maintain the avidity of a bivalent antibody as well as have a molecular weight exceeding the renal clearance threshold [14••].
Other bispecific antibodies have an IgG-like structure with binding sites on each arm along with an Fc region. These antibodies can be engineered to optimize specific Fc-related effector functions and can possess an extended half-life similar to native immunoglobulins and can facilitate manufacturing and purification of the antibody [15, 16]. Currently, at least nine different bispecific antibodies are in clinical trials for the treatment of AML (Table 1).
Challenges in the Treatment of AML with Bispecific Antigens
The development of antibody-based therapies in AML to date has been limited by the lack of suitable tumor-associated antigens. The ideal target antigen in AML would be highly expressed on leukemic cells including the most primitive leukemic stem cells, but would spare normal hematopoietic cells and other non-hematopoietic tissues. In addition, an ideal target antigen would be crucial for the survival of the leukemic cell and not undergo downregulation in response to binding. In acute lymphoblastic leukemia, blinatumomab targets CD19 which is ubiquitously expressed on B cells. Even though CD19 lacks specificity for leukemic lymphoblasts and is also expressed on normal B lymphocytes, the resulting lymphopenia and hypogammaglobulinemia are manageable side effects in patients with ALL. Similarly, in AML, there is significant overlap of antigen expression patterns between leukemic cells and normal hematopoietic cells. This overlap predicts on-target off-tumor toxicity to the hematopoietic compartment and prolonged cytopenias including severe neutropenia which clinically is more likely to result in life-threatening complications when compared to B lymphopenia. Currently, the most frequently used targets for AML immunotherapy are CD33 and CD123. While initial studies used single-valent monoclonal antibodies, antibody-drug conjugates, T cell redirecting bispecific antibodies, and chimeric antigen receptor T cells targeting these antigens are all currently under clinical development in AML.
A common toxicity associated with T cell redirecting therapy including both bispecific antibodies and chimeric antigen receptor T cells is the cytokine release syndrome (CRS) [17, 18]. CRS is a systemic inflammatory response that is associated with cytokine elevation as part of the in vivo activation of the effector immune cells [19]. The syndrome is associated with high level of inflammatory cytokines such as C-reactive protein (CRP), ferritin, IL-6, INF-γ, TNF-α, and IL-2 [17, 20, 21]. Clinically, symptoms range from mild flu-like symptoms to high fever, capillary leak, vasodilatory shock, and end-organ dysfunction. CRS may cause life-threatening complications including ARDS, renal failure, hepatic failure, and cardiac dysfunction. Due to the massive immune activation, CRS may be associated with macrophage activation syndrome or hemophagocytic lymphohistiocytosis [22]. Neurotoxicity can range from mild confusion, headaches, and word-finding difficulties to severe encephalopathy, seizures, aphasia, delirium, and cerebral edema [17, 23, 24].
Development of CRS is in part due to the magnitude of T cell activation which are influenced by factors including the target antigen, antibody construct, antibody dose, and high disease burden and/or antigen load. Treatment of CRS ranges from interrupting the infusion of the drug and antipyretics and in more severe cases vasopressors, corticosteroids, and anti-IL-6 therapy using tocilizumab and siltuximab.
CD33 Targeted Bispecific Antibodies
CD33, also known as sialic acid-binding Ig-like lectin 3 (Siglec-3) is a 67-kDa trans-membrane cell surface glycoprotein, which is commonly expressed on acute myeloid leukemia blasts in > 90% of patients as well as leukemic stem and progenitor cells [25, 26]. It is estimated that about 30% of healthy bone marrow myeloid progenitors express CD33, which leads to on-target off-tumor toxicity [27, 28, 29••]. Multiple therapeutic concepts have targeted CD33 since the early 2000s including monoclonal antibodies, antibody-drug conjugates, and retargeting of T cell with bispecific antibodies [30, 31]. The anti-CD33 antibody-drug conjugate gemtuzumab ozogamicin (Mylotarg) was initially approved for treatment of relapsed AML in 2000, later withdrawn from the market after the failure of a large randomized phase III study conducted in the USA and then reapproved in 2017 following the results of four European randomized studies which demonstrated survival benefit for favorable and intermediate-risk AML patients. [32, 33]. Concern was raised as to whether antibodies against CD33 would affect human CD34+ hematopoietic stem cells (HSCs). In one experiment, freshly isolated T cells were incubated with autologous CD34+ HSCs in the presence or absence of bispecific CD33xCD3 antibodies. In vitro analysis of the hematopoietic potential in colony-forming unit assays did not reveal any difference to the control groups. The same cells were injected into sub-lethally irradiated NOD/SCID IL2Ry mice and developed normal chimerism, showing that the hematopoietic potential of these cells was not affected [34, 35].
AMG330 is a human BiTE tandem single-chain antibody with the N-terminal specific for human CD33 and its C-terminal is directed toward CD3ε [29••]. In vitro studies showed activation of T cells manifested as expression of CD69 and CD25 as well as release of IFN-γ, TNF-α, interleukin-2, IL-10, and IL-6 [36••]. Ex vivo, AMG330 showed a potent dose- and effector to target cell ratio-dependent activity against human AML cell lines [29, 37]. Daily intravenous administration of AMG330 significantly prolonged the survival of immune-deficient mice adoptively transferred with human MOLM-13 AML cells and human T cells [36••]. A first in human phase I study in which AMG330 is administered as a 14–28-day continuous intravenous infusion in patients with relapsed/refractory AML began recruiting in August 2015 (NCT02520427). AMG673 is a related newer anti-CD33 × anti-CD3 BiTE antibody construct developed by Amgen. AMG673 fuses an Fc domain extending the half-life of the antibody to 7 days which permits weekly dosing of the agent [11]. The phase 1 study began recruitment in September 2017 (NCT03224819).
AMV564 is a tetravalent anti-CD33 × anti-CD3 tandem diabody construct that is comprised of two VH and VL chains that form antigen-binding single-chain variable fragments (scFvs). The protein forms a homodimer creating two binding sites for each epitope and increases the avidity of the antibody to its targets. It also increases the molecular weight to approximately 106 kDa avoiding first pass renal clearance and resulting in a longer half-life in comparison to BiTEs [14••]. Preclinical data with cryopreserved human AML specimens demonstrated in vitro activity across the spectrum of disease stage, cytogenetic risk, and CD33 expression levels [14••]. AMV564 is currently studied in a first-in-human phase I trial in patients with relapsed or refractory AML in which the agent is dosed as a 14-day continuous infusion (NCT03144245) every 28 days. Preliminary results presented at the European Hematology Association in June 2018 showed evidence of T cell activation evident by increased cytokine levels and antigen markers of T cell activation. There was evidence of biologic activity with 13–38% reductions in the bone marrow blasts in 10/16 patients. The treatment was generally well tolerated with no grade 3–4 toxicities attributed to the study treatment with manageable grade 2 CRS observed at the 50 μg/day dose in a single patient [38•].
GEM333 is another CD3xCD33 humanized bispecific antibody currently in phase I clinical trials in Germany for relapsed or refractory AML (NCTT03516760). GEM333 antibody is a humanized single-chain bispecific antibody with variable and light and heavy chains to both CD3 and CD33 are arranged in a tandem format using a novel linker [39]. In preclinical studies, the construct was capable of redirecting human T cells efficiently toward CD33+ AML blasts as demonstrated by killing of AML cell lines and patient-derived AML blasts both in vitro and in NSG mice. Notably, the antibody had no effect on normal human CD34+ hematopoietic stem and progenitor cells in both colony-forming assays or in NSG repopulating experiments [34].
Hepatotoxicity and in particular sinusoidal obstruction syndrome in patients undergoing allogeneic hematopoietic cell transplant have been observed with CD33 antibody-drug conjugates gemtuzumab ozogamicin (Mylotarg) and vadastuximab talirine (SGN-CD33A). As CD33 is expressed on liver Kupffer cells and sinusoidal endothelial cells, this has raised concern that the hepatotoxicity may be result of an on-target effect to CD33 [40,41,42]. However, it is known that the CD22 antibody drug conjugate, inotuzumab, has had similar concerns with sinusoidal obstructive syndrome [43] indicating that this may be more specific to the drug conjugate, calicheamicin, suggesting a potential advantage to the bispecific antibody format.
Recently, it has been reported that a single nucleotide polymorphism (SNP) in the CD33 splice enhancer region regulates the expression of a splicing variant of CD33 which lacks IgV domain. This domain serves as the antibody binding site for both gemtuzumab ozogamicin and CD33 diagnostic immunophenotyping panels. Approximately 50% of patients treated on a Children’s Oncology Group study carried this SNP which was associated with a lack of benefit to gemtuzumab [44]. The CD33-binding scFv of AMG 330 has similarly been mapped to the IgV domain of CD33 [36••]. Although a similar influence of this SNP on AMG330 activity might be predicted, preclinical studies of AMG 330-induced cytotoxicity using sublines of cells transduced with mutant forms of the CD33 SNPs did not detect any difference in the activity of AMG 330 [37].
CD123 Targeted Bispecific Antibodies
CD123 (interleukin-3 receptor alpha chain) is the low affinity binding subunit of the IL3 receptor. CD123 is expressed on myeloid progenitors, plasmacytoid dendritic cells, monocytes, and basophils [45]. Binding of IL-3 triggers CD123 hetero-dimerization with the β-subunit shared by the granulocyte macrophage-colony-stimulating factor and IL5 receptor complex inducing hematopoietic progenitor cell differentiation and proliferation by phosphorylation of Janus kinase, activation of PI3 kinase, and upregulation of anti-apoptotic proteins [46, 47]. CD123 was initially described as a putative marker of leukemic stem cells with increased expression in the CD34+/CD38− primitive leukemic blasts which contained the NSG repopulating activity of AML samples [48]. Elevated expression of CD123 on AML blasts is associated with higher blast counts and poorer prognosis with lower CR rates [49]. Early studies with anti-CD123 antibodies showed that inhibiting CD123 led to prolonged survival of AML xenografted mice [50]. Clinical trials are in process of evaluating anti-CD123 monoclonal antibodies, antibody-drug conjugates, and bispecific antibodies.
Flotetuzumab (MGD-006, Macrogenics) is a CD3xCD123 DART that was shown to recognize CD123+ leukemia cells and induce T cell activation with resulting cytotoxic effect. MGD006 induced a dose-dependent killing of AML cell lines and primary AML blasts in vitro and in NOD SCIDγ mice injected with human AML [51••]. Animal models in cynomolgus macaques showed that MGD006 caused marked depletion of circulating CD123+ cells and was generally well tolerated, paving the way to human trials [52, 53]. A first-in-human phase I dose escalation trial in patients with AML and high-risk MDS is currently recruiting (NCT02152956). The study enrolls patients with relapsed or refractory AML or hypomethylating agent refractory MDS with > 10% marrow blasts. Preliminary results showed that of 14 patients treated at the maximum tolerated dose of ≥ 500 ng/kg/day, anti-leukemic activity was observed in 57% of the patients with 28% of the patients achieving CR. Anti-leukemic activity was seen even in high-risk patients with duration of response ranging 1–5.8 months. Immunohistochemistry studies showed an increase in the density of T cells in the bone marrow of AML patients treated with flotetuzumab. The most common side effects being infusion-related reactions and cytokine release syndrome, as well as fever and nausea. Only 15.8% of the patients had grade ≥ 3 toxicity [54•]. Cytokine release syndrome was felt to be manageable using a step-up dose escalation scheme in which two-step lead in dose of 30 ng/kg/day for 3 days followed by 100 ng/kg/day for 4 days in cycle one prior to receiving the 500 mg/kg/day phase II dose. Enrollment to the phase II portion of the study is ongoing.
JNJ-63709178 (Janssen Pharmaceuticals) is a CD3xCD123 bispecific IgG1 antibody generated using a process called controlled Fab-arm exchange also known as Genmab DuoBody® technology. The method involves the separate expression of two parental IgG1 monoclonal antibodies that each contains a single matched point mutation at the CH3-CH3 domain interface. During controlled reduction of disulfide bonds in which the parental antibodies separate into half molecules, the matched CH3 mutation drives the reassembly of heterozygous binding arms producing the bispecific IgG1 antibody [15]. In contrast to BiTEs and DARTs, these IgG-like bispecific antibodies retain their Fc region and its associated effector functions and in vivo stability. The antibody was shown to recruit T cells to CD123 expressing tumor cells inducing cytolytic effect in vitro. In AML murine xenograft models, the compound was able to suppress tumor growth and induce tumor regression in the presence of human peripheral blood T cells [55]. A phase I clinical trial in relapsed and refractory AML patients was initiated but placed on hold in March 2016 due to grade 3 events (NCT02715011). No further clinical information has been released by the sponsor.
Other CD123 bispecifics include XmAb14045 (Xencor Inc.). This antibody possesses a unique Fc domain and spontaneously forms stable heterodimers to facilitate manufacturing of the antibody. The Fc domain was also engineered to abolish binding to FcγR to reduce the potential for nonselective T cell activation but preserve binding to human FcRn and maintain long serum half-life. In contrast to non-Fc domain-containing bispecific formats, XmAb14045 had a prolonged serum half-life in mice of 6.2 days. In vitro, XmAb14045 stimulated T cell-mediated killing of CD123+ KG-1a and TF-1 AML cell lines with an EC50 < 1 ng/ml (8 pM). In cynomolgus monkeys, XmAb14045 strongly activated T cells and stimulated depletion of over 99% of circulating CD123+ cells including basophils and plasmacytoid dendritic cells within 1 h of administration [56]. XmAb14045 entered a phase I clinical trial in June 2016 (NCT02730312).
Concerns of hematologic toxicity with T cell redirecting therapy against CD123 have been raised during development of CART cells directed against CD123. CD123 CART cells could eradicate AML in NSG mice, but also were found to eradicate normal human myelopoiesis in cord blood-derived CD34+ engrafted NSG mice [57]. For these reasons, CART cell approaches with CD123 are being utilized as a bridge to hematopoietic cell transplant. In contrast, trials with anti-CD123 monoclonal and bispecific antibody-based therapies conducted thus far have not encountered similar issues with hematopoietic toxicity.
Alternative Bispecific Antibody Approaches
MCLA-117 is a human full-length IgG1 bispecific antibody targeting CLEC12A (also known as CLL-1) and CD3. CLEC12A is a myeloid differentiation antigen expressed on ~ 90% of newly diagnosed and relapsed AML. CLEC12A is selectively expressed on leukemic stem cells, but not on normal early hematopoietic progenitors including hematopoietic stem cells [58, 59]. The antibody was designed with a low affinity anti-CD3 Fab arm for controlled T cell activation, as well as high affinity anti-CLEC12A arm for efficient tumor targeting. It also encompasses a silenced Fc region to prevent binding to FcγR+ cells while retaining a long half-life. Analysis of normal bone marrow demonstrated that MCLA-117 binds to the granulocyte-macrophage progenitor, however not to the CD34+CD38− compartment that includes hematopoietic stem cells. In vitro studies showed efficient CLEC12A antigen-dependent T cell activation and tumor target cell lysis in HL-60 cell line, as well as normal monocytes. MCLA-117 was able to induce T cell-mediated lysis of AML blasts in an ex vivo culture system, despite low effective effector-to-target (E:T) ratios [60]. MCL-117 is currently being investigated in a phase I clinical study (NCT03038230) to evaluate the safety, tolerability, and preliminary efficacy in adult AML patients.
In addition to cytotoxic T cells, there in an established role for NK cells in the exertion of graft versus leukemia effect. Clinical data show that alloreactive NK cell from mismatched stem cell grafts are associated with a potent GVL effect [61]. Haploidentical NK cell infusions have been shown to improve outcomes in patients with refractory AML [62]. CD16 is a potent NK cell activating receptor that showed promising results as an effector target for the treatment of lymphoid tumors [63, 64]. A new anti-CD33xCD16 bispecific killer cell engager (BiKE) was created. This BiKE was shown to induce NK cell activation and degranulation in response to CD33+ HL60 cells. It also induced target cell death as measured by chromium release assay in all E:T ratios. The BiKE induced NK cell activation against AML blasts from a patient with de novo AML [65]. Addition of IL-15 cross-linker to promote NK cell activation and proliferation, termed a trispecific killer engager (TriKE), improves NK function against AML cell lines [66]. NCT03214666 is a single-center phase I/II clinical trial of CD16/IL-15/CD33 tri-specific killer cell engager (TriKE) for treatment of CD33-expressing high-risk MDS, relapsed/refractory AML, and systemic mastocytosis, estimated to open to accrual in November 2018.
An alternative approach for bispecific antibodies is to target antiphagocytic markers expressed by leukemic cells. CD47 is an integrin-associated protein which is commonly overexpressed on cancer cells including AML blasts. CD47 functions as a “don’t eat me” signal by serving as the ligand to signal regulatory protein alpha (SIRPα) on macrophages and inhibiting phagocytosis [67]. Blocking monoclonal antibodies to CD47 enables the engagement of phagocytic effector immune cells and elimination of primary human AML specimens engrafted in immunodeficient mice [68]. Since CD47 is ubiquitously expressed on normal cells, it may function as an antigen sink preventing effective targeting of CD47 with monoclonal antibodies in humans. To overcome this challenge, bispecific antibodies which target CD47 blockade to tumor cells are being developed [69]. A bispecific CD47xCD33 IgG-like antibody, HMBD004 (Hummingbird Bioscience), has been demonstrated to decrease AML tumor burden in immunodeficient mice with human studies in AML currently being planned [70].
Finally, ongoing studies are exploring novel approaches to enhance the activity of bispecific antibodies. In preclinical studies, AMG330 showed significantly higher cytotoxicity in specimens from patients with newly diagnosed AML than those with relapsed/refractory disease, despite similar levels of CD33 on the myeloblasts [71]. A potential mechanism for this finding was obtained using AML cell lines engineered to overexpress individual T cell ligands. Expression of inhibitory ligands such as PD-L1 and PD-L2 reduced the cytolytic activity of AMG330, whereas expression of activating ligands such as CD80 and CD86 augmented the cytotoxic activity [72]. PD-L1 was not constitutively expressed on AML cells at the time of primary diagnosis; however, it was upregulated on primary AML cells upon addition of AMG330 to ex vivo cultures conferring resistance to AMG330 activity. Furthermore, it was shown that inflammatory cytokines alone, such as IFN-γ and TNF-α, can induce upregulation of PD-L1 expression on AML cells, suggesting that the inflammatory milieu itself may be causing the upregulation of PD-L1 on AML blasts. Antibody-mediated blockade of the PD-1/PD-L1 interaction led to AMG330-mediated cytolysis [73]. These findings support the future evaluation of checkpoint inhibitors in conjunction with bispecific antibodies to enhance their activity in AML.
Conclusions
Immunotherapy of cancer has become a rapidly progressing field over the past years, becoming standard of care in many solid tumor and hematologic malignancies. The development of novel immunotherapeutic agents for treatment of AML has been delayed due to difficulties in findings leukemia-specific target molecules that will decrease on-target off-tumor toxicities. Despite these difficulties, many new bispecific antibodies are currently being developed clinically with encouraging preliminary results. Future trials may test the combination of bispecific antibodies with other forms of immunotherapy such as co-stimulation of signal pathways and targeting of immune evasion mechanisms to further increase the efficacy and clinical benefit of these treatments.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Perez P, Hoffman RW, Shaw S, Bluestone JA, Segal DM. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature. 1985;316(6026):354–6.
Staerz UD, Kanagawa O, Bevan MJ. Hybrid antibodies can target sites for attack by T-cells. Nature. 1985;314(6012):628–31. https://doi.org/10.1038/314628a0.
Kantarjian H, Stein A, Gokbuget N, Fielding AK, Schuh AC, Ribera JM, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836–47. https://doi.org/10.1056/NEJMoa1609783.
Loffler A, Gruen M, Wuchter C, Schriever F, Kufer P, Dreier T, et al. Efficient elimination of chronic lymphocytic leukaemia B cells by autologous T cells with a bispecific anti-CD19/anti-CD3 single-chain antibody construct. Leukemia. 2003;17(5):900–9. https://doi.org/10.1038/sj.leu.2402890.
Brennan M, Davison PF, Paulus H. Preparation of bispecific antibodies by chemical recombination of monoclonal immunoglobulin G1 fragments. Science. 1985;229(4708):81–3.
Staerz UD, Bevan MJ. Hybrid hybridoma producing a bispecific monoclonal antibody that can focus effector T-cell activity. Proc Natl Acad Sci U S A. 1986;83(5):1453–7.
• Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discov Today. 2015;(7):20, 838–847. https://doi.org/10.1016/j.drudis.2015.02.008 Review summarizing different bispecific antibody formats.
Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7(9):715–25. https://doi.org/10.1038/nri2155.
Sheridan C. Despite slow progress, bispecifics generate buzz. Nat Biotechnol. 2016;34(12):1215–7. https://doi.org/10.1038/nbt1216-1215.
Rathi C, Meibohm B. Clinical pharmacology of bispecific antibody constructs. J Clin Pharmacol. 2015;55(Suppl 3):S21–8. https://doi.org/10.1002/jcph.445.
Arvedson T. Possibility for once-weekly dosing with an anti-CD33 half-life extended BiTE. AACR Annual Meeting 2017; April 1–5, 2017; Washington, DC;. 2017.
Johnson S, Burke S, Huang L, Gorlatov S, Li H, Wang W, et al. Effector cell recruitment with novel Fv-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B-cell depletion. J Mol Biol. 2010;399(3):436–49. https://doi.org/10.1016/j.jmb.2010.04.001.
Moore PA, Zhang W, Rainey GJ, Burke S, Li H, Huang L, et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood. 2011;117(17):4542–51. https://doi.org/10.1182/blood-2010-09-306449.
•• Reusch U, Harrington KH, Gudgeon CJ, Fucek I, Ellwanger K, Weichel M, et al. Characterization of CD33/CD3 tetravalent bispecific tandem diabodies (TandAbs) for the treatment of acute myeloid leukemia. Clin Cancer Res. 2016;22(23):5829–38. https://doi.org/10.1158/1078-0432.CCR-16-0350 Preclinical characterization of AMV564.
Labrijn AF, Meesters JI, de Goeij BE, van den Bremer ET, Neijssen J, van Kampen MD, et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc Natl Acad Sci U S A. 2013;110(13):5145–50. https://doi.org/10.1073/pnas.1220145110.
Moore GL, Bautista C, Pong E, Nguyen DH, Jacinto J, Eivazi A, et al. A novel bispecific antibody format enables simultaneous bivalent and monovalent co-engagement of distinct target antigens. MAbs. 2011;3(6):546–57. https://doi.org/10.4161/mabs.3.6.18123.
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. https://doi.org/10.1126/scitranslmed.3008226.
Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121(26):5154–7. https://doi.org/10.1182/blood-2013-02-485623.
Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–95. https://doi.org/10.1182/blood-2014-05-552729.
Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–20. https://doi.org/10.1182/blood-2011-10-384388.
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–18. https://doi.org/10.1056/NEJMoa1215134.
Billiau AD, Roskams T, Van Damme-Lombaerts R, Matthys P, Wouters C. Macrophage activation syndrome: characteristic findings on liver biopsy illustrating the key role of activated, IFN-g-producing lymphocytes and IL-6- and TNF-alpha-producing macrophages. Blood. 2005;105(4):1648–51. https://doi.org/10.1182/blood-2004-08-2997.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (London, England). 2015;385(9967):517–28. https://doi.org/10.1016/s0140-6736(14)61403-3.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–17. https://doi.org/10.1056/NEJMoa1407222.
Dinndorf PA, Andrews RG, Benjamin D, Ridgway D, Wolff L, Bernstein ID. Expression of normal myeloid-associated antigens by acute leukemia cells. Blood. 1986;67(4):1048–53.
Hauswirth AW, Florian S, Printz D, Sotlar K, Krauth MT, Fritsch G, et al. Expression of the target receptor CD33 in CD34+/CD38-/CD123+ AML stem cells. Eur J Clin Investig. 2007;37(1):73–82. https://doi.org/10.1111/j.1365-2362.2007.01746.x.
Nguyen DH, Ball ED, Varki A. Myeloid precursors and acute myeloid leukemia cells express multiple CD33-related Siglecs. Exp Hematol. 2006;34(6):728–35. https://doi.org/10.1016/j.exphem.2006.03.003.
Ehninger A, Kramer M, Rollig C, Thiede C, Bornhauser M, von Bonin M, et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014;4:e218. https://doi.org/10.1038/bcj.2014.39.
•• Krupka C, Kufer P, Kischel R, Zugmaier G, Bogeholz J, Kohnke T, et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell-engaging antibody AMG 330. Blood. 2014;123(3):356–65. https://doi.org/10.1182/blood-2013-08-523548 Preclincial studies of the BiTE, AMG330.
Walter RB, Appelbaum FR, Estey EH, Bernstein ID. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood. 2012;119(26):6198–208. https://doi.org/10.1182/blood-2011-11-325050.
Aigner M, Feulner J, Schaffer S, Kischel R, Kufer P, Schneider K, et al. T lymphocytes can be effectively recruited for ex vivo and in vivo lysis of AML blasts by a novel CD33/CD3-bispecific BiTE antibody construct. Leukemia. 2013;27(5):1107–15. https://doi.org/10.1038/leu.2012.341.
Burnett AK, Hills RK, Milligan D, Kjeldsen L, Kell J, Russell NH, et al. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: results of the MRC AML15 trial. J Clin Oncol. 2011;29(4):369–77. https://doi.org/10.1200/jco.2010.31.4310.
Appelbaum FR, Bernstein ID. Gemtuzumab ozogamicin for acute myeloid leukemia. Blood. 2017;130(22):2373–6. https://doi.org/10.1182/blood-2017-09-797712.
Arndt C, von Bonin M, Cartellieri M, Feldmann A, Koristka S, Michalk I, et al. Redirection of T cells with a first fully humanized bispecific CD33-CD3 antibody efficiently eliminates AML blasts without harming hematopoietic stem cells. Leukemia. 2013;27(4):964–7. https://doi.org/10.1038/leu.2013.18.
Dutour A, Marin V, Pizzitola I, Valsesia-Wittmann S, Lee D, Yvon E, et al. In vitro and in vivo antitumor effect of anti-CD33 chimeric receptor-expressing EBV-CTL against CD33 acute myeloid leukemia. Advances in hematology. 2012;2012:683065. https://doi.org/10.1155/2012/683065.
•• Friedrich M, Henn A, Raum T, Bajtus M, Matthes K, Hendrich L, et al. Preclinical characterization of AMG 330, a CD3/CD33-bispecific T-cell-engaging antibody with potential for treatment of acute myelogenous leukemia. Mol Cancer Ther. 2014;13(6):1549–57. https://doi.org/10.1158/1535-7163.MCT-13-0956. Article describing preclinical studies of AMG330 for AML.
Laszlo GS, Gudgeon CJ, Harrington KH, Dell'Aringa J, Newhall KJ, Means GD, et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood. 2014;123(4):554–61. https://doi.org/10.1182/blood-2013-09-527044.
• Westervelt P, Roboz, GJ et al. Phase 1 first-in-human Trial of AMV564, a bivalent bispecific (2x2) CD33/CD3 T-cell engager, in patients with relapsed/refractory acute myeloid leukemia (AML). Presented at the 23rd Congress of the European Hematology Association (EHA), June 14-17, Stockholm, Sweden; 2018. Initial clinical results of AMV564.
Stamova S, Cartellieri M, Feldmann A, Arndt C, Koristka S, Bartsch H, et al. Unexpected recombinations in single chain bispecific anti-CD3-anti-CD33 antibodies can be avoided by a novel linker module. Mol Immunol. 2011;49(3):474–82. https://doi.org/10.1016/j.molimm.2011.09.019.
McKoy JM, Angelotta C, Bennett CL, Tallman MS, Wadleigh M, Evens AM, et al. Gemtuzumab ozogamicin-associated sinusoidal obstructive syndrome (SOS): an overview from the research on adverse drug events and reports (RADAR) project. Leuk Res. 2007;31(5):599–604. https://doi.org/10.1016/j.leukres.2006.07.005.
Maniecki MB, Hasle H, Bendix K, Moller HJ. Is hepatotoxicity in patients treated with gemtuzumabozogamicin due to specific targeting of hepatocytes? Leuk Res. 2011;35(6):e84–6. https://doi.org/10.1016/j.leukres.2011.01.025.
Robinson B. Seattle genetics discontinues phase 3 CASCADE trial of vadastuximab talirine (SGN-CD33A) in frontline acute myeloid leukemia. 2018. http://investor.seattlegenetics.com/phoenix.zhtml?c=124860&p=irol-newsArticle&ID=2281531.
Kantarjian HM, DJ DA, Advani AS, Stelljes M, Kebriaei P, Cassaday RD, et al. Hepatic adverse event profile of inotuzumab ozogamicin in adult patients with relapsed or refractory acute lymphoblastic leukaemia: results from the open-label, randomised, phase 3 INO-VATE study. Lancet Haematol. 2017;4(8):e387–e98. https://doi.org/10.1016/S2352-3026(17)30103-5.
Lamba JK, Chauhan L, Shin M, Loken MR, Pollard JA, Wang YC, et al. CD33 splicing polymorphism determines gemtuzumab ozogamicin response in de novo acute myeloid leukemia: report from randomized phase III children’s oncology group trial AAML0531. J Clin Oncol. 2017;35(23):2674–82. https://doi.org/10.1200/JCO.2016.71.2513.
Munoz L, Nomdedeu JF, Lopez O, Carnicer MJ, Bellido M, Aventin A, et al. Interleukin-3 receptor alpha chain (CD123) is widely expressed in hematologic malignancies. Haematologica. 2001;86(12):1261–9.
Reddy EP, Korapati A, Chaturvedi P, Rane S. IL-3 signaling and the role of Src kinases, JAKs and STATs: a covert liaison unveiled. Oncogene. 2000;19(21):2532–47. https://doi.org/10.1038/sj.onc.1203594.
Blalock WL, Weinstein-Oppenheimer C, Chang F, Hoyle PE, Wang XY, Algate PA, et al. Signal transduction, cell cycle regulatory, and anti-apoptotic pathways regulated by IL-3 in hematopoietic cells: possible sites for intervention with anti-neoplastic drugs. Leukemia. 1999;13(8):1109–66.
Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14(10):1777–84.
Testa U, Riccioni R, Militi S, Coccia E, Stellacci E, Samoggia P, et al. Elevated expression of IL-3Ralpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood. 2002;100(8):2980–8. https://doi.org/10.1182/blood-2002-03-0852.
Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5(1):31–42. https://doi.org/10.1016/j.stem.2009.04.018.
•• Al-Hussaini M, Rettig MP, Ritchey JK, Karpova D, Uy GL, Eissenberg LG, et al. Targeting CD123 in acute myeloid leukemia using a T-cell-directed dual-affinity retargeting platform. Blood. 2016;127(1):122–31. https://doi.org/10.1182/blood-2014-05-575704 Preclinical studies of MGd-006, flotetuzumab for AML.
Chichili GR, Huang L, Li H, Burke S, He L, Tang Q, et al. A CD3xCD123 bispecific DART for redirecting host T cells to myelogenous leukemia: preclinical activity and safety in nonhuman primates. Sci Transl Med. 2015;7(289):289ra82. https://doi.org/10.1126/scitranslmed.aaa5693.
Campagne O, Delmas A, Fouliard S, Chenel M, Chichili GR, Li H, et al. Integrated pharmacokinetic/pharmacodynamic model of a bispecific CD3xCD123 DART molecule in nonhuman primates: evaluation of activity and impact of immunogenicity. Clin Cancer Res. 2018;24(11):2631–41. https://doi.org/10.1158/1078-0432.Ccr-17-2265.
• Uy GL, et al. Preliminary results of a phase 1 study of flotetuzumab, a CD123 x CD3 bispecific Dart® protein, in patients with relapsed/refractory acute myeloid leukemia and myelodysplastic syndrome. Blood. 2017;130(Suppl 1):637 Initial clinical results from phase 1 dose escalation study of flotetuzumab.
Gaudet FNJ, et al. Development of a CD123xCD3 bispecific antibody (JNJ-63709178) for the treatment of acute myeloid leukemia (AML). Blood. 2016;128(22):2824.
Chu SY, Pong E, Chen H, Phung S, Chan EW, Endo NA, et al. Immunotherapy with long-lived anti-CD123 × anti-CD3 bispecific antibodies stimulates potent T cell-mediated killing of human AML cell lines and of CD123+ cells in monkeys: a potential therapy for acute myelogenous leukemia. Blood. 2014;124(21):2316.
Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123(15):2343–54. https://doi.org/10.1182/blood-2013-09-529537.
van Rhenen A, van Dongen GA, Kelder A, Rombouts EJ, Feller N, Moshaver B, et al. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood. 2007;110(7):2659–66. https://doi.org/10.1182/blood-2007-03-083048.
Taussig DC, Pearce DJ, Simpson C, Rohatiner AZ, Lister TA, Kelly G, et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106(13):4086–92. https://doi.org/10.1182/blood-2005-03-1072.
Van Loo PF, Doornbos R, Dolstra H, Shamsili S, Bakker L. Preclinical evaluation of MCLA117, a CLEC12AxCD3 bispecific antibody efficiently targeting a novel leukemic stem cell associated antigen in AML. Blood. 2015;126(23):325.
Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295(5562):2097–100. https://doi.org/10.1126/science.1068440.
Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105(8):3051–7. https://doi.org/10.1182/blood-2004-07-2974.
Gleason MK, Verneris MR, Todhunter DA, Zhang B, McCullar V, Zhou SX, et al. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol Cancer Ther. 2012;11(12):2674–84. https://doi.org/10.1158/1535-7163.Mct-12-0692.
Bruenke J, Barbin K, Kunert S, Lang P, Pfeiffer M, Stieglmaier K, et al. Effective lysis of lymphoma cells with a stabilised bispecific single-chain Fv antibody against CD19 and FcgRIII (CD16). Br J Haematol. 2005;130(2):218–28. https://doi.org/10.1111/j.1365-2141.2005.05414.x.
Wiernik A, Foley B, Zhang B, Verneris MR, Warlick E, Gleason MK, et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin Cancer Res. 2013;19(14):3844–55. https://doi.org/10.1158/1078-0432.Ccr-13-0505.
Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU, et al. IL15 trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin Cancer Res. 2016;22(14):3440–50. https://doi.org/10.1158/1078-0432.CCR-15-2710.
Oldenborg PA, Gresham HD, Lindberg FP. CD47-signal regulatory protein alpha (SIRPalpha) regulates Fcg and complement receptor-mediated phagocytosis. J Exp Med. 2001;193(7):855–62.
Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD Jr, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138(2):286–99. https://doi.org/10.1016/j.cell.2009.05.045.
Dheilly E, Moine V, Broyer L, Salgado-Pires S, Johnson Z, Papaioannou A, et al. Selective blockade of the ubiquitous checkpoint receptor CD47 is enabled by dual-targeting bispecific antibodies. Mol Ther. 2017;25(2):523–33. https://doi.org/10.1016/j.ymthe.2016.11.006.
Boyd-Kirkup J, Thakkar D, Brauer P, Zhou J, Chng W-J, Ingram PJ. HMBD004, a novel anti-CD47xCD33 bispecific antibody displays potent anti-tumor effects in pre-clinical models of AML. Blood. 2017;130(Suppl 1):1378.
Harrington KH, Gudgeon CJ, Laszlo GS, Newhall KJ, Sinclair AM, Frankel SR, et al. The broad anti-AML activity of the CD33/CD3 BiTE antibody construct, AMG 330, is impacted by disease stage and risk. PLoS One. 2015;10(8):e0135945. https://doi.org/10.1371/journal.pone.0135945.
Laszlo GS, Gudgeon CJ, Harrington KH, Walter RB. T-cell ligands modulate the cytolytic activity of the CD33/CD3 BiTE antibody construct, AMG 330. Blood cancer journal. 2015;5:e340. https://doi.org/10.1038/bcj.2015.68.
Krupka C, Kufer P, Kischel R, Zugmaier G, Lichtenegger FS, Kohnke T, et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: reversing a T-cell-induced immune escape mechanism. Leukemia. 2016;30(2):484–91. https://doi.org/10.1038/leu.2015.214.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Geoffrey Uy reports personal fees from Glycomimetics, personal fees from Pfizer, personal fees from Curis, personal fees from Jazz, and personal fees from Novartis, outside the submitted work. Daniel Guy declares that he has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Acute Myeloid Leukemias
Rights and permissions
About this article

Cite this article
Guy, D.G., Uy, G.L. Bispecific Antibodies for the Treatment of Acute Myeloid Leukemia. Curr Hematol Malig Rep 13, 417–425 (2018). https://doi.org/10.1007/s11899-018-0472-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11899-018-0472-8